

Abstract
Background & Aims:
Interferon regulatory factor 3 (IRF3) is a transcription factor mediating anti-viral responses, yet recent evidence indicates that IRF3 also has critical non-transcriptional functions, including activating RIG-I-like receptors-induced IRF-3-mediated pathway of apoptosis (RIPA) and restricting activity of NFκB Using a novel murine model expressing only non-transcriptional IRF3 activity (Irf3S1/S1), we tested the hypothesis that non-transcriptional functions of IRF3 modulate innate immune responses in the Gao-binge (acute on chronic) model of alcohol-related liver disease.
Methods:
C57BL/6, Irf3−/− and Irf3S1/S1 were exposed to Gao-binge ethanol-induced liver injury. IRF3-mediated RIPA was investigated in cultured macrophages.
Results:
Phospho-IRF3 and IRF3-mediated signals were elevated in livers of patients with alcoholic hepatitis. In C57BL/6 mice, Gao-binge ethanol exposure activated IRF3 signaling and resulted in hepatocellular injury. Indicators of liver injury were differentially impacted by Irf3 genotype. Irf3−/−, but not Irf3S1/S1, mice were protected from steatosis, ALT/AST and inflammatory cytokine expression. In contrast, neutrophil accumulation and ER stress were independent of genotype. Protection from Gao-binge injury in Irf3−/− mice was associated with an increased ratio of Ly6Clow (restorative) to Ly6Chigh (inflammatory) cells compared to C57BL/6 and Irf3S1/S1 mice. Reduced ratios of Ly6Clow/Ly6Chigh in C57BL/6 and Irf3S1/S1 mice were associated with an increased apoptosis in the Ly6Clow population in response to Gao-binge. Activation of primary cultures of macrophages with Poly (I:C) induced translocation of IRF3 to mitochondria, association with Bax and activation of Caspases 3 and 9, processes indicative of activation of the RIPA pathway.
Conclusions:
Taken together, these data identify important contributions of the non-transcriptional function of IRF3 in modulating the innate immune environment in response to Gao-binge ethanol exposure via regulation of immune cell apoptosis.
Keywords: Alcoholic liver disease, IRF3, ER stress, neutrophils, apoptosis, restorative monocytes
Lay Summary:
Activation of the innate immune system contributes to inflammation in the progression of alcoholic liver disease, as well as resolution of injury. Here we show that a nontranscriptional activity of IRF3 modulates the innate immune environment of the liver in a mouse model of alcoholic hepatitis by increasing the apoptotic cell death of immune cells that promote the resolution of injury.
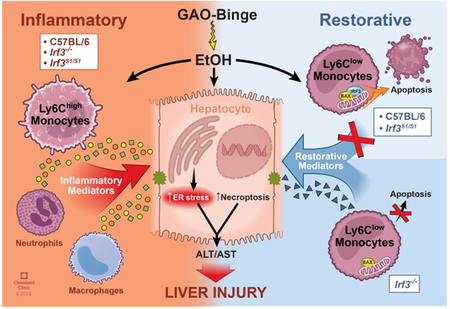
Graphical Abstract
Alcohol consumption is a leading cause of preventable morbidity and mortality worldwide (1). The pathogenesis of alcoholic liver disease (ALD) is initially characterized by steatosis, progressing in some individuals to fibrosis and cirrhosis. Alcoholic hepatitis (AH), a severe inflammatory condition, with extensive infiltration of leukocytes and hepatocellular injury, can occur at any stage of disease progression; 28-day mortality rates range from 25–35 percent (1) (2). The development of AH is a complex process involving both parenchymal and nonparenchymal cells resident in the liver, as well as the recruitment of immune cells to the liver in response to damage and inflammation (3). Current therapies, focusing on suppressing inflammation, are ineffective in many patients with severe AH and outcomes remain poor (4).
