

Abstract
Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) are commonly used to control pain, inflammation, and limit the cardinal signs of injury in humans. However, prolonged use of NSAIDs increases the risk of heart attack (myocardial infarction; MI) and the subsequent risk of heart and renal failure. The molecular and cellular mechanism of action for this adverse effect, particularly along the cardiorenal network, is incomplete. To define the mechanism, carprofen (CAP), an NSAID was administered at the dose of 5 mg/kg to C57BL/6 male mice for two weeks. After last dose of CAP treatment mice were subjected to permanent occlusion of coronary artery that induces irreversible cardiac remodeling while maintaining naive and MI-controls. After MI, cardiac pathology and dysfunction were confirmed, along with additional measurements of kidney function, histology, and injury markers, such as plasma creatinine. CAP treatment increased plasma creatinine levels and subsequently, myocardial structural disorganization increased. Kidney neutrophil gelatinase associated lipocalin (NGAL) and protein expression were increased post-MI. After two weeks CAP treatment, the expression of pyrogenic pro-inflammatory cytokines TNF-α and IL-1β were increased compared to non-CAP treated mice, indicative of amplified inflammatory response. There was also evidence that renal injury of both the post-CAP treatment controls and post-CAP MI were much greater than the non-CAP treated naïve controls, as serum creatinine and NGAL levels were elevated along with obvious structural impairment of the glomerulus. Therefore, CAP treatment tampers with the acute inflammatory response that promotes cardiorenal syndrome and non-resolving inflammation post-MI in acute heart failure.
Keywords: renal inflammation, cytokines, carprofen, anti-inflammatory, non-resolving inflammation
Graphical Abstract:
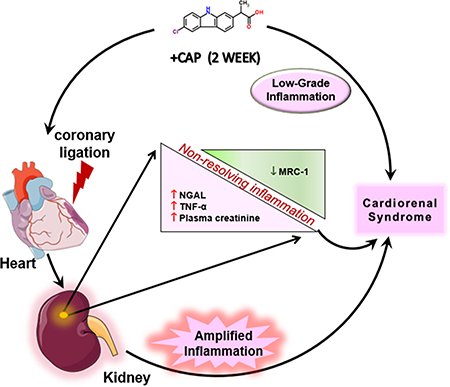
Sketch showing the subacute CAP pretreatment impact on the inflammatory and resolving process post-MI.
Introduction
In response to injury, initiation of leukocytes-directed intense inflammation is part of the innate immune response of the body and plays a vital role in resolution, and tissue repair as the response eliminates damaged tissue and cell debris (Frangogiannis, 2014, Halade et al., 2018c, Kain et al., 2014). While leukocyte-directed acute inflammation is a necessary process, it can lead to undesirable and collateral damage to healthy tissues if initiation of resolution of inflammation is delayed, such as in the setting of obesity and aging (Halade and Kain, 2017, Halade et al., 2016, Lopez et al., 2015). In the clinical setting of myocardial infarction (MI; heart attack), an immediate inflammatory response is activated by narrowing of the coronary artery, leading to the demise of cardiomyocytes, cardiorenal dysfunction and heart failure (Damman et al., 2014, Halade and Kain, 2017). Prolonged and uncontrolled inflammation after cardiac injury or ischemia is involved in the development of chronic heart and renal failure, accompanied by ventricular remodeling (Cho et al., 2013, Damman et al., 2014, Frangogiannis, 2014, Lopez et al., 2015).
Often, cardiac injury, such as MI, advances a cardiorenal inflammatory response with infiltration of leukocytes; which includes with the entry of first responder neutrophils and increased expression of inflammatory cytokines and chemokines. Post-MI, expressions of pyrogenic pro-inflammatory markers in the kidney, like tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), have been shown to increase significantly in a rat model (Cho et al., 2013). In addition to these increased inflammatory markers, there was also evidence of acute kidney injury (AKI), as the rodents with the MI had significantly increased levels of neutrophil gelatinase-associated lipocalin (NGAL) in the plasma and urine (Cho et al., 2013).
Traditionally, one of the most common ways to control inflammation and inflammation-related pain is to use Non-Steroidal Anti-Inflammatory Drugs (NSAIDs). The primary mechanism of action of NSAIDs is to limit the biosynthesis of prostaglandins, which are cardinal signaling molecules that are produced in response to infection or injury. Biosynthesis of prostaglandins facilitates in the dilation of the renal arterioles (Dixit et al., 2010). NSAIDs lead to an increase in plasma leukotriene levels by shunting arachidonic acid to react with lipoxygenase enzymes (LOX). Arachidonic acid-derived leukotriene B4 assists the entry of neutrophils into the tissue by upregulation of integrins on the neutrophil surface demonstrating that NSAIDs can initiate neutrophils entry into tissue before the injury. NSAIDs can also impair the macrophage functions, as they induce DNA damage in monocytes (Ribas et al., 2014). As a result, presented outcome revealed that the pre-treatment of NSAIDs, specifically carprofen (CAP), impaired the initiation of an acute response that may decrease the ability of immune cells to resolve inflammation and impair tissue repair process, specifically macrophages-neutrophil interaction, to produce pro-resolving mediators.
Methods
Animal compliance
All surgery protocols involving animals were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Revised 2015) and were approved by the Animal Care and Use Committee of the University of Alabama at Birmingham (Halade et al., 2018a).
