

Abstract
ER-PM junctions are subcellular sites where the endoplasmic reticulum (ER) and the plasma membrane (PM) are kept in close appositions, providing a platform for inter-organelle contact. These membrane contact sites are important for many physiological functions in mammalian cells, including excitation-contraction coupling, store-operated Ca2+ entry, and non-vesicular transfer of lipids between the ER and the PM. Here we review recent insights into the 3D structure and spatial organization of ER-PM junctions in mammalian cells as well as molecular mechanisms underlying the formation and functions of mammalian ER-PM junctions.
Keywords: ER-PM junctions, membrane contact sites, Ca2+ signaling, non-vesicular lipid transfer, cholesterol transport
Introduction
The endoplasmic reticulum (ER) is an extensive membrane organelle important for Ca2+ storage, biosynthesis of proteins and lipids, and protein folding. Communication between the ER and the plasma membrane (PM) is important for cells to properly respond to extracellular stimulation and maintain cellular homeostasis. Close appositions between the ER and the PM at ER-PM junctions, also referred to as ER-PM contacts or ER-PM contact sites, enable direct contacts between these two membrane compartments for signaling and molecular trafficking [1].
ER-PM junctions were first observed using electron microscopy (EM) in muscle cells and referred to as dyads and triads [2]. Dyads and triads are found to be essential for excitation-contraction (E-C) coupling underlying muscle contraction [3]. At dyads and triads, depolarization of voltage-gated Ca2+ channels at the PM triggers the juxtaposed ryanodine receptors at the ER to release stored Ca2+ stimulating contraction of muscle cells. Junctophilins (JPHs) enriched at dyads and triads are important for the formation of these ER-PM junctions and E-C coupling [4]. Deficiency in JPHs leads to cardiac arrests and lethality in mice.
Nearly five decades after, ER-PM junctions were identified as the spatial platform of the ubiquitous store-operated Ca2+ entry (SOCE) pathway for Ca2+ signaling and homeostasis [5, 6]. Following a decrease in ER Ca2+, SOCE is activated by translocation of the ER membrane protein STIM1 to ER-PM junctions, enabling its interaction with the PM Ca2+ channel Orai1 [7]. SOCE is essential for numerous physiological processes such as the immune response, secretion, cell migration, and tissue development. Humans deficient in SOCE suffer severe combined immunodeficiency, skeletal myopathy, and ectodermal dysplasia [8].
More recently, it was found that lipid exchange at ER-PM junctions is pivotal to the phosphatidylinositol (PI) cycle, a fundamental process first observed in 1953 in receptor-stimulated pancreas slices [9]. The PI cycle involves transfer of phosphatidic acid (PA), which is converted from diacylglycerol (DAG) generated by stimulation-induced breakdown of PI 4,5-bisphosphate (PI(4,5)P2), from the PM to the ER where it can be converted to PI and transferred back to the PM for PI(4,5)P2 re-synthesis to sustain cell signaling [10, 11]. The production of PA following receptor stimulation induces translocation of the cytosolic PI/PA transfer protein Nir2 to ER-PM junctions to mediate the PI cycle [12-14]. The PI cycle is not only important for sustaining receptor-induced signaling, but also required to maintain numerous PI(4,5)P2-dependent cellular functions such as ion transport, membrane trafficking, and cytoskeleton dynamics.
The known functions of mammalian ER-PM junctions continue to expand. Additional proteins that constitutively or dynamically enriched at ER-PM junctions have been identified, including extended synaptotagmins (E-Syts), TMEM24, Kv2 voltage-gated K+ channels, oxysterol-binding protein (OSBP)-related proteins (ORP) 5 and ORP8, and GRAM domain-containing proteins (GRAMDs). Moreover, recent developments in imaging technologies and tools enable the visualization of the 3D structure of ER-PM junctions and their spatial organization in mammalian cells, advancing our understanding of these membrane junctions. The aim of this review is to discuss recent insights into this fast growing field of research on the structure, functions, and regulation of mammalian ER-PM junctions, complementing several comprehensive reviews on this topic [1, 10, 15, 16].
3D structure and spatial organization of mammalian ER-PM junctions
Early EM studies on the structure of mammalian ER-PM junctions were mostly performed using muscle and neuronal cells. ER-PM junctions in muscle cells were observed in specific regions of sarcomere, the contraction unit of the muscle [3]. The gap distance between the ER and the PM at these junctions is kept at 9 to 12 nm. Correlations between the localization, number, and gap distance of ER-PM junctions with the contraction speed of different types of muscles were observed, consistent with an important role of ER-PM junctions in E-C coupling and muscle contraction. In neurons, ER-PM junctions were observed and described as close appositions between the PM and the subsurface cisterns (SSC) with a narrow ER lumen [17]. These SSC-PM junctions can be several microns long with a 5-8 nm gap between the SSC and the PM.
