

Abstract
Surgical trauma and ischemia reperfusion injury (IRI) are unavoidable aspects of any solid organ transplant procedure. They trigger a multifactorial antigen-independent inflammatory process that profoundly affects both the early and long-term outcomes of the transplanted organ. The injury associated with donor organ procurement, storage, and engraftment triggers innate immune activation that inevitably results in cell death, which may occur in many different forms. Dying cells in donor grafts release damage-associated molecular patterns (DAMPs), which alert recipient innate cells, including macrophages and dendritic cells (DCs), through the activation of the complement cascade and toll-like receptors (TLRs). The long-term effect of inflammation on innate immune cells is associated with changes in cellular metabolism that skew the cells towards aerobic glycolysis, resulting in innate immune cell activation and inflammatory cytokine production. The different roles of proinflammatory cytokines in innate immune activation have been described, and these cytokines also stimulate optimal T-cell expansion during allograft rejection. Therefore, early innate immune events after organ transplantation determine the fate of the adaptive immune response. In this review, we summarize the contributions of innate immunity to allograft rejection and discuss recent studies and emerging concepts in the targeted delivery of therapeutics to modulate the innate immune system to enhance allograft survival.
Subject terms: Allotransplantation, Innate immunity
Introduction
Activation of the innate immune system is induced early during donor organ procurement and engraftment, and these events are associated with tissue damage and the death of donor cells in transplanted organs. Cell death is a highly regulated process that contributes to multiple aspects of innate immune activation, including graft inflammation, tissue damage, and the elimination of damaged cells in the graft, as well as the repair of damaged tissue following transplantation. The best-studied form of cell death is apoptosis, a process of programmed cell death that is tightly controlled and requires the activation of caspases. Recently, however, it has been shown that various other forms of cell death also exist, and they are collectively called regulated necrotic cell death, which includes necroptosis, ferroptosis, and pyroptosis. Depending on the conditions in which donor cells are induced to die, cell death pathways can be activated by different mechanisms or when the classic process of apoptosis is not possible (i.e., caspase inhibition).1 In essence, apoptosis results in the engulfment of dying cells by macrophages, leading to the rapid clearance of the dying cells without activation of the immune system. Thus, apoptosis is generally considered noninflammatory or even protolerogenic. However, apoptotic cells can also be inflammatory if they are not phagocytosed in a timely manner.2 In contrast to apoptosis, ferroptosis is a highly inflammatory process that contributes to organ damage during ischemic injury. In this process, cell death coincides with cell swelling and plasma membrane rupture. Pyroptosis is a type of inflammatory cell death in which dying cells release potent inflammatory contents before death. Therefore, the underlying mechanisms that control cell death during ischemia reperfusion in a donor organ are critical components of the innate immune response to the transplanted allograft (Figs. 1, 2).
Fig. 1.
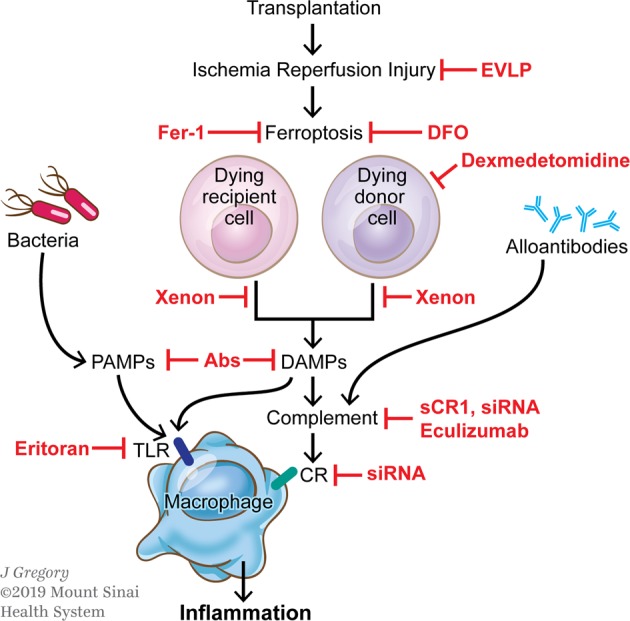
Pathways linking transplantation-associated tissue damage to graft inflammation. Transplantation-associated ischemia reperfusion injury (IRI) causes inflammatory cell death (e.g., ferroptosis) in host and donor cells, leading to the release of damage-associated molecular patterns (DAMPs). DAMPs bind to Toll-like receptors (TLRs) or activate the complement cascade by creating products that bind to complement receptors (CR), resulting in the activation of inflammatory functions in macrophages. In addition, bacteria that translocate into the tissue during surgery and release pathogen-associated molecular patterns (PAMPs) or alloantibodies, activating the complement system, can also activate macrophages. Inhibitors of this multistep process are described in red. Ex vivo lung perfusion (EVLP) attenuates the inflammatory response to IRI. Ferrostatin-1 (Fer-1) and desferrioxamine (DFO) inhibit ferroptosis. Dexmedetomidine increases cell survival. Xenon gas inhibits the release of DAMPs. Monoclonal antibodies (Abs) specific for PAMPs or DAMPs prevent binding to TLRs. Soluble complement receptor 1 (sCR1) or siRNA decrease the expression of complement factors or receptors, and eculizumab interferes with the complement cascade. Eritoran is a TLR4 antagonist
Fig. 2.
