

Abstract
Human muscarinic receptor, M2 is one of the five subtypes of muscarinic receptors belonging to the family of G protein-coupled receptors. Muscarinic receptors are targets for multiple neurodegenerative diseases. The challenge has been designing subtype selective ligands against one of the five muscarinic receptors. We report high resolution structures of a thermostabilized mutant M2 receptor bound to a subtype selective antagonist AF-DX 384 and a non-selective antagonist NMS. The thermostabilizing mutation S110R in M2 was predicted using a theoretical strategy previously developed in our group. Comparison of the crystal structures and pharmacological properties of the M2 receptor shows that the Arg in the S110R mutant mimics the stabilizing role of the sodium cation, that is known to allosterically stabilize inactive state(s) of class A GPCRs. Molecular Dynamics simulations reveal that tightening of the ligand-residue contacts in M2 receptor compared to M3 receptor leads to subtype selectivity of AF-DX 384.
Keywords: G-protein-coupled receptor, muscarinic receptor, rationally thermostabilized mutant, X-ray crystallography, subtype-selective ligand
Graphical Abstract
Introduction
Muscarinic acetylcholine receptors (mAChRs) are members of the class A G-protein–coupled receptors (GPCRs) and are activated by the neurotransmitter acetylcholine 1. The mAChRs play an important role in multiple functions of the central and peripheral nervous systems. The five subtypes M1 to M5 of the mAChR family are expressed in diverse regions of the central nervous system and are implicated as major drug targets for Alzheimer’s disease, Parkinson’s disease, depression, and schizophrenia2,3. Several drugs, including drugs selective for specific mAChR subtypes, have already been developed to treat these diseases 2,4. In the past few years, the structures of human muscarinic M1–M4 receptors bound to various ligands have been determined 5–9. Although these structures are immensely useful for structure based ligand design, they are uninformative in understanding the structural basis for subtype selectivity of ligands.
Very recently, we applied a novel theoretical strategy to identify mutation positions in class A GPCRs that would lead to increased thermostability compared to the wild type. The details of this strategy are described elsewhere10. We predicted that substitution at position 3.39 (Ballesteros-Weinstein residue numbering system used for class A GPCRs)11 on transmembrane helix 3, with arginine or lysine would enhance the thermostability in all the receptors studied. The residues at positions 3.39 and 2.50 are known to be involved in binding a sodium ion which plays an important role in stabilizing the inactive state conformation, as well as in the transition to the active conformation 12,13.
In this work we have shown that the enhanced thermostability resulting from mutating position 3.39 to a cationic amino acid such as Arg, exhibits a similar role as sodium ion in stabilizing the inactive state of the M2 receptor. Using the rationally designed thermostabilized mutant S110R, we determined the structures of the M2 receptor bound to a non-selective inverse agonist NMS (N-methyl scopolamine) 14 and an M2/M4-selective antagonist AF-DX 38415,16; the latter compound has been widely used to map the distribution of M2 and M4 receptors in the brain and to study their involvement in the development and treatment of Alzheimer’s disease, dementia, schizophrenia, and mood disorders 17–21. We compared the crystal structure of the S110R mutant bound to NMS with that of the wild type and performed pharmacological analyses. The results indicate that the side chain of the arginine at 3.39 in the mutant mimics the role of the allosteric sodium ion. Furthermore, molecular dynamics (MD) simulations starting from our crystal structures, provide deep insights into the subtype selectivity of AF-DX 384 and the structural impact of the thermostabilizing mutation.
RESULTS
Structural role of the S110R mutation in thermostabilization
In this study we replaced the T4Lysozyme (T4L) used previously in M2-T4L (PDB ID: 3UON) construct, by BRIL to generate M2-BRIL and selected the S110R3.39 mutation predicted using our theoretical method, to further stabilize M2-BRIL10. In Sf9 cells, the functional expression level of the S110R mutant was 3-fold greater than the wild-type M2-BRIL (Supplementary Fig. 1a), yielding about 1 mg of purified S110R-BRIL per liter of cell culture.
The apparent melting temperatures (Tm) of the wild-type and S110R proteins bound to NMS were 60.5°C and 65.5°C, respectively (Supplementary Fig. 1b and 1c) as measured using using a tryptophan fluorescence-detection size-exclusion chromatography–based thermostability (FSEC-TS) assay 22. Both S110R-mutant and wild-type M2-BRIL were crystallized using the lipidic cubic phase method in the presence of NMS and alcuronium, a positive allosteric modulator of NMS 23 (Supplementary Fig. 2). The structures of wild-type and S110R-mutant M2-BRIL (S110R-BRIL/NMS) were determined at resolutions of 3.0 Å and 2.3 Å, respectively (Fig. 1a, Supplementary Tables 1 and 2). As shown in Fig. 1a, these two structures are highly similar, with a root-mean-square deviation (RMSD) of 0.3 Å in the coordinates of 271 Cα atoms (residues 20–214, 383–458). The geometry of TMs in M2-BRIL/NMS was also very similar to that in M3-mT4L (PDB ID: 4U16) (Supplementary Fig. 3a and 3b). The electron density was observed at the allosteric site of S110R-BRIL/NMS structure, but the model of alcuronium did not fit the electron density and was compatible with PEG300. Indeed, previous research reported the electron density of PEG300 in the allosteric site in crystal structures of M1, M3 and M4 muscarinic receptors8,9.
Figure 1.

Structures of M2-BRIL and the S110R mutant bound to NMS. (a) Superposition of M2-BRIL (cyan) and S110R mutant (gray) bound to NMS. (b) R1103.39 and D692.50 of the S110R mutant form a salt bridge interaction. R1103.39 also interacts with S4437.46. Superposition of (c) M2-BRIL or the (d) S110R mutant bound to NMS with A2A receptor bound to ZM 241385 (PDB ID: 4EIY, blue).
Futhermore, we also determined the structure of QNB bound S110R-BRIL (Supplementary Table 2). We observed subtle differences in structures of TM1, TM5 and TM6 between QNB bound M2-T4L and NMS bound S110R mutant (Supplementary Fig 3c and 3d). Since the structures of S110R mutants bound with QNB or NMS were almost identical (Supplementary Fig. 3e and 3f), this difference could be caused by the mutation and not the ligand. Details of the ligand-binding mode of these structures and structural comparisons with known tiotropium-bound muscarinic receptor subtypes are provided in Supplementary Fig. 4. The orientation of the side chain of D3.32, which is highly conserved throughout the biogenic amine GPCRs, varied depending on each ligand-bound muscarinic receptor subtype. Among them, the side chains of D3.32 in tiotropium bound M4-mT4L and NMS bound S110R mutant were most distant from the amine group of the ligand, which suggested that this could affect the ligand affinity.
