

Abstract
G protein-coupled receptors (GPCRs) play key roles in intercellular signaling in the brain. Their effects on cellular function have been largely studied in neurons, but their functional consequences on astrocytes are less known. Using both endogenous and chemogenetic approaches with DREADDs, we have investigated the effects of Gq and Gi/o GPCR activation on astroglial Ca2+-based activity, gliotransmitter release, and the functional consequences on neuronal electrical activity. We found that while GqGPCR activation led to cellular activation in both neurons and astrocytes, Gi/oGPCR activation led to cellular inhibition in neurons and cellular activation in astrocytes. Astroglial activation by either Gq or Gi/o protein-mediated signaling stimulated gliotransmitter release, which increased neuronal excitability. Additionally, activation of Gq and Gi/o DREADDs in vivo increased astrocyte Ca2+ activity and modified neuronal network electrical activity. Present results reveal additional complexity of the signaling consequences of excitatory and inhibitory neurotransmitters in astroglia-neuron network operation and brain function.
Keywords: astrocytes, G protein-coupled receptors, DREADDs, gliotransmission, astrocyte Ca2+
INTRODUCTION
Intercellular chemical communication in the brain is based on neurotransmitters released by neurons and neurotransmitter receptors in the target cells, which upon activation can enhance or inhibit cellular activity. Two major classes of neurotransmitter receptors are responsible for transducing the intercellular chemical signaling – ligand-gated channels with ionotropic function that directly change membrane ionic permeability, and G protein-coupled receptors (GPCRs) with metabotropic function that activate intracellular signaling pathways. G proteins can be classified into four major families (i.e. Gs, Gq, Gi/o, G12/13) according to the downstream signaling triggered by the activation of their α subunit. Briefly, some of the main signaling pathways are as follows: Gq activates phospholipase C (PLC), Gi/o decreases cyclic AMP levels through inhibition of adenylyl cyclase, Gs stimulates adenylyl cyclase, and G12/13 regulates small GTPases (Huang & Thathiah, 2015; Stewart & Fisher, 2015). Although there are a multitude of effects downstream of G protein activation, the classical view of Gq and Gi/o protein signaling in neurons is excitation and inhibition, respectively (Huang & Thathiah, 2015). Whether these functional consequences of different G protein-mediated signaling occurs in other brain cells, like astrocytes, remains largely unknown.
Astrocytes are emerging as important cells in brain function by exchanging signaling with neurons through their participation in the tripartite synapse (Araque, Parpura, Sanzgiri, & Haydon, 1999). Rather than exhibiting electrical excitability like neurons, astrocytes display a form of excitability based on changes in intracellular Ca2+ concentration (Araque, Carmignoto, & Haydon, 2001; Araque et al., 2014; Di Castro et al., 2011; Haydon & Carmignoto, 2006; Mariotti et al., 2018; Navarrete & Araque, 2010; Panatier et al., 2011; Perea & Araque, 2005; Perea et al., 2016; Perea, Navarrete, & Araque, 2009; Verkhratsky & Kettenmann, 1996; Volterra, Liaudet, & Savtchouk, 2014; Volterra & Meldolesi, 2005). Upon activation, astrocytes can release their own signaling molecules, termed gliotransmitters, that regulate neuronal excitability, synaptic transmission, and plasticity (Araque et al., 2014; Bezzi et al., 2004; Halassa & Haydon, 2010; Rusakov, 2015; Volterra & Meldolesi, 2005) see however (Fiacco & McCarthy, 2018). A major route of inducing Ca2+ elevations is through activation of Gq GPCRs that results in the release of Ca2+ from internal stores through activation of PLC and IP3 receptors (IP3R) (Di Castro et al., 2011; Panatier et al., 2011; Pasti, Volterra, Pozzan, & Carmignoto, 1997; Perea & Araque, 2005; Porter & McCarthy, 1996). The effects of Gi/o GPCR activation in astrocytes are less known. Reports indicate that neurotransmitter receptors typically coupled to Gi/o GPCRs, such as CB1 and GABAB receptors, can increase Ca2+ levels and stimulate gliotransmission (Covelo & Araque, 2018; Kang, Jiang, Goldman, & Nedergaard, 1998; Mariotti, Losi, Sessolo, Marcon, & Carmignoto, 2016; Meier, Kafitz, & Rose, 2008; Navarrete & Araque, 2008; Perea et al., 2016). While CB1 receptors have been shown to promiscuously couple to Gq proteins (Lauckner, Hille, & Mackie, 2005; Navarrete & Araque, 2008), whether Gi/o protein activation inhibits cellular activity in astrocytes, like in neurons, remains unclear.
Here, we have investigated the functional consequences of selective activation of Gq and Gi/o GPCRs on astrocytic and neuronal activity using endogenous and chemogenetic (i.e. Designer Receptors Exclusively Activated by Designer Drugs, DREADDs) approaches. Combining electrophysiological and Ca2+ imaging techniques in hippocampal mouse brain slices, we found that Gq GPCR activation in both neurons and astrocytes led to cellular activation. Hereafter, cellular activation is defined as calcium increases in both neurons and astrocytes and depolarization in neurons. In contrast, Gi/o GPCR activation inhibited neurons, but led to astrocyte activation. Furthermore, Gq or Gi/o GPCR activation in astrocytes stimulated the release of glutamate, which enhanced neuronal excitability. Activation of both Gq and Gi/o DREADDs in astrocytes of the primary somatosensory cortex in vivo similarly led to the enhancement of both astrocyte Ca2+ elevations and cortical delta rhythms. These results indicate that while activation of different GPCR pathways in neurons led to either excitation or inhibition, in astrocytes they led only to activation, suggesting that inhibitory signaling is a particular property of neurons and not astrocytes.
METHODS
Experimental model and subject details
Hippocampal coronal slices were obtained from both male and female 6–12 weeks old C57BL/6, GCaMP3 (GLAST-CreE RT2 x R26-lsl-GCaMP3; (Paukert et al., 2014)), GCaMP6f (Tg(Slc1a3-cre/ERT)1Nat/J (JAX:012586) x Ai95(RCL-GCaMP6f)-D (JAX:028865); (Agarwal et al., 2017) and IP3R2−/− (Li, Zima, Sheikh, Blatter, & Chen, 2005) mice. GCaMP3 mice received 8 daily doses (57mg/kg) of tamoxifen injections two weeks prior to experimentation. Mice were housed in a 14/10 light/dark cycle with ad libitum food and water. All experiments were in compliance with the Animal Care and Use Committee at the University of Minnesota.
Stereotaxic surgery
4–6-week-old mice were anesthetized with ketamine/xylazine (10 ml/kg) and underwent stereotaxic surgery of AAV5-CaMKIIα-hM3Dq or AAV5-CaMKIIα-hM4Di to assess Gq and Gi/o signaling in neurons, respectively, or AAV8-GFAP-hM3Dq or AAV8-GFAP-hM4Di to assess Gq and Gi/o signaling in astrocytes, respectively. Stereotaxic coordinates used for all hippocampal injections were (relative to Bregma in mm) −2.65 A-P, ±2.25 M-L and −1.75 D-V. Stereotaxic coordinates used for all in vivo cortical injections were (relative to Bregma in mm): −−2.00 A-P, ±2.00 M-L and −1.00, −0.80, and −0.60 D-V. For in vivo control experiments, mice underwent stereotaxic surgery of AAV8-GFAP-mCherry. Experiments were performed 2–6 weeks after surgery. All DREADDs viruses were purchased from the UNC Vector Core, while the plasmid to generate the AAV8-GFAP-mCherry was purchased from Addgene (Plasmid #58909) and was generated by the University of Minnesota Viral Vector and Cloning Core.
Hippocampal slice preparation
The brain was removed quickly after decapitation and placed in ice-cold artificial cerebrospinal fluid (ACSF). Coronal hippocampal slices (350um thick) were made with a vibratome and incubated (>1 h) in a holding chamber at room temperature (21–24 degrees C) in ACSF containing (in mM): NaCl 124, KCl 2.69, KH2PO4 1.25, MgSO4 2, NaHCO3 26, CaCl2 2, ascorbic acid 0.4, and glucose 10, and continuously bubbled with carbogen (95% O2 and 5% CO2) (pH 7.3). Slices were transferred to an immersion recording chamber and superfused at 2mL/min with gassed magnesium-free ACSF containing (in mM): NaCl 124, KCl 2.69, KH2PO4 1.25, NaHCO3 26, CaCl2 4, glucose 10, and glycine 4. Tetrodotoxin (TTX, 1μM) was included in the perfusion system for all slice experiments to block action potential-mediated neurotransmission, except for experiments in Figure 7. To isolate GABAB receptor-mediated events in the GABA application experiments, picrotoxin (GABAA receptor antagonist, 50 μM) was added to the perfusion system. In experiments to block Gq-PLC signaling, the PLC inhibitor U73122 (4 μM) was included in the perfusion system. In experiments to block Gi/o signaling, slices were incubated in ACSF containing the Gi/o inhibitor pertussis toxin (PTX; 7.5 μg/ml) for 3–4 hours prior to experiments. In experiments to block muscarinic ACh receptors, atropine (50 μM) was included in the perfusion system. In experiments to block GABAB receptors, CGP54626 (1 mM) was included in the perfusion system. Cells were visualized under 40x water immersion objective using differential interface contrast (DIC) in an Olympus microscope.
Figure 7. Astrocytic Gq and Gi/o DREADD activation increases neuronal action potential firing.
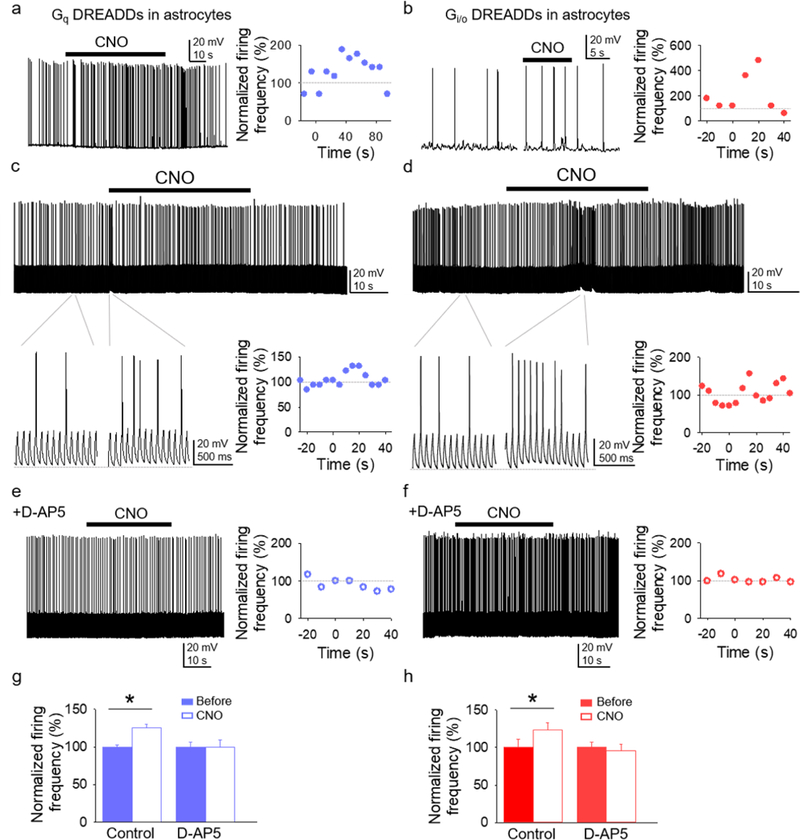
(A and B) Representative trace of spontaneous action potential firing of CA1 pyramidal neuron before and after CNO application to GqDREADD-expressing astrocytes (A) and Gi/oDREADD-expressing astrocytes (B), and the normalized (from basal) firing frequency vs. time. CNO application was at t=0 (as in the other panels). (C and D) Representative trace (top) of action potential firing induced by short depolarizing pulses before and after CNO application to GqDREADD-expressing astrocytes (C) and Gi/oDREADD-expressing astrocytes (D), expanded traces (bottom left panels), and normalized firing frequency vs. time (bottom right panels). (E and F) Response to CNO in the presence of D-AP5. (G and H) Normalized firing frequency before and after CNO application to GqDREADD-expressing astrocytes (G) and GqDREADD-expressing astrocytes (H) in control and D-AP5. Data are represented as mean ± SEM. P < 0.05 (*).