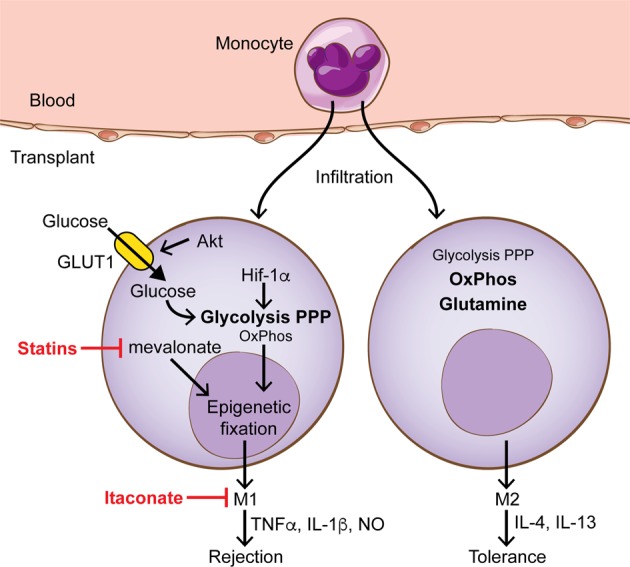
Targeting immunometabolism in macrophages to prevent graft rejection. Monocytes from the circulation enter a transplant and acquire either proinflammatory functions (M1) that contribute to graft rejection or immunoregulatory functions (M2) that promote graft tolerance. M2 macrophages generate energy mainly through oxidative phosphorylation (OxPhos) and glutamine metabolism, while M1 macrophages increase metabolic flux through the pentose phosphate pathway (PPP) and glycolysis. HIF-1α and Akt increase glycolysis by upregulating the expression of glycolytic enzymes and the glucose transporter GLUT1, respectively. The cholesterol pathway intermediate mevalonate, which can be blocked using statins, is involved in the epigenetic fixation of the proinflammatory phenotype. Itaconate is an anti-inflammatory metabolite whose expression is upregulated upon macrophage activation
In transplanted organs, the vascular tissue damage associated with transplant surgery promotes the recruitment of recipient inflammatory monocytes that infiltrate the allograft in a chemokine-dependent manner.3 Graft-infiltrating monocytes detect the presence of multiple stimuli that result from ischemia-reperfusion injury (IRI), including dying cells. This detection results in strong inflammatory responses induced by molecules derived from dead allogeneic donor cells, which are recognized by the innate immune system as pathogen-associated molecular patterns (PAMPs). This ancient mechanism of immunological defense is also triggered by self-derived damage-associated molecular patterns (DAMPs) following sterile inflammatory stimuli during the surgical anastomosis involved in the organ transplant procedure. Both PAMPs and DAMPs are recognized through pattern recognition receptors (PRRs) on the cell surface and in the cytoplasm of myeloid cells, and this recognition affects both the early and long-term function of the allograft.4–6 Here, we review the signaling pathways mediated by complement receptors and toll-like-receptors (TLRs), which represent two of the main types of PRRs that are triggered by danger molecules released from dead cells in the context of organ transplantation.7,8
Danger signals released from dying cells during ischemia and reperfusion, such as high-mobility group box 1 (HMGB1) and ATP, induce metabolic changes in innate immune cells when released into the extracellular compartment.9,10 It has become evident that cellular metabolism is intimately linked with immune function, and the concept of immunometabolic research is gaining interest among transplant immunologists.11–13 In fact, activated immune cells need to redirect their metabolic flux because their bioenergetic and biosynthetic demands are very distinct from those of resting cells. Since metabolic changes are a prerequisite for differentiation and therefore cellular immune functions such as cytokine production, this offers the opportunity for manipulation for the prevention and treatment of inflammatory diseases as well as for the prevention of transplant rejection.14 Notably, because this intervention approach is based on the distinct metabolic requirements of the cells, it allows selective targeting of the differentiation and function of certain cells. In this review, we summarize how metabolic interventions in cells of the innate immune system can effectively tailor immune responses towards allograft protection, and this metabolic-focused approach shows great promise for improving the standard of care for transplant patients.
Cell death
Because of tissue injury during organ transplantation, a significant number of cells in the donor organs are committed to cell death. In the case of kidney transplants, most kidneys transplanted now experience certain forms of damage even before resuming their functions in the recipient due to ischemia reperfusion injury (IRI). Recent studies have shown that renal tubular epithelial cells can die by ferroptosis, a type of cell death with poorly defined mechanisms.15,16 Consequently, the donor organ parenchyma releases DAMPs following cell death. Donor DAMPs are sensed by host monocytes and macrophages, leading to the activation of these cells, which causes graft injury.17,18 However, solid organ transplantation is one of the few situations in which cell death can be anticipated, allowing therapeutic targeting of cell death pathways. As mentioned above, ferroptosis has been implicated in compromised graft function and graft rejection.19,20 In models of islet transplantation, some mediators of islet oxidative stress, such as glutathione peroxidase-4 (GPX4), have been identified as inhibitors of ferroptosis. Thus, mechanisms affecting GPX4 function may impact islet function and viability. In an in vitro study, human islet viability and function were found to be compromised in the presence of erastin or RSL3, which are both ferroptosis-inducing agents (FIA). Furthermore, these effects were counteracted with pretreatment with ferroptosis inhibitors, such as ferrostatin-1 (Fer-1) or desferrioxamine (DFO). The pretreatment of islets with inhibitors did not have a deleterious effect on engraftment in an immunodeficient mouse transplant model.21 As human islet transplantation has been held back by the donor cell death associated with the islet preparation procedure, ferroptosis inhibition could be a useful option to improve graft viability and function.
Another strategy to improve long-term allograft survival is to suppress innate immunity events, referred to as the “time-restricted therapeutic window”, and this strategy includes treatment of the donor organ during harvest and the recipient during allograft reperfusion.22 In this respect, ex vivo lung perfusion (EVLP) has been proposed to prevent the detrimental effects of the DAMP release associated with IRI. Preclinical studies in lung transplantation have demonstrated that normothermic EVLP improves lung function after transplantation.23,24 The combination of an adenosine A2A agonist and EVLP was shown to attenuate the acute inflammatory response to IRI and improve pulmonary function.24 An additional therapeutic option is gene delivery during organ preservation.25 Machuca et al. noted that the administration of adenoviral interleukin-10 gene therapy (AdhIL-10) during EVLP reduced IRI and improved posttransplantation lung function over EVLP alone.26 IL-10 is an anti-inflammatory cytokine that downregulates the activity of the innate immune system, specifically suppressing the ability of myeloid cells to present antigens to T cells.27 Ex vivo IL-10 gene therapy in injured human donor lungs induces a significant shift from a proinflammatory to an anti-inflammatory cytokine profile, which is associated with improved lung function.28 In addition, the administration of α1-anti-trypsin (A1AT) during EVLP to porcine lung donors also attenuates IRI and improves graft function after transplantation.29,30 Mechanistically, A1AT has antiapoptotic and anti-inflammatory properties and can attenuate the activation of macrophages and neutrophils in addition to inducing tolerogenic dendritic cells.29 These data suggest that the induction of different forms of cell death leads to compromised graft function, which can be attenuated in the presence of inhibitors.