Compared with their counterparts in muscles and neurons, ER-PM junctions in non-excitable cells are of low abundance and more difficult to detect using thin-section EM. The average size of ER-PM junctions in Jurkat T cells is 150 nm with a gap of 10-25 nm [6]. In adherent HeLa cells, ER-PM junctions are 100-200 nm in size and preferentially localize at the basal/adhesion surface of the cell [18]. Interestingly, the number, but not the size, of ER-PM junctions increases during Ca2+ signaling [6].
Recently, a genetically-encoded ER-PM junctional marker named MAPPER was developed based on STIM1 [1, 12]. While thin-section transmission EM (TEM) reveals the gap distance of ER-PM junctions, MAPPER enables visualization of the whole contacting area of ER-PM junctions in living cells using light microscopy techniques such as confocal and total internal reflection fluorescence (TIRF) imaging (Figure 1). In addition, MAPPER greatly increases the throughput in analyzing ER-PM junctions by simultaneously visualizing hundreds of ER-PM junctions in a typical adherent mammalian cell. Analysis of ER-PM junctions using MAPPER in HeLa cells suggest that in addition to an increase in the number, a decrease in the gap distance also occurs during Ca2+ signaling [12]. This decrease is regulated by translocation of E-Syt1 to ER-PM junctions following receptor-induced increase in cytosolic Ca2+, and facilitates Nir2 targeting to ER-PM junctions to mediate the PI cycle. Consistently, a decrease in gap distance from 21.8 nm to 14.8 nm was detected using cryo-electron tomography in E-Syt1-overepxressing COS-7 cells during Ca2+ signaling [19].
Figure 1. Structure and spatial organization of ER-PM junctions.

(A) A TEM image of an ER-PM junction (red arrow head) in a MAPPER-expressing HeLa cell. (B-C) Images of the adherent surface of a MAPPER-expressing HeLa cell acquired using TIRF microscopy (TIRFM) and PALM. (D) A close-up from (C). (E) TIRF-SIM image of a HeLa cell with ER-PM junctions labeled by MAPPER (green) and stained with AF568-phalloidin for F-actin (orange). (F) A close-up from (E). (G) Diagram depicting spatial distribution of single ER-PM junctions and cortical actin. Scale bars: 1 (B), 0.2 (D), 10 (E) and 2 (F) μm. Figure 1A and 1B-F were originally published in [10] and [20].
The size and shape of the contact area as well as the spatial organization of mammalian ER-PM junctions were examined using MAPPER and three super-resolution imaging techniques. Stimulated emission depletion (STED) microscopy was used to track the same ER-PM junctions in live HeLa cells, revealing that the size of individual ER-PM junctions remains the same during Ca2+ signaling [12]. Photoactivated localization microscopy (PALM), which provides superior lateral resolution, determined ER-PM junctions in HeLa cells to be oblong-shaped with the dimensions of ~120nm × ~80nm [20]. Based on these measurements, a single ER-PM junction is predicted to accommodate up to several hundred proteins. Lastly, TIRF-structured illumination microscopy (TIRF-SIM) revealed that cortical actin contributes to the spatial distribution of ER-PM junctions, with most ER-PM junctions in close proximity to F-actin [20]. Disruption of cortical actin results in decreased stability of ER-PM junctions, manifested as increased lateral motility and reduced number of junctions, suggesting intimate interactions between ER-PM junctions and actin cytoskeleton proximal to the PM.
A recent study using focused ion beam scanning electron microscopy (FIB-SEM) revealed the 3D architecture of ER-PM junctions in neuronal cells [21]. The size of neuronal ER-PM junctions was found to be generally larger than those observed in other cells. Notably, ER-PM junctions are larger in cell bodies than those in dendrites and axons. In addition, ER-PM junctions were shown to be highly abundant, representing over 10% of the PM in neurons, compared with 2~4% in non-excitable cells [1].