In the S110R-BRIL/NMS structure, the side chain of the arginine residue at position 3.39 forms a salt bridge with the aspartate at 2.50 (D692.50), as well as a hydrogen bond with the serine at 7.46 (S4337.46) (Fig. 1b and Supplementary Fig. 5). The newly formed polar interaction network among these residues stabilizes the receptor core structure, presumably contributing to the higher-resolution diffraction of the S110R mutant crystals. D2.50 is highly conserved among class A GPCRs and is important for binding the allosteric sodium ion 12,24. Notably, superposition of the structures of the A2A receptor with the sodium ion (PDB ID: 4EIY) and S110R-BRIL/NMS reveals that the positive charge of the arginine side chain in S110R-BRIL/NMS replaces the allosteric sodium ion (Fig. 1c and 1d).
The S110R mutation increases the affinity for M2R and antagonists
We performed ligand binding assays to characterize the pharmacological properties of the wild-type and S110R-mutant receptors, using several antagonists. The affinity of the S110R mutant for QNB was unchanged, although the Bmax was increased. In the case of NMS, the affinity decreased slightly, with Kd values of 1.3 nM and 6.4 nM for the wild type and S110R mutant, respectively (Supplementary Table 3). The Bmax of NMS was almost the same in both proteins (Fig. 2a). Similar results were observed in the inverse agonist tiotropium (Supplementary Fig. 6a and Supplementary Table 4) that has a similar chemical structure as QNB and NMS (Supplementary Fig. 6e). More importantly, the M2 receptor–selective antagonist AF-DX 384, which has a lower affinity than QNB or NMS (pKi = 8.2) 25 (Supplementary Fig. 6b, Supplementary Table 4) showed an order of magnitude increase in binding affinity to the S110R mutant compared to the wild type from a Ki of 76.4 nM (wild type) to a Ki of 2.0 nM (S110R) (Fig. 2b), indicating that the S110R mutant favors AF-DX 384 over NMS, QNB and tiotropium. The competition experiment with M1 selective antagonist, pirenzepine20, with a chemical structure similar to AF-DX 384 also resulted in improved affinity for the S110R mutant (Supplementary Fig. 6c, 6f, and Supplementary Table 4).
Figure 2.

Binding assay of M2 variants with agonist and antagonists. (a) Effects of mutation at position 3.39 on equilibrium binding of the indicated radioligands. Saturation curves for binding of the inverse agonist QNB to M2-BRIL (filled circles) or the S110R mutant (filled triangles), and of the inverse agonist NMS to M2-BRIL (open squares) or the S110R mutant (open triangles). (b) Inhibition of [3H]NMS binding with M2-BRIL (filled circles) and the S110R mutant (filled square) by AF-DX 384. (c and d) Analysis of sodium ion–dependent binding of (c) NMS and (d) AF-DX 384 to M2-BRIL. (e) Inhibition of [3H]NMS binding with the M2 receptor_delta_ICL3 (open circle) and S110R mutant (open square) by the agonist iperoxo. (f) Inhibition of [3H]NMS binding with the M2-BRIL by the agonist iperoxo with (closed circle) or without (closed square) sodium ions. In a and c, error bars, s. e. m. (n = 3 separate binding assays on the same cell-membrane stock). In b, d, e and f, error bars, s. e. m. (n = 3 separate competition assays on the same cell-membrane stock).
Comparison of the crystal structures suggests that the side chain of the arginine at 3.39 in the S110R mutant mimics the role of the allosteric sodium ion. To confirm this idea, we performed a ligand-binding experiment in the presence or absence of sodium ions. We found that the binding affinity of the M2 receptor for AF-DX 384 was sodium-dependent, whereas its affinity for NMS was not. Addition of sodium ions increased the affinity of the M2 receptor to AF-DX 384, altering the Ki from 129.5 nM (no sodium ions) to 21.9 nM (150 mM sodium ions) (Fig. 2c and 2d, and Supplementary Table 3). These results suggest that binding of AF-DX 384 is cooperatively regulated by the allosteric sodium ion, or an equivalent charge, at the core of the M2 receptor, whereas binding of NMS, QNB, or tiotropium is not. On the other hand, the affinity of the agonist iperoxo in S110R mutant reduced almost 1000-fold relative to the wild type (Fig. 2e and Supplementary Table 3) while this difference was not observed in the wild type M2 in the presence of sodium ions (Fig. 2f and Supplementary Table 3). Similar results were obtained with the partial agonist Xanomeline 26(Supplementary Fig. 6d, 6g, and Supplementary Table 4). Taken together these results indicate that the permanent presence of the positive charge at the site of the allosteric sodium ion in the S110R mutant can either stabilize the inactive receptor or hamper the transition to the active conformation 10,24.
Conformational changes of the ligand-binding site
We attempted to crystallize both wild-type and S110R-mutant M2-BRIL in the presence of AF-DX 384, but only succeeded with the S110R mutant. The crystal structure of AF-DX 384 bound to mutant S110R M2-BRIL (S110R/AF-DX 384) was determined at a resolution of 2.95 Å (Supplementary Table 2). AF-DX 384 is a larger molecule than QNB and NMS and while the major parts of these ligands overlap in their crystal structures in M2 receptor, the 2-(piperidin-1-yl)ethyl group and the N,N-dipropylaminomethyl group of AF-DX 384 protrudes towards TM3 more than QNB and NMS (Fig. 3a and 3b). While most of the interacting residues are shared between AF-DX 384 and QNB, the N,N-dipropyl aminomethyl group of AF-DX 384 forms additional contacts with W993.28 and L1003.29 in TM3 (Fig. 3b, and Supplementary Table 5). The most striking feature of the binding mode of AF-DX 384 in M2 is the movement of Y1043.33, Y4267.39, and Y4036.51, which constitute the “tyrosine lid” (Fig. 3b and 3c). In the structures of muscarinic receptor bound to QNB and iperoxo, the orthosteric ligand is almost completely occluded from the solvent, with the tyrosine lid located directly above the ligand 5,7. In the structure of the M2 receptor bound with NMS, the orthosteric site is blocked by the tyrosine lid as seen in the extracellular view shown in Fig. 3c left. By contrast, in the AF-DX 384 bound S110R structure, two of these tyrosine residues, Y1043.33 and Y4267.39, open outward (Fig. 3b and 3c right) thus allowing the two propyl groups of AF-DX 384 extend from the orthosteric pocket towards the extracellular surface (Fig. 3a, 3b, and 3c right). One of them points toward TM3 and interacts with W993.28 and L1003.29, whereas the other points toward the center of the tyrosine lid, apparently triggering the movement of Y1043.33 and Y4267.39 (Fig. 3b). As a result, the tyrosine lid opens up, and the orthosteric site becomes more accessible from the extracellular space (Fig. 3b and 3c right).