The complement cascade
The complement cascade is an important contributor to innate immunity and is composed of a tightly regulated network of proteins that play a critical role in inflammation and host defense.31 Complement activation results in the clearance of immune complexes, injured cells, and invading pathogens.32 More than 50 genes encoding proteins of complement components, including membrane receptors and regulators, have been identified.33 The complement proteins are primarily produced in the liver,34 although macrophages and epithelial cells in the gastrointestinal and urinary tracts also synthesize significant amounts of complement proteins.35
There are three different pathways that activate the complement system, the classical, alternative, and lectin pathways, and the pathways involve proteins that mostly exist as inactive zymogens.31 Upon activation, complement proteins interact in a highly targeted and strictly regulated enzymatic cascade that generates proteolytic fragments, which mediate numerous biological functions.36 The classical pathway can be activated by antigen-antibody immune complexes, apoptotic or necrotic cells, or by acute phase proteins, including serum amyloid P protein and C-reactive protein.32,37 After binding IgM or IgG antibodies to a cognate antigen, the Fc portion of an antibody interacts with C1q, leading to the sequential activation of C1r and C1s. Activated C1s cleaves C4 into C4a and C4b and C2 into C2a and C2b. Then, C4b binds to C2a, forming the C4bC2a complex. C4bC2a is an enzymatic complex that cleaves the abundant plasma protein C3 into the anaphylatoxin C3a and C3b.32 In the alternative pathway, serum C3 can spontaneously hydrolyze to generate C3(H2O), which binds to the surface of foreign cells and factor B.37 Once factor B binds to C3(H2O), it can be cleaved by the constitutively active serum protease factor D, generating the C3 convertase complex C3(H2O)Bb, which cleaves native C3 molecules into C3a and C3b.38 The binding of carbohydrates or acetylated residues present on specific glycocalyx patterns expressed by apoptotic and necrotic cells to pattern-recognition molecules, such as mannose-binding lectin (MBL), collectin 11 (CL-K1), or ficolins, may trigger the lectin pathway.37,39
All three of these pathways merge on the proteolytic activation of C3 (cleavage of C3 by C3 convertases) to produce C3a and C3b.40 Once C3b binds to C3 convertase, the C5 convertase (C4bC2aC3b), which can cleave C5 to C5a and C5b, forms. The anaphylatoxins C3a and C5a can bind to their receptors, which are expressed on a large number of cell subsets, including neutrophils, eosinophils, mast cells, monocytes/macrophages, dendritic cells, endothelial cells, astrocytes, and microglia.41 The anaphylatoxins C3a and C5a can enhance inflammation, leukocyte recruitment, cytokine and chemokine release, and oxygen radical production and increase blood vessel permeability.42 In contrast, C3b molecules can opsonize immune complexes and enhance phagocytosis,43 while the sequential binding of C5b to C6, C7, C8, and 10–16 C9 molecules leads to the formation of a macromolecular structure called the terminal membrane attack complex C5b-9 (MAC).44 This complex can form holes in the cell membrane, resulting in the leakage of intracellular contents and cell destruction.43
Mounting evidence from basic and clinical research supports the role of the complement cascade in several complications during allograft transplantation. The activation of the complement cascade and the innate immune system can occur shortly after cardiac arrest or brain death in deceased organ donors.45 In addition, the complement system can be activated by donor-specific alloantibodies46 or IRI.42,47 Complement cascade activation amplifies the expression of the C3a and C5a proteins, which recruit and activate neutrophils and monocytes.45,48 When neutrophils and monocytes are recruited from the peripheral circulation into an allograft, they become activated and release proinflammatory cytokines, chemokines, and reactive oxygen species, causing cell apoptosis and necrosis.49 The formation of the membrane attack complex C5b-9, which causes non-receptor-mediated cell activation and cytotoxicity, represents another element of complement cascade activation during IRI following organ transplantation.6
Accumulating experimental and clinical evidence has demonstrated that the inhibition of complement activation can be a promising target for therapeutic intervention in organ transplantation.50 To date, numerous therapeutic agents, such as monoclonal antibodies, small molecules, and small interfering RNA (siRNA) agents, have been developed to block complement cascade activation. The administration of soluble complement receptor-1 antagonist (sCR1), which inactivates the C3 and C5 convertases, results in reduced neutrophil migration into grafts and reduced posttransplantation reperfusion edema in a swine lung allotransplantation model.51 The clinical trial testing sCR1, TP-10, showed 90% complement inhibition in IRI for 24 h after lung transplantation.52 Gueler et al. showed that treating recipient mice with a C5aR antagonist before transplantation remarkably enhances graft survival and reduces monocyte/macrophage infiltration.53 In a recent phase I/II clinical study, the effect of C1 esterase inhibitor (C1INH) on the prevention of delayed graft function in kidney transplant recipients was studied. Significantly fewer dialysis sessions were observed at 2-4 weeks post-transplantation, and better renal function was observed at 1 year in C1INH-treated patients.54 The other suggested approach to attenuate IRI in organ transplantation is silencing the C5a receptor (C5aR) gene using siRNA. To this end, a study showed that mice that received C5aR siRNA two days before the induction of ischemia have reduced expression of the proinflammatory cytokine TNF-α and chemokines MIP-2 and KC, resulting in reductions in neutrophil influx and cell necrosis in the kidneys.55 One of the first FDA-approved therapeutics to block complement cascade activation was eculizumab. Eculizumab is a humanized monoclonal antibody that binds to C5 and inhibits its cleavage to C5a, thereby preventing the formation of the MAC (C5b-9).56 A few clinical case reports have shown promising results for eculizumab in patients with atypical hemolytic uremic syndrome undergoing kidney transplantation.57,58 Another suggested strategy to inhibit complement cascade activation is silencing C3 using siRNA. C3 is the central component of complement cascade activation where all three pathways converge.40 The systemic administration of a C3-specific siRNA in a mouse kidney model of IRI diminished renal C3 synthesis, resulting in less renal injury and mouse mortality.55
Toll-like receptors (TLRs)