Molecular mechanisms underlying formation and functions of mammalian ER-PM junctions
The ER and the PM are kept in close apposition at ER-PM junctions by protein-protein and protein-lipid interactions between the two membrane compartments. Many proteins have been reported to support the formation and functions of mammalian ER-PM junctions. Here we discuss seven groups of proteins that harbor well-characterized dual targeting mechanisms for the ER and the PM, enabling their constitutive or dynamic enrichment at ER-PM junctions (Figure 2).
Figure 2. Proteins enriched at mammalian ER-PM junctions.
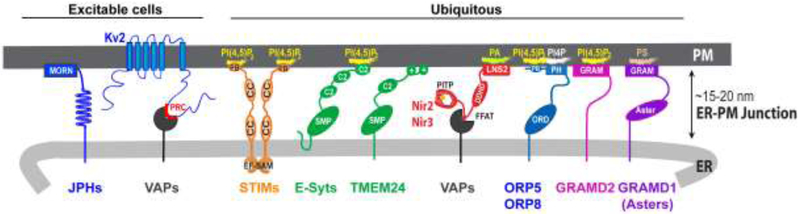
JPH, Junctophilin; MORN, membrane occupation and recognition nexus motif; PRC, proximity restriction and clustering domain; EF-SAM, EF hand and sterile alpha motif; CC, coiled-coil domain; PB, polybasic domain; SMP, synaptotagmin-like mitochondrial-lipid binding protein domain; PITP, phosphatidylinositol transfer protein domain; FFAT, two phenylalanines (FF) in an acidic tract motif; DDHD, domain characterized by these conserved residues; LNS2, Lipin/Ned1/Smp2 domain; ORD, OSBP-related domain; PH, pleckstrin homology domain; GRAM, glucosyltransferases, Rab-like GTPase activators and myotubularins domain; ASTER, START (StAR-related lipid-transfer)-like domain.
JPHs:
JPHs are four ER membrane proteins highly expressed in excitable mammalian cells [4]. JPH1 and JPH2 are selectively expressed in muscles, whereas JPH3 and JPH4 are highly expressed in neural tissues. JPH contains multiple cytosolic membrane occupation and recognition nexus (MORN) motifs that mediate their localization to ER-PM junctions by binding to phospholipids at the PM. JPH1 knockout mice died shortly after birth with reduced number of triad junctions in skeletal muscles [22], demonstrating the essential role of ER-PM junctions in E-C coupling as described in the introduction. JPH2 knockout mice exhibited embryonic lethality with cardiac arrest and reduced number of ER-PM junctions in cardiac myocytes [23]. JPH3 and JPH4 double knockout mice exhibited impaired motor coordination and learning [24]. These findings demonstrate the important role of JPHs in the formation of ER-PM junctions and functions of muscle and neuronal cells.
Kv2 channels:
Kv2.1 and Kv2.2, also known as KCNB1 and KCNB2, are PM voltage-gated K+ channels abundantly expressed in mammalian brain and also found in other excitable tissues including muscles and pancreatic islets. Unique among the 7 groups of ER-PM junctional proteins discussed here, Kv2 channels are currently the only known PM proteins mediating ER-PM tethering. Kv2.1 and Kv2.2 were initially observed to localize in large clusters in the PM at proximal dendrites, axon initial segments, and with particular abundance, in the soma of neurons [25]. These clusters were later shown to localize at ER-PM junctions via the binding of the proximity restriction and clustering (PRC) domain in the cytosolic region to the ER [26, 27]. Notably, Kv2.1 and Kv2.2 enrichment is independent of ion conductance [28], and clustered Kv2.1 channels do not efficiently conduct K+ [29].
Recently, two independent groups employed proteomic analyses and identified the ER membrane VAMP-associated proteins (VAPs), VAPA and VAPB, as interactors of Kv2.1 [30, 31]. VAPs bind double phenylalanine in an acid tract (FFAT) motif-containing proteins, and localize them to junctions between the ER and other membrane compartments such as the PM, Golgi, and endosomes [32]. Interestingly, VAP-Kv2 interaction is mediated by a noncanonical phosphorylation-dependent FFAT motif in the PRC domain of Kv2.1 [30, 31]. Conversely, Ca2+-dependent dephosphorylation induced by excessive excitatory stimulation by exposure to noxious conditions such as hypoxia result in Kv2 cluster dispersal [33]. While the ER-PM targeting mechanism has been identified, the biological function of Kv2 channels localizing at ER-PM junctions remains to be addressed.