Slice electrophysiology
Whole-cell electrophysiological recordings were performed in pyramidal neurons of the CA1 region of the hippocampus. Patch pipettes were pulled from thick-walled borosilicate glass (1.5 mm outer diameter) on a Sutter Instruments P-1000 puller. Pipettes (3–8 MΩ) were filled with the internal solution that contained (in mM): K-Gluconate 135, KCl 10, HEPES 10, MgCl2 1, ATP-Na2 2 (pH = 7.3 adjusted with KOH; osmolality 280–290 mOsm/L). Recordings were obtained and filtered (1KHz) by PC-ONE amplifiers (Dagan Instruments, Minneapolis, MN). Signals were fed to a Pentium-based PC through a DigiData 1440A interface board. The pCLAMP 10.2 (Axon Instruments) software was used for stimulus generation, data display, acquisition and storage. For neuronal DREADDs experiments, only mCherry-positive CA1 neurons were patched, and mCherry-negative neurons were recorded for sham conditions. For neuronal slow inward current (SIC) and action potential firing responses after astrocytic DREADDs activation, only mCherry-positive slices were used, and mCherry-negative slices were used for sham conditions. ACh- and CNO-evoked neuronal currents and SICs were recorded in voltage-clamp at a holding potential of −70mV and GABA-mediated currents were recorded at a holding potential of −40mV. To mimic excitatory inputs and observe changes in action potential firing over time (Figure 7), a continuous train of depolarizing current pulses (20ms, ~200 pA, delivered every 50ms) was applied. SICs were defined as currents with a τon > 5 and lasting > 40ms. Experiments were performed at room temperature (21–24 °C).
Agonist application
For all slice agonist application experiments, pipettes (3–8 MΩ) were filled with ACSF plus 1 mM ACh or 1 mM GABA or 1 mM CNO, or 2mM (S)-3,5-Dihydroxyphenylglycine (DHPG). Pipettes were positioned over the pyramidal layer to assess neuronal responses to the agonist (Figures 1, 3), or over the stratum radiatum to assess astrocytic Ca2+ response to agonist application, SICs, and firing modulation (Figures 2, 4-7). To illustrate the time course of the agonist effects on SIC frequency in Figure 6, the number of SICs were grouped in 10s bins. To compare the agonist effects, the mean SIC frequency recorded 1 minute before and after agonist application was calculated. Agonists were delivered using a Dagan PMI-100 Pressure Micro-injector (15 PSI, 2–5s for all experiments except for the following: 30s for chemogenetic SIC experiments and firing modulation experiments).
Figure 1. Gq signaling induces neuronal activation.
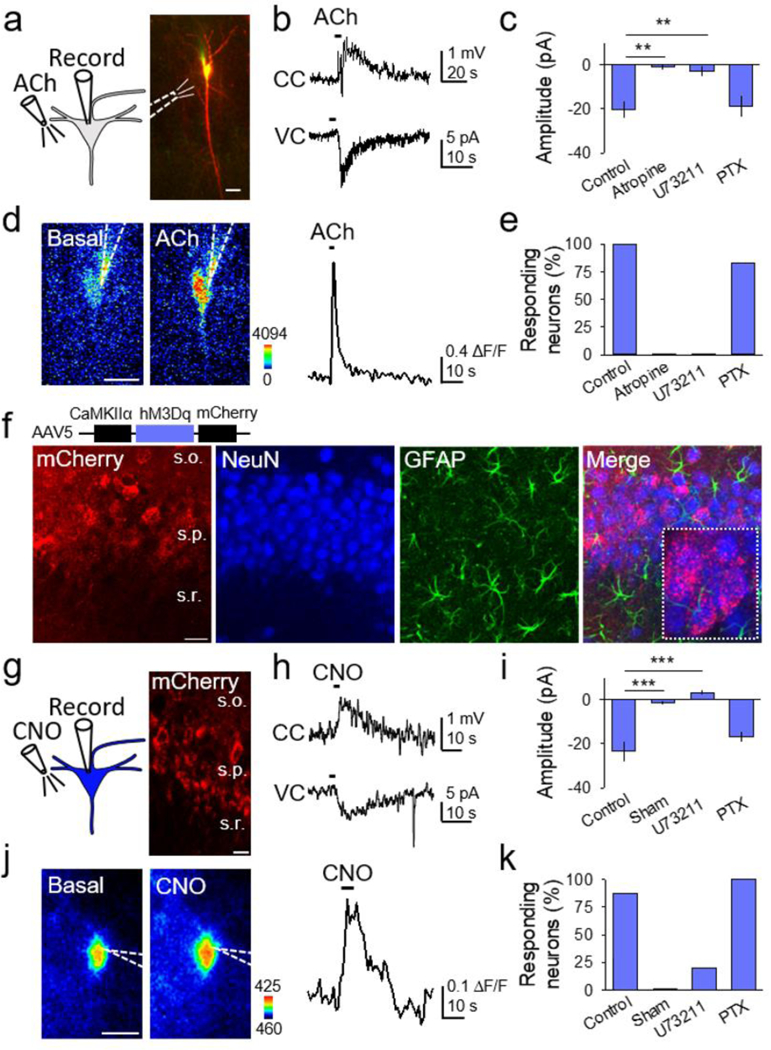
(A) Scheme of neuronal Gq GPCR activation by ACh, and TexasRed and Fluo4-filled CA1 neuron (scale bar, 20µm). (B) Representative traces showing effects of local ACh application in current clamp (CC) and voltage clamp (VC). (C) ACh-induced current amplitude in different conditions (Kruskal-Wallis One-way ANOVA, p < 0.001). (D) Pseudocolor Fluo4 fluorescence images before and after ACh application in the neuron depicted in (A) (scale bar, 20µm), and corresponding fluorescent Ca2+ trace. (E) Percentage of neurons that responded with a Ca2+ increase to ACh in different conditions. (F) Immunohistochemical images of hippocampus injected with AAV5-CaMKIIα-hM3Dq-mCherry. From left to right: mCherry, NeuN, GFAP and merge (scale bar, 25 μm; s.o., stratum oriens; s.p., stratum pyramidale; s.r., stratum radiatum). (G) Scheme of neuronal chemogenetic GqDREADD activation and image showing mCherry-expressing CA1 neurons (scale bar, 15 µm). (H) Representative traces showing effects of local CNO application in CC and VC. (I) CNO-induced current amplitude in different conditions (Kruskal-Wallis One-way ANOVA, p < 0.001). (J) Pseudocolor Fluo4 fluorescence image before and after CNO application (scale bar, 10µm), and corresponding fluorescent Ca2+ trace. (K) Percentage of neurons that responded with a Ca2+ increase to CNO in different conditions. Data are represented as mean ± SEM. P < 0.01 (**), and P < 0.001 (***).
Figure 3. Gi/o activation in neurons is inhibitory.
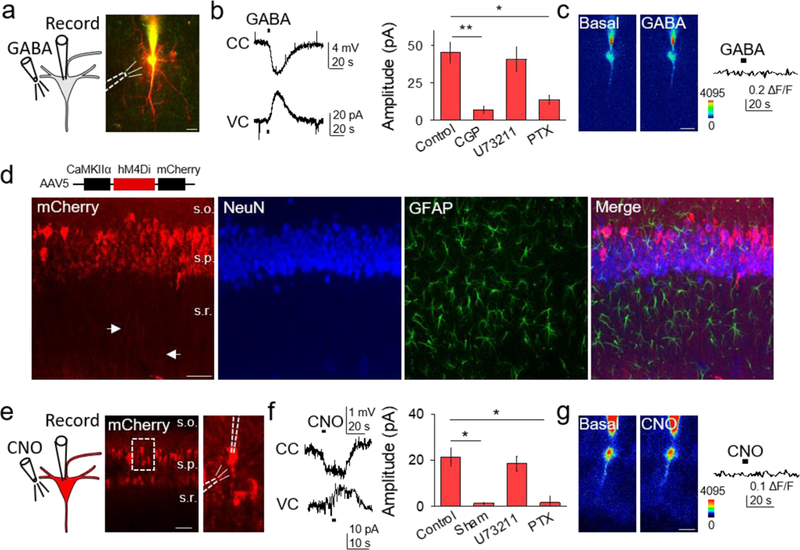
(A) Scheme of neuronal Gi/o activation by GABA, and TexasRed and Fluo4-filled CA1 neuron (scale bar, 20μm). (B) Representative traces showing GABA-induced responses in current clamp (CC) and voltage clamp (VC), and GABA-induced current amplitude in different conditions (Kruskal-Wallis One-way ANOVA, p < 0.01, p < 0.05). (C) Pseudocolor Fluo4 images before and after GABA application from the neuron depicted in (A) (scale bar, 20µm), and the corresponding fluorescent Ca2+ trace. (D) Immunohistochemical images of AAV5-CaMKIIα-hM4Di-mCherry expression in hippocampus. From left to right: mCherry, NeuN, GFAP and merge (scale bar, 50 μm; s.o., stratum oriens; s.p., stratum pyramidale; s.r., stratum radiatum). (E) Scheme of neuronal chemogenetic Gi/oDREADD activation and fluorescent image showing mCherry-expressing neurons (scale bar, 50µm). (F) Representative traces showing CNO-evoked responses in CC and VC, and CNO-induced current amplitude in different conditions (Kruskal-Wallis One-way ANOVA, p < 0.05). (G) Pseudocolor Fluo4 images before and after CNO application (scale bar, 20µm), and corresponding fluorescent Ca2+ trace. Data are represented as mean ± SEM. P < 0.05 (*) and P < 0.01 (**).
Figure 2. Gq activation in astrocytes elevates their Ca2+ levels.
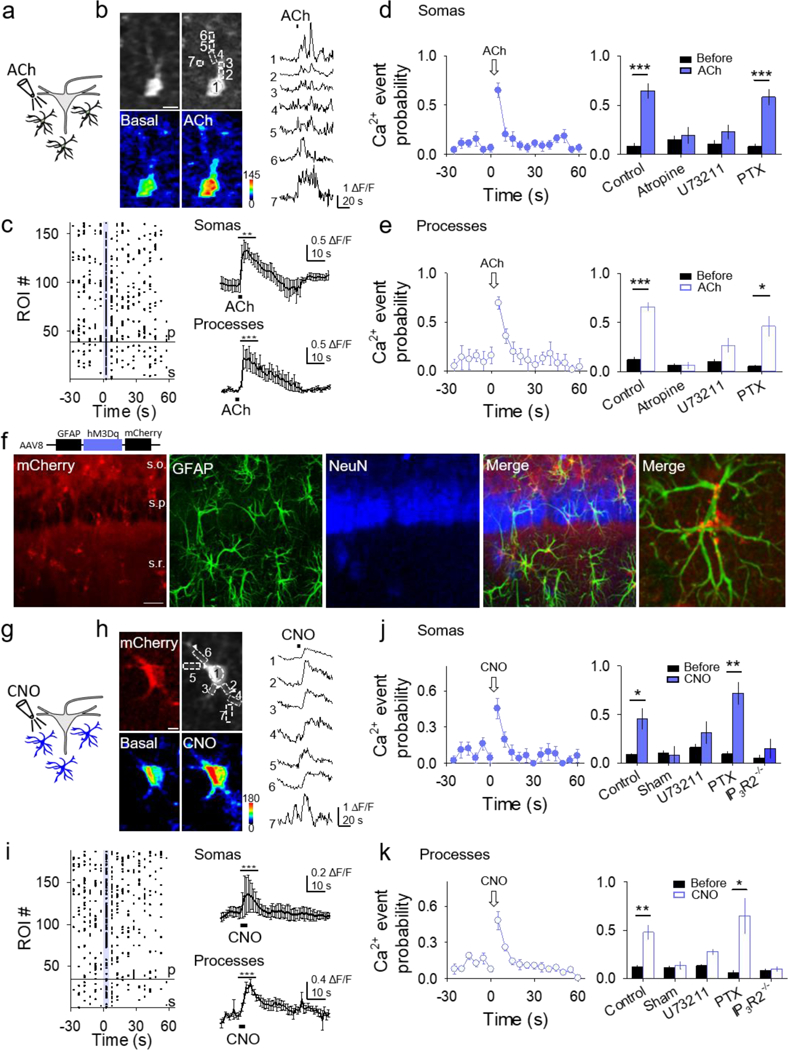
(A) Scheme of Gq GPCR activation in astrocytes by ACh locally applied over stratum radiatum astrocytes. (B) Fluorescence images of an astrocyte showing selected domains in soma and processes (top), pseudocolor Ca2+ images before (basal) and after local application of ACh (bottom) (scale bar, 5 μm), and Ca2+ traces from domains shown in top right panel. (C) Left: Raster plot showing Ca2+ events for each ROI (somas (s) below reference line, processes (p) above reference line). Right: Average Ca2+ traces from responding somas (top) and processes (bottom). T-test compares 10s before and 10s after agonist. (D and E) Ca2+ event probability in somas (D) and processes (E) vs. time, and maximum values in different conditions. (F) Immunohistochemical images of AAV8-GFAP-hM3Dq-mCherry expression in the hippocampus. From left to right: Expression of mCherry, GFAP, NeuN, a merge of all three, and a higher magnification merge image showing colocalization of GFAP and mCherry (25x; scale bar, 20 μm; s.o., stratum oriens; s.p., stratum pyramidale; s.r., stratum radiatum). (G-K) as (A-E) but with local application of CNO to GqDREADD expressing astrocytes instead of ACh. Data are represented as mean ± SEM. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
Figure 4. Gi/o activation in astrocytes elevates their Ca2+ levels.