Carprofen (CAP) treatment protocol
The mice were randomly divided into four groups: Group 1) -CAPNo-MI, Group 2) -CAP+MI-d1, Group 3) +CAP-No-MI, Group 4) +CAP+MI-d1. CAP was administered for two weeks at the dose of 5 mg/kg, subcutaneously indicating subacute duration of CAP with less rapid change and then the mice in the +MI groups were subjected to coronary artery ligation (Halade et al., 2018a).
Coronary artery surgery to induce MI
This study had a total of 33 C57BL/6 mice weighing between 25–35 g. The mice in Groups 3 and 4 were subjected to surgery to induce an MI. The surgery entailed a ligation of the left anterior descending coronary artery, 1 mm distal to the left atrium. During the surgery, mice were anesthetized with a 2% isoflurane and oxygen mix as noted previously (Halade et al., 2018a, Halade et al., 2013, Lopez et al., 2015, Zamilpa et al., 2013).
Transthoracic echocardiography
Echocardiography images of the mice were acquired using VisualSonics Vevo 3100 and analyzed using Vevo LAB 3.1.0 as previously described (Halade et al., 2018a).
Necropsy
All four groups were anesthetized under a 1:1 of 2% isoflurane and 100% oxygen. The lungs, LV, spleen, plasma, and kidneys were harvested and weighed, and the tibia was collected for the standardization of weights. The samples were kept in 10% zinc formalin for immunohistochemistry or flash frozen for molecular analysis (Halade et al., 2018a, Halade et al., 2018b, Halade et al., 2013, Lopez et al., 2015, Zamilpa et al., 2013).
Hematoxylin and eosin staining
Longitudinal sections of the left ventricle and kidney from each group were acquired and embedded in paraffin wax. Different slides were stained with hematoxylin and eosin (H&E) and a BX43 microscope with an attached Olympus DP73 camera was used to acquire images of the heart and kidney. The glomeruli of samples were subjectively evaluated for signs of mesangial hyper-cellularity and disorganization. The images of the glomeruli were scored from 0 to 10, where 0 indicated a sample is having no glomerular impairment and 10 indicated a sample with the most significant pathological changes. A total of 4 images were analyzed for each representative mouse (Halade et al., 2018a, Halade et al., 2013, Lopez et al., 2015, Zamilpa et al., 2013).
COX-2 activity
The COX activity assay kit (Cayman Chemical, 760151) was used according to the manufacturer’s instructions to determine COX-2 activity in the kidney tissue (Halade et al., 2018b).
Periodic Acid Schiff Reagent (PAS) staining
For no-MI and CAP treated mice, the kidney samples were embedded in paraffin wax for preservation. The wax was dissolved using a CitriSolv (Fisher Scientific) solution. The samples were then stained with a periodic acid-Schiff reagent and the renal tissue examined for evidence of structural disorganization (Halade et al., 2016).
Neutrophil staining:
For neutrophil assessment, the slides were deparaffinized and placed in a pressure cooker for heat-mediated antigen retrieval. Rabbit serum was used for blocking, and the slides were incubated with rat anti-mouse neutrophils at 4°C overnight. A Vectastain Elite ABC kit (Vector) was then used to stain the neutrophils (Halade et al., 2013, Lopez et al., 2015).
Glomerular filtration rate (GFR)
Three CAP-treated female animals and four non-treated female animals were selected to determine GFR. These mice were anesthetized using 1.5 – 2% isoflurane. A small portion of hair was removed from the dorsal side of each mouse, and a monitor was attached using adhesive and medical tape. A solution of 15mg/mL of FITC-sinistrin dissolved in 0.9% sterile saline was injected intravenously. The mice were then returned to their cages and monitored for two hours. Data were analyzed using elimination kinetics (Black et al., 2018, Schock-Kusch et al., 2012).
Kidney RNA isolation and real-time quantitative PCR
RNA was extracted from various kidney tissues, including kidney tissues from the splenocardiac project, using the Trizol method. cDNA was synthesized using 2 μg of RNA and a SuperScript Vilo cDNA Synthesis Kit (Invitrogen). Quantitative PCR for the TNF-α (Mm00443258_m1), IL-1β(Mm01336189_m1), MRC-1 (Mm00485172_m1), NGAL(Mm01324470_m1), KIM-1 (Mm00506686_m1), Nephrin (Mm01176615_g1), COX-1 (Mm00477214_m1), and COX-2 (Mm00478374_m1) genes were performed using TaqMan (Applied Biosystems, CA, USA) probes as done previously (10). Gene expression was normalized with hypoxanthine phosphoribosyltransferase-1 (Hprt-1). The results were reported as 2^-ΔCT. RT-PCR experiments were performed in duplicates with n= 5 mice per group (Halade et al., 2018b).
TUNEL, vimentin, and NGAL confocal microscopy
Immunofluorescence studies were conducted to determine the level of NGAL and Vimentin in the kidney tissue. Slides of kidney tissue were fixed, permeated, and blocked with an antibody against Vimentin (Abcam, ab92547) and NGAL (MAB1757, R&D Systems) overnight. The sections were stained with a secondary antibody: anti-mouse for the Vimentin, anti-goat for the NGAL, and Alexa 555, a fluorochrome (A21422, Molecular Probes), for 1 hour. Confocal microscopy was performed as described previously (Halade et al., 2018a).
Serum creatinine
10 μl of plasma ([2H3] creatinine) was added to an aliquot of mice serum. The protein was then removed from the mixture by adding solvent, drying under nitrogen, and exchanging the solvent. Creatinine was separated on a short, hydrophilic LC-column and detected using multiple reaction monitoring (MRM). The resulting ion peaks were converted to a concentration using an external calibration curve (Halade et al., 2018a, Takahashi et al., 2007, Young et al., 2007).