STIMs:
STIM1 and its family member STIM2, are ubiquitously expressed singlepass transmembrane ER proteins important for SOCE as described in the introduction. Following ER Ca2+ depletion triggered by receptor stimulation, dissociation of Ca2+ from the EF-SAM domain in the luminal region induces STIM1 oligomerization and conformational change [5, 34]. An exposed C-terminal polybasic (PB) motif in activated STIM1 binds to PI(4,5)P2 at the PM, resulting in STIM1 enrichment at ER-PM junctions [35]. Consistently, significant reduction of PM PI(4,5)P2 level abrogated STIM1 localization to ER-PM junctions, limiting SOCE in Ras-association domain family 4 (RASSF4) knockdown cells [36]. Activated STIM1 at ER-PM junctions interacts with the PM Ca2+ channel Orai1, leading to Orai1 enrichment at ER-PM junctions and SOCE for propagation of Ca2+ signaling and refill of ER Ca2+ store [6]. Interestingly, STIM1 dynamically tracks microtubule plus ends by binding to the EB1 protein via its TRIP motif near the C-terminus [37]. EB1 binding limits STIM1 targeting to ER-PM junctions, providing a mechanism that shapes local Ca2+ signaling in cellular regions with growing microtubules [38].
E-Syts and TMEM24:
E-Syt1, E-Syt2, and E-Syt3 are ER membrane proteins with a cytosolic region containing a synaptotagmin-like mitochondrial-lipid binding protein (SMP) domain followed by multiple C2 domains [39]. E-Syt2 and E-Syt3 constitutively enrich at ER-PM junctions with their C2 domains interacting with PM PI(4,5)P2, whereas E-Syt1 dynamically translocates to ER-PM junctions following a significant increase of cytosolic Ca2+ that releases its auto-inhibition [12, 39, 40]. Knockdown of all three E-Syts or a decrease in PM PI(4,5)P2, as in RASSF4-knockdown cells, resulted in a significant reduction of ER-PM junctions [36, 39]. Structural and biochemical studies indicated that the SMP domain of E-Syts can transfer glycerolipids and DAG [41, 42]. Nevertheless, mice deficient in all E-Syts exhibited no obvious defects, suggesting redundancy in the functions of these ER-PM tethering lipid transfer proteins [43, 44]. A recent study identified TMEM24, another SMP domain-containing ER membrane protein with a preference to transfer PI [45]. TMEM24 localizes at ER-PM junctions by binding to the PM via its positively-charged C-terminal region. It was proposed that Ca2+-dependent phosphorylation and dephosphorylation of TMEM24 result in oscillatory localization of TMEM24 at ER-PM junctions, regulating pulsatile insulin secretion in insulinoma cells.
Nir2 and Nir3:
Nir2 and Nir3, also known as PITPNM1 and PITPNM2, are widely expressed cytosolic lipid transfer proteins that mediate the PI cycle as described in the introduction. Nir2 and Nir3 contain an N-terminal PI transfer protein (PITP) domain, an FFAT motif that can bind VAPs in the ER, a DDHD domain with unknown functions, and a C-terminal LNS2 domain that can bind PA. The production of PA at the PM following receptor-induced PI(4,5)P2 hydrolysis leads to recruitment of Nir2 and Nir3 to ER-PM junctions within a minute [12-14], Interestingly, the Nir2 mutant with mutations in the FFAT and LNS2 domains failed to mediate PI(4,5)P2 replenishment following the hydrolysis [13], indicating that enrichment at ER-PM junctions is important for Nir2 to mediate the PI cycle. Nir2 knockout mice were embryonic lethal [46], consistent with its role in maintaining the level of PM PI(4,5)P2 important for numerous cellular processes.
ORP5 and ORP8:
ORP5 and ORP8 are ER integral membrane proteins containing an N-terminal pleckstrin homology (PH) domain and an OBSP-related domain (ORD). It was proposed that ORP5 and ORP8 enrich at ER-PM junctions by binding to PI 4-phosphate (PI4P) at the PM via the PH domain, and mediate transport of PM PI4P to the ER and phosphatidylserine (PS) from the ER to the PM via the ORD [47]. Recent studies revealed that PM binding of ORP5 and ORP8 involves both the N-terminal PB region and the PH domain [48-50]. In addition, structural studies uncovered that the PH domain of ORP5 and ORP8 binds PI(4,5)P2, and enrichment of ORP5 and ORP8 at ER-PM junctions depends on the level of PM PI(4,5)P2 [48, 50], Notably, ORP5 constitutively localizes at ER-PM junctions, whereas OPR8 enriches at ER-PM junctions when PM PI(4,5)P2 level is elevated. Knocking down both ORP5 and ORP8 resulted in elevated PM PI(4,5)P2. Based on these findings, a rheostat model was proposed in which ORP5 and ORP8 countertransport PS and PI4P between the ER and the PM at ER-PM junctions to maintain the homeostasis of PM PI(4,5)P2 by limiting its precursor, PI4P [50].