TLRs, which have central roles in the initiation of allograft inflammation and development of acute and chronic allograft rejection, represent another component of innate immunity.59 TLRs are germline-encoded pattern-recognition receptors (PRRs) expressed primarily on antigen-presenting cells.60 TLRs are also expressed on endothelial and stromal cells, including the epithelial cells lining the respiratory, intestinal and urogenital tracts.4 PRRs can recognize microbe-specific molecular structures, known as pathogen-associated molecular patterns (PAMPs), and damage-associated molecular patterns (DAMPs), which are released by damaged/dying cells in the body.61 Upon recognition of any PAMP or DAMP, PRRs activate downstream signaling pathways that not only trigger innate immune responses by producing proinflammatory cytokines/chemokines but can also upregulate costimulatory molecule expression and amplify the antigen-processing and antigen presentation capacities of innate immune cells, leading to the initiation of antigen-specific adaptive immune responses.62–65
Organ transplantation surgery can cause low-grade penetration of recipient bacteria, donor commensal bacteria in the case of lung or intestinal transplantation, or translocation of intestinal bacteria because of surgical stress.66 In addition, IRI can also induce the expression of endogenous PRRs.66 As a result, the ligation of donor and recipient TLRs by both microbial molecular patterns and stress/damage-associated endogenous ligands after allograft transplantation is a risk factor. The inevitable period of ischemia reperfusion during allograft transplantation causes a number of local and systemic cellular and biochemical changes, leading to the upregulation or induction of the expression of the endogenous ligands of TLRs, including heparin sulfate, fibrinogen, hyaluronan, high-mobility group box chromosomal protein 1 (HMGB1), and heat shock protein (HSP), especially HSP60 and HSP70, in injured and necrotic cells.64,67 Consequently, TLR signaling pathways are upregulated by Toll/IL-1 resistance (TIR) domain-containing adapter molecules, such as myeloid differentiation factor 88 (MyD88), TIRAP/Mal, toll/interferon response factor (TRIF), and TRAM.68 While TLR1, TLR2, TLR5, TLR6, TLR7, TLR8, and TLR9 initiate MyD88-dependent signaling pathways, TLR3 is TRIF-dependent, and TLR4 can signal through both MyD88 and TRIF pathways.69 MyD88 activates transcription factors, such as nuclear factor kappa B (NF-κB), activator protein 1 (AP-1), and mitogen-activated protein kinases (MAPK), leading to the production of proinflammatory chemokines and cytokines.69 In addition, this signaling allows antigen-presenting cells to mature, migrate to draining lymph nodes and activate naive T cells.70 The TRIF signaling pathway stimulates cytokine production through interferon regulatory factor 3 (IRF3), NF-κB and AP-1, leading to proinflammatory responses.71 TLRs can also cause direct apoptosis via the Fas-associated death domain protein (FADD) and caspase 8 pathways.67
Considering the pathological role of TLRs in allograft rejection, inhibitors that target TLR signaling could open novel avenues to improve long-term transplantation outcomes. Mounting evidence in the literature demonstrates that the generation of DAMPs during IRI activates TLR4 and TLR2, and this activation results in the production of proinflammatory cytokines and chemokines, facilitating leukocyte migration and infiltration.72,73 Several studies have reported significantly lower levels of proinflammatory mediators and cellular infiltration associated with preserved graft function in TLR4-knockout and TLR2-knockout mice compared to wild-type mice.73–75 In an elegant study, Kaczorowski et al. demonstrated that TLR4 signaling is dominant in both the systemic and intragraft inflammatory responses that occur after cold ischemia reperfusion in the setting of organ transplantation.76 The authors performed syngeneic heart transplants in TLR4-deficient mice and observed lower levels of serum TNF-α, IL-6, monocyte chemoattractant protein 1 (MCP1), IL-1β, and troponin I as well as intragraft TNF-α, IL-1β, IL-6, ICAM-1, and inducible nitric oxide synthase (iNOS) mRNA levels in the knockout to knockout group compared to the wild-type to wild-type group.76 Clinical studies have also demonstrated the roles of TLR2 and TLR4 signaling in acute allograft rejection. Palmer et al. suggested that the activation of innate immunity through TLR4 in a donor kidney contributes to the development of acute rejection after renal transplantation.77 Deng et al. reported higher expression of TLR2 and TLR4 on circulating monocytes from conditioned liver transplant recipients with acute rejection than on those from patients with normal liver function.60 Testro et al. noted higher expression of TLR4 in patients who experienced rejection after liver transplantation than in patients who did not experience rejection.78 Similar results have been reported in patients with acute and chronic renal transplant rejection.79,80 Therefore, inhibitors or antagonists that target TLR or downstream signaling components may attenuate IRI and enhance allograft survival. Blocking antibodies against TLR signaling pathway molecules have shown promising results in animal models. For instance, the administration of an anti-HMGB1 neutralizing antibody to wild-type mice attenuated kidney IRI, as evidenced by reduced apoptosis in tubular epithelial cells, less infiltration of neutrophils and macrophages, and lower levels of IL-6, TNF-α, and MCP1.81 Li et al. noted similar effects following the blockade of extracellular HMGB1 in a mouse model of IRI.82 The authors observed reduced ischemia reperfusion-induced renal dysfunction and suppressed inflammation and tubular apoptosis upon administration of a rabbit anti-mouse HMGB1 antibody before renal ischemia.82 Furthermore, neutralizing HMGB1 reduced the number of inflammatory CD11b+Ly6Chigh myeloid cells in the allograft and the spleen in a cardiac transplantation model.83 Another suggested strategy to diminish the cytoplasmic translocation of HMGB1 and subsequently reduce IRI is using a noble gas (xenon or argon) to treat either the donor organ or the recipient.84 In vitro experiments demonstrated that xenon treatment could attenuate HMGB-1 translocation and NF-κB activation in human lung epithelial and proximal tubular cells.85,86 Zhao et al. demonstrated that donor exposure to xenon before graft retrieval or recipient exposure after engraftment decreased caspase-3 expression, localized HMGB1 within the nucleus and prevented TLR-4/NF-κB activation in tubular cells in a syngeneic rat model of kidney transplantation.72