Figure 3.

Conformational changes of the tyrosine lid. (a, left) The amino acid residues that interact with the ligand are shown by the number by enclosing the residue numbers with ellipses of the same color as the background of each part of AF-DX 384. Labeled AF-DX 384 substructures: the 5,11-dihydro-6H-pyrido[2,3-b]-[1,4]benzodiazepin-6-one group (red), N-ethylamide (or ethylaminocarbonyl) group (blue), the 2-(piperidin-1-yl)ethyl group (yellow), and N,N-dipropylaminomethyl group (green). (a, right) Supeposition of QNB (green), NMS (gray), and AF-DX 384 (magenta) from structures of M2-T4L bound with QNB (PDB ID:3UON), S110R-BRIL bound with NMS, and S110R-BRIL bound with AF-DX 384, respectively.
(b) Conformational changes within the ligand-binding pocket, shown from the extracellular side. Among the amino acid residues constituting the tyrosine lid, Y1043.33 and Y4267.39 have moved significantly in S110R bound to AF-DX 384. Superposition of the S110R mutant bound to NMS (gray) and AF-DX 384 (magenta) onto M2-T4L bound to QNB (green) (PDB ID: 3UON). Surface and cartoon representation (c) of the S110R mutant structure bound to NMS (c left) and AF-DX 384 (c right), respectively. The side chains of the residues of the tyrosine lid are colored in orange.
Binding mode of M2 receptor to AF-DX 384
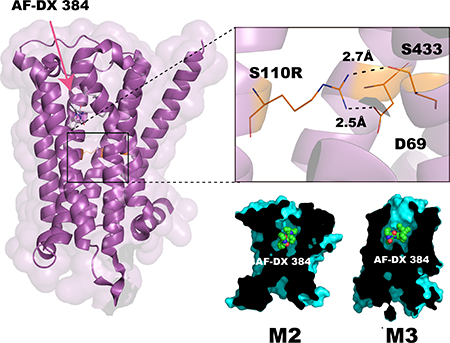
The structure of S110R/AF-DX 384 complex differs slightly from the NMS-bound structure by an RMSD of 0.7 Å for the 275 Cα atoms (residues 17–214, 380–456) superimposed in Figure 4a. The extracellular tip of TM5 in the AF-DX 384–bound structure is pushed out by about 3.5 Å relative to its position in the NMS-bound structure, due to the bulky 5,11-dihydro-6H-pyrido[2,3-b]-[1,4]benzodiazepin-6-one group of the antagonist (Fig. 4a). D1033.32, which is highly conserved among the biogenic amine GPCRs, forms ionic bonds with two tertiary amine moieties of AF-DX 384 (Fig. 3a, left and 4b). S1073.36 and N4046.52 form hydrogen bonds with the nitrogen atom of N-ethylamide and the oxygen and nitrogen atoms of the 5,11-dihydro-6H-pyrido[2,3-b]-[1,4]benzodiazepin-6-one group, respectively. Both Y1043.33 and Y4036.51 form hydrogen bonds with the oxygen atom of N-ethylamide (Fig. 3a, left and 4c). The remaining associations between the M2 receptor and AF-DX 384 are mediated by van der Waals interactions. On the other hand, the arginine residue at position 3.39 does not directly contribute to the binding affinity enhancement of AF-DX 384, because this residue does not directly interact with this ligand (Fig. 4d). AF-DX 384 is an M2/M4-selective antagonist, i.e., it has a higher affinity for M2/M4 than for M1/M3 (Ki values of 15.8 nM and 154.9 nM for M2 and M3, respectively) (Supplementary Fig. 6b and Supplementary Table 4) 15,16. Besides the 12 residues of the M2 receptor that interact with NMS, eight more residues form additional contacts with AF-DX 384 that are expected to contribute to the antagonist’s selectivity. However, these residues are completely conserved among muscarinic receptors (Supplementary Table 5). It is possible that selectivity is a consequence of not only the static interaction seen in the crystal structure, but also the protein dynamics. Hence, we investigated the selectivity mechanism using molecular dynamics simulations.
Figure 4.

Binding mode of the M2 receptor to AF-DX 384. (a) Superposition of the S110R mutant bound to NMS (gray) and AF-DX 384 (magenta). The extracellular end of TM5 in the AF-DX 384–bound structure is 3.5 Å away from its position in the NMS-bound structure. The side chains of the residues in the AF-DX 384–bound structure are colored in orange. (b) In the AF-DX 384–bound structure, D1033.32 interacts with two nitrogen atoms of AF-DX 384. (c) Both Y1043.33 and Y4036.51 form hydrogen bonds with the oxygen atom of N-ethylamide. (d) The distance between AF-DX 384 and the arginine residue at the position 3.39.
Molecular dynamics simulations of the S110R mutant
Crystal structures and pharmacological analysis of the M2-BRIL variant revealed that the mutation at position 3.39 stabilized the inactive conformation of the receptor and increased the affinity for the subtype-selective antagonist AF-DX 384. Despite extensive efforts to crystallize the wild-type receptor/AF-DX 384, we never obtained crystals; consequently, it was not clear from the S110R/AF-DX 384 crystal structure alone how this mutation affects the affinity and also confers subtype selectivity over M3 receptor. It is also possible that the affinity enhancement in S110R was a consequence of protein dynamics that could not be observed in the crystal structures27. Hence, we performed molecular dynamics (MD) simulations to investigate the molecular mechanism underlying the thermostability of S110R mutant and also the subtype selectivity of AF-DX 384 in M2 and M3 receptors.