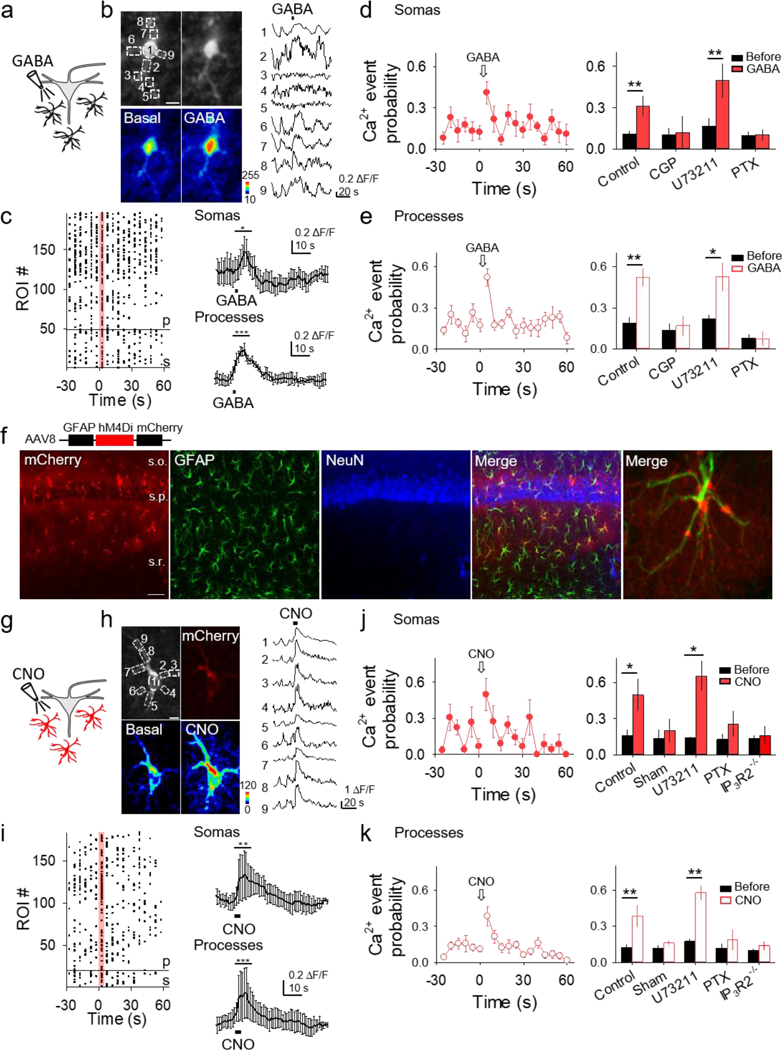
(A) Scheme of endogenous astrocyte Gi/o GPCR activation by GABA. (B) Fluorescence images of an astrocyte showing selected domains in soma and processes (top row), pseudocolor Ca2+ images before and after local application of GABA (bottom row; scale bar, 5 μm), and Ca2+ traces from domains shown in top left panel (right). (C) Left: Raster plot showing Ca2+ events for each ROI (somas (s) below reference line, processes (p) above reference line). Right: Average Ca2+ trace from responding somas (top) and processes (bottom). T-test compares 10s before and 10s after agonist. (D and E) Ca2+ event probability in somas (D) and processes (E) vs. time, and maximum values in different conditions. (F) Immunohistochemical images of AAV8-GFAP-hM4Di-mCherry expression in the hippocampus. From left to right: mCherry, GFAP, NeuN, a merge of all three, and a higher magnification merge image showing colocalization of GFAP and mCherry (25x; scale bar, 40 μm; s.o., stratum oriens; s.p., stratum pyramidale; s.r., stratum radiatum). (G-K) as (A-E) but with local application of CNO to Gi/oDREADD-expressing astrocytes instead of GABA. Data are represented as mean ± SEM. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
Figure 6. Astrocyte activation via Gq and Gi/o DREADDs induces slow inward currents in neurons.
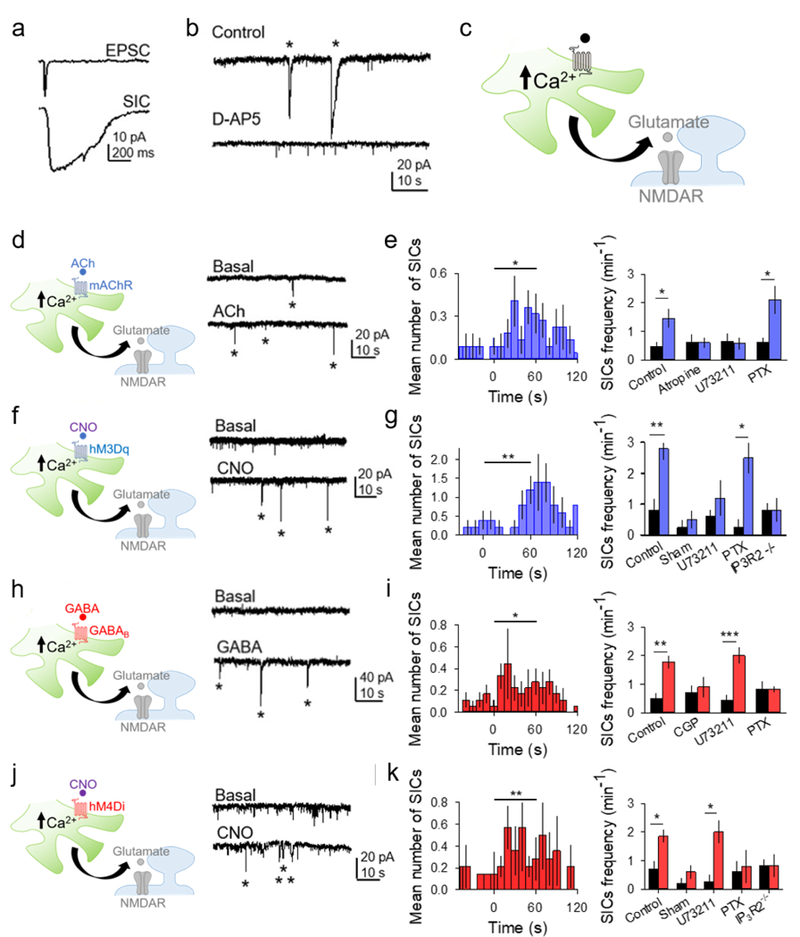
(A) Representative excitatory postsynaptic current (EPSC) and slow inward current (SIC) recorded from a CA1 pyramidal neuron. (B) Representative traces showing SICs (asterisks) in control and in D-AP5. (C) Scheme of neuronal-astroglial synaptic elements and gliotransmission. (D) Scheme and representative traces showing SICs (asterisks) before and after ACh application. (E) Left: Mean number of SICs vs. time, binned at 10s (ACh was applied at t=0). T-test compares one minute before and one minute after agonist. Right: SIC frequency (per min) one minute before and one minute after ACh in different conditions. Statistical significance was determined by the Student’s t-test comparing mean values one minute before and after agonist application. (F-G) as in (D-E) but with CNO application to GqDREADD-expressing astrocytes. (H-I) as in (D-E) but with GABA application instead of ACh. (J-K) as in (D-E) but with CNO application to Gi/oDREADD-expressing astrocytes. Data are represented as mean ± SEM. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
Ca2+ imaging
Ca2+ levels in astrocytes were monitored by two-photon microscopy (Leica DM6000 CFS upright multiphoton microscope with TCS SP5 MP laser) using the genetically encoded Ca2+ indicator mice GCaMP3 and GCaMP6 expressed in GLAST-positive cells. Some (n=199/512) soma Ca2+ responses were monitored by an Olympus BX51W1 epifluorescent microscope using Fluo-4-AM (5 μM in 0.01% of pluronic, 45min incubation at room temperature). Astrocytic Ca2+ levels were recorded from the astrocyte cell body and processes, and Ca2+ variations were estimated as changes in the fluorescence signal over the baseline (∆F/F0). A Ca2+ signal was considered a Ca2+ event when F/F0 >3 standard deviations greater than baseline fluorescence. Our measurement of Ca2+ event probability was calculated as the number of Ca2+ elevations grouped in 5s bins recorded from the astrocytes in the field of view (3–6 astrocytes per analyzed region), and mean values were obtained by averaging each different experiment. References to “n” in the text correspond to the number of experiments performed for each condition. Ca2+ analysis was performed only on astrocyte soma and processes within the field of view that showed fluctuations in Ca2+ levels throughout the recording. A domain was only selected along an active process length if its activity pattern differed from neighboring regions, so as to avoid oversampling of an active process. The number of process domains per experiment was 16.6 ± 0.48. Ca2+ levels in neurons were monitored using bulk loading of Oregon Green BAPTA-1 (5 μM in 0.01% of pluronic, 1hr incubation at room temperature) or adding Fluo4 (50 μM) to the recording pipette. A change in fluorescence (∆F/F0 > 3 standard deviations from baseline fluorescence) occurring within 5s of agonist application was defined as a neuronal Ca2+ response.