GRAMDs:
GRAMD1a-c and GRAMD2a and b comprise a family of mammalian proteins with structural similarity to the yeast Ltc/Lam ER membrane proteins [51-53], GRAMDs contain an N-terminal GRAM domain—found in glucosyltransferases, Rab-like GTPase activators, and myotubularins—that resembles a PH domain, and a single C-terminal ER-anchoring transmembrane domain. Both GRAMDIa and GRAMD2a require the GRAM domain for ER-PM junction localization [54]. Additionally, GRAMD2a localizes at ER-PM junctions in a PI(4,5)P2-dependent manner. In a recent study, Sandhu et al. redefined the exon-intron topology of Gramd1a-c, characterized a central domain resembling steroidogenic acute regulatory protein (STAR)-related lipid transfer (START) domain, and renamed the protein products Aster-A, -B, and -C [55]. Asters bind cholesterol in vitro, and the crystal structure of the central domain in Aster-A reveals a 7-stranded beta-sheet pocket for cholesterol occupancy. Aster-B is recruited to ER-PM junctions in response to cholesterol and the GRAM domain of Aster-B is sufficient for cholesterol induced recruitment to the PM. Interestingly, the Aster-B GRAM domain pelleted with liposomes containing PS independent of cholesterol content. Lastly, mice lacking Aster-B exhibited altered adrenal sterol homeostasis, consistent with a role of Asters in facilitating cholesterol transport from the PM to the ER downstream of the high-density lipoprotein receptor. These findings implicate GRAMD/Aster proteins in sterol transport via ER-PM junctions, yet the relationship linking PS and sterol levels at the PM is currently unknown.
Perspectives
Recent findings indicate that mammalian ER-PM junctions are membrane contact sites supported by cortical actin and multiple families of proteins at a given time. Since the enrichment of these proteins at ER-PM junctions can be regulated by various factors such as ER/cytosolic Ca2+ levels, PM PA/PS/PI4P/PI(4,5)P2/cholesterol levels, or phosphorylation, it is likely that the composition of single ER-PM junctions is heterogeneous and controlled by the local environment and stimulated state of a cell. Also, it is difficult to completely disrupt mammalian ER-PM junctions, given many different proteins contribute to the formation and stability of these structures. It is of interest to investigate the crosstalk among these proteins at ER-PM junctions, determine the factors that control the size and shape of ER-PM junctions, characterize the molecules that mediate the close association between cortical actin and ER-PM junctions, examine the distribution of ER-PM junctions during actin reorganization and cell migration, and continue to identify new tethers and functions of ER-PM junctions.
Arguably, mammalian ER-PM junctions are the best characterized membrane contact sites with many insights into their structure, regulation, and physiological functions. The knowledge derived from studying mammalian ER-PM junctions may be applied to understand inter-organelle signaling at other membrane contact sites.