First, to investigate the mechanism underlying the thermostability of S110R compared to the wild type, we compared the flexibility of three receptor structures from the MD simulations: the S110R mutant, and the wild type M2 receptor with and without sodium ion (In the following, they are written as S110R, M2WT+Na+, M2WT-Na+, respectively). The thermal B-factors calculated from the root mean square fluctuation (RMSF) from an average structure for each residue, obtained from MD simulations is shown as a heat map in Figure 5a. This figure shows that the S110R was the least flexible in the TM regions followed by M2WT+Na+ and M2WT-Na+. Supplementary Figure 7 shows the relative flexibility of residues in each TM helix for the three systems. This plot shows that the residues in helices TM1, TM6, and TM7 were highly flexible in the wild type, whereas fluctuations of these TM regions were remarkably suppressed in the S110R mutant. The mobility of the ligand AF-DX 384 in S110R and M2WT+Na+ is reduced compared to M2WT-Na-+(Supplementary Fig. 8a). The relative flexibility of the residues in the extracellular loop 2 (ECL2) in S110R is lower than the corresponding region in M2WT+Na+ (Supplementary Fig. 8b). We have used the changes in inter-residue distances in the intracellular region between TM3–TM6 and TM3–TM7 to assess the extent of conformational changes sampled during the MD simulations in the M2 receptor. It should be noted that these distances are not used for measuring the receptor activation since we are simulating the conformational ensemble just around the inactive states of the M2 receptor in this study. MD simulations of S110R revealed a decrease in the TM3–TM6 distance compared to the starting crystal structure of the mutant, indicating tightening of these two helices relative to M2WT+Na+ or M2WT-Na+ (Fig. 5b). It is also notable that the conformational sampling was significantly tighter in S110R than in the wild type, as demonstrated by the sharper peak. S110R engages in stronger interhelical interactions than M2WT+Na+ or M2WT-Na+: specifically, TM3 in S110R engages in more polar interactions with residues in TM2, TM6, and TM7. The increase in interhelical packing interactions comes mainly from intracellular regions of the TM domain, and is likely responsible for the lower flexibility of S110R (Supplementary Fig. 9). Such tightening of the interhelical packing in the TM bundle in thermostable mutants has been observed previously in other GPCRs 28.
Figure 5.

Analysis of structural stabilization of the S110R M2 mutant compared to the wild type M2 using MD simulations. (a) The residue based thermal B-factor calculated from the MD simulations from the RMSF of the wild-type M2 receptor with and without sodium ion, and the S110R mutant are shown as a heat map on the representative conformation extracted from the most populated conformation cluster. The loop regions and the N-terminus of the receptor on TM1 shown in grey are omitted in the representation for clarity (b) The conformational ensemble sampled during the MD simulations of S110R mutant compared to the wild type with and without Na+ ion for M2 receptor. These two inter-residue distances between TM3-TM6 and TM3-TM7 are used as measures to assess the extent of the conformational changes in the three systems. The crystal structures of inactive and fully active states of M2 are indicated as 3UON for and 4MQS in the figure just for reference. (c) Changes in the average ligand residue distances during the MD simulations (left) from M2 wild-type without sodium ions to M2 wild-type with sodium ions and to S110R M2 mutant, and (right) from M3 wild-type without sodium ions to M3 wild-type with sodium ions.
We have also used the MD simulations to rationalize the molecular mechanism underlying the increased binding affinity of AF-DX 384 to S110R compared to the wild type. We calculated the changes in the contact distances between ligand and residues in the ligand binding site for those contacts that stay sustained for over 40% of the simulation time. As shown in Figure 5c left, the ligand receptor residue contacts contract in going from M2WT-Na+ to M2WT+Na+ and to S110R. It is seen that the average ligand residue distances shrink during the MD simulations in S110R and in M2WT+Na+ compared to M2WT-Na+. The residues Y802.61, W993.28, L1003.29, and Y1043.33 from TM2 and TM3 make close interactions with AF-DX 384 in the S110R mutant structure, but these contacts are above 4.5Å in M2WT-Na+ (Fig. 5c left). These are the helices among which the S110R mutation tightens the inter-helical interactions in the intracellular region (Fig. 1b–d). Binding free energy calculations using the Bennett acceptance ratio29,30, a free-energy perturbation method, showed a 5.9 kcal/mol increase in the binding free energy of AF-DX 384 in S110R relative to M2WT-Na+. Therefore, we concluded that the binding affinity was elevated due to an increase in the number of amino acids interacting with the ligand and by tightening of ligand residue distances in the ligand binding site, triggered by tight TM bundle formation. As observed in the M2 receptor, the number of amino acids interacting with AF-DX 384 in the presence of sodium ion also increased at the M3 receptor (Fig. 5c right).
Finally, we performed MD simulations to investigate the molecular mechanism of the subtype selectivity of AF-DX 384 between the M2 and M3 receptors in the presence of sodium ion. Because the amino acid residues in the orthosteric sites of M2 and M3 receptor are highly conserved, the residues responsible for subtype selectivity cannot be identified based on structural information alone. Focusing on the ligand-residue distances in the presence of sodium ion, most of the sustained ligand contacts with residues in the TM2 and TM3 tighten in M2WT+Na+ compared to M3 wild type with sodium ion (Fig. 6a). This could lead to contraction of the binding site of AF-DX 384 in M2 receptor compared to M3 receptor as shown in Fig. 6b.
Figure 6.

Analysis of subtype selectivity of AF-DX 384 using MD simulation. (a) Changes in the average ligand residue distances during the MD simulations from M3 wild-type with sodium ions to M2 wild-type with sodium ions. (b) Cross-sectional views of AF-DX 384 binding site in M2 and M3 structures in the presence of sodium ion.
DISCUSSION
Stabilizing a membrane protein for structural studies is a time- and resource-intensive process, especially for proteins with high plasticity, such as GPCRs. In this study, we demonstrated that a mutation predicted using our novel method could indeed stabilize the M2 muscarinic acetylcholine receptor. Using this mutant, we were able to determine the crystal structure of M2 receptor bound to subtype-selective and non-selective antagonists at resolutions higher than could be achieved with the wild-type protein. In addition to thermostabilization, the S110R mutation significantly increased affinity for the subtype-selective antagonist AF-DX 384 15,16, enabling its co-crystallization, whereas the wild type could not be crystallized with this ligand despite extensive efforts. The structural information also revealed that the arginine residue of the S110R mutant did not directly interact with AF-DX 384. However, the MD simulations revealed that the S110R mutation improved affinity with AF-DX 384 by stabilizing the conformation of the whole receptor through tighter inter-helical interactions. The 3.39 position is important for binding of an allosteric sodium ion, which is thought to play a role in stabilizing the inactive state of some class A GPCRs 12. In the high-resolution structure of the adenosine A2A receptor, the conserved residues D2.50 and S3.39, interact with the allosteric sodium ion 31. In the structure of the S110R mutant, the positive charge of the side chain of the arginine at 3.39 mimics the allosteric sodium ion and the resultant ionic interaction among TM2, TM3, and TM7 stabilizes the receptor core. Our FSEC-TS data confirmed that the overall structural stability of the S110R mutant was higher than that of the wild type. Recent studies have elucidated the allosteric role of sodium ion binding site in GPCR activation.13,32 These studies showed that replacing the aspartic acid residue at the position 2.50 in A2A adenosine receptor with uncharged asparagine residue showed reduced G-protein signaling. The S110R M2 mutant on the other hand retains the agonist binding affinity and stabilizes the antagonist binding state.