Statistical analysis
Data are presented as mean ± SEM. GraphPad prism 7 was used to conduct statistical analysis. Two-way ANOVA’s were used to determine the significance of the values compared to the -CAP -MI group (*p<0.05) and the +CAP -MI group ($p<0.05). For glomerulus structure analysis, 20 glomeruli were scored to rate their cellular organization, 0 being the most structured and 10 being the most disorganized.
Results
Subacute CAP treatment increased serum creatinine in mice, indicative of renal injury.
After two weeks of CAP treatment, plasma serum creatinine levels were determined using mass spectrometry. Gene expression levels of inflammatory cytokines and renal injury markers were measured in the kidney and compared to +/−CAP naïve controls (Figure 1A; study design). After 2 weeks CAP treatment and 24 hours post-MI, the +CAP+MI-d1 mice were compared with -CAP+MI-d1 mice to determine whether the inflammation and resolution response was impaired by CAP treatment in kidney. Post-MI, cytokines were measured, and histology was examined to evaluate the effect of CAP treatment on the initiation and resolution of inflammation in the setting of MI (Figure 1B). CAP pretreatment elevated plasma creatinine levels compared with -CAP-no-MI mice, indicating that CAP treatment, without MI, triggered renal injury (Figure 1C). The CAP pretreatment increased renal damage, suggestive of an adverse effect on kidney hemodynamics and inflammation. The decrease in plasma creatinine +CAP+MI-d1 compared to +CAP-no-MI may suggest a decrease in the integrity of the glomerular filtration barrier (Figure 1C). The glomerular filtration studies revealed that there were no significant differences in GFR between -CAP-no-MI and +CAP-no-MI mice (Figure 1D), demonstrating that the CAP treatment does not lead to renal dysfunction, but does induce molecular and cellular renal injury.
Figure 1. Subacute CAP treatment increases the level of renal injury markers pre-MI.

A. Study design describes a two-week subacute treatment of carprofen (CAP) to mice with pre and post myocardial infarction (MI) to study the effects of CAP on cardiorenal inflammation. B. A list of biological measures used to evaluate the effect of the CAP and measure renal injury markers. C. Plasma creatinine (*p<0.05 vs. –CAP No-MI; analyzed by two-way ANOVA); values are means ± SEM; n= 4 mice/group. and D. Representative and average glomerular filtration rate (GFR); values are means ± SEM; n= 3 mice/group.
MI-induced heart dysfunction confirmed using echocardiography for pathological remodeling.
Dysfunction of the heart after coronary artery ligation was confirmed using ultrasound (Vevo 3100) and data was analyzed using Vevo LAB software 3.1 (Figure 2; Table 1). Compared to control groups, mice treated with CAP showed subtle contractile dysfunction prior to MI. This is evidenced by the shorter vector lines (green) during systole when arresting the heart in mid-systole (Figure 2A). Both -CAP and +CAP mice that were subjected to MI surgery were shown to have significantly reduced left ventricular fractional shortening, indicative of impaired overall contractile function as shown in (Figure 2 A and C). Additionally, mice that were subjected to coronary artery ligation surgery displayed, posterior wall-thinning, impaired global circumferential strain, and regional dyssynchronicity (Figure 2 A, B, and D; Table 1). Furthermore, these mice displayed greater end systolic and end diastolic dimension values than control mice, suggestive of MI-induced pathological remodeling.
Figure 2: Short axis B Mode, circumferential three-dimensional strain, left ventricle wall trace M mode, and segmental short axis synchronicity indicates changes in ventricle size accompanied by impaired left ventricle systolic function following transmural myocardial infarction.

A. Green vector and left ventricle wall tracing suggestive of MI-indcued contractile dysfunction comapred with No-MI controls presented as short axis B-mode in mid-systole. B. Left ventricle M-mode wall trace of representative subjects from each experimental group. C. Three dimensional representation of short axis circumferencial strain. D. Traces of MI-indcued left ventricle circumferential dyssynchronicity compared with No-MI controls.
Table 1.
Echocardiography parameters before and after subacute CAP treatment in acute cardiac heart failure compared to naïve controls
| Echo Parameters | −CAP no-MI (n=7) | +CAP no-MI (n=6) | −CAP MI-d1 (n=5) | +CAP MI-d1 (n=5) |
|---|---|---|---|---|
| Heart rate (bpm) | 479 ± 8 | 460 ± 12 | 469 ± 21 | 479 ± 46 |
| EDD (mm) | 4 ± 0.08 | 4 ± 0.06 | 4 ± 0.20 | 4 ± 0.16 |
| ESD (mm) | 2 ± 0.11 | 3 ± 0.09 | 4 ± 0.18 | 4 ± 0.23 |
| Fractional Shortening (%) | 40 ± 2 | 21 ± 0.01* | 8 ± 2*$ | 8 ± 4*$ |
| IVSd (mm) | 0.73 ± 0.03 | 0.78 ± 0.04 | 0.59 ± 0.06* | 0.81 ± 0.02 |
| PWTd (mm) | 0.76 ± 0.04 | 0.76 ± 0.05 | 0.68 ± 0.08 | 0.76 ± 0.06 |
| IVSs (mm) | 1.10 ± 0.08 | 1.06 ± 0.07 | 0.64 ± 0.07*$ | 0.86 ± 0.04 |
| PWTs (mm) | 0.98 ± 0.10 | 1.03 ± 0.05 | 0.68 ± 0.06*$ | 0.77 ± 0.09 |
| GLS | −19 ± 0.3 | −14 ± 1* | −5 ± 1*$ | −5 ± 2*$ |
Abbreviations: Values are mean±SEM. N indicates sample size; bpm, beats per minute; mm, millimeter; EDD, End diastolic dimension; ESD, End systolic dimension; IVSd, Inter ventricular septum diastole; PWTd, Posterior wall thickness diastole; IVSs, inter ventricular septum systole; PWTs, posterior wall thickness systole; GLS, global longitudinal strain
p< 0.05 vs −CAP-no-MI
p<0.05 vs +CAP-no-MI groups
Subacute CAP treatment impaired the initiation of inflammatory response post-MI.