In vivo experiments and analysis
Two- to six-month-old mice were anesthetized using 1.8 mg/kg urethane injected intraperitoneally, faux tears were applied, and body temperature was maintained at 37°C. Once anesthetized, animals were placed in a stereotaxic frame and a midline incision was made along the scalp. A 3mm diameter craniotomy was performed centered over the primary somatosensory cortex (relative to Bregma in mm: −2.00 A-P, −2.00 M-L). A 0.25mm tungsten wire was placed over the exposed cortex to measure electrocorticography (ECoG), and agar was applied before a glass coverslip was cemented atop the craniotomy. A screw soldered to wire was inserted over the cerebellum to act as the reference and another screw was placed over the ipsilateral frontal plate. A 3D printed frame was mounted to the exposed skull with dental acrylic and fastened to a holder. The holder and animal were then placed underneath a Leica SP5 two-photon microscope for imaging. ECoG was recorded by connecting the lead to the exposed wire over the cortical surface, and reference and ground were attached to the wire soldered to the screw placed over the cerebellum. Prior to data acquisition, a needle was fed into the intraperitoneal cavity connected to a syringe with CNO. ECoG was sent to an AM Systems 3000 AC/DC differential amplifier sampled at 10kHz, filtered at 1Hz-3kHz and digitized using an Axon Digidata 1550 Acquisition System connected to a Dell Optiplex 7010 PC. In this arrangement, ECoG was recorded simultaneously with astrocyte Ca2+ monitored in cortical layers 2/3 (i.e. 100–300 µm below the cortical surface). Ca2+ imaging was obtained at 1 frame per second through a 25X objective with an additional 1.7X digital zoom. Following 20 minutes of baseline, 2–3 mg/kg CNO was injected intraperitoneally. Following CNO injections, measurements were obtained for 90 minutes. In a subset of mice, ECoG was measured in the absence of Ca2+ imaging. Raw ECoG data was lowpass filtered at 300 Hz and analyzed using a custom MATLAB program where a short-time Fourier transform was done using a hamming window to measure spectral content every minute. Normalized ECoG power spectra were created by using min-max normalization with respect to the baseline. In vivo Ca2+ data was analyzed using a custom MATLAB program to detect Ca2+ events via change in fluorescence, or the derivative of the fluorescence, when the derivative of fluorescence was > 2 standard deviations of the baseline speed over the average baseline speed. After an event was detected, the program detected the end of the event when the trace decayed back to the onset amplitude; from this the duration of the event was quantified, and the amplitude was calculated by taking the maximum value between onset and decay and subtracting it from the amplitude of the onset.
Immunohistochemistry
Anesthetized mice were perfused through the left cardiac ventricle with 4% paraformaldehyde in 0.1M PBS. The brains were removed, postfixed with paraformaldehyde and cut into 50 µm slices using a Leica VT 1000S vibratome. Brain sections were blocked with phosphate-buffered saline (PBS) containing 10% normal goat serum (NGS) or normal donkey serum (NDS) and 0.2% Triton X-100 and incubated overnight with the primary antibody diluted in blocking solution. Appropriate fluorochrome-labeled secondary antibodies (Life Technologies, Waltham, MA) were used for detection. An antibody against glial fibrillary acidic protein (GFAP, 1:1000; Sigma-Aldrich) was used as a marker for astrocytes. An antibody against neuron-specific nuclear protein (NeuN, 1:500; Millipore) was used as a marker for neurons. Fluorescent stained sections were mounted with Vectashield mounting medium (Vector Laboratories). The stained sections were visualized with a Nikon NiE C2 or an Olympus FluoView FV1000 upright confocal microscope and analyzed for colocalization of DREADDs with either neurons or astrocytes using ImageJ software (NIH, Bethesda, MD).
Quantification and statistical analyses
Data are expressed as mean ± standard error of the mean (SEM). Data normal distribution was assessed using a Shapiro-Wilk Normality test. Ca2+ event probability, Ca2+ fluorescence, SIC frequency, and in vivo Ca2+ events were analyzed using a two-tailed paired Student’s t-test (α = 0.05) comparing baseline to post-stimulus. In vivo delta power was analyzed using a Wilcoxon signed-rank test (α = 0.05) comparing baseline to post-stimulus. A one-way ANOVA was used with Holm-Sidak posthoc test to compare treatment groups against the control group for neuronal electrophysiology response data. When the Shapiro-Wilk Normality test failed, a Kruskal-Wallis ANOVA with a Dunn’s posthoc was used. Statistical differences were established with P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
RESULTS
Gq GPCR activation in neurons
We first investigated the effects on neurons of endogenous Gq GPCR activation by acetylcholine (ACh), which activates muscarinic ACh receptors (mAChRs). Type 1, 3 and 5 mAChRs, known to be coupled to Gq proteins, are highly expressed in hippocampal neurons (Berkeley et al., 2001; Park & Spruston, 2012; Scheiderer et al., 2008). We performed whole-cell electrophysiological recordings from hippocampal CA1 pyramidal neurons, monitored neuronal Ca2+ levels, and locally applied ACh (Figure 1A). CA1 neurons responded to ACh with an inward current in voltage clamp conditions, a transient depolarization in current clamp, and Ca2+ elevations (n = 9; Figures 1B–1E). These responses were abolished by the mAChR antagonist atropine (50 μM; n = 5; Figures 1C and 1E). Furthermore, ACh-evoked responses were prevented by the PLC inhibitor U73122 (4 μM; n = 7), but present after incubation with the Gi/o inhibitor pertussis toxin (7.5 μg/ml; PTX; n = 6; Figures 1C and 1E), which is consistent with the canonical Gq signaling pathway (Huang & Thathiah, 2015; Stewart & Fisher, 2015). These results indicate that ACh-induced activation of Gq GPCR signaling evokes neuronal responses (i.e. inward currents and Ca2+ increases) associated with neuronal excitation.
To confirm the effects of Gq signaling in neurons, we selectively expressed GqDREADDs in hippocampal CA1 pyramidal neurons using the viral vector AAV5-CaMKIIα-hM3Dq-mCherry, which contains the GqDREADD and the fluorescent reporter mCherry under the CaMKIIα promoter, which is highly expressed in CA1 pyramidal neurons (Erondu & Kennedy, 1985; Schulman & Lou, 1989). The selective expression of GqDREADDs in CA1 pyramidal neurons was confirmed by immunohistochemistry (Figure 1F) and the electrophysiological properties of the recorded neurons expressing mCherry (Gasparini & Magee, 2006; Magee & Carruth, 1999). Local application of the DREADD agonist Clozapine-N-oxide (CNO) evoked transient depolarizations, inward currents, and Ca2+ increases in GqDREADD-expressing neurons (n = 8; Figures 1G–1K), but not in neurons lacking GqDREADDs (n = 6; Figures 1I and 1K). CNO-evoked responses were diminished by U73122 (n = 5), but not by PTX (n = 6; Figures 1I and 1K). These results indicate that Gq activation by endogenous or chemogenetic means induces neuronal responses associated with cellular excitation.
Gq GPCR activation in astrocytes
Hippocampal astrocytes respond with Ca2+ elevations to ACh through activation of mAChRs (Araque, Martin, Perea, Arellano, & Buno, 2002; Shelton & McCarthy, 2000), but the signaling pathway activated remains undefined. Thus, we locally applied ACh and monitored Ca2+ levels in the soma and processes of astrocytes located in the CA1 stratum radiatum using transgenic mice that expressed the genetically-encoded Ca2+ indicators GCaMP3 or GCaMP6 under the astroglial GLAST promoter (Figure 2A). ACh transiently elevated Ca2+ in both astrocyte somas and processes (Figures 2B-E) (Araque et al., 2002; Navarrete et al., 2012; Perea & Araque, 2005; Shelton & McCarthy, 2000; Takata et al., 2011), as quantitatively shown by the increase of the Ca2+ event probability (36 astrocyte somas, n = 6; 122 process domains, n = 5; Figures 2D–2E). ACh-induced Ca2+ increases were blocked by the mAChR antagonist atropine (18 astrocyte somas, n = 6; 78 process domains, n = 6; Figures 2D and 2E) and by the PLC inhibitor U73122 (27 astrocyte somas, n = 7; 106 process domains, n = 7; Figures 2D and 2E), but were present in slices incubated with the Gi/o inhibitor PTX (54 astrocyte somas, n = 9; 83 process domains, n = 7; Figures 2D and 2E). These results indicate that mAChRs in astrocytes activate Gq proteins that increase intracellular Ca2+ levels through stimulation of PLC.
To selectively stimulate Gq-linked GPCRs in astrocytes, we targeted stratum radiatum hippocampal astrocytes with the viral vector AAV8-GFAP-hM3Dq-mCherry, which contains the GqDREADD and the fluorescent reporter mCherry under the control of the astroglial GFAP promoter (Figure 2F). CNO application mimicked the ACh effects, elevating Ca2+ in both somas and processes of GqDREADD-expressing astrocytes (35 astrocyte somas, n = 6; 153 process domains, n = 6; Figures 2G–2K), but not in astrocytes lacking GqDREADD expression (18 astrocyte somas, n = 6; 100 process domains, n = 6; Figures 2J and 2K). CNO-evoked responses in both somas and processes were blocked by U73122 (20 astrocyte somas, n = 5; 83 process domains, n = 6), but were still present in slices incubated with PTX (18 astrocyte somas, n = 5; 53 process domains, n = 5; Figures 2J and 2K), indicating that Gq GPCR signaling pathways increase Ca2+ in astrocytes by activation of PLC. To further test this idea and to determine the source of the Ca2+ elevation and the intracellular signaling downstream of GqDREADD activation, we performed the experiment in IP3R2−/− mice (Li et al., 2005), which lack type 2 IP3 receptors, the main receptor subtype responsible for astrocytic Ca2+ mobilization from internal stores (Di Castro et al., 2011; Gomez-Gonzalo et al., 2015; Martin-Fernandez et al., 2017; Martin, Bajo-Graneras, Moratalla, Perea, & Araque, 2015; Navarrete et al., 2012; Petravicz, Fiacco, & McCarthy, 2008). CNO application in GqDREADD-expressing slices from IP3R2−/− mice showed no significant increase in Ca2+ event probability (14 astrocyte somas, n = 5; 46 process domains, n = 5; Figures 2J and 2K), indicating that Gq-GPCR activation induces astrocyte Ca2+ mobilization from internal stores. Taken together, these results indicate that stimulating Gq GPCR signaling evokes astrocyte responses (i.e. Ca2+ increases) associated with astrocyte activation.
Gi/o GPCR activation in neurons
To investigate Gi/o signaling cascade effects on neurons, we used GABA to activate endogenous GABAB receptors, which are known to be coupled to Gi/o proteins (Logothetis, Kurachi, Galper, Neer, & Clapham, 1987; North, 1989; Wickman et al., 1994). GABAB-mediated responses were isolated by blocking GABAA receptors with picrotoxin (50 μM). In CA1 pyramidal neurons, GABA application evoked a hyperpolarization in current-clamp conditions, and an outward current in voltage-clamp conditions (n = 8; Figures 3A and 3B), with no changes in Ca2+ levels (n = 6; see Figure 3C). The outward currents were blocked by the GABAB receptor antagonist CGP54626 (1 mM; n = 5), as well as by the Gi/o inhibitor PTX (n = 5), but were unchanged by the PLC inhibitor U73122 (n = 5; Figure 3B). These data indicate that Gi/o intracellular signaling cascades evoke effects (i.e. outward currents and absence of Ca2+ increases) consistent with neuronal inhibition.
We further investigated the effects of neuronal Gi/o-linked GPCRs using the Gi/oDREADD (Armbruster, Li, Pausch, Herlitze, & Roth, 2007), which was specifically expressed in CA1 pyramidal neurons with the viral vector AAV5-CaMKIIα-hM4Di-mCherry (Figure 3D and 3E). CNO application hyperpolarized and evoked outward currents in Gi/oDREADD-expressing neurons (n = 7), but not in neurons lacking Gi/oDREADD expression (n = 5; Figure 3F). These effects were absent in PTX-incubated slices (n = 5), but were similar to control in the presence of U73122 (n = 5; Figure 3F). Like GABA, CNO did not modify Ca2+ levels (n = 6; see Figure 3G). These data indicate that Gi/o signaling evoked by GABA or DREADDs has inhibitory actions on neurons.
Gi/o GPCR activation in astrocytes
We then tested the effects of Gi/oGPCR stimulation on astrocyte Ca2+ activity. Local application of GABA increased the Ca2+ event probability in both somas and processes (49 astrocyte somas, n = 10; 147 process domains, n = 7; Figures 4A–4E). These effects were not observed in PTX-treated slices (57 astrocyte somas, n = 9; 69 processes domains, n = 5; Figures 4D and 4E) or in the presence of the GABAB receptor antagonist CGP54626 (26 astrocyte somas, n = 5; 61 process domains, n = 6; Figures 4D and 4E), but were still present in U73122 conditions (38 astrocyte somas, n = 8; 161 process domains, n = 5; Figures 4D and 4E), indicating that, unlike in neurons, stimulation of Gi/oGPCR signaling in astrocytes leads to cellular activation.