Acknowledgements
We thank the Liou Laboratory members for valuable discussions. This work was supported by National Institutes of Health grant GM113079, Welch Foundation Grant I- 1789, and National Institutes of Health Cell and Molecular Biology Training Program T32 GM008203. J. Liou is a Sowell Family Scholar in Medical Research. The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Chang CL, Chen YJ, Liou J: ER-plasma membrane junctions: Why and how do we study them? Biochim Biophys Acta Mol Cell Res 2017, 1864:1494–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Porter KR, Palade GE: Studies on the endoplasmic reticulum. III. Its form and distribution in striated muscle cells. J Biophys Biochem Cytol 1957, 3:269–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franzini-Armstrong C: The relationship between form and function throughout the history of excitation-contraction coupling. J Gen Physiol 2018, 150:189–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeshima H, Hoshijima M, Song LS: Ca(2)(+) microdomains organized by junctophilins. Cell Calcium 2015, 58:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., Meyer T: STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 2005, 15:1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu MM, Buchanan J, Luik RM, Lewis RS: Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 2006, 174:803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prakriya M, Lewis RS: Store-Operated Calcium Channels. Physiol Rev 2015, 95:1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lacruz RS, Feske S: Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci 2015, 1356:45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hokin MR, Hokin LE: Enzyme secretion and the incorporation of P32 into phospholipides of pancreas slices. J Biol Chem 1953, 203:967–977. [PubMed] [Google Scholar]
- 10.Chang CL, Liou J: Homeostatic regulation of the PI(4,5)P2-Ca(2+) signaling system at ER-PM junctions. Biochim Biophys Acta 2016, 1861:862–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michell RH: Inositol phospholipids and cell surface receptor function. Biochim Biophys Acta 1975, 415:81–47. [DOI] [PubMed] [Google Scholar]
- 12.Chang CL, Hsieh TS, Yang TT, Rothberg KG, Azizoglu DB, Volk E, Liao JC, Liou J: Feedback regulation of receptor-induced Ca2+ signaling mediated by E-Syt1 and Nir2 at endoplasmic reticulum-plasma membrane junctions. Cell Rep 2013, 5:813–825. [DOI] [PubMed] [Google Scholar]
- 13.Chang CL, Liou J: Phosphatidylinositol 4,5-bisphosphate Homeostasis Regulated by Nir2 and Nir3 at Endoplasmic Reticulum-Plasma Membrane Junctions. J Biol Chem 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YJ, Guzman-Hernandez ML, Wisniewski E, Balla T: Phosphatidylinositol-Phosphatidic Acid Exchange by Nir2 at ER-PM Contact Sites Maintains Phosphoinositide Signaling Competence. Dev Cell 2015, 33:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carrasco S, Meyer T: STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annu Rev Biochem 2011, 80:973–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saheki Y, De Camilli P: Endoplasmic Reticulum-Plasma Membrane Contact Sites. Annu Rev Biochem 2017, 86:659–684. [DOI] [PubMed] [Google Scholar]
- 17.Rosenbluth J: Subsurface cisterns and their relationship to the neuronal plasma membrane. J Cell Biol 1962, 13:405–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orci L, Ravazzola M, Le Coadic M, Shen WW, Demaurex N, Cosson P: From the Cover: STIM1-induced precortical and cortical subdomains of the endoplasmic reticulum. Proc Natl Acad Sci U S A 2009, 106:19358–19362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Busnadiego R, Saheki Y, De Camilli P: Three-dimensional architecture of extended synaptotagmin-mediated endoplasmic reticulum-plasma membrane contact sites. Proc Natl Acad Sci U S A 2015, 112:E2004–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh TS, Chen YJ, Chang CL, Lee WR, Liou J: Cortical actin contributes to spatial organization of ER-PM junctions. Mol Biol Cell 2017, 28:3171–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Using MAPPER and the super-resolution microscopy techniques SIM and PALM, the authors define the morphological and spatial features of ER-PM junctions. Additionally, they demonstrate cortical actin places a crucial role in defining the spatial distribution and stability of junctions impacting Nir2-mediated PI(4,5P)2 homeostasis.
- 21.Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF, De Camilli P: Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci U S A 2017, 114:E4859–E4867. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Using the advanced focused ion beam-scanning EM, the authors created 3D reconstructions of organelles in contact with the ER in distinct compartments of adult mouse neuronal cells.
- 22.Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, Kitamura K, Takeshima H: Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol 2001, 154:1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K: Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 2000, 6:11–22. [DOI] [PubMed] [Google Scholar]
- 24.Moriguchi S, Nishi M, Komazaki S, Sakagami H, Miyazaki T, Masumiya H, Saito SY, Watanabe M, Kondo H, Yawo H, et al. : Functional uncoupling between Ca2+ release and afterhyperpolarization in mutant hippocampal neurons lacking junctophilins. Proc Natl Acad Sci U S A 2006, 103:10811–10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trimmer JS: Subcellular localization of K+ channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron 2015, 85:238–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim ST, Antonucci DE, Scannevin RH, Trimmer JS: A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 2000, 25:385–397. [DOI] [PubMed] [Google Scholar]
- 27.Fox PD, Haberkorn CJ, Akin EJ, Seel PJ, Krapf D, Tamkun MM: Induction of stable ER-plasma-membrane junctions by Kv2.1 potassium channels. J Cell Sci 2015, 128:2096–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirmiz M, Palacio S, Thapa P, King AN, Sack JT, Trimmer JS: Remodeling neuronal ER-PM junctions is a conserved nonconducting function of Kv2 plasma membrane ion channels. Mol Biol Cell 2018, 29:2410–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]; * The authors demonstrate that remodeling of neuronal ER-PM junctions is a conserved function of Kv2 PM Ion channels and DKO of Kv2.1 and Kv2.2 in mice alters brain neuron ER-PM junctions. Kv2-mediated remodeling is independent of ion conductance and instead dependent on a conserved PRC domain.