Examination of H&E stained slides of the LV showed clear evidence that the MI-induced LV is dilated in the +CAP+MI-d1 groups compared with -CAP+MI-d1 groups. The myocardium is injured with necrosis and apoptosis, and the fibers have lost their structural integrity allowing the nuclei to no longer be anchored in the muscle fibers in CAP treated MI-d1 groups (Figure 3A). The neutrophil stained sections also showed that there was an increase in neutrophils in the +CAP-MI-d1 group compared to the -CAP+MI-d1 group (Figure 3B and C). IL-1β levels increased significantly in the +CAP No-MI naïve controls when compared to the -CAP-No-MI naïve controls, however TNF-α displayed an insignificant upregulation in these groups (Figure 3D and E). These changes in the LV pathology along with the increases in these pro-inflammatory cytokines indicated that CAP treatment primed the immune system for an inflammatory response pre-MI. The simultaneous increase in the expression of MRC-1, a key reparative inflammatory marker, further indicated that the reparative inflammatory response was increased before injury due to the CAP-induced pre-activation of immune cells. In the -CAP+MI-d1group, there was an insignificant increase in the levels of TNF-α and IL-1β, suggestive of MI-triggered initiation of an inflammatory response (Figure 3D and E). However, the levels of gene expression for TNF-α and IL-1β were decreased post-MI in the +CAP+MI-d1 group, indicative of compromised immune responses after CAP treatment (Figure 3D and E). The expression of MRC-1 was significantly decreased in the +CAP+MI-d1 group when compared to the +CAP-No-MI group, suggestive of an impaired inflammatory-resolution response due to CAP in the naïve control group (Figure 3F). The statistical analysis of cytokines is presented in the (Figure 3G). These data suggest that CAP treatment amplified the inflammatory response and impaired initiation of the acute inflammation-resolution response after cardiac injury.
Figure 3. Subacute treatment of CAP suppresses the innate immune response in the event of an MI.

A1. Representative hematoxylin and eosin (H&E) stained (40×) images of the left ventricle (LV) of the –CAP-no-MI, +CAP-no-MI, -CAP+MI-d1, and +CAP+MI-d1 groups. B. Representative neutrophil stained (40×) images of the infarcted left ventricle (LV) of the –CAP+MI-d1 and +CAP+MI-d1. C. Bar graph representing percentage neutrohil density in -CAP and +CAP groups at d1 post-MI. mRNA expression of pro-inflammatory markers. D. TNF-α and E. IL-1β. mRNA expression of anti-inflammatory markers. F. MRC-1. G. Table is representing CAP effect and MI impact by two-way ANOVA. +CAP and –CAP treated pre-MI and post-MI groups. mRNA levels are normalized with hypoxanthine phosphoribosyltransferase 1 (HPRT-1). Values are means ± SEM; n= 5 mice/group *p< 0.05 vs –CAP-no-MI; $p < 0.05 vs +CAP-no-MI groups (analyzed by two-way ANOVA).
Subacute CAP treatment amplified MI-mediated renal injury.
The histology analysis of H&E stained sections of the renal medulla and glomerulus showed an increase in neutrophil count, inflammation of tubules and thinning of the basement membrane of Bowman’s capsule in +CAP+MI-d1 mice (Figure 4A). The pretreatment of CAP to mice resulted in proximal convoluted tubule swelling and swelling of the glomerular capillaries (Figure 4A) (Halade et al., 2013). Analysis of gene expression levels showed that neutrophil NGAL was expressed at higher levels in the -CAP+MI-d1 mice when compared to the naïve control, indicating that neutrophil infiltration is part of the initial immune response to tissue injury. CAP treatment significantly elevated NGAL expression post-MI when compared to the -CAP+MI-d1 mice suggestive of amplified neutrophil infiltration and renal tissue injury due to CAP (Figure 4B). Another marker of renal injury, KIM-1, which also increased in the -CAP+MI-d1 mice as compared to the -CAP-no-MI control mice (Figure 4C), though this increase was not significant. However, CAP treated both +CAP-no-MI and +CAP+MI-d1 group did not displayed any significant changes (Figure 4C) indicating suppressed initiation of the host response. Nephrin, an integral part of the renal filtration barrier, had decreased expression levels post-MI, indicating that the MI does induce some renal injury (Figure 4D). However, in the +CAP-no-MI control mice, the expression levels of nephrin were already lowered, indicating that the CAP treatment altered kidney morphology. The levels of nephrin in the -CAP+MI-d1 mice were similar to the expression levels of the +CAP-MI-d1 mice, indicating that CAP and MI separately induced similar amounts of renal injury (Figure 4C). The levels of nephrin expression were the lowest in the +CAP+MI-d1 mice, again showing that CAP pretreatment, when followed by an MI, can trigger excessive amounts of damage in the kidney.