To further test this hypothesis, Gi/oDREADDs were specifically expressed in astrocytes using the viral vector AAV8-GFAP-hM4Di-mCherry (Figure 4F). CNO application transiently increased the Ca2+ event probability in somas and processes of Gi/oDREADD-expressing astrocytes (21 astrocyte somas, n = 5; 163 process domains, n = 8), but not in non-expressing astrocytes (19 astrocyte somas, n = 5; 71 process domains, n = 5; Figures 4G–4K). These Ca2+ responses were present in U73122 (20 astrocyte somas, n = 5; 160 process domains, n = 6), but were absent in PTX-treated slices (20 astrocyte somas, n = 6; 144 process domains, n = 6), and in slices from IP3R2−/− mice (22 astrocyte somas, n = 5; 48 process domains, n = 5; Figures 4J and 4K). Taken together, these results indicate that Gi/o signaling evoked by endogenous and chemogenetic approaches resulted in astrocyte activation manifested as Ca2+ increases.
Simultaneous activation of Gq and Gi/o GPCR signaling in astrocytes
We further tested the hypothesis that Gq and Gi/o GPCR signaling in astrocytes operates through distinct intracellular pathways. We hypothesized that two agonists acting on similar GPCR signaling pathways would interact producing a non-linear response, whereas two agonists activating different GPCR signaling would evoke a linear response. We first tested two agonists that are known to activate Gq GPCR signaling, ACh and group I mGluR agonist (S)-3,5-Dihydroxyphenylglycine (DHPG). We either applied them separately or both simultaneously and monitored the Ca2+ responses. We found that the observed Ca2+ response elicited by the simultaneous ACh and DHPG application was lower than the response expected if there was no interaction; i.e., the linear summation of the peak Ca2+ elevation evoked by each agonist applied separately (n = 26; Figures 5A, 5B, and 5E) (Perea & Araque, 2005). This relative reduction of the response evoked by these agonists indicates an interaction of the signaling pathways activated and suggests that they operated through the same intracellular pathways. Next, we applied separately ACh, GABA, or both simultaneously to measure the relative contribution of Gq and Gi/o GPCR signaling, respectively, to the astrocyte Ca2+ response. We found that the observed amplitude of the Ca2+ response to simultaneous ACh and GABA application was similar to that of the expected response; i.e., the linear summation of the responses evoked independently (n = 29; Figures 5C, 5D, and 5E). Moreover, the peak amplitude of the Ca2+ response could be further increased by increasing the concentration of ACh (peak fluorescence evoked by 1 mM ACh: 39.4 ± 4.8%, peak fluorescence evoked by 10 mM ACh: 64.6 ± 8.3%, p = 0.040, n = 4 videos, 13 astrocytes), indicating that the receptors and signaling pathways were not saturated. The fact that the Ca2+ response linearly summated with simultaneous Gq and Gi/o GPCR agonist application suggests that distinct intracellular pathways were activated, indicating that the actions of Gi/o and Gq GPCRs do not occlude and that the detected Ca2+ signal is not saturated. Furthermore, these results indicate that two distinct intracellular signaling mechanisms are activated downstream of Gq and Gi/o GPCR signaling in astrocytes.
Figure 5. Gi/o and Gq GPCRs stimulate different signaling pathways that do not occlude.
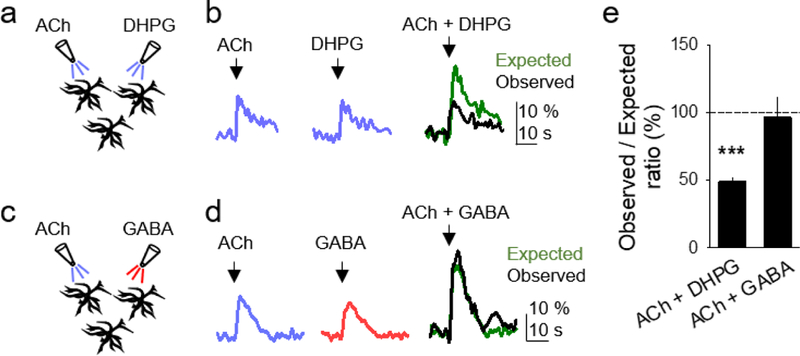
(A and B) Scheme of experimental set-up, and astrocyte Ca2+ responses to ACh (left), DHPG (middle), and both simultaneously (right), showing the observed (black trace) and expected (green trace) responses. Expected response corresponds to the linear summation of the responses evoked by independent application of ACh and DHPG. (C and D) as in (A and B), but with GABA instead of DHPG. (E) Histogram of the ratio of the observed and expected Ca2+ responses evoked by simultaneous application of GPCR agonists in the different pharmacological conditions. Expected responses were considered the linear summation of the responses elicited by independent stimulation of the agonists. Data are represented as mean ± SEM. P < 0.001 (***).
Downstream effects of astrocytic Gq and Gi/o GPCR activation on neurons
Next, we investigated whether activation of Gq and Gi/o protein signaling in astrocytes can lead to gliotransmitter release and regulation of neuronal excitability. Astrocyte Ca2+ elevations have been shown to stimulate the release of glutamate, which evokes slow inward currents (SICs) mediated by activation of neuronal NMDA receptors (Araque, Parpura, Sanzgiri, & Haydon, 1998; Araque et al., 1999; Araque, Sanzgiri, Parpura, & Haydon, 1998; Fellin et al., 2004; Mariotti et al., 2016; Martin et al., 2015; Navarrete & Araque, 2008; Perea & Araque, 2005; Perea, Sur, & Araque, 2014). SICs can be distinguished from synaptic currents by their different time courses and their sensitivity to the NMDA receptor antagonist D-AP5 (50 µM; average SIC amplitude = 19.7 pA ± 1.6 pA; n = 182; Figures 6A–6C). We used SICs as a biological assay to detect astrocytic glutamate release. ACh application, which increased astrocyte Ca2+ via Gq protein activation (see Figure 2), transiently increased the frequency of SICs in CA1 pyramidal neurons (n = 11; Figures 6D and 6E). In correspondence with the ACh-evoked effects on Ca2+, this effect was blocked by atropine (n = 5) and U73122 (n = 7), but was present in PTX-treated slices (n = 5; Figure 6E). Similarly, CNO application in slices with GqDREADD-expressing astrocytes transiently increased SIC frequency (n = 5), an effect that remained in PTX-treated slices (n = 4; Figures 6F–6G). This effect was absent in the presence of U73122 (n = 5), in sham conditions with no expression of DREADDs (n = 4), and in slices from IP3R2−/− mice (n = 8; Figures 6G). These data indicate that Gq-linked GPCR activation in astrocytes stimulates the release of glutamate that activates neuronal NMDA receptors.
We then quantified the effects of Gi/o GPCR activation on SIC frequency. GABA transiently increased the frequency of SICs (n = 9; Figures 6H and5I). As in the case of GABA-evoked Ca2+ responses, SIC frequency increases were abolished by CGP54626 (n = 5) and PTX (n = 6), but were detected in slices treated with U73122 (n = 9; Figure 6I). Likewise, CNO application in slices with Gi/oDREADD-expressing astrocytes increased SIC frequency in control conditions (n = 7) and in the presence of U73122 (n = 4), but this effect was absent in PTX-incubated slices (n = 5), in sham conditions (n = 5), and in slices from IP3R2−/− mice (n = 8; Figures 6J and 6K). Taken together, Gq and Gi/o GPCR activation in astrocytes led to increases in Ca2+, which were associated with an increase in the number of SICs in nearby neurons.
SICs are proposed to enhance neuronal excitability and regulate neuronal synchronization (Angulo, Kozlov, Charpak, & Audinat, 2004; Fellin et al., 2004). We directly tested this idea by investigating the effects of Gq and Gi/o GPCR stimulation in astrocytes on action potential firing in nearby neurons (Figure 7). We activated Gq- and Gi/oDREADD-expressing astrocytes while recording hippocampal CA1 pyramidal neurons in current-clamp conditions. In a first approach, the neuronal membrane potential was slightly depolarized from resting membrane potential by injecting a continuous current to facilitate firing of spontaneous action potentials (Figures 7A and 7B). In a second approach, a continuous train of depolarizing current pulses (20ms, ~200 pA, delivered every 50ms) was applied to observe any potential changes in action potential firing (Figures 7C and 7D). In both conditions, CNO application to either Gq- or Gi/oDREADD-expressing astrocytes enhanced the frequency of action potential firing (Figures 7A–7D, and 7G-7H), which agrees with the gliotransmitter-evoked enhancement of neuronal excitability (see Figure 6). Indeed, the astrocyte Gq- and Gi/oDREADD effects on action potential firing were accompanied by small, but conspicuous, transient depolarizations (insets in Figures 7C and 7D), which appear consistent with the SICs recorded in voltage-clamp (see Figures 6F and 6J), although experimental limitations cannot provide direct evidence that SICs (recorded in voltage-clamp) are responsible for these depolarizations (recorded in current-clamp). Nevertheless, activation of both Gq- and Gi/oDREADDs in the presence of the NMDA receptor antagonist D-AP5 failed to evoke the previously observed transient depolarizations and increase in action potential firing (Figures 7E–7H), supporting the idea that these phenomena were due to neuronal NMDAR activation by astrocytic glutamate. Taken together, these results indicate that activation of both Gq and Gi/o proteins activated astrocytes in the form of a Ca2+ elevation. This cellular activation in astrocytes subsequently stimulated gliotransmitter release that led to the enhancement of neuronal excitability.
Effects of astrocytic Gq and Gi/o GPCR signaling in vivo
Finally, we investigated whether similar phenomena occurred in vivo. We injected the viral vectors AAV8-GFAP-hM3Dq-mCherry or AAV8-GFAP-hM4Di-mCherry into the primary somatosensory cortex to selectively express in cortical astrocytes Gq or Gi/o DREADDs, respectively (Figure 8A). Two weeks after injection, we monitored astrocyte Ca2+ activity and cortical local field potentials in anesthetized mice, in basal conditions and 1 hour after intraperitoneal injection of CNO (2–3 mg/kg). In mice (n = 6) with GqDREADD-expressing astrocytes, CNO increased the frequency (from 1.30 ± 0.06 to 2.70 ± 0.28 events/min; n = 21 astrocytes), amplitude (to 218.9 ± 31.2 % from basal values between each astrocyte), and duration of the Ca2+ events (from 2.84 ± 0.17 to 6.00 ± 0.69 s) (Figures 8B-8G). Similarly, CNO injection to mice (n = 6) with Gi/oDREADD-expressing astrocytes also increased the frequency (from 1.21 ± 0.08 to 2.00 ± 0.23 events/min; n = 24 astrocytes), amplitude (to 229.5 ± 32.724 % from basal values of each astrocyte), and duration of Ca2+ events (from 4.20 ± 0.79 to 6.62 ± 0.80 s) (Figures 8B-8G). Introducing saline into mice (n = 3) with GqDREADD-expressing astrocytes did not increase the frequency (from 1.35 ± 0.05 to 1.30 ± 0.05 events/min in 37 astrocytes), amplitude (to 98.1 ± 6.0 % from basal values between astrocytes), or duration of the Ca2+ events (from 2.81 ± 0.11 to 2.80 ± 0.12 s). Similarly, introducing saline in mice (n = 3) with Gi/o-DREADD-expressing astrocytes did not increase frequency (from 1.30 ± 0.04 to 1.34 ± 0.06 events/min; n = 53), amplitude (to 114.8 ± 10.3 % from basal values between astrocytes), or duration (from 2.80 ± 0.10 to 2.83 ± 0.13 s) of the Ca2+ events. Additionally, introducing CNO in mice (n = 3) expressing mCherry in astrocytes did not increase the frequency (from 1.32 ± 0.04 to 1.44 ± 0.06 events/min; n = 77), amplitude (to 118.7 ± 9.3 % from baseline between astrocytes), or duration of the Ca2+ events (from 2.78 ± 0.09 to 3.10 ± 0.16 s).