- 29.O'Connell KM, Loftus R, Tamkun MM: Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc Natl Acad Sci U S A 2010, 107:12351–12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson B, Leek AN, Sole L, Maverick EE, Levine TP, Tamkun MM: Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc Natl Acad Sci U S A 2018, 115:E7331–E7340. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors identify VAPs as interactors of Kv2.1 at ER-PM junctions in neuronal cells through a proximity-based biotinylation approach. Kv2.1-VAP binding is mediated by a noncanonical VAP-binding motif on the C-terminus of Kv2.1.
- 31.Kirmiz M, Vierra NC, Palacio S, Trimmer JS: Identification of VAPA and VAPB as Kv2 Channel-Interacting Proteins Defining Endoplasmic Reticulum-Plasma Membrane Junctions in Mammalian Brain Neurons. J Neurosci 2018, 38:7562–7584. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors identify VAPA and VAPB as interactors of Kv2.1 in brain neurons, and demonstrate that Kv2 channels colocalize with VAPs at ER-PM junctions. VAPA KO is sufficient to reduce Kv2.1 clustering and VAP-KV2.1 interaction is mediated by a noncanonical phosphorylation-dependent FFAT motif in the PRC domain of Kv2.1.
- 32.Murphy SE, Levine TP: VAP, a Versatile Access Point for the Endoplasmic Reticulum: Review and analysis of FFAT-like motifs in the VAPome. Biochim Biophys Acta 2016, 1861:952–961. [DOI] [PubMed] [Google Scholar]
- 33.Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS: Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci 2004, 7:711–718. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Y, Srinivasan P, Razavi S, Seymour S, Meraner P, Gudlur A, Stathopulos PB, Ikura M, Rao A, Hogan PG: Initial activation of STIM1, the regulator of store-operated calcium entry. Nat Struct Mol Biol 2013, 20:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liou J, Fivaz M, Inoue T, Meyer T: Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci U S A 2007, 104:9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen YJ, Chang CL, Lee WR, Liou J: RASSF4 controls SOCE and ER-PM junctions through regulation of PI(4,5)P2. J Cell Biol 2017, 216:2011–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]; * The authors identify RASSF4 as a novel regulator of SOCE and ER-PM junctions by altering PM PI(4,5)P2 levels. This is achieved by RASSF4 interaction and regulation of ARF6, an upstream regulator of PIP5Ks.
- 37.Grigoriev I, Gouveia SM, van der Vaart B, Demmers J, Smyth JT, Honnappa S, Splinter D, Steinmetz MO, Putney JW Jr., Hoogenraad CC, et al. : STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol 2008, 18:177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang CL, Chen YJ, Quintanilla CG, Hsieh TS, Liou J: EB1 binding restricts STIM1 translocation to ER-PM junctions and regulates store-operated Ca(2+) entry. J Cell Biol 2018, 217:2047–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Using a synthetic biology approach, the authors reveal that EB1 binding to STIM1 regulates SOCE by delaying STIM1 translocation to ER-PM junctions. The authors propose a trapping mechanism in which STIM1-EB1 interaction spatiotemporally tunes SOCE amplitude and kinetics in regions with microtubule growing ends.
- 39.Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P: PI(4,5)P2-Dependent and Ca(2+)-Regulated ER-PM Interactions Mediated by the Extended Synaptotagmins. Cell 2013, 153:1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bian X, Saheki Y, De Camilli P: Ca(2+) releases E-Syt1 autoinhibition to couple ER-plasma membrane tethering with lipid transport. EMBO J 2018, 37:219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]; * The authors propose an auto inhibitory mechanism underlying the Ca2+ mediated E-Syt1 tethering and lipid transport at ER-PM junctions. Ca2+ binding to the C2C domain enables E-Syt1 tethering via C2E-PI(4,5)P2 binding, while Ca2+ binding to the C2A domain releases auto inhibition of the SMP domain and enables lipid transport.