Figure 4: Subacute carprofen (CAP) exposure intensified inflammatory response in the cortex, medulla, and glomerulus of kidney post-MI.

A. Representative hematoxylin and eosin (H&E) stained (10X) images of the cortex, medulla, and glomerulus for –CAP and +CAP treated no-MI and post-MI groups. mRNA expression of kidney injury markers. B. NGAL, C. KIM-1, and D. Nephrin. mRNA levels are normalized with hypoxanthine phosphoribosyltransferase 1 (HPRT-1). Values are means±SEM; n= 5 mice/group *p< 0.05 vs – CAP-no-MI; $p < 0.05 vs +CAP-no-MI groups (analyzed by two-way ANOVA).
Subacute CAP treatment increased fibrosis and neutrophil infiltration in the kidney pre-MI.
Vimentin, a marker of fibrosis, can be used to detect impairment with the healing process. Immunofluorescence images of mice kidneys from the four groups showed that Vimentin expression was increased in the event of an MI, indicating that an MI triggered the fibrotic healing process (Figure 5A). Vimentin expression also increased with CAP treatment in the +CAP+MI-d1 group, demonstrating that CAP treatment, without MI, triggered renal injury (Figure 5A). The expression of vimentin was more localized in the -CAP+MI-d1 group, whereas the +CAP+MI-d1 group showed evidence of generalized vimentin expression. Potentially, this shows that the MI prompted less renal disorganization than the CAP treatment (Figure 5A). The +CAP+MI-d1 group displayed higher levels of vimentin expression than the -CAP+MI-d1 group, demonstrating that CAP amplified the structural disorganization post-MI (Figure 5A). The +CAP+MI-d1 group also had more vimentin than the +CAP-MI-d1 group, showing that the MI and CAP treatment have an additive, deleterious effect along the cardiorenal axis. NGAL serves as a marker of the initial, innate immune response to injury. When comparing NGAL expression levels in the -CAP+MI-d1 and -CAP+MI-d1 groups, there was a greater amount of NGAL in the -CAP+MI-d1 group, indicating that neutrophil infiltration is part of the typical immune response to tissue injury (Figure 5B). However, the +CAP+MI-d1 group also showed increased levels of NGAL as compared to the naïve control, demonstrating that CAP treatment pre-activated the immune system and tampered with the initiation of the immune response (Figure 5B). The +CAP+MI-d1 group had higher levels of NGAL expression than the -CAP+MI-d1 and the +CAP+MI-d1 groups (Figure 5B). Again, the increased NGAL expression demonstrates that the CAP treatment, combined with an MI, led to an amplified acute inflammatory response as shown by neutrophil swarming in the +CAP+MI-d1 group.
Figure 5. Subacute CAP treatment increases the expression of vimentin and NGAL in kidney tissue.

A. Immunofluorescence images are showing an increase in vimentin expression (red) and nuclei (blue) 24 hours after MI following 2 week CAP treatment, in both the -CAP+MI-d1 and +CAP+MI-d1 groups. B. Immunofluorescence images are depicting an increase in neutrophil gelatinase associated lipocalin (NGAL) expression (red) and nuclei (blue) 24 hours after MI following a 2 week CAP treatment, in both the -CAP+MI and +CAP+MI groups.
CAP treatment increased tubular necrosis and amplified inflammatory response in the event of an MI.
The expression of cardinal inflammation triggering enzymes COX-1 and COX-2 were also examined. Cyclooxygenase (COX) mRNA, typically inhibited by NSAIDs, was measured in renal tissue of each mouse. The +CAP had significantly upregulated levels of COX-2 when compared to the –CAP groups with no significant changes in COX-1 (Figure 6A and B) indicating that the combination of the CAP treatment and MI amplified renal tissue inflammation. However, COX-2 activity was decreased in +CAP treated groups (Figure 6C). Fluorescence studies with terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick-end labeling (TUNEL) staining gave evidence of renal disorganization in +CAP+MI-d1 mice. The -CAP+MI-d1 group, in comparison to the -CAP-no-MI group, had more apoptotic cells (green) about the intact nuclei (red), demonstrating that the MI triggered some apoptotic renal injury (Figure 6D). In the +CAP+MI-d1 group, there was evidence of tubular apoptosis, showing that CAP caused renal inflammation and apoptosis before cardiac injury (Figure 6D and 6E). Both the -CAP+MI-d1 and +CAP+MI-d1 groups showed excessive renal damage, as the staining highlighted several apoptotic cells, demonstrating that the MI and CAP treatment both caused accelerated tubular apoptosis (Figure 6D).
Figure 6. Subacute COXAP treatment increases tubular apoptosis and adversely affect the inflammatory reparative response post-MI.

mRNA expression levels of pro-inflammatory enzymes A. COX-1 and B. COX-2. mRNA levels are normalized with hypoxanthine phosphoribosyltransferase 1 (HPRT-1) C. Bar graph representing COX-2 activity in the kidney of –CAP and +CAP treated no-MI and post- MI groups. (*p<0.05 vs –CAP No-MI; $p<0.05 vs –CAP MI-d1; analyzed by two-way ANOVA). Values are means ± SEM; n= 3 mice/group. D. TUNEL stained images of the kidney showing that CAP treatment resulted in greater amounts of tubular apoptosis pre and post MI. E. Bar grpah representing number of apoptotic cells per section analyzed in –CAP and +CAP treated no-MI and post-MI groups. *p<0.05 vs –CAP No-MI; $p<0.05 vs –CAP MI-d1; analyzed by two-way ANOVA; values are means ± SEM; n= 3 mice/group.