Figure 8. In vivo activation of both Gq and Gi/o DREADDs in astrocytes elevates their Ca2+ levels and regulates neuronal electrical activity.
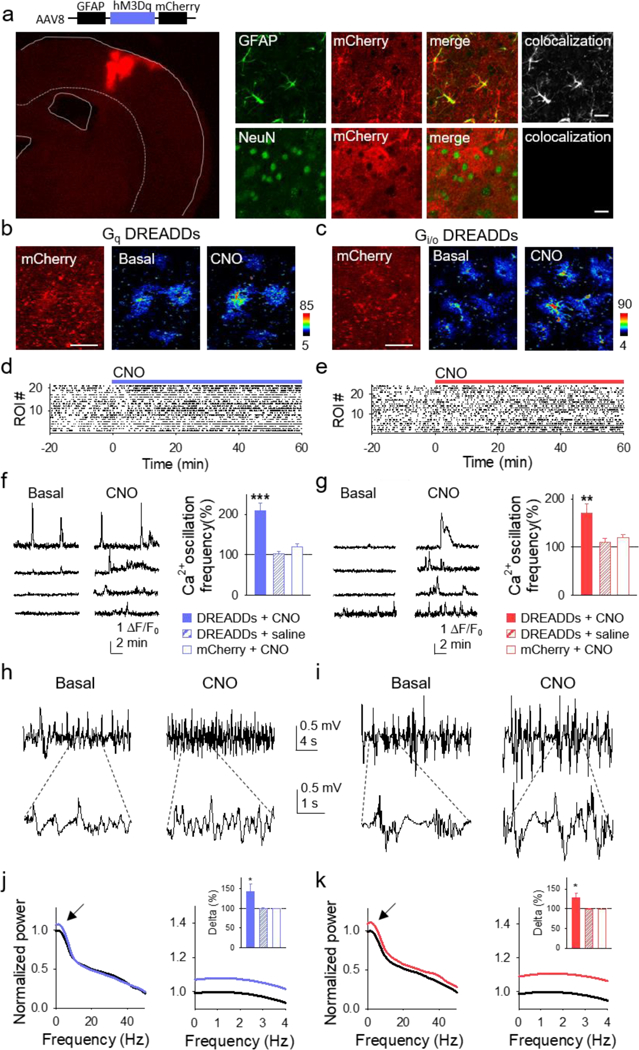
(A) Immunohistochemical representative image of GqDREADDs virus injected into the primary somatosensory cortex staining for neurons (NeuN; blue), astroglia (GFAP; green), the reporter mCherry (red), and colocalization of mCherry with GFAP (top) and NeuN (bottom; scale bar, 20 μm). (B and C) Left: Representative images of mCherry in astrocytes reporting expression of GqDREADDs and Gi/oDREADDs (scale bar, 50 μm). Pseudocolor Ca2+images before (middle) and after (right) intraperitoneal injection (i.p.) of CNO. (D and E) Raster plots of Ca2+ events before and after i.p. injection of CNO in mice with astrocytes expressing Gq and Gi/oDREADDs, respectively. (F and G) Representative Ca2+ traces during basal (left) and after i.p. CNO injection (right) in GqDREADD- (F) and Gi/oDREADD-injected (G) mice. Ca2+ oscillation frequency normalized to baseline for DREADDs-infected cortex with i.p. CNO injections (solid bars), for DREADDs-infected cortex with i.p. saline injections (hashed bars), and for mCherry control-infected cortex with i.p. CNO injections (unfilled bars) for GqDREADDs- (F) and Gi/oDREADDs-injected (G) mice. (H and I) Representative cortical local field potential recordings prior to CNO (left) and after (right) in GqDREADDs- (H) and Gi/oDREADDs-injected (I) mice. Lowpass filtered at 40Hz. (J and K) Normalized power spectrum of cortical local field potentials before (black) and after CNO in GqDREADDs- (J: blue) and Gi/oDREADDs-injected (K: red) mice. Normalized power spectrum of slow-wave delta (0–4 Hz) activity for DREADDs-infected cortex with i.p. CNO injections (solid bars), DREADDs-infected cortex with i.p. saline injections (hashed bars), and mCherry control-infected cortex with i.p. CNO injections for GqDREADDs- (J) and Gi/oDREADDs-injected (K) mice. Error bars are SEM. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***).
Associated with the CNO-evoked astrocyte activation, CNO altered the electrical network activity manifested as an enhancement of the slow-wave delta activity (0–4 Hz) in mice with astrocytes expressing either Gq or Gi/oDREADDs (n = 6 mice for each case; Figures 8H-8K). These effects were not observed when mice with either Gq or Gi/oDREADDs-expressing astrocytes were injected with saline (n=3 mice for each case; Figures 8F, 8G, 8J, and 8K), or when CNO was delivered to mice infected with the control virus GFAP-mCherry that lacks DREADDs (n=3 mice; Figures 8F, 8G, 8J, and 8K). These results indicate that in vivo activation of either Gq or Gi/o GPCR signaling in astrocytes similarly led to the activation of astrocytes, and that these effects were associated with alterations of the neural delta network activity.
DISCUSSION
The present results show that Gq GPCR activation in neurons and astrocytes activated both cell types. In contrast, while neuronal Gi/o GPCR activation inhibited cellular activity, Gi/o GPCR stimulation in astrocytes enhanced their Ca2+-based cellular activity. We found that activation of endogenous Gq or Gi/o GPCRs or DREADDs in astrocytes induced Ca2+ increases in astrocyte somas and processes and stimulated the release of the gliotransmitter glutamate, which led to an increase in SIC frequency and action potential firing in hippocampal neurons, and impacted neuronal network activity in vivo. Hence, astrocyte G protein-mediated signaling, whether Gq or Gi/o, activated astrocytes by way of an evoked Ca2+ response. Therefore, neurotransmitters acting on Gi/o GPCRs proteins directly inhibit neurons but activate astrocytes, which then can feed-forward excite neurons. Consistent with this idea, we have found that astrocyte specific Gq or Gi/o GPCR activation enhanced astrocyte Ca2+ events and delta activity in vivo. These results add further complexity to the signaling mechanisms and effects of GPCR-mediated signaling in the brain, revealing important and unexpected functional consequences on the actions of inhibitory neurotransmitters in astroglial-neuronal networks.
Our results show that neuronal Gq GPCR activation either with ACh or GqDREADDs increase neuronal excitability, both inducing inward currents, membrane depolarizations, and Ca2+ elevations through PLC-mediated intracellular signaling. These results are in agreement with previous studies that reported that electrophysiological and Ca2+ signal effects of ACh on neurons are mediated by PLC activation (Aiken, Lampe, Murphy, & Brown, 1995; Brown & Yu, 2000; Dasari & Gulledge, 2011; Suh & Hille, 2002; Zhang et al., 2003). GqDREADD activation has also been shown to induce neuronal Ca2+ increases (Alexander et al., 2009; Armbruster et al., 2007). Present data further show that GqDREADD activation also evokes similar electrophysiological responses as ACh, and that the Ca2+ elevations are mediated by the PLC signaling pathway. In contrast, we found that endogenous or chemogenetic Gi/o GPCR activation elicited neuronal outward currents and hyperpolarizations (Armbruster et al., 2007; Logothetis et al., 1987; North, 1989; Wickman et al., 1994; Zhu et al., 2014), and no Ca2+ changes. Regarding Ca2+ signaling, the βγ subunit dissociation after Gi/o activation can activate either PLC (Singer, Brown, & Sternweis, 1997) or the IP3R directly (Zeng et al., 2003), leading to Ca2+mobilization from internal stores. It was interesting that we did not see this increase in neuronal Ca2+, suggesting either that this signaling pathway was not activated, or that we were not able to detect it. This finding contrasts with results from Gi/oDREADD signaling in astrocytes (see below), where Gi/oDREADD activation led to Ca2+ increases. Taken together, these data are in agreement with and expand upon previous literature showing that Gi/o activation is inhibitory in neurons.
In contrast to the differential effects of Gq and Gi/o GPCR signaling in neurons, both types of G proteins led to cellular activation in astrocytes in the form of Ca2+ increases. Our results show that ACh and GqDREADD activation similarly increases Ca2+ in astrocyte somas and processes through PLC- and IP3R2-mediated signaling pathways. Our data expand upon existing literature regarding astrocyte responsiveness to ACh (Araque et al., 2002; Chen et al., 2012; Navarrete et al., 2012; Perea & Araque, 2005; Shelton & McCarthy, 2000; Takata et al., 2011). Since previous studies used either a nonselective mAChR antagonist or techniques to manipulate Ca2+ release from internal stores, the specific GPCR pathway mediating the Ca2+ responses was not known. Here we used PLC and Gi/o inhibitors to block Gq- and Gi/o-mediated effects, respectively. Since Gi/o GPCR signaling has also been reported to activate PLC through the βγ subunit (Singer et al., 1997), U73122 treatment could also block any potential Gi/o-induced activation of PLC. Additionally, the βγ subunits that dissociate from Gi/o GPCR activation could directly activate the IP3R (Zeng et al., 2003). Accordingly, there could be residual Ca2+ elevation with U73122 treatment alone, due to βγ activation of the IP3R, as well as with PTX treatment alone, due to PLC-activated production of IP3. However, we observed that the ACh-induced Ca2+ responses were largely mediated through Gq-PLC-IP3 than through Gi/o-βγ-IP3R2 signaling.
GqDREADDs have been previously used to specifically activate astrocytes (Adamsky et al., 2018; Agulhon et al., 2013; Bonder & McCarthy, 2014; Bull et al., 2014; Chai et al., 2017; Martin-Fernandez et al., 2017; Scofield et al., 2015), but the Ca2+ dynamics and intracellular signaling pathways involved were not fully characterized. We show that GqDREADD activation stimulated Ca2+ increases in a manner akin to endogenous Ca2+ dynamics evoked by an endogenous stimulus (i.e., ACh; see Figure 2), and through the canonical signaling of Gq GPCRs, i.e., PLC activation and IP3-mediated Ca2+ mobilization. These results indicate that astrocyte activation via GqDREADDs closely mimics physiological stimuli.
Unlike in neurons, endogenous and chemogenetic Gi/o GPCR signaling led to astrocyte activation in the form of Ca2+ increases in a PTX-sensitive manner, demonstrating that the responses were indeed mediated by the Gi/o type of G protein. This is in line with other studies showing Ca2+ elevations in astrocyte somas upon Gi/o-coupled GABAB receptor activation (Covelo & Araque, 2018; Kang et al., 1998; Meier et al., 2008; Navarrete & Araque, 2008; Perea et al., 2016; Serrano, Haddjeri, Lacaille, & Robitaille, 2006). Here, we show GABAB activation also induces Ca2+ increases in astrocytic processes. It seems highly likely that other endogenous neurotransmitters coupled to Gi/o GPCRs may also activate astrocytes, but further studies are required to test this hypothesis.
Boddum et al. (2016) have recently reported that activation of astrocyte GABA transporters lead to astrocytic Na+ concentration elevations and consequent astrocytic Ca2+ increases through Na+/Ca2+ exchange (Boddum et al., 2016). Present results indicate that the GABA effects on astrocyte Ca2+ are mediated by GABAB receptor activation because they are blocked by the receptor antagonist CGP. These results are consistent with two different and complementary mechanisms mediated by GABA transporters and receptors that are revealed by different experimental conditions. Indeed, Boddum et al. used long (≥ 5min) bath applications of GABA, which led to astrocytic Na+ concentration raise, consequent increase in astrocytic Ca2+ through Na+/Ca2+ exchange and downstream effects with slow time course (on a temporal scale of minutes). In our study, we used local puff application of GABA (2s) and monitored the short-term effects on astrocyte Ca2+ (on a temporal scale of seconds). Therefore, GABA signaling in astrocytes may occur through two complementary mechanisms with different time courses mediated by GABA transporters and receptors.