The thermostabilized mutant, S110R M2 had a significantly lower affinity for the agonist iperoxo than the wild type, whereas the affinity was unchanged or slightly lower for the inverse agonists (QNB, tiotropium and NMS) (Fig. 2a, e, f, Supplementary Fig. 6a, Supplementary Table 3 and 4). This result was consistent with the finding that an elevated sodium concentration has a negative allosteric effect on the binding of agonists to the adenosine A2A receptor, but little or no effect on the binding of antagonists 31. The contrasting effects of the S110R mutation on the binding of antagonists, AF-DX 384 15,16 and pirenzepine, versus the inverse agonists 14, suggest that antagonists bind the M2 receptor in an allosteric sodium ion– dependent manner, whereas inverse agonists do not. In contrast to these ligands, AF-DX 384 had a remarkably higher affinity for the S110R mutant, and this effect was recapitulated using a ligand-binding assay in the presence of sodium ions. This result is consistent with the observation that the side chain of the arginine at 3.39 in the S110R mutant takes the position of the allosteric sodium ion (Fig. 1b and d, Supplementary Table 3). MD simulations confirmed that the structure of the S110R mutant was stabilized in the inactive form in comparison with the wild type (Fig. 5b). The flexibility of the ligand-binding pocket also reduced, and the ligand-residue contacts in the binding site of S110R mutant were tighter to that of the wild type (Fig. 5c). These results suggest that the number of amino acids interacting with AF-DX 384 is increased in the mutant, leading to improved affinity for the ligand.
Due to the enhanced affinity and higher thermostability of the S110R mutant, we were able for the first time to crystallize a muscarinic receptor bound to a subtype-selective antagonist. Although the ligand-interacting residues are highly conserved among muscarinic GPCRs (Supplementary Table 5), the tyrosine lid has a distinct configuration when bound to AF-DX 384 in comparison with the NMS- or QNB-bound states, creating an open vestibule to the extracellular space (Fig. 3c). This open tyrosine lid may hamper the tight binding of AF-DX 384 and could have prevented crystallization of the receptor with this ligand in the absence of the stabilizing S110R mutation. In contrast to the conserved orthosteric binding site, the allosteric binding site of each muscarinic receptor is unique 33. Using MD simulations on the wild type M2 and M3 receptors with and without sodium ion, we have shown that the presence of sodium ion tightens the ligand-receptor contact distances with residues in the ligand binding site in both M2 and M3 receptors. Replacement of sodium ion with the S110R mutant in M2, showed further tightening of the ligand-receptor contact distances that explains the increased binding affinity of AF-DX 384 in M2 receptor. We observed that the ligand-receptor contacts are tighter with the residues Y2.61, L3.29 Y3.33, Y6.51 in TM2, TM3 and TM6 in M2 receptor with sodium ion than in the M3 receptor with sodium ion. The tightening of these residue contacts and the differential interactions of the ligand with ECL2 residues are possible causes of selectivity of AF-DX 384 to M2 over M3 receptor.
In this study, we experimentally validated the high expression and thermostability of the S110R mutant of the human muscarinic M2 acetylcholine receptor, a prototypical GPCR. Because the serine residue at position 3.39 is highly conserved in a large number of class A GPCRs, the same mutation is expected to stabilize and facilitate crystallization of other GPCRs. In our hands, mutations at the same position improve thermostability in two other class A GPCRs, adenosine A2a receptor and prostaglandin E receptor EP4 10. As demonstrated by the results presented here, receptors that bind to pharmacologically important ligands with modest affinity can be crystallized as complexes by taking advantage of a stabilizing mutation. Our approach represents a general stabilization method that will facilitate the structural study of GPCRs.
METHODS
Methods, including statement availability and any associated accession codes and references, are available in the online methods.
ONLINE METHODS
Expression and purification.
The M2-BRIL fusion construct was generated using synthetic DNA (GeneScript). The S110R mutant was constructed by PCR-mediated mutagenesis. C-terminally His-tagged M2-BRIL fusion protein was cloned into the pFastBac1 (Invitrogen) baculovirus expression vector with the hemagglutinin (HA) signal sequence followed by an N-terminal FLAG tag. We inserted two 3C protease digestion sites: between the M2 receptor and the His tag sequence, and between the M2 receptor and the HA signal sequence. BRIL was substituted into residues 221–376 of the M2 receptor’s ICL3. The constructs were expressed in Sf9 insect cells using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Cells were infected at a density of 3–4 × 106 cells/mL and grown for 48 h at 27°C. Cells were harvested by centrifugation and stored at −80°C.
Sf9 cells were lysed by osmotic shock in the presence of 10 μM atropine (Sigma-Aldrich). The subsequent purification steps were the same as those of previously reported orexin 2 receptor 34. Appropriate ligand was added at 10 μM from the step using FLAG antibody column.
Crystallization.
Purified M2-BRIL or the S110R mutant was mixed with 1.5 parts (w/w) of monoolein with 10% (w/w) cholesterol (Sigma) using the syringe reconstitution method 35. The lipidic cubic phase mix was dispensed in 30-nL droplets onto glass plates and overlaid with 600 nL of precipitant solution using a Gryphon (Art Robbins Instruments) or NT8 (Formulatrix). Crystallization experiments were carried out in 96-well glass sandwich plates (Molecular Dimensions), and the crystallization plates were incubated at 20°C. Crystals of M2-BRIL or the S110R mutant bound to NMS or QNB appeared after 24 h and matured to full size in 4–5 days in precipitant solution consisting of 50 mM MES-NaOH pH 6.2–7.0, 26–32% PEG300, 300–500 mM ammonium fluoride, 1% 1,2,3-heptanetriol, 0.5 mM NMS or QNB, and 5% DMSO. Crystals of the S110R mutant bound to AF-DX 384 appeared after 24 h and matured to full size in 1 week in precipitant solution consisting of 50 mM MES-NaOH pH 6.2–7.0, 18% PEG300, 100 mM magnesium acetate, 1% 1,2,3-heptanetriol, 0.5 mM AF-DX 384, and 5% DMSO.
Structure determination and refinement.