In summary, the subacute CAP treatment by itself caused subtle low-grade inflammation, as evidenced by the increase in mRNA levels of pro-inflammatory cytokines as well as the increase in neutrophil infiltration when compared to the naïve control. When the prolonged CAP treatment was assessed in the setting of an MI, there was evidence of more inflamed kidney as the expression of pro-inflammatory cytokines and renal injury markers were higher in the +CAP+MI mice. Therefore, CAP treatment followed by an MI resulted in an impaired acute inflammatory response that amplified inflammation MI-induced cardiorenal syndrome.
Discussion
The inflammatory response, while associated with pain and swelling is vital in responding to tissue injury as it aids in the clearance of dead cells and cell debris (Frangogiannis, 2014, Halade and Norris, 2018, Kain et al., 2014). Tissue injury triggers a reparative inflammatory response which is characterized by an increase in pro-inflammatory cytokines, leukocytes infiltration (neutrophil and monocytes/macrophage) into the injured tissue and eventual resolution of the injury (Frangogiannis, 2014, Kain et al., 2014). A myocardial infarction (MI) not only induces ischemia in the cardiac tissue but also inflicts significant injury on the kidney, as shown by the increase in both plasma and urine NGAL levels post-MI in a rat model (Cho et al., 2013, Damman et al., 2014). The renal pathology induced by the MI results in the development of cardiorenal syndrome, which describes the pathological changes when cardiac or renal failure triggers bidirectional feedback. Prolonged NSAID usage has been shown to elevate the risk of cardiac and renal failure events (Pawlosky, 2013, Silverman and Pfeifer, 1987). Use of NSAIDs has also been shown to trigger renal damage. While the renal impairment is usually reversible, prolonged NSAID use can result in abnormal renal function for a more extensive period (Dixit et al., 2010). Our investigation elucidated the molecular and cellular mechanism of CAP-mediated adverse effects after subacute treatment. CAP treatment 1) triggered an increase of kidney injury marker; 2) impaired initiation of acute inflammatory responses post-MI; and, 3) had an adverse additive effect when combined with an MI which led to marked dysregulation of cellular and molecular markers. CAP-triggered changes in renal structure even without the presence of the MI. Again, the MI and CAP treatment seemed to have had an additive effect, where the CAP treatment alone triggered subtle low-grade inflammation, but the combination of the MI and CAP triggered dysregulation of an acute inflammatory response. Prolonged CAP treatment with MI amplified this inflammation and amplified cardiorenal syndrome. Non-resolving inflammation provides a cautionary note regarding NSAID use in patients at risk for cardiovascular events. The three molecular and cellular actions mentioned above are detailed below.
First, the data presented reveal that CAP treatment triggers renal inflammation even prior to the MI surgery. Plasma creatinine levels, a biomarker of renal injury, increased in the mice subjected to CAP treatment, indicating that renal inflammation occurs because of the NSAID treatment alone (Horl, 2010). In addition to this preliminary marker of renal pathology, the levels of NGAL and KIM-1, two biomarkers of renal injury, increase after subacute CAP treatment, which is consistent with previous studies (Vaidya et al., 2008). The renal inflammation done by the CAP increase is also shown in the renal histology where the proximal convoluted tubules are inflamed, and the glomerular structural integrity is compromised (Markowitz et al., 2015). As such, the CAP treatment triggered renal inflammation without MI, demonstrating that NSAID treatment causes kidney injury (Dixit et al., 2010).
In addition to triggering baseline renal inflammation, CAP treatment also tampers with two aspects of the reparative inflammatory response: the initiation and the resolution of inflammation. The initiation of the inflammatory response is characterized by the expression of pro-inflammatory cytokines, neutrophil infiltration, biosynthesis of immunoresolvents and production of chemo-attractants (Serhan, 2014, 2017). In this study, CAP treatment increased the expression levels of pyrogenic TNF-α and IL-1β levels before the MI. CAP treatment also increased the expression of COX-2 pre-MI. COX-2 is an enzyme that is induced in the onset of the inflammation process and produces chemo-attractants such as prostaglandin E2 (PGE2), which serves to recruit immune cells to the site of injury (Khan et al., 2007). The increase in COX-2 in +CAP -MI mice, indicates that the inflammatory response has already begun, even without tissue injury (Khan et al., 2007). These findings corroborate previous studies where celecoxib, a COX-2 inhibitor, treatment resulted in increased TNF-α levels and ibuprofen. Also a COX-2 selective inhibitor in mice triggered renal impairment (Choudhury and Ahmed, 2006, Dixit et al., 2010, Khan et al., 2007). The initiation and resolution of inflammation are two distinct but unified and overlapping processes (Halade and Norris, 2018, Sansbury and Spite, 2016, Sugimoto et al., 2016). MRC-1 is involved in the resolution of inflammation as it helps clear the glycoproteins that participate in the ascendency of the inflammatory response (Gazi and Martinez-Pomares, 2009). The CAP treated mice show increased levels of MRC-1 even prior to the injury, when compared to the naïve control, indicating that that the onset of the resolution phase was also initiated prior to the tissue injury, like the pro-inflammatory cytokines (Sansbury and Spite, 2016, Sugimoto et al., 2016).