There is a growing appreciation of a dynamic and complex role of the innate immune system in the progression of ALD, as well as the resolution of hepatocellular injury. Both fluid-phase elements, such as complement (5), and cellular components of the innate immune system contribute to progression and resolution of ethanol-induced liver injury (3). This dynamic interplay between injury and repair is mediated, at least in part, by the tremendous plasticity of resident tissue macrophages and infiltrating monocytes; the phenotype of these innate immune cells is rapidly modulated in response to signals within their microenvironment (6). Pathogen associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs) are key signals of injury in ALD (3). Increased exposure of Kupffer cells, the resident hepatic macrophages, to gut-derived LPS during chronic ethanol (7) activates TLR4-dependent production of inflammatory mediators (3). In response to these initial inflammatory signals, circulating monocytes and neutrophils infiltrate the liver (3). Depending on the stage of injury/repair, infiltrating monocytes can acquire multiple phenotypes, exhibiting pro-inflammatory, anti-inflammatory and/or pro-resolution/remodeling activity (6).
The precise molecular mechanisms controlling the heterogeneity of innate immune cells in the liver in response to ethanol exposure is not well understood. However, it is likely that regulation of recruitment and phenotypic maturation, as well as maintaining an appropriate balance between pro-survival and pro-death pathways is critical to the ability of innate immune cells to rapidly respond to the demands of maintaining liver homeostasis in the face of ethanol-induced injury (8). While there is a good appreciation of how ethanol regulates the expression of chemokines and subsequent recruitment of immune cells to the liver (6), the potential mechanisms regulating immune cell death in the liver are particularly understudied.
Interferon-regulatory factor 3 (IRF3) is an important regulator of anti-viral activity. Upon activation, IRF3 is phosphorylated and pIRF3 acts as a transcription factor essential for the induction of interferon-β (IFN-β) and antiviral genes. Absence of these anti-viral functions makes Irf3−/− mice susceptible to a wide range of viral infections (9). In addition to its transcriptional functions, IRF3 directly triggers a pro-apoptotic pathway, termed RIG-I-like receptors (RLR)-induced IRF-3-mediated pathway of apoptosis (RIPA) via a non-transcriptional mechanism (10). In RIPA, IRF3 is activated by linear ubiquitination on two lysine residues, resulting in the interaction of IRF3 with the pro-apoptotic protein Bax. The IRF3/BAX complex then translocates to the mitochondria where it triggers apoptosis (10,11).
Recent data suggest that IRF3 plays a critical role in the progression of ALD (12,13), as well as NAFLD/NASH (14,15) and fibrosis (16). However, it is not known if the transcriptional and/or non-transcriptional functions of IRF3 contribute to ethanol-induced liver injury. Since regulated apoptosis is an important mechanism for the resolution of inflammation, we hypothesized that the exacerbated inflammatory responses to AH involves an inappropriate utilization of the IRF3-mediated RIPA pathway.
To test this hypothesis, we exposed a novel knock-in mouse, in which the wild-type Irf3 was replaced by a mutant Irf3 gene encoding a protein lacking key phosphorylation sites (SS388/390AA) required for translocation of IRF3 to the nucleus (10,11), to the Gao-binge (acute on chronic) model of alcohol-related liver disease. While Irf3−/− mice, lacking both the transcriptional and non-transcriptional functions of IRF3, exhibited robust neutrophil accumulation and ER stress in response to Gao-binge ethanol exposure, they were protected from increased inflammatory cytokine expression and hepatocellular injury. Irf3−/− mice accumulated more Ly6Clow (restorative) monocytes in the liver, associated with a decrease in apoptosis of this population of Ly6Clow monocytes after recruitment. In contrast, C57BL/6 and the Irf3S1/S1 mice were sensitive to Gao-binge ethanol exposure, associated with an increased proportion of apoptotic Ly6Clow monocytes and a lower ratio of Ly6Clow/Ly6Chigh monocytes. These data delineate highly novel functions for the non-transcriptional functions of IRF3 in ethanol-induced liver injury, identifying for the first time, that, in addition to the anti-viral function of IRF3-mediated RIPA, the non-transcriptional activity of IRF3 also plays an important metabolic function in maintaining innate immune homeostasis in the liver.