Diffraction data were collected at the SPring-8 beamline BL32XU in Hyogo, Japan. The M2-BRIL/NMS dataset was produced by merging several datasets manually collected using the MX225HS detector (Rayonix), each spanning 20–420°. The datasets of S110R-BRIL/NMS, mercury-bound S110R-BRIL/NMS, S110R-BRIL/AF-DX 384, and S110R-BRIL/QNB were collected using the automated system ZOO (manuscript in preparation). The small wedge datasets (3– 5° each) were collected from automatically recognized crystal positions based on raster scan results using SHIKA 36. Exposure conditions were automatically adjusted using KUMA 37 such that each crystal absorbed a dose of 7–12 MGy. For S110R-BRIL/QNB data, the EIGER X 9M detector (DECTRIS) was used. All datasets were collected at a wavelength of 1.0 Å under a cryostream operating at 100 K with beam sizes from 10 × 8 to 20 × 8 μm2. All collected datasets were processed automatically using KAMO (https://github.com/keitaroyam/yamtbx/blob/master/doc/kamo-en.md)38. The datasets were indexed and integrated using XDS 39. The datasets, indexed with consistent unit cell parameters, were subjected to hierarchical cluster analysis based on correlation coefficients of observed intensities (M2-BRIL/NMS) or unit cell similarity using BLEND 40. The clusters of datasets with an expected completeness and multiplicity of more than 90% and 2, respectively, were separately merged using XSCALE 39, with outlier rejections implemented in KAMO. The structures of M2-BRIL and the S110R mutant bound to several kinds of antagonists were solved by molecular replacement using Phaser 41 in Phenix 42. First, the structure of M2-BRIL bound to NMS was determined using the M2-T4L structure without T4L and the structure of BRIL in A2A-BRIL (PDB ID: 4EIY) as search models. The structures of the S110R mutant bound to NMS, QNB, or AF-DX 384 were determined using the structure of M2-BRIL bound to NMS as a search model. The resultant solution was improved by manual iterative building in Coot 43, followed by refinement with phenix.refine in the Phenix program suite 42. Initial coordinates and restraint parameters for NMS, QNB, and AF-DX 384 were prepared using the Grade and PRODRG web servers 44,45. The electron density of BRIL of the S110R/bound with NMS was very weak. To obtain experimental phase information, SAD phasing was performed using the mercury-bound S110R-BRIL/NMS dataset. Heavy-atom search, phasing, and phase improvement were carried out using the SHELX C/D/E programs 46. However, the experimental phase by mercury also did not help improve the electron density around BRIL. In the Ramachandran plots of M2-BRIL bound to NMS and the S110R mutant bound to NMS, QNB, or AF-DX 384, the residues were in favored regions in 97.8%, 98.9%, 98.9%, and 97.6% of cases, respectively, and the rest were in allowed regions.
Ligand-binding assay.
Ligand-binding assays were performed using a radiolabeled antagonist, [3H]-NMS (PerkinElmer). M2 receptor variants (M2-BRIL and S110R-BRIL) were expressed in Sf9. M2 receptor variants expressed on the membrane was prepared as described above. For single-point binding assays, 40 nM [3H]-NMS was incubated for 1 h on ice in 100 μL of buffer (20 mM HEPES-NaOH pH 7.4, 150 mM NaCl, 5 mM MgCl2) containing 2.5 μg of membrane proteins. M2R_delta_ICL3 and M2R(S110R)_delta_ICL3 where most of ICL3 (233–359) were deleted were expressed in Sf9 cells. Membrane homogenates were prepared and re-suspended in a binding buffer (20 mM HEPES-NaOH [pH 7.5], 100 mM NaCl, 5 mM MgCl2, 0.1% BSA), and then incubated for 2.5 hours at room temperature (24 C°) in serial dilutions of iperoxo in the presence of a fixed concentration of [3H]-NMS. To separate bound and free radiolabeled antagonist, incubations were terminated by rapid vacuum filtration through Whatman GF/B filters pre-soaked in 0.3% polyethylenimine. The retained radioactivity was measured on an LCS-5100 liquid scintillation counter (ALOKA) in Clear-sol I scintillation liquid (Nacalai Tesque). For binding assays using full length M2 and M3 receptor expressed in HEK293, measurements was carried out by the same method as the above measurement using Sf9 membrane. For sodium ion–dependent binding assays, 20 mM potassium phosphate (pH 7.0) with or without 150 mM NaCl was used. Radioligand binding assays were carried out with membranes prepared from Sf9 cells as described34. Membrane homogenates were prepared and re-suspended in binding buffer (20 mM HEPES-NaOH [pH 7.5], 100 mM NaCl, 5 mM MgCl2, 0.1% BSA), and then incubated in serial dilutions of iperoxo in the presence of a fixed concentration of [3H]-NMS. Reactions were stopped by rapid filtration through GF/C filters. Radioactivity on the filters was measured by liquid scintillation counting. Data were analyzed using the Prism 6.0 software (GraphPad Software).
Fluorescence-detection size-exclusion chromatography–based thermostability (FSEC-TS) assay.
One microgram of M2-BRIL or the S110R mutant was incubated in 200 μL of buffer (20 mM HEPES-NaOH pH 7.5, 0.1 M NaCl, 0.01% [w/v] L-MNG, and 0.001% CHS [w/v]) at the indicated temperature using a thermostatic bath. The samples were then centrifuged at 123,000 ɡ for 30 min. Fifty microliters of supernatant was loaded onto an SEC column (Superdex 200 5/150 increase, GE Healthcare) at 4°C at a flow rate of 0.45 mL/min. Tryptophan fluorescence (Em 280 nm/ Ex 325 nm) was monitored.
M2 and M3 receptor structure preparation.
Molecular dynamics (MD) simulations were performed on four systems: (1) M2 receptor with the S110R mutation bound to antagonist AF-DX 384, (2) wild-type M2 receptor with AF-DX 384, (3) wild-type M2 receptor with sodium ion and AF-DX 384, and (4) wild-type M3 receptor with AF-DX 384. Simulations were conducted using the GROMACS MD simulation 47 package with the GROMOS force field 48. The starting structures for all four simulations were generated from their respective crystal structures. The S110R-M2 receptor crystal structure bound to AF-DX 384, solved in this study, was used for S110R-mutant simulations. The starting structure for the wild-type M2 receptor was generated from the S110R-M2 mutant crystal structure by mutating R110 back to Ser using Maestro9 49. For simulations of wild-type M2 and M3 containing Na+ ion in the sodium binding pocket, the starting structure was generated by superimposing the wild-type M2 structure onto the A2AR crystal structure (PDB ID: 4EIY), in which the Na+ ion is resolved 31, using Visual Molecular Dynamics (VMD). The starting conformation of wild-type M3 was the crystal structure of the M3 acetylcholine receptor (pdb ID: 4DAJ) 6 and the sodium ion was transferred using a similar procedure described for M2 above. In all MD simulations, the BRIL present in the crystallization constructs was removed, and the resultant amino and carboxy termini were capped with acetyl and N-methyl groups, respectively, at the end of TM5 and the start of TM6.