When the pre-treatment of CAP was followed by the MI, the levels of pro-inflammatory cytokines TNF-α and IL-1β decreased, rather increased, in response to tissue injury (Frangogiannis, 2014, Kain et al., 2014). This lack of initiation of the inflammatory response can be traced back to the pre-treatment of CAP, which increases the TNF-α and IL-1β levels prior to injury; as such, tissue injury no longer serves to initiate the inflammatory response. The KIM-1 marker remained similar pre and post-MI in the CAP treatment group, rather than increasing as it should in the event of kidney injury, potentially indicating that the markers used to alert the body to tissue injury are non-functional (Shukla et al., 2017, Vaidya et al., 2008). KIM-1 also contributes to the production of pro-inflammatory cytokines (Brooks and Bonventre, 2015). As KIM-1 does not increase post CAP treatment and post MI, the optimal reparative inflammatory response in the kidney is not triggered.
Additionally, KIM-1 also serves to phagocytose apoptotic cellular debris, and as the TUNEL staining shows, there are more apoptotic cells in the +CAP+MI mice compared to control groups. This increase indicates that the CAP induced lack of KIM-1 prevented the normal clearance process, magnifying the inflammation post-MI (Brooks and Bonventre, 2015). MRC-1 is also a surface marker of M2 macrophages, which are responsible for wound healing and tissue repair (Jablonski et al., 2015, Mills, 2012). Post-MI, the expression of MRC-1 drops from pre-MI levels to the same amount of MRC-1 seen in the -CAP+MI-d1 mice, showing that CAP not only tampered with the initiation of the resolution process but impaired to promote resolution and reparative response.
The adverse effects that CAP treatment has on the immune system are mirrored in the amplified renal tissue injury seen post-MI. The myocardium of the +CAP+MI-d1 mice is excessively edematous when compared to the other groups, and the structural integrity of the fibers is compromised (Garcia-Dorado et al., 2012). Furthermore, the glomerulus shows more obvious pathology and the protein expression levels of NGAL and vimentin are increased post-CAP and post-MI (Cao et al., 2015, Markowitz et al., 2015, Vaidya et al., 2008). In summary, when CAP treatment prolonged before MI, then there is an increase in tissue disorganization, and impaired natural endogenous reparative inflammatory response is suppressed.
Limitations and perspective
This study has limitations to its application to a human subject as there are differences between mice and human physiology. Furthermore, the study only assessed results 24 hours post-MI mainly in acute heart failure in young mice and future studies will have to examine more time points to elucidate the complete mechanism of CAP’s effects on the cardiorenal syndrome. The CAP treatment interferes with the initiation and resolution phases of the inflammatory response, increasing the amount of tissue injury caused by an MI. The specific mechanism of this interference as well as whether CAP affects macrophages is not clear and requires further studies. From a clinical standpoint, there are also a few viable options other than NSAIDs to relieve inflammation-related pain, but the effects of CAP demonstrate that more research is warranted to develop effective alternatives.
Conclusion
Prolonged treatment of carprofen induced renal injury markers and amplified non-resolving inflammation following cardiac injury. Subacute treatment of pain-killer carprofen altered cellular and molecular events after cardiac injury and intensified acute inflammation in cardiac healing. Presented results are indicative of immune suppressive and impaired resolution of inflammation mechanisms due to the subacute treatment of pain-killer carprofen thereby cardiorenal syndrome in acute heart failure.
ACKNOWLEGMENTS
We acknowledge support from the UAB-UCSD O’Brien Core Center for Acute Kidney Injury Research (NIH P30-DK079337) for this project.
FUNDING SUPPORT:
Authors acknowledge the funding support from National Institutes of Health [HL132989] to G.V.H. and American Heart Association postdoctoral fellowship [POST31000008] to V. K. Authors Veena Krishnan Department of Chemistry, Birmingham-Southern College, Birmingham, Alabama, Gabrielle Cunningham, Jefferson State Community College, Birmingham, Alabama, and David Booker, UAB second year medical student acquired and analyzed data with of help of other co-authors and corresponding author.