Eritoran is a TLR4 antagonist that has been tested in allograft transplantation. Eritoran is a synthetic structural analog of the lipid A portion of lipopolysaccharide (LPS) that blocks LPS from binding at the cell surface to the MD2-TLR-4 receptor and terminates MD2/TLR4-mediated signaling.87,88 Eritoran treatment induced less monocyte infiltration, lower levels of TNF-α, IL-1β, IL-6, and MCP1, and prolonged survival in a rat transplantation model.89 It is possible that eritoran blocks the HMGB1-TLR4 interaction and attenuates the TLR4-dependent release of HMGB1, as suggested in a mouse model of liver IRI.90 Dexmedetomidine has also been demonstrated to have cytoprotective and anti-inflammatory effects in a renal IRI mouse model.91 Mechanistically, dexmedetomidine activates pro-cell survival pAKT signals via α2 adrenoceptors, resulting in reduced cell death and HMGB1 release and subsequently inhibiting TLR4 signaling.91
Innate immunometabolism
We have recently identified macrophages as key players in the regulation of transplant tolerance.92 Depending on environmental factors, macrophages can be polarized to different functional states: M1 macrophages secrete proinflammatory cytokines and produce antimicrobial substances, while M2 macrophages produce IL-4 and IL-13 and are involved in tissue repair and immune regulation.93 M1 and M2 macrophages are distinct in their metabolic processes: proinflammatory M1 macrophages have to redirect their metabolic flux towards glycolysis and the pentose phosphate pathway and simultaneously suppress oxidative phosphorylation,93–97 while M2 macrophages use oxidative phosphorylation and glutamine metabolism for ATP generation.98,99 Furthermore, M1 and M2 macrophages also differ in their arginine metabolism. While M1 macrophages express the enzyme inducible nitric oxide synthase (iNOS), which metabolizes arginine into nitric oxide (NO) and citrulline for antimicrobial functions, M2 macrophages express the enzyme arginase-1, which metabolizes arginine into ornithine and urea, providing building blocks for cellular proliferation and tissue repair.100 While certain metabolic molecules are produced as a result of macrophage polarization, specific metabolites also regulate macrophage differentiation. For example, itaconate is one of the most highly induced metabolites in macrophages upon activation with lipopolysaccharide, and this molecule regulates succinate levels, mitochondrial respiration and inflammatory cytokine production.101,102 In a model of IRI, itaconate prevented proinflammatory activation, demonstrating that the manipulation of cellular metabolism is a promising strategy to modify the immune response.102
Changes in metabolic pathways during macrophage polarization depend on factors that integrate environmental signals, such as Akt, mTOR, and AMPK.103 For example, signaling through Akt increases the cell surface expression of the glucose transporter GLUT1 as well as the phosphorylation of hexokinase; these events markedly increase glycolytic flux in macrophages.104,105 Reprogramming cellular metabolism also depends on transcriptional regulators that control the expression of key enzymes that orchestrate switches among different metabolic pathways. For example, monocytes that are deficient in nuclear receptor Nur77 (Nr4a1) are arrested in the S phase of the cell cycle and undergo apoptosis in the bone marrow, leading to low numbers of Ly6Clo monocytes in the circulation.106 In addition, Nur77-deficient macrophages fail to switch to their mitochondrial metabolism upon stimulation and therefore accumulate higher levels of succinate and produce more nitric oxide and proinflammatory cytokines in a succinate dehydrogenase (SDH)-dependent manner, leading to the exacerbation of chronic inflammatory diseases.107
Another important transcriptional regulator in the control of metabolic adaptation during macrophage activation is hypoxia-induced factor (HIF)-1α. HIF-1α is one of the master regulators of glycolysis, which is often activated upon stimulation with LPS.108 The deletion of the HIF-1α gene in macrophages inhibits the metabolic switch to glycolysis, thus resulting in reduced antimicrobial function. Interestingly, HIF-1α-mediated increases in aerobic glycolysis and the accumulation of intermediates of the tricarboxylic acid (TCA) cycle, such as fumarate and glutamate, control chromatin remodeling including histone methylation (histone 3 lysine 4 trimethylation, H3K4me3) and acetylation (histone 3 lysine 27 acetylation, H3K27ac). This process results in the epigenetic reprogramming of innate immune cells, and upon restimulation, these cells exhibit enhanced production of proinflammatory cytokines, a process that has recently been called “trained immunity”.109–112 Of note, trained immunity is a term used to differentiate enhanced innate responses from the classic “memory recall” of adaptive immune cells. Importantly, macrophages that have acquired trained immunity in the context of transplantation play an important role in promoting allograft rejection.113 Thus, interfering with the metabolic processes that form the basis of long-lasting epigenetic reprogramming, i.e., preventing trained immunity in macrophages, is a promising therapeutic strategy for the prevention of allograft rejection.
Apart from glycolysis, other metabolic pathways have been identified to be crucial for trained immunity in macrophages and represent targets for intervention. A recent study showed that mevalonate, a metabolite of the cholesterol synthesis pathway, promotes trained immunity in macrophages.114 Accordingly, myeloid cells from patients with hyper-IgD syndrome (HIDS) who accumulate mevalonate as a result of a mevalonate kinase deficiency display a trained immunity phenotype in their macrophages. Statins prevent mevalonate production and are used clinically to inhibit the cholesterol synthesis pathway. Interestingly, statins can also effectively prevent trained immunity. These data provide proof that targeting metabolic pathways can have long-lasting effects on macrophage functions to promote tolerance.
Concluding remarks
One of the major goals in organ transplantation is the establishment of donor-specific tolerance. The induction of transplant tolerance is a challenging task as donor organs are subjected to tissue injury related to ischemia reperfusion processes. In addition to the IRI damage of the allograft, the recipient innate immune system initiates highly inflammatory processes as a result of the vascular injury associated with transplant surgery. As a result, proinflammatory monocytes that infiltrate the allograft sense danger molecules through complement receptors and TLRs, reprogram their metabolic pathways and become inflammatory macrophages. They upregulate costimulatory molecule expression, secrete proinflammatory cytokines and consequently activate innate and adaptive immunity, leading to organ rejection. Therefore, protocols that modulate cell death pathways in donor organs or interfere with the PRRs of host innate immune cells and regulate the immunometabolism of these cells represent promising therapeutic approaches that may be used synergistically with current immunosuppressive agents for the induction of immune tolerance.
Competing interests
The authors declare no competing interests.