MD simulation details.
All MD simulations of M2 and M3 muscarinic receptors were embedded in a hydrated palmitoyl–oleoyl– phosphatidyl–choline (POPC) lipid bilayer with periodic boundary conditions and SPC water molecules 50 using the GROMACS v5.1 package. The inflateGRO module in GROMACS was used for packing of POPC molecules around the receptor. The SETTLER 51 and LINCS 29 algorithms were used for bonds and angles for water and all other bonds, allowing a time step of 2 fs. A cut-off distance of 12 Å for non-bond interactions was introduced, and the PME (Particle Mesh Ewald) method 30,52 was used for long-range van der Waals interactions. Snapshot coordinates were saved every 20 ps.
All four systems (M2-WT, M2-WT-Na+, M2-S110R, and M3-WT, M3-WT-Na+) were first equilibrated using 500 ns of constant volume and temperature (NVT) ensemble MD simulations at 310 K. To pack the lipid bilayer around the receptor, the protein and ligand were restrained at this stage using a harmonic restraining force with a force constant of 1,000 kJ/mol/nm2. Then, the system was equilibrated using a constant pressure and temperature (NPT) ensemble by gradually decreasing the harmonic position restraint force constants from 5 to 1 kcal/mol/Å2, applied to all heavy atoms of the protein and ligand AF-DX 384, over 5 ns. In the final NPT equilibration run, all positional restraints were released and the simulation was run for 10 ns. Production simulations were initiated from the final snapshot of the NPT equilibration run. After equilibration to the expected temperature and pressure, we performed ten different production runs with different initial velocities; each run was 200 ns. We performed MD simulations on all four systems, each 2 μs long. The M3 wild type receptor with sodium ion was simulated for a total 1 μs with 5 production runs 200ns each.
Methods used for the analysis of MD simulation trajectories.
For trajectory analysis, we analyzed all 2 μs trajectories from MD simulations using GROMACS tools. VMD and Pymol programs were used for visualization of trajectories and VMD-based Tcl scripts for conformational investigation of MD trajectories. The interhelical hydrogen bond contacts were calculated using the gmx hbond utility in GROMACS, with a distance cutoff of 3.5 Å and angle cutoff of 30°. The number of ligand–receptor hydrogen bonds and interhelical hydrogen bonds in the receptor were calculated based on occupancy of these hydrogen bonds for more than 50% of occupancy in the 2 μs trajectories using a Tcl script called contactFreq.
The ligand-binding free energy (Δ) simulations were carried out using the Bennett Acceptance Ratio (BAR) 52,53 method in GROMACS v5.1. BAR combines the information normally used for forward and reverse free energy perturbations, and can be expressed a function of a coupling parameter, λ, which indicates the level of change that has taken place between two states (bound and unbound), as well as the extent to which the Hamiltonian has been perturbed and the system has been transformed. Simulations conducted at different values of λ allow us to plot a ∂H/∂λ curve, from which Δ is derived. Transformations from ligand-bound (λ = 0) to ligand-free (λ =1) were performed in equidistant λ spacing of 0.05 (Δλ = 0.05) from 0 to 1 for decoupling of Coulombic and van der Waals interactions.
For the calculation of root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF), RMSD in coordinates and RMSF for every residue were calculated using the g_rms and g_rmsf modules of GROMACS. These quantities allowed us to compare the flexibility of the wild type with and without Na+ bound to the S110R-mutated M2 receptor. We performed clustering analysis of the ensemble based on RMSD (Root-Mean-Square-Deviation) in the coordinates of main chain atoms of transmembrane helices. We used the gmx cluster module in GROMACS. We used 1.5Å of RMSD cut-off on MD trajectories and snapshots were taken every 20 ps. The representative structure is the snapshot with the smallest RMSD to the center of the most populated conformational cluster.
Data availabvility.
The atomic coordinates and structure factors for the reported crystal structures have been deposited in the Protein Data Bank under accession codes 5ZK8 (M2-BRIL/NMS), 5ZKC (S110R-BRIL/NMS), 5ZKB (S110R-BRIL/AF-DX 384), 5ZK3 (S110R-BRIL/QNB), 5YC8(S110R-BRIL/NMS:Hg). Raw diffraction images have been also deposited in Zenodo data repository (https://doi.org/10.5281/zenodo.1172266 for S110R-BRIL/NMS:Hg, https://doi.org/10.5281/zenodo.1094808 for others)
Supplementary Material
Acknowledgments
We acknowledge support from the Research Acceleration Program of the JST (S.I.); the Platform Project for Supporting Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT); the JSPS-NSF International Collaboration in Chemistry (ICC) (T.K. and B.K.); the Takeda Science Foundation (T.K. [Kyoto University] and R.S.); the Japan Agency for Medical Research and Development (AMED) (T.K. and T.M.); JSPS KAKENHI (Grant No. 15K08268 to R.S.; 15H06862 to K.Y.); and the ImPACT Program of the Council for Science, Technology and Innovation (Cabinet Office, Government of Japan) (T.M.). The MD simulations performed by S.L. and N.V. were supported by NIH R01-GM097261 (N.V.). We thank the beamline staff at SPring-8 for data collection and processing, and Dr. Takaaki Sumiyoshi for providing the information about the ligands. The X-ray crystallography data collection was performed at SPring-8 (Proposal No. 2013A1379, 2013B1092, 2013B1184, 2014A1301, 2014B1273, 2014B1355, 2015A1044, 2015A1080, 2015B2044, and 2015B2080). DNA sequencing analysis was performed at the Medical Research Support Center, Graduate School of Medicine, Kyoto University. Tomoya Nakagita made a diagram of the structure formula of ligands.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Fredriksson R, Lagerström MC, Lundin L-G & Schiöth HB The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol 63, 1256–72 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Wess J, Eglen RM & Gautam D Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat. Rev Drug Discov 6, 721–733 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Kruse AC, Hu J, Kobilka BK & Wess J Muscarinic acetylcholine receptor X-ray structures: Potential implications for drug development. Curr. Opin. Pharmacol 16, 24–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caulfield MP & Birdsall NJM International union of pharmacology. XVII. classification of muscarinic acetylcholine receptors. Pharmacol. Rev 50, 279–290 (1998). [PubMed] [Google Scholar]
- 5.Haga K et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482, 547–551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kruse AC et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kruse AC et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thal DM et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 531, 335–340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorsen TSS, Matt R, Weis WII & Kobilka BKK Modified T4 Lysozyme Fusion Proteins Facilitate G Protein-Coupled Receptor Crystallogenesis. Structure 22, 1657–1664 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasuda S et al. Hot-Spot Residues to be Mutated Common in G Protein-Coupled Receptors of Class A: Identification of Thermostabilizing Mutations Followed by Determination of Three-Dimensional Structures for Two Example Receptors. J. Phys. Chem. B 121, 6341–6350 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ballesteros Juan A., H. W. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 (1995). [Google Scholar]