Materials and Methods
Gao-binge ethanol feeding
All animals received humane care and all procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Breeding colonies of Irf3−/− and Irf3S1/S1 on a C57BL/6 background (10,11) were maintained at the Cleveland Clinic. Eight to ten week old female C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were allowed free access to a Lieber-DeCarli liquid diet (Dyets, Bethlehem, PA; Cat#710260) containing ethanol at 5% (v/v) or 28% of total calories or pair-fed control diet that isocalorically substituted maltose dextrin for ethanol for 10 days (17). On the final day of the experiment, pair-fed mice were gavaged with 5g/kg maltose and ethanol-fed mice were gavaged with 5g/kg ethanol in water. Mice were euthanized 6h after gavage. Additional details of the feeding protocol and tissue collection can be found in Supplemental Materials.
Patient liver samples from the Early Transplant Tissue Repository:
De-identified samples from five livers explanted from severe AH patients during liver transplantation or five wedge biopsies from healthy donor livers were snap frozen in liquid nitrogen and stored at −80°C. Samples were provided by the Clinical Resource for Alcoholic Hepatitis Investigations at Johns Hopkins University. Written informed consent was obtained from each patient included in the study and the study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the Institutional Review Boards at Johns Hopkins Medical Institutions. Descriptive biochemical and clinical data for this cohort have been reported previously (18).
Biochemical assays, Immunohistochemistry and Flow cytometry
Detailed methods can be found in Supplemental Materials and CTAT table.
Statistical Analysis
Values shown in all figures represent the means ± SEM. Analysis of variance was performed using the general linear models procedure (SAS, Carey, IN). Data were log-transformed as necessary to obtain a normal distribution. Follow-up comparisons were made by least square means testing. P values of less than 0.05 were considered significant.
RESULTS
IRF3 is implicated in the progression of ethanol-induced liver injury. Phosphorylation of IRF3 and expression of STING were increased in livers of patients with alcoholic hepatitis undergoing liver transplant compared to liver explants from healthy controls (Figure 1A). Cleavage of caspase-9, implicated in IRF3-mediated RIPA (10,11), and capase-3 was also higher in livers from patients with AH compared to healthy controls (Figure 1A). IRF3 was also phosphorylated in livers of mice in response to Gao-binge ethanol exposure and expression of STING increased (Figure 1B), consistent with the increase in phospho-IRF3 observed in response to chronic ethanol feeding (12,13).
Figure 1: IRF3 phosphorylation, ubiquitination and transcriptional function in livers of patients with alcoholic hepatitis and mice after Gao-binge ethanol exposure.

A) Livers from patients undergoing liver transplant for severe alcoholic hepatitis (AH) or liver explants from healthy controls (HC) were analyzed by Western blots for phospho-IRF3, IRF3, STING, caspase-9 and caspase 3. GAPDH was used as a loading control. Images are representative of 5 healthy controls and 5 patients. B) C57BL/6 mice were exposed to the Gao-binge model of ethanol-induced liver injury with 10 day feeding of a 5% (vol/vol) Lieber-DeCarli liquid diet, followed by a challenge with 5g/kg ethanol by gavage. Control mice were pair-fed liquid diets substituting maltose-dextrins for ethanol and challenged with a gavage of maltose. Livers were lysed and phospho-IRF3 and STING assessed by Western blot. HSC70 was used as a loading control. C) Expression of mRNA for IRFβ and IFIT3 genes in liver of patients with AH and healthy controls was assessed by qRT-PCR and normalized to 18S rRNA. D/E/F/G) C57BL/6, Irf3−/− and Irf3S1/S1 mice were exposed to the Gao-binge model of ethanol-induced liver injury. Expression of mRNA for D) IRF3, E) IFIT1 and F) IFIT3 were assessed in whole liver and G) IFMβ assessed in isolated non-parenchymal cells by qRT-PCR and normalized to 18S rRNA. H) Liver lysates from C57BL/6 and Irf3S1/S1 mice were immunoprecipitated with anti-Ubiquitin antibody and probed with anti-IRF3 and anti-HSC70 antibody. (PF:Pair-fed; GB: Gao-binge ethanol). Pull-downs are representative of 4 mice per diet group. For human data, values represent means ± SEM, n=5 per group, *p<0.05, assessed by Student’s t-test. For murine data, values represent means ± SEM, n=4 per group. Values with different superscripts are significantly different from each other, p<0.05, assessed by ANOVA.