References
- 1.Tait SW, Ichim G, Green DR. Die another way—non-apoptotic mechanisms of cell death. J. Cell Sci. 2014;127:2135–2144. doi: 10.1242/jcs.093575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Juncadella IJ, et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493:547–551. doi: 10.1038/nature11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia MR, et al. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J. Clin. Invest. 2010;120:2486–2496. doi: 10.1172/JCI41628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boros P, Bromberg JS. New cellular and molecular immune pathways in ischemia/reperfusion injury. Am. J. Transplant. 2006;6:652–658. doi: 10.1111/j.1600-6143.2005.01228.x. [DOI] [PubMed] [Google Scholar]
- 5.Boros P, Bromberg JS. De novo autoimmunity after organ transplantation: targets and possible pathways. Hum. Immunol. 2008;69:383–388. doi: 10.1016/j.humimm.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Sacks SH, Chowdhury P, Zhou W. Role of the complement system in rejection. Curr. Opin. Immunol. 2003;15:487–492. doi: 10.1016/s0952-7915(03)00100-6. [DOI] [PubMed] [Google Scholar]
- 7.Kataoka H, Kono H, Patel Z, Kimura Y, Rock KL. Evaluation of the contribution of multiple DAMPs and DAMP receptors in cell death-induced sterile inflammatory responses. PLoS One. 2014;9:e104741. doi: 10.1371/journal.pone.0104741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat. Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iyer SS, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl Acad. Sci. USA. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 11.Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nat. Rev. Immunol. 2011;11:81. doi: 10.1038/nri2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016;16:553–565. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Degauque N, Brosseau C, Brouard S. Regulation of the immune response by the inflammatory metabolic microenvironment in the context of allotransplantation. Front Immunol. 2018;9:1465. doi: 10.3389/fimmu.2018.01465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanimine N, Turka LA, Priyadharshini B. Navigating T-cell immunometabolism in transplantation. Transplantation. 2018;102:230–239. doi: 10.1097/TP.0000000000001951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linkermann A, et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl Acad. Sci. USA. 2014;111:16836–16841. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller T, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol. Life Sci. 2017;74:3631–3645. doi: 10.1007/s00018-017-2547-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oberbarnscheidt MH, et al. Non-self recognition by monocytes initiates allograft rejection. J. Clin. Invest. 2014;124:3579–3589. doi: 10.1172/JCI74370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarhan M, von Massenhausen A, Hugo C, Oberbauer R, Linkermann A. Immunological consequences of kidney cell death. Cell Death Dis. 2018;9:114. doi: 10.1038/s41419-017-0057-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruni A, et al. Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis. 2018;9:595. doi: 10.1038/s41419-018-0506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zschiedrich S, et al. An update on ABO-incompatible kidney transplantation. Transpl. Int. 2015;28:387–397. doi: 10.1111/tri.12485. [DOI] [PubMed] [Google Scholar]
- 21.Halloran PF, Reeve JP, Pereira AB, Hidalgo LG, Famulski KS. Antibody-mediated rejection, T cell-mediated rejection, and the injury-repair response: new insights from the Genome Canada studies of kidney transplant biopsies. Kidney Int. 2014;85:258–264. doi: 10.1038/ki.2013.300. [DOI] [PubMed] [Google Scholar]
- 22.Land WG. Innate immunity-mediated allograft rejection and strategies to prevent it. Transplant. Proc. 2007;39:667–672. doi: 10.1016/j.transproceed.2007.01.052. [DOI] [PubMed] [Google Scholar]
- 23.Cypel M, et al. Normothermic ex vivo lung perfusion in clinical lung transplantation. N. Engl. J. Med. 2011;364:1431–1440. doi: 10.1056/NEJMoa1014597. [DOI] [PubMed] [Google Scholar]
- 24.Emaminia A, et al. Adenosine A2A agonist improves lung function during ex vivo lung perfusion. Ann. Thorac. Surg. 2011;92:1840–1846. doi: 10.1016/j.athoracsur.2011.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guibert EE, et al. Organ preservation: current concepts and new strategies for the next decade. Transfus. Med. Hemother. 2011;38:125–142. doi: 10.1159/000327033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Machuca TN, et al. Safety and efficacy of ex vivo donor lung adenoviral IL-10 gene therapy in a large animal lung transplant survival model. Human. Gene Ther. 2017;28:757–765. doi: 10.1089/hum.2016.070. [DOI] [PubMed] [Google Scholar]
- 27.Mittal SK, Roche PA. Suppression of antigen presentation by IL-10. Curr. Opin. Immunol. 2015;34:22–27. doi: 10.1016/j.coi.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cypel M, et al. Functional repair of human donor lungs by IL-10 gene therapy. Sci. Transl. Med. 2009;1:4ra9–4ra9. doi: 10.1126/scitranslmed.3000266. [DOI] [PubMed] [Google Scholar]
- 29.Lin H, et al. α1-Anti-trypsin improves function of porcine donor lungs during ex-vivo lung perfusion. J. Heart Lung Transplant. 2018;37:656–666. doi: 10.1016/j.healun.2017.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Iskender I, et al. Human α1-antitrypsin improves early post-transplant lung function: pre-clinical studies in a pig lung transplant model. J. Heart Lung Transplant. 2016;35:913–921. doi: 10.1016/j.healun.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 31.Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011;343:227–235. doi: 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noris M, Remuzzi G. Overview of complement activation and regulation. Semin. Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marrón-Liñares GM, et al. Polymorphisms in genes related to the complement system and antibody-mediated cardiac allograft rejection. J. Heart Lung Transplant. 2018;37:477–485. doi: 10.1016/j.healun.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 34.Merle, N. S., Church, S. E., Fremeaux-Bacchi, V. & Roumenina, L. T. Complement system part I—molecular mechanisms of activation and regulation. Front. Immunol.6 (2015). [DOI] [PMC free article] [PubMed]
- 35.Varela JC, Tomlinson S. Complement: an overview for the clinician. Hematology. 2015;29:409–427. doi: 10.1016/j.hoc.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atkinson, J. P. et al. (eds). Clinical Immunology (Fifth Edition). 299–317 (London, 2019).