- 41.Saheki Y, Bian X, Schauder CM, Sawaki Y, Surma MA, Klose C, Pincet F, Reinisch KM, De Camilli P: Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat Cell Biol 2016, 18:504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schauder CM, Wu X, Saheki Y, Narayanaswamy P, Torta F, Wenk MR, De Camilli P, Reinisch KM: Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature 2014, 510:552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sclip A, Bacaj T, Giam LR, Sudhof TC: Extended Synaptotagmin (ESyt) Triple Knock-Out Mice Are Viable and Fertile without Obvious Endoplasmic Reticulum Dysfunction. PLoS One 2016, 11:e0158295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tremblay MG, Moss T: Loss of all 3 Extended Synaptotagmins does not affect normal mouse development, viability or fertility. Cell Cycle 2016, 15:2360–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lees JA, Messa M, Sun EW, Wheeler H, Torta F, Wenk MR, De Camilli P, Reinisch KM: Lipid transport by TMEM24 at ER-plasma membrane contacts regulates pulsatile insulin secretion. Science 2017, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors uncover a mechanism by which TMEM24 regulates insulin secretion. TMEM24 is an ER-anchored protein that reversibly localizes to ER-PM junctions and facilitates PI transfer via an SMP domain. Elevation of cytosolic Ca2+ triggers phosphorylation of TMEM24 and removal from ER-PM junctions, limiting lipid transfer and PI(4,5)P2 resynthesis and altering insulin secretion.
- 46.Lu C, Peng YW, Shang J, Pawlyk BS, Yu F, Li T: The mammalian retinal degeneration B2 gene is not required for photoreceptor function and survival. Neuroscience 2001, 107:35–41. [DOI] [PubMed] [Google Scholar]
- 47.Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P: INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 2015, 349:428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghai R, Du X, Wang H, Dong J, Ferguson C, Brown AJ, Parton RG, Wu JW, Yang H: ORP5 and ORP8 bind phosphatidylinositol-4, 5-biphosphate (PtdIns(4,5)P 2) and regulate its level at the plasma membrane. Nat Commun 2017, 8:757. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors demonstrate that the OSBP-related domain of ORP8 can transport multiple phosphoinositides, and provide evidence that recruitment of ORP5 and ORP8 to ER-PM junctions is mediated by binding of the PH domain to PI(4,5)P2. They also show that knock down of both ORP5 and ORP8 results in increased PM PI(4,5)P2.
- 49.Lee M, Fairn GD: Both the PH domain and N-terminal region of oxysterol-binding protein related protein 8S are required for localization to PM-ER contact sites. Biochem Biophys Res Commun 2018, 496:1088–1094. [DOI] [PubMed] [Google Scholar]
- 50.Sohn M, Korzeniowski M, Zewe JP, Wills RC, Hammond GRV, Humpolickova J, Vrzal L, Chalupska D, Veverka V, Fairn GD, et al. : PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER-PM contact sites. J Cell Biol 2018, 217:1797–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors demonstrate that ORP5 and ORP8 are differentially recruited to the PM via binding of their PH domains to PI4P and PI(4,5)P2. While ORP5 requires normal levels of these lipids, ORP8 recruitment requires elevated PI(4,5)P2. A model was proposed linking PI4P, PI(4,5)P2 and PS metabolism regulated by ORP5 and ORP8.
- 51.Elbaz-Alon Y, Eisenberg-Bord M, Shinder V, Stiller SB, Shimoni E, Wiedemann N, Geiger T, Schuldiner M: Lam6 Regulates the Extent of Contacts between Organelles. Cell Rep 2015, 12:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gatta AT, Wong LH, Sere YY, Calderon-Norena DM, Cockcroft S, Menon AK, Levine TP: A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. Elite 2015, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA, Nunnari J: Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J Cell Biol 2015, 209:539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Besprozvannaya M, Dickson E, Li H, Ginburg KS, Bers DM, Auwerx J, Nunnari J: GRAM domain proteins specialize functionally distinct ER-PM contact sites in human cells. Elite 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors show that the human family members of yeast Ltc/Lam sterol transportors, GRAMD1a and GRAMD2a, localize at distinct ER-PM junctions.
- 55.Sandhu J, Li S, Fairall L, Pfisterer SG, Gurnett JE, Xiao X, Weston TA, Vashi D, Ferrari A, Orozco JL, et al. : Aster Proteins Facilitate Nonvesicular Plasma Membrane to ER Cholesterol Transport in Mammalian Cells. Cell 2018, 175:514–529 e520. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors describe three ER-resident proteins, Aster-A, -B, -C, that bind and remove cholesterol from the plasma membrane via a central domain resembling the sterol-binding fold in StARD proteins. Aster recruitment is mediated by an N-terminal PS-binding domain, and mice lacking Aster-B have altered sterol metabolism.