It has been reported that GABAB receptor-induced Ca2+ elevations can be attributed to PLC signaling (Hirono, Yoshioka, & Konishi, 2001; New, An, Ip, & Wong, 2006) and PTX-sensitive Ca2+ mobilization through the IP3R2 (Mariotti et al., 2016). Gi/o GPCR activation can result in PLC-induced Ca2+ mobilization through Gi/o-βγ-PLC signaling (Singer et al., 1997), however, we found no effect of U73122 in GABA- and CNO-induced Ca2+ signaling in astrocytes, and instead found that PTX treatment alone abolished the Ca2+ increase. Moreover, the lack of CNO-evoked Ca2+ increases in IP3R2−/− mice suggests that both the Gq and Gi/o protein-induced Ca2+ increases require IP3R2 activation. These data suggest that Gi/oGPCR-induced Ca2+ elevations in astrocytes may have been mediated via βγ subunits directly binding the IP3R2 (Zeng et al., 2003) and not through PLC activation. Future experiments, for example using βγ inhibitors, are necessary to determine the specific signaling cascade leading to Ca2+ elevations. Taken together, both GPCR pathways appear to converge on the IP3R2, but differ in the exact signaling mechanisms leading to IP3R2 activation. Lastly, we found in vivo that activation of either Gq or Gi/o DREADDs targeted to cortical astrocytes induced an increase in Ca2+ events following CNO injection. These data provide evidence that this chemogenetic approach to selectively activate astrocytes remains feasible in a more intact preparation.
Both Gq- and Gi/o-induced astrocyte Ca2+ elevations, whether elicited by endogenous receptors or DREADDs, stimulated glutamate release from astrocytes, which was detected as an increase in the frequency of SICs. SICs, which are known to be mediated by activation of neuronal NMDA receptors, have been found to enhance neuronal excitability and increase neuronal synchrony (Angulo et al., 2004; Araque, Sanzgiri, et al., 1998; Fellin et al., 2004; Perea & Araque, 2005; Shigetomi, Bowser, Sofroniew, & Khakh, 2008). While a recent study using GqDREADDs in astrocytes failed to detect significant changes in SIC frequency upon CNO stimulation (Chai et al., 2017), we observed robust effects. Although the origin of these discrepant results is unclear, it may be due to different sensitivity in detecting SICs, as indicated by our relatively lower mean SIC amplitude.
In addition to an increase in SIC frequency in nearby neurons, selective astrocyte activation via either GqDREADD or Gi/oDREADD signaling led to an increase in action potential firing of hippocampal neurons. While most reports of neuronal-glial interactions have shown synaptic transmission regulation by astrocytes (Araque et al., 2014), present results add to recent studies showing that astrocytes can also regulate neuronal firing and network activity (Lee et al., 2014; Poskanzer & Yuste, 2011, 2016; Shen, Nikolic, Meunier, Pfrieger, & Audinat, 2017; Tan et al., 2017). Indeed, the observed increase in astrocyte Ca2+ activity upon CNO injection in vivo co-occurred with an upregulated delta range of the slow-wave activity. This is consistent with previous studies showing the contribution of gliotransmission to in vivo network function (Fellin et al., 2009; Poskanzer & Yuste, 2011). Interestingly, our findings show that stimulating either the Gq or Gi/o protein pathways in astrocytes led to increases in astrocyte Ca2+ and slow-wave activity in the delta range in vivo, suggesting both pathways play similar regulatory roles in the living brain. The absence of effects of CNO administration in slice and in vivo in mice injected with the control virus AAV8-GFAP-mCherry indicates that the observed effects of CNO are not due to off-target effects but rather direct activation of astrocytes. It is important to note that mechanistic correlations should not be made between the results obtained in situ with those obtained in vivo, as these two regions have distinct neuronal-glial network interactions. Rather, we aimed to expand our results in vivo to test the hypothesis that activating astrocytes either via Gq or Gi/o protein signaling pathways influenced neuronal network activity. Taken together, astrocytes have multiple modulatory roles, controlling neuronal information processing via synaptic transmission as well as directly regulating neuronal output and network activity.
The excitation/inhibition balance is important for proper brain function, and its dysfunction may lead to brain disorders, such as epilepsy and autism. In addition to the importance of excitatory transmission in brain communication, inhibitory transmission controls synaptic transmission and neuronal firing frequency (Buzsaki & Chrobak, 1995; Engel et al., 2001; Salinas & Sejnowski, 2001). Beyond the direct effects on cell signaling reduction, inhibition is essential in the operation of neural networks, contributing to the generation of rhythmic activity by synchronizing the discharge of principal cell populations (Bartos, Vida, & Jonas, 2007; Kullmann, 2011; Pouille & Scanziani, 2001). Present findings indicate that inhibition is a specific property of neurons and may be fundamentally different between neurons and astrocytes. While inhibitory neurotransmitters have direct inhibitory effects on neuronal activity through direct activation of neuronal receptors, present data indicate that they may also have an indirect excitatory effect on neurons through activation of astrocytes. Hence, the same neurotransmitters that directly inhibit neurons may also activate astrocytes, which then feed-forward excite neurons (Perea et al., 2016). Therefore, the present results reveal additional complexity of the signaling consequences of excitatory and inhibitory neurotransmitters in network operation and brain function.
Main points.
Gq signaling in neurons and astrocytes leads to cellular activation, but while Gi/o signaling in neurons leads to cellular inhibition, it leads to activation in astrocytes and downstream gliotransmission that modulates neuronal activity.
Acknowledgements
We would like to thank Ruth Quintana, Stephanie Nistler, and Heidi Busch for technical support; Mario Martin, and Michelle Corkrum for helpful suggestions; Mark Sanders, Guillermo Marques and Jason Mitchell at the University of Minnesota – University Imaging Centers for assistance using the Leica SP5 Multiphoton Confocal Upright Microscope; The MnDRIVE Optogenetics Core at the University of Minnesota for technical support; Viral vectors used in this study were prepared by the University of Minnesota Viral Vector and Cloning Core; Ju Chen for generously donating the IP3R2−/− mice; Dwight Bergles and Amit Agarwal for generously donating the GLAST-CreERT2xR26-lsl-GCaMP3 mice; and the UNC Vector Core for providing the DREADDs viruses. This work was supported by NIH-NINDS (R01NS097312–01) and Human Frontier Science Program (Research Grant RGP0036/2014) to A.A and NIH-NINDS (5 F31 NS 93751–3) to C.A.D and NIH-NIA (1 F31 AG057155–01A1) to J.L.. The authors declare no competing financial interests.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adamsky A, Kol A, Kreisel T, Doron A, Ozeri-Engelhard N, Melcer T, . . . Goshen I (2018). Astrocytic Activation Generates De Novo Neuronal Potentiation and Memory Enhancement. Cell. doi: 10.1016/j.cell.2018.05.002 [DOI] [PubMed] [Google Scholar]
- Agarwal A, Wu PH, Hughes EG, Fukaya M, Tischfield MA, Langseth AJ, . . . Bergles DE (2017). Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron, 93(3), 587-605 e587. doi: 10.1016/j.neuron.2016.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agulhon C, Boyt KM, Xie AX, Friocourt F, Roth BL, & McCarthy KD (2013). Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein-coupled receptor activation in vivo. J Physiol, 591(22), 5599–5609. doi: 10.1113/jphysiol.2013.261289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken SP, Lampe BJ, Murphy PA, & Brown BS (1995). Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol, 115(7), 1163–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, . . . Roth BL. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron, 63(1), 27–39. doi: 10.1016/j.neuron.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, & Audinat E (2004). Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci, 24(31), 6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, & Haydon PG (2001). Dynamic signaling between astrocytes and neurons. Annu Rev Physiol, 63, 795–813. doi: 10.1146/annurev.physiol.63.1.795 [DOI] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, & Volterra A (2014). Gliotransmitters travel in time and space. Neuron, 81(4), 728–739. doi: 10.1016/j.neuron.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Martin ED, Perea G, Arellano JI, & Buno W (2002). Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J Neurosci, 22(7), 2443–2450. doi:20026212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, & Haydon PG (1998). Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci, 10(6), 2129–2142. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, & Haydon PG (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci, 22(5), 208–215. [DOI] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V, & Haydon PG (1998). Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci, 18(17), 6822–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, & Roth BL (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A, 104(12), 5163–5168. doi: 10.1073/pnas.0700293104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, & Jonas P (2007). Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci, 8(1), 45–56. doi: 10.1038/nrn2044 [DOI] [PubMed] [Google Scholar]
- Berkeley JL, Gomeza J, Wess J, Hamilton SE, Nathanson NM, & Levey AI (2001). M1 muscarinic acetylcholine receptors activate extracellular signal-regulated kinase in CA1 pyramidal neurons in mouse hippocampal slices. Mol Cell Neurosci, 18(5), 512–524. doi: 10.1006/mcne.2001.1042 [DOI] [PubMed] [Google Scholar]
- Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhauser C, Pilati E, & Volterra A (2004). Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci, 7(6), 613–620. doi: 10.1038/nn1246 [DOI] [PubMed] [Google Scholar]
- Boddum K, Jensen TP, Magloire V, Kristiansen U, Rusakov DA, Pavlov I, & Walker MC (2016). Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat Commun, 7, 13572. doi: 10.1038/ncomms13572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder DE, & McCarthy KD (2014). Astrocytic Gq-GPCR-linked IP3R-dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex in vivo. J Neurosci, 34(39), 13139–13150. doi: 10.1523/JNEUROSCI.2591-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BS, & Yu SP (2000). Modulation and genetic identification of the M channel. Prog Biophys Mol Biol, 73(2–4), 135–166. [DOI] [PubMed] [Google Scholar]
- Bull C, Freitas KC, Zou S, Poland RS, Syed WA, Urban DJ, . . . Bowers MS (2014). Rat nucleus accumbens core astrocytes modulate reward and the motivation to self-administer ethanol after abstinence. Neuropsychopharmacology, 39(12), 2835–2845. doi: 10.1038/npp.2014.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, & Chrobak JJ (1995). Temporal structure in spatially organized neuronal ensembles: a role for interneuronal networks. Curr Opin Neurobiol, 5(4), 504–510. [DOI] [PubMed] [Google Scholar]
- Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, . . . Khakh BS (2017). Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron, 95(3), 531-549 e539. doi: 10.1016/j.neuron.2017.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Sugihara H, Sharma J, Perea G, Petravicz J, Le C, & Sur M (2012). Nucleus basalis-enabled stimulus-specific plasticity in the visual cortex is mediated by astrocytes. Proc Natl Acad Sci U S A, 109(41), E2832–2841. doi: 10.1073/pnas.1206557109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covelo A, & Araque A (2018). Neuronal activity determines distinct gliotransmitter release from a single astrocyte. Elife, 7. doi: 10.7554/eLife.32237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari S, & Gulledge AT (2011). M1 and M4 receptors modulate hippocampal pyramidal neurons. J Neurophysiol, 105(2), 779–792. doi: 10.1152/jn.00686.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, . . . Volterra A. (2011). Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci, 14(10), 1276–1284. doi: 10.1038/nn.2929 [DOI] [PubMed] [Google Scholar]