Footnotes
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Black LM, Lever JM, Traylor AM, Chen B, Yang Z, Esman SK, et al. Divergent effects of AKI to CKD models on inflammation and fibrosis. American journal of physiology Renal physiology. 2018;315:F1107–f18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CR, Bonventre JV. KIM-1/TIM-1 in proximal tubular cell immune response. Oncotarget. 2015;6:44059–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao YH, Lv LL, Zhang X, Hu H, Ding LH, Yin D, et al. Urinary vimentin mRNA as a potential novel biomarker of renal fibrosis. American journal of physiology Renal physiology. 2015;309:F514–22. [DOI] [PubMed] [Google Scholar]
- Cho E, Kim M, Ko YS, Lee HY, Song M, Kim MG, et al. Role of inflammation in the pathogenesis of cardiorenal syndrome in a rat myocardial infarction model. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2013;28:2766–78. [DOI] [PubMed] [Google Scholar]
- Choudhury D, Ahmed Z. Drug-associated renal dysfunction and injury. Nature clinical practice Nephrology. 2006;2:80–91. [DOI] [PubMed] [Google Scholar]
- Damman K, Tang WH, Testani JM, McMurray JJ. Terminology and definition of changes renal function in heart failure. European heart journal. 2014;35:3413–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit M, Doan T, Kirschner R, Dixit N. Significant Acute Kidney Injury Due to Non-steroidal Anti-inflammatory Drugs: Inpatient Setting. Pharmaceuticals (Basel, Switzerland). 2010;3:1279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nature reviews Cardiology. 2014;11:255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Dorado D, Andres-Villarreal M, Ruiz-Meana M, Inserte J, Barba I. Myocardial edema: a translational view. Journal of molecular and cellular cardiology. 2012;52:931–9. [DOI] [PubMed] [Google Scholar]
- Gazi U, Martinez-Pomares L. Influence of the mannose receptor in host immune responses. Immunobiology. 2009;214:554–61. [DOI] [PubMed] [Google Scholar]
- Halade GV, Kain V. Obesity and Cardiometabolic Defects in Heart Failure Pathology. Comprehensive Physiology. 2017;7:1463–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halade GV, Kain V, Black LM, Prabhu SD, Ingle KA. Aging dysregulates D- and E-series resolvins to modulate cardiosplenic and cardiorenal network following myocardial infarction. Aging. 2016;8:2611–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halade GV, Kain V, Ingle KA. Heart functional and structural compendium of cardiosplenic and cardiorenal networks in acute and chronic heart failure pathology. American journal of physiology Heart and circulatory physiology. 2018a;314:H255–H67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halade GV, Kain V, Wright GM, Jadapalli JK. Subacute treatment of carprofen facilitate splenocardiac resolution deficit in cardiac injury. Journal of leukocyte biology. 2018b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halade GV, Ma Y, Ramirez TA, Zhang J, Dai Q, Hensler JG, et al. Reduced BDNF attenuates inflammation and angiogenesis to improve survival and cardiac function following myocardial infarction in mice. American journal of physiology Heart and circulatory physiology. 2013;305:H1830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halade GV, Norris PC. Splenic leukocytes define the resolution of inflammation in heart failure. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halade GV, Norris PC, Kain V, Serhan CN, Ingle KA. Splenic leukocytes define the resolution of inflammation in heart failure. Science signaling. 2018c;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horl WH. Nonsteroidal Anti-Inflammatory Drugs and the Kidney. Pharmaceuticals (Basel, Switzerland). 2010;3:2291–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, et al. Novel Markers to Delineate Murine M1 and M2 Macrophages. PloS one. 2015;10:e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kain V, Prabhu SD, Halade GV. Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic research in cardiology. 2014;109:444. [DOI] [PubMed] [Google Scholar]
- Khan AA, Iadarola M, Yang HY, Dionne RA. Expression of COX-1 and COX-2 in a clinical model of acute inflammation. The journal of pain: official journal of the American Pain Society. 2007;8:349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez EF, Kabarowski JH, Ingle KA, Kain V, Barnes S, Crossman DK, et al. Obesity superimposed on aging magnifies inflammation and delays the resolving response after myocardial infarction. American journal of physiology Heart and circulatory physiology. 2015;308:H269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz GS, Bomback AS, Perazella MA. Drug-induced glomerular disease: direct cellular injury. Clinical journal of the American Society of Nephrology: CJASN. 2015;10:1291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Critical reviews in immunology. 2012;32:463–88. [DOI] [PubMed] [Google Scholar]
- Pawlosky N Cardiovascular risk: Are all NSAIDs alike? Canadian pharmacists journal: CPJ = Revue des pharmaciens du Canada: RPC. 2013;146:80–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas JL, da Silva CA, de Andrade L, Galvan GL, Cestari MM, Trindade ES, et al. Effects of anti-inflammatory drugs in primary kidney cell culture of a freshwater fish. Fish & shellfish immunology. 2014;40:296–303. [DOI] [PubMed] [Google Scholar]
- Sansbury BE, Spite M. Resolution of Acute Inflammation and the Role of Resolvins in Immunity, Thrombosis, and Vascular Biology. Circulation research. 2016;119:113–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schock-Kusch D, Shulhevich Y, Xie Q, Hesser J, Stsepankou D, Neudecker S, et al. Online feedback-controlled renal constant infusion clearances in rats. Kidney Int. 2012;82:314–20. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2017;31:1273–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, Rai MK, Prasad N, Agarwal V. Short-Term Non-Steroid Anti-Inflammatory Drug Use in Spondyloarthritis Patients Induces Subclinical Acute Kidney Injury: Biomarkers Study. Nephron. 2017;135:277–86. [DOI] [PubMed] [Google Scholar]
- Silverman HS, Pfeifer MP. Relation between use of anti-inflammatory agents and left ventricular free wall rupture during acute myocardial infarction. The American journal of cardiology. 1987;59:363–4. [DOI] [PubMed] [Google Scholar]
- Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Frontiers in immunology. 2016;7:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Boysen G, Li F, Li Y, Swenberg JA. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int. 2007;71:266–71. [DOI] [PubMed] [Google Scholar]
- Vaidya VS, Ferguson MA, Bonventre JV. Biomarkers of acute kidney injury. Annual review of pharmacology and toxicology. 2008;48:463–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S, Struys E, Wood T. Quantification of creatine and guanidinoacetate using GC-MS and LC-MS/MS for the detection of cerebral creatine deficiency syndromes. Current protocols in human genetics. 2007;Chapter 17:Unit 17.3. [DOI] [PubMed] [Google Scholar]
- Zamilpa R, Zhang J, Chiao YA, de Castro Bras LE, Halade GV, Ma Y, et al. Cardiac Wound Healing Post-myocardial Infarction: A Novel Method to Target Extracellular Matrix Remodeling in the Left Ventricle. Methods in molecular biology (Clifton, NJ). 2013;1037:313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]