- 12.Katritch V et al. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci 39, 233–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White KL et al. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 1–11 (2018). doi: 10.1016/j.str.2017.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Ligt RA, Kourounakis AP & IJzerman AP Inverse agonism at G protein-coupled receptors: (patho)physiological relevance and implications for drug discovery. Br. J. Pharmacol 130, 1–12 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller JH, and M VAG M. Binding of AF-DX 384 to Cloned and native muscarinic receptors. J. Pharmacol. Exp. Ther 259, 601–607 (1991). [PubMed] [Google Scholar]
- 16.Niederursel B, Tränkle C, Andresen I, Lambrecht G & Mohr K M2 Receptor Binding of the Selective Antagonist AF-DX 384: Possible Involvement of the Common Allosteric Site. Mol. Pharmacol 53, 304–312 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Zavitsanou K, Katsifis A, Yu Y, Feng X & Huang XF M2/M4 muscarinic receptor binding in the anterior cingulate cortex in schizophrenia and mood disorders. Brain Res. Bull 65, 397–403 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Teaktong T et al. Muscarinic M2 and M4 receptors in anterior cingulate cortex: Relation to neuropsychiatric symptoms in dementia with Lewy bodies. Behav. Brain Res 161, 299–305 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Kitaichi K & Day JC Short communication A novel muscarinic M 4 receptor antagonist provides further evidence of an autoreceptor role for the muscarinic M 2 receptor sub-type. Eur. J. Pharmacol 383, 53–56 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Piggott M et al. Comparati v e distribution of binding of the muscarinic receptor ligands pirenzepine, AF-DX 384, ( R, R ) -I-QNB and ( R, S ) -I-QNB to human brain. J. Chem. Neuroanat 24, 211–223 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Gibbons AS, Scarr E, Mclean C, Sundram S & Dean B Decreased muscarinic receptor binding in the frontal cortex of bipolar disorder and major depressive disorder subjects. J. Affect. Disord 116, 184–191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hattori M, Hibbs RE & Gouaux E A fluorescence-detection size-exclusion chromatography-based thermostability assay for membrane protein precrystallization screening. Structure 20, 1293–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gregory KJ, Sexton PM & Christopoulos A Allosteric modulation of muscarinic acetylcholine receptors. Curr. Neuropharmacol 5, 157–167 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vickery ON et al. Intracellular Transfer of Na+in an Active-State G-Protein-Coupled Receptor. Structure 26, 171–180.e2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heitz F et al. Site-directed mutagenesis of the putative human muscarinic M2 receptor binding site. Eur. J. Pharmacol 380, 183–195 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Jakub1 J & El-fakahany EE Differences in Kinetics of Xanomeline Binding and Selectivity of Activation of G Proteins at M 1 and M 2 Muscarinic Acetylcholine Receptors. 70, 656–666 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Bhattacharya S, Lee S, Grisshammer R, Tate CG & Vaidehi N Rapid Computational Prediction of Thermostabilizing Mutations for G Protein-Coupled Receptors. (2014). doi: 10.1021/ct500616v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaidehi N, Grisshammer R & Tate CG How Can Mutations Thermostabilize G-Protein-Coupled Receptors? Trends Pharmacol. Sci 37, 37–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hess B, Bekker H, Berendsen HJC & Fraaije JGEM LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 18, 1463–1472 (1997). [Google Scholar]
- 30.Darden T, York D & Pedersen L Particle mesh Ewald: An N□log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089 (1993). [Google Scholar]
- 31.Liu W et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eddy MT et al. Allosteric Coupling of Drug Binding and Intracellular Signaling in the A 2A Adenosine Receptor. Cell 172, 68–80.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dror RO et al. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A 108, 13118–13123 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suno R et al. Crystal Structures of Human Orexin 2 Receptor Bound to the Subtype-Selective Antagonist EMPA. Structure 26, 7–19.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Caffrey M & Porter C Crystallizing membrane proteins for structure determination using lipidic mesophases. Natuer Protoc. 4, 706–731 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ueno G et al. Remote access and automation of SPring-8 MX beamlines. in AIP Conference Proceedings 1741, (2016). [Google Scholar]
- 37.Hirata K, Foadi J, Evans G, Hasegawa K, Z. O. Structural Biology with Microfocus Beamlines. Adv. Methods Struct. Biol. Springer Protoc 241–273 (2016). [Google Scholar]
- 38.Yamashita K, Hirata K & Yamamoto M KAMO: towards automated data processing for microcrystals. Acta Crystallogr. Sect. D Struct. Biol 74, 441–449 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W Xds. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foadi J et al. Clustering procedures for the optimal selection of data sets from multiple crystals in macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 69, 1617–1632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.OS, S. grade, version 1.102. (2011). Available at: http://www.globalphasing.com.
- 45.Schüttelkopf AW & Van Aalten DMF PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr 60, 1355–1363 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Sheldrick GM Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 479–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hess B, Kutzner C, van der Spoel D & Lindahl E GROMACS 4: algorithms for highly efficient, load balanced, and scalable molecular simulations. J. Chem. Theory Comput 4, 435–447 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Oostenbrink C, Villa A, Mark AE & Van Gunsteren WF A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem 25, 1656–1676 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Schrödinger L : N. Y. Maestro, version 9.0. (2009).
- 50.Berendsen HJC, Postma JPM, Gunsteren WF Van & Hermans J Interaction models for water in relation to protein hydration. Intermol. Forces 331–342 (1981). doi: 10.1007/978-94-015-7658-1_21 [DOI] [Google Scholar]
- 51.Miyamoto S & Kollman PA Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem 13, 952–962 (1992). [Google Scholar]
- 52.Bennett CH Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys 22, 245–268 (1976). [Google Scholar]
- 53.Shirts MR & Pande VS Comparison of efficiency and bias of free energies computed by exponential averaging, the Bennett acceptance ratio, and thermodynamic integration. J. Chem. Phys 122, 1–16 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.