- Engel D, Pahner I, Schulze K, Frahm C, Jarry H, Ahnert-Hilger G, & Draguhn A (2001). Plasticity of rat central inhibitory synapses through GABA metabolism. J Physiol, 535(Pt 2), 473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erondu NE, & Kennedy MB (1985). Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J Neurosci, 5(12), 3270–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Halassa MM, Terunuma M, Succol F, Takano H, Frank M, . . . Haydon PG. (2009). Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc Natl Acad Sci U S A, 106(35), 15037–15042. doi: 10.1073/pnas.0906419106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, & Carmignoto G (2004). Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron, 43(5), 729–743. doi: 10.1016/j.neuron.2004.08.011 [DOI] [PubMed] [Google Scholar]
- Fiacco TA, & McCarthy KD (2018). Multiple Lines of Evidence Indicate That Gliotransmission Does Not Occur under Physiological Conditions. J Neurosci, 38(1), 3–13. doi: 10.1523/JNEUROSCI.0016-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini S, & Magee JC (2006). State-dependent dendritic computation in hippocampal CA1 pyramidal neurons. J Neurosci, 26(7), 2088–2100. doi: 10.1523/JNEUROSCI.4428-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, Navarrete M, Perea G, Covelo A, Martin-Fernandez M, Shigemoto R, . . . Araque A. (2015). Endocannabinoids Induce Lateral Long-Term Potentiation of Transmitter Release by Stimulation of Gliotransmission. Cereb Cortex, 25(10), 3699–3712. doi: 10.1093/cercor/bhu231 [DOI] [PubMed] [Google Scholar]
- Halassa MM, & Haydon PG (2010). Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol, 72, 335–355. doi: 10.1146/annurev-physiol-021909-135843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG, & Carmignoto G (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev, 86(3), 1009–1031. doi: 10.1152/physrev.00049.2005 [DOI] [PubMed] [Google Scholar]
- Hirono M, Yoshioka T, & Konishi S (2001). GABA(B) receptor activation enhances mGluR-mediated responses at cerebellar excitatory synapses. Nat Neurosci, 4(12), 1207–1216. doi: 10.1038/nn764 [DOI] [PubMed] [Google Scholar]
- Huang Y, & Thathiah A (2015). Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett, 589(14), 1607–1619. doi: 10.1016/j.febslet.2015.05.007 [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, & Nedergaard M (1998). Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci, 1(8), 683–692. doi: 10.1038/3684 [DOI] [PubMed] [Google Scholar]
- Kullmann DM (2011). Interneuron networks in the hippocampus. Curr Opin Neurobiol, 21(5), 709–716. doi: 10.1016/j.conb.2011.05.006 [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, & Mackie K (2005). The cannabinoid agonist WIN55,212–2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A, 102(52), 19144–19149. doi: 10.1073/pnas.0509588102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Ghetti A, Pinto-Duarte A, Wang X, Dziewczapolski G, Galimi F, . . . Heinemann SF. (2014). Astrocytes contribute to gamma oscillations and recognition memory. Proc Natl Acad Sci U S A, 111(32), E3343–3352. doi: 10.1073/pnas.1410893111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zima AV, Sheikh F, Blatter LA, & Chen J (2005). Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res, 96(12), 1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ, & Clapham DE (1987). The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature, 325(6102), 321–326. doi: 10.1038/325321a0 [DOI] [PubMed] [Google Scholar]
- Magee JC, & Carruth M (1999). Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J Neurophysiol, 82(4), 1895–1901. [DOI] [PubMed] [Google Scholar]
- Mariotti L, Losi G, Lia A, Melone M, Chiavegato A, Gomez-Gonzalo M, . . . Carmignoto G (2018). Interneuron-specific signaling evokes distinctive somatostatin-mediated responses in adult cortical astrocytes. Nat Commun, 9(1), 82. doi: 10.1038/s41467-017-02642-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti L, Losi G, Sessolo M, Marcon I, & Carmignoto G (2016). The inhibitory neurotransmitter GABA evokes long-lasting Ca(2+) oscillations in cortical astrocytes. Glia, 64(3), 363–373. doi: 10.1002/glia.22933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fernandez M, Jamison S, Robin LM, Zhao Z, Martin ED, Aguilar J, . . . Araque A (2017). Synapse-specific astrocyte gating of amygdala-related behavior. Nat Neurosci, 20(11), 1540–1548. doi: 10.1038/nn.4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R, Bajo-Graneras R, Moratalla R, Perea G, & Araque A (2015). Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science, 349(6249), 730–734. doi: 10.1126/science.aaa7945 [DOI] [PubMed] [Google Scholar]
- Meier SD, Kafitz KW, & Rose CR (2008). Developmental profile and mechanisms of GABA-induced calcium signaling in hippocampal astrocytes. Glia, 56(10), 1127–1137. doi: 10.1002/glia.20684 [DOI] [PubMed] [Google Scholar]
- Navarrete M, & Araque A (2008). Endocannabinoids mediate neuron-astrocyte communication. Neuron, 57(6), 883–893. doi: 10.1016/j.neuron.2008.01.029 [DOI] [PubMed] [Google Scholar]
- Navarrete M, & Araque A (2010). Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron, 68(1), 113–126. doi: 10.1016/j.neuron.2010.08.043 [DOI] [PubMed] [Google Scholar]
- Navarrete M, Perea G, Fernandez de Sevilla D, Gomez-Gonzalo M, Nunez A, Martin ED, & Araque A (2012). Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol, 10(2), e1001259. doi: 10.1371/journal.pbio.1001259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- New DC, An H, Ip NY, & Wong YH (2006). GABAB heterodimeric receptors promote Ca2+ influx via store-operated channels in rat cortical neurons and transfected Chinese hamster ovary cells. Neuroscience, 137(4), 1347–1358. doi: 10.1016/j.neuroscience.2005.10.033 [DOI] [PubMed] [Google Scholar]
- North RA (1989). Twelfth Gaddum memorial lecture. Drug receptors and the inhibition of nerve cells. Br J Pharmacol, 98(1), 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Vallee J, Haber M, Murai KK, Lacaille JC, & Robitaille R (2011). Astrocytes are endogenous regulators of basal transmission at central synapses. Cell, 146(5), 785–798. doi: 10.1016/j.cell.2011.07.022 [DOI] [PubMed] [Google Scholar]
- Park JY, & Spruston N (2012). Synergistic actions of metabotropic acetylcholine and glutamate receptors on the excitability of hippocampal CA1 pyramidal neurons. J Neurosci, 32(18), 6081–6091. doi: 10.1523/JNEUROSCI.6519-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, & Carmignoto G (1997). Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci, 17(20), 7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paukert M, Agarwal A, Cha J, Doze VA, Kang JU, & Bergles DE (2014). Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron, 82(6), 1263–1270. doi: 10.1016/j.neuron.2014.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, & Araque A (2005). Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci, 25(9), 2192–2203. doi: 10.1523/JNEUROSCI.3965-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Gomez R, Mederos S, Covelo A, Ballesteros JJ, Schlosser L, . . . .Araque A (2016). Activity-dependent switch of GABAergic inhibition into glutamatergic excitation in astrocyte-neuron networks. Elife, 5. doi: 10.7554/eLife.20362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M, & Araque A (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci, 32(8), 421–431. doi: 10.1016/j.tins.2009.05.001 [DOI] [PubMed] [Google Scholar]
- Perea G, Sur M, & Araque A (2014). Neuron-glia networks: integral gear of brain function. Front Cell Neurosci, 8, 378. doi: 10.3389/fncel.2014.00378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petravicz J, Fiacco TA, & McCarthy KD (2008). Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J Neurosci, 28(19), 4967–4973. doi: 10.1523/JNEUROSCI.5572-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, & McCarthy KD (1996). Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci, 16(16), 5073–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poskanzer KE, & Yuste R (2011). Astrocytic regulation of cortical UP states. Proc Natl Acad Sci U S A, 108(45), 18453–18458. doi: 10.1073/pnas.1112378108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poskanzer KE, & Yuste R (2016). Astrocytes regulate cortical state switching in vivo. Proc Natl Acad Sci U S A, 113(19), E2675–2684. doi: 10.1073/pnas.1520759113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, & Scanziani M (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science, 293(5532), 1159–1163. doi: 10.1126/science.1060342 [DOI] [PubMed] [Google Scholar]
- Rusakov DA (2015). Disentangling calcium-driven astrocyte physiology. Nat Rev Neurosci, 16(4), 226–233. doi: 10.1038/nrn3878 [DOI] [PubMed] [Google Scholar]
- Salinas E, & Sejnowski TJ (2001). Correlated neuronal activity and the flow of neural information. Nat Rev Neurosci, 2(8), 539–550. doi: 10.1038/35086012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiderer CL, Smith CC, McCutchen E, McCoy PA, Thacker EE, Kolasa K, . . . McMahon LL. (2008). Coactivation of M(1) muscarinic and alpha1 adrenergic receptors stimulates extracellular signal-regulated protein kinase and induces long-term depression at CA3-CA1 synapses in rat hippocampus. J Neurosci, 28(20), 5350–5358. doi: 10.1523/JNEUROSCI.5058-06.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman H, & Lou LL (1989). Multifunctional Ca2+/calmodulin-dependent protein kinase: domain structure and regulation. Trends Biochem Sci, 14(2), 62–66. [DOI] [PubMed] [Google Scholar]
- Scofield MD, Boger HA, Smith RJ, Li H, Haydon PG, & Kalivas PW (2015). Gq-DREADD Selectively Initiates Glial Glutamate Release and Inhibits Cue-induced Cocaine Seeking. Biol Psychiatry, 78(7), 441–451. doi: 10.1016/j.biopsych.2015.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, & Robitaille R (2006). GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci, 26(20), 5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton MK, & McCarthy KD (2000). Hippocampal astrocytes exhibit Ca2+-elevating muscarinic cholinergic and histaminergic receptors in situ. J Neurochem, 74(2), 555–563. [DOI] [PubMed] [Google Scholar]
- Shen W, Nikolic L, Meunier C, Pfrieger F, & Audinat E (2017). An autocrine purinergic signaling controls astrocyte-induced neuronal excitation. Sci Rep, 7(1), 11280. doi: 10.1038/s41598-017-11793-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, & Khakh BS (2008). Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci, 28(26), 6659–6663. doi: 10.1523/JNEUROSCI.1717-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer WD, Brown HA, & Sternweis PC (1997). Regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu Rev Biochem, 66, 475–509. doi: 10.1146/annurev.biochem.66.1.475 [DOI] [PubMed] [Google Scholar]
- Stewart A, & Fisher RA (2015). Introduction: G Protein-coupled Receptors and RGS Proteins. Prog Mol Biol Transl Sci, 133, 1–11. doi: 10.1016/bs.pmbts.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Suh BC, & Hille B (2002). Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron, 35(3), 507–520. [DOI] [PubMed] [Google Scholar]
- Takata N, Mishima T, Hisatsune C, Nagai T, Ebisui E, Mikoshiba K, & Hirase H (2011). Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J Neurosci, 31(49), 18155–18165. doi: 10.1523/JNEUROSCI.5289-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Liu Y, Xi W, Lou HF, Zhu L, Guo Z, . . . .Duan S. (2017). Glia-derived ATP inversely regulates excitability of pyramidal and CCK-positive neurons. Nat Commun, 8, 13772. doi: 10.1038/ncomms13772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, & Kettenmann H (1996). Calcium signalling in glial cells. Trends Neurosci, 19(8), 346–352. [DOI] [PubMed] [Google Scholar]
- Volterra A, Liaudet N, & Savtchouk I (2014). Astrocyte Ca(2)(+) signalling: an unexpected complexity. Nat Rev Neurosci, 15(5), 327–335. doi: 10.1038/nrn3725 [DOI] [PubMed] [Google Scholar]
- Volterra A, & Meldolesi J (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci, 6(8), 626–640. doi: 10.1038/nrn1722 [DOI] [PubMed] [Google Scholar]
- Wickman KD, Iniguez-Lluhl JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, . . . Clapham DE (1994). Recombinant G-protein beta gamma-subunits activate the muscarinic-gated atrial potassium channel. Nature, 368(6468), 255–257. doi: 10.1038/368255a0 [DOI] [PubMed] [Google Scholar]
- Zeng W, Mak DO, Li Q, Shin DM, Foskett JK, & Muallem S (2003). A new mode of Ca2+ signaling by G protein-coupled receptors: gating of IP3 receptor Ca2+ release channels by Gbetagamma. Curr Biol, 13(10), 872–876. [DOI] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, & Logothetis DE (2003). PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron, 37(6), 963–975. [DOI] [PubMed] [Google Scholar]
- Zhu H, Pleil KE, Urban DJ, Moy SS, Kash TL, & Roth BL (2014). Chemogenetic inactivation of ventral hippocampal glutamatergic neurons disrupts consolidation of contextual fear memory. Neuropsychopharmacology, 39(8), 1880–1892. doi: 10.1038/npp.2014.35 [DOI] [PMC free article] [PubMed] [Google Scholar]