- 37.Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J. Immunol. 2006;176:1305–1310. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 38.Merle, N. S., Noe, R., Halbwachs-Mecarelli, L., Fremeaux-Bacchi, V. & Roumenina, L. T. Complement system part II: role in immunity. Front. Immunol.6 (2015). [DOI] [PMC free article] [PubMed]
- 39.Beltrame, M. H., Catarino, S. J., Goeldner, I., Boldt, A. B. W. & de Messias-Reason, I. J. The lectin pathway of complement and rheumatic heart disease. Front. Pediatr.2 (2015). [DOI] [PMC free article] [PubMed]
- 40.Angioi A, et al. Diagnosis of complement alternative pathway disorders. Kidney Int. 2016;89:278–288. doi: 10.1016/j.kint.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 41.Mathern DR, Heeger PS. Molecules great and small: the complement system. Clin. J. Am. Soc. Nephrol. 2015;10:1636–1650. doi: 10.2215/CJN.06230614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cernoch M, Viklicky O. Complement in kidney tTransplantation. Front. Med. 2017;4:66–66. doi: 10.3389/fmed.2017.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kahr WHA. Complement halts angiogenesis gone wild. Blood. 2010;116:4393–4394. doi: 10.1182/blood-2010-08-297648. [DOI] [PubMed] [Google Scholar]
- 44.Makrides SC. Therapeutic inhibition of the complement system. Pharmacol. Rev. 1998;50:59–88. [PubMed] [Google Scholar]
- 45.Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat. Rev. Nephrol. 2016;12:383. doi: 10.1038/nrneph.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stegall MD, Chedid MF, Cornell LD. The role of complement in antibody-mediated rejection in kidney transplantation. Nat. Rev. Nephrol. 2012;8:670. doi: 10.1038/nrneph.2012.212. [DOI] [PubMed] [Google Scholar]
- 47.Chun N, et al. Complement dependence of murine costimulatory blockade-resistant cellular cardiac allograft rejection. Am. J. Transplant. 2017;17:2810–2819. doi: 10.1111/ajt.14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arumugam TV, Shiels IA, Woodruff TM, Granger DN, Taylor SM. The role of the complement system in ischemia-reperfusion injury. Shock. 2004;21:401–409. doi: 10.1097/00024382-200405000-00002. [DOI] [PubMed] [Google Scholar]
- 49.Danobeitia JS, Djamali A, Fernandez LA. The role of complement in the pathogenesis of renal ischemia-reperfusion injury and fibrosis. Fibrogenesis Tissue Repair. 2014;7:16. doi: 10.1186/1755-1536-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheerin NS. Should complement activation be a target for therapy in renal transplantation? J. Am. Soc. Nephrol. 2008;19:2250–2251. doi: 10.1681/ASN.2008101064. [DOI] [PubMed] [Google Scholar]
- 51.Schmid RA, et al. Effect of soluble complement receptor type 1 on reperfusion edema and neutrophil migration after lung allotransplantation in swine. J. Thorac. Cardiovasc Surg. 1998;116:90–97. doi: 10.1016/S0022-5223(98)70246-6. [DOI] [PubMed] [Google Scholar]
- 52.Keshavjee S, Davis RD, Zamora MR, de Perrot M, Patterson GA. A randomized, placebo-controlled trial of complement inhibition in ischemia-reperfusion injury after lung transplantation in human beings. J. Thorac. Cardiovasc Surg. 2005;129:423–428. doi: 10.1016/j.jtcvs.2004.06.048. [DOI] [PubMed] [Google Scholar]
- 53.Gueler F, et al. Complement 5a receptor inhibition improves renal allograft survival. J. Am. Soc. Nephrol. 2008;19:2302–2312. doi: 10.1681/ASN.2007111267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jordan SC, et al. A phase I/II, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Am. J. Transplant. 2018;18:2955–2964. doi: 10.1111/ajt.14767. [DOI] [PubMed] [Google Scholar]
- 55.Zheng X, et al. Gene silencing of complement C5a receptor using siRNA for preventing ischemia/reperfusion injury. Am. J. Pathol. 2008;173:973–980. doi: 10.2353/ajpath.2008.080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat. Biotechnol. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weitz M, Amon O, Bassler D, Koenigsrainer A, Nadalin S. Prophylactic eculizumab prior to kidney transplantation for atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2011;26:1325. doi: 10.1007/s00467-011-1879-9. [DOI] [PubMed] [Google Scholar]
- 58.Barnett ANR, et al. The use of eculizumab in renal transplantation. Clin. Transplant. 2013;27:E216–E229. doi: 10.1111/ctr.12102. [DOI] [PubMed] [Google Scholar]
- 59.Braza F, Brouard S, Chadban S, Goldstein DR. Role of TLRs and DAMPs in allograft inflammation and transplant outcomes. Nat. Rev. Nephrol. 2016;12:281. doi: 10.1038/nrneph.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng JF, et al. The role of toll-like receptors 2 and 4 in acute allograft rejection after liver transplantation. Transplant. Proc. 2007;39:3222–3224. doi: 10.1016/j.transproceed.2007.02.102. [DOI] [PubMed] [Google Scholar]
- 61.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front. Immunol. 2014;5:461–461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alegre ML, Chong A. Toll-like receptors (TLRs) in transplantation. Front. Biosci. (Elite Ed.) 2009;1:36–43. doi: 10.2741/e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Medzhitov R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001;1:135. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 64.Leventhal JS, Schröppel B. Toll-like receptors in transplantation: sensing and reacting to injury. Kidney Int. 2012;81:826–832. doi: 10.1038/ki.2011.498. [DOI] [PubMed] [Google Scholar]
- 65.Sheen JH, et al. TLR-induced murine dendritic cell (DC) activation requires DC-intrinsic complement. J. Immunol. 2017;199:278–291. doi: 10.4049/jimmunol.1700339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alegre ML, Goldstein DR, Chong AS. Toll-like receptor signaling in transplantation. Curr. Opin. Organ Transplant. 2008;13:358–365. doi: 10.1097/MOT.0b013e3283061149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patel H, et al. Toll-like receptors in ischaemia and its potential role in the pathophysiology of muscle damage in critical limb ischaemia. Cardiol. Res. Pract. 2012;2012:121237–121237. doi: 10.1155/2012/121237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takeda K, Akira S. Toll-like receptors in innate immunity. Int. Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 69.Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000prime Rep. 2014;6:97–97. doi: 10.12703/P6-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goldstein DR, Tesar BM, Akira S, Lakkis FG. Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J. Clin. Investig. 2003;111:1571–1578. doi: 10.1172/JCI17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang S, et al. Recipient toll-like receptors contribute to chronic graft dysfunction by both MyD88- and TRIF-dependent signaling. Dis. Models. 2010;3:92–103. doi: 10.1242/dmm.003533. [DOI] [PubMed] [Google Scholar]
- 72.Zhao H, Perez JS, Lu K, George AJT, Ma D. Role of toll-like receptor-4 in renal graft ischemia-reperfusion injury. Am. J. Physiol. 2014;306:F801–F811. doi: 10.1152/ajprenal.00469.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arslan F, Keogh B, McGuirk P, Parker AE. TLR2 and TLR4 in ischemia reperfusion injury. Mediat. Inflamm. 2010;2010:704202–704202. doi: 10.1155/2010/704202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leemans JC, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Investig. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pulskens WP, et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One. 2008;3:e3596–e3596. doi: 10.1371/journal.pone.0003596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaczorowski DJ, et al. Toll-like receptor 4 mediates the early inflammatory response after cold ischemia/reperfusion. Transplantation. 2007;84:1279–1287. doi: 10.1097/01.tp.0000287597.87571.17. [DOI] [PubMed] [Google Scholar]
- 77.Palmer SM, et al. Donor polymorphisms in Toll-like receptor-4 influence the development of rejection after renal transplantation. Clin. Transplant. 2006;20:30–36. doi: 10.1111/j.1399-0012.2005.00436.x. [DOI] [PubMed] [Google Scholar]
- 78.Testro AG, et al. Acute allograft rejection in human liver transplant recipients is associated with signaling through toll-like receptor 4. J. Gastroenterol. Hepatol. 2011;26:155–163. doi: 10.1111/j.1440-1746.2010.06324.x. [DOI] [PubMed] [Google Scholar]
- 79.Kwon J, Park J, Lee D, Kim YS, Jeong HJ. Toll-like receptor expression in patients with renal allograft dysfunction. Transplant. Proc. 2008;40:3479–3480. doi: 10.1016/j.transproceed.2008.06.073. [DOI] [PubMed] [Google Scholar]
- 80.Braudeau C, et al. Contrasted blood and intragraft toll-like receptor 4 mRNA profiles in operational tolerance versus chronic rejection in kidney transplant recipients. Transplantation. 2008;86:130–136. doi: 10.1097/TP.0b013e31817b8dc5. [DOI] [PubMed] [Google Scholar]
- 81.Wu L. A Flt3L encounter: mTOR signaling in dendritic cells. Immunity. 2010;33:580–582. doi: 10.1016/j.immuni.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 82.Li J, et al. Neutralization of the extracellular HMGB1 released by ischaemic damaged renal cells protects against renal ischaemia-reperfusion injury. Nephrol. Dial. Transplant. 2011;26:469–478. doi: 10.1093/ndt/gfq466. [DOI] [PubMed] [Google Scholar]
- 83.Zou H, et al. HMGB1 is involved in chronic rejection of cardiac allograft via promoting inflammatory-like mDCs. Am. J. Transplant. 2014;14:1765–1777. doi: 10.1111/ajt.12781. [DOI] [PubMed] [Google Scholar]
- 84.Irani Y, et al. Noble gas (Argon and Xenon)-saturated cold storage solutions reduce ischemia-reperfusion injury in a rat model of renal transplantation. Nephron Extra. 2011;1:272–282. doi: 10.1159/000335197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao H, et al. Xenon treatment protects against remote lung injury after kidney transplantation in rats. Anesthesiology. 2015;122:1312–1326. doi: 10.1097/ALN.0000000000000664. [DOI] [PubMed] [Google Scholar]
- 86.Zhao H, et al. Xenon treatment protects against cold ischemia associated delayed graft function and prolongs graft survival in rats. Am. J. Transplant. 2013;13:2006–2018. doi: 10.1111/ajt.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barochia A, Solomon S, Cui X, Natanson C, Eichacker PQ. Eritoran tetrasodium (E5564) treatment for sepsis: review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol. 2011;7:479–494. doi: 10.1517/17425255.2011.558190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Opal SM, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309:1154–1162. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 89.Liu M, et al. Protective effects of Toll-like receptor 4 inhibitor eritoran on renal ischemia-reperfusion injury. Transplant. Proc. 2010;42:1539–1544. doi: 10.1016/j.transproceed.2010.03.133. [DOI] [PubMed] [Google Scholar]
- 90.McDonald KA, et al. Toll-like receptor 4 (TLR4) antagonist eritoran tetrasodium attenuates liver ischemia and reperfusion injury through inhibition of high-mobility group box protein B1 (HMGB1) signaling. Mol. Med. 2015;20:639–648. doi: 10.2119/molmed.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gu J, et al. Dexmedetomidine provides renoprotection against ischemia-reperfusion injury in mice. Crit. Care. 2011;15:R153. doi: 10.1186/cc10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Conde P, et al. DC-SIGN(+) macrophages control the induction of transplantation tolerance. Immunity. 2015;42:1143–1158. doi: 10.1016/j.immuni.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baardman J, et al. A defective pentose phosphate pathway reduces inflammatory macrophage responses during hypercholesterolemia. Cell Rep. 2018;25:2044–2052 e2045. doi: 10.1016/j.celrep.2018.10.092. [DOI] [PubMed] [Google Scholar]
- 95.Krawczyk CM, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016;213:15–23. doi: 10.1084/jem.20151570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang F, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine. 2018;30:303–316. doi: 10.1016/j.ebiom.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vats D, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jha AK, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 100.Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532. doi: 10.3389/fimmu.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yu, X. H., Zhang, D. W., Zheng, X. L. & Tang, C. K. Itaconate: an emerging determinant of inflammation in activated macrophages. Immunol. Cell Biol.97, 134–141 (2018). [DOI] [PubMed]
- 102.Lampropoulou V, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24:158–166. doi: 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Byles V, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013;4:2834. doi: 10.1038/ncomms3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Everts B, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014;15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hanna RN, et al. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat. Immunol. 2011;12:778–785. doi: 10.1038/ni.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Koenis DS, et al. Nuclear receptor Nur77 limits the macrophage inflammatory response through transcriptional reprogramming of mitochondrial metabolism. Cell Rep. 2018;24:2127–2140. doi: 10.1016/j.celrep.2018.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cramer T, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sohrabi Y, Godfrey R, Findeisen HM. Altered cellular metabolism drives trained immunity. Trends Endocrinol. Metab. 2018;29:602–605. doi: 10.1016/j.tem.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 110.Netea MG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Arts RJ, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24:807–819. doi: 10.1016/j.cmet.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cheng SC, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Braza MS, et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity. 2018;49:819–828. doi: 10.1016/j.immuni.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bekkering S, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. 2018;172:135–146. doi: 10.1016/j.cell.2017.11.025. [DOI] [PubMed] [Google Scholar]