While expression of IRF3 mRNA was not affected by AH (Figure 1C), expression of the IRF3-dependent genes, IFNβ and IFIT3, was higher in livers from patients with AH compared to healthy controls (Figure 1C). Similarly, IRF3 mRNA was not affected by Gao-binge ethanol exposure (Figure 1D), but expression of IFIT1 and IFIT3 (Figure 1E/F) was increased in whole liver and IFNβ expression increased in isolated non-parenchymal cells (Figure 1G). As expected, IRF3-mediated gene expression was not induced in Irf3−/− or Irf3S1/S1 mice, which lack IRF3-mediated transcriptional activity (Figure 1E–G).
Since the non-transcriptional activity of IRF3 is dependent on ubiquitination, we next assessed whether Gao-binge ethanol exposure also increased ubiquitination of IRF3. Ubiquitinated proteins were pulled down from liver lysates of C57BL/6 and Irf3S1/S1 and then probed for IRF3. The quantity of IRF3 pulled down with anti-ubiquitin was higher in ethanol-fed mice compared to pair-fed (Figure 1H). Culture of Huh7 hepatocytes or RAW264.7 macrophages with ethanol also increased the ubiquitination of transfected V5-tagged IRF3 compared to cells cultured without ethanol (Supplemental Figure 1). Taken together, these data indicate that IRF3 is activated in liver in response to ethanol via both phosphorylation and ubiquitination.
In order to determine if the transcriptional and/or non-transcriptional activity of IRF3 contributed to ethanol-induced liver injury, C57BL/6, Irf3−/− and Irf3S1/S1 mice were exposed to the Gao-binge ethanol and measures of inflammation and hepatocellular injury assessed 6 h post-binge. Gao-binge ethanol exposure increased indicators of liver injury in C57BL/6 mice, characterized by increased ALT and AST (Figure 2A) and elevated liver triglycerides (Figure 2B and Supplemental Figure 2), as well as increased expression of mRNA for inflammatory cytokines and chemokines (Figure 2C). Irf3−/− mice were protected from these indicators of ethanol-induced liver injury, similar to the protection reported by Petrasek, et al., in response to chronic ethanol feeding for 4 weeks (13). If this protection was due to the absence of the transcriptional activity of IRF3, then Irf3S1/S1 mice, expressing only the non-transcriptional activity of IRF3, should also be protected from Gao-binge ethanol exposure. Importantly, the Irf3S1/S1 mice were sensitive to the injurious effects of acute on chronic ethanol exposure (Figure 2), indicating that it was a non-transcriptional function of IRF3 that contributed to injury.
Figure 2: The non-transcriptional activity of IRF3 contributes to ethanol-induced liver injury in the Gao-binge model of ethanol-induced liver injury.

C57BL/6, Irf3−/− and Irf3S1/S1 mice were exposed to the Gao-binge model as in Figure 1. Measures of hepatocellular injury and steatosis were assessed 6 h post-binge. A) ALT and AST were measured in plasma and expressed as U/ml activity relative to pair-fed mice within genotype. B) Hepatic steatosis was assessed by Oil Red O staining and semi-quantified. C) Expression of mRNA for inflammatory cytokines and chemokines was assessed by qRT-PCR and normalized to 18S rRNA. Values represent means ± SEM, n=4 per group. Values with different superscripts are significantly different from each other, p<0.05, assessed by ANOVA..
Interestingly, in the Gao-binge model of ethanol-induced liver injury, very little hepatocyte apoptosis was detected by M30 or TUNEL staining in C57BL/6 mice (data not shown), consistent with earlier reports (19,20). Gao-binge ethanol exposure also impairs the barrier function of the intestine (21), but portal endotoxin concentrations in response to Gao-binge were independent of genotypes (Supplemental Figure 3). Further, liver injury in response to Gao-binge ethanol exposure is also associated with ER stress (19) and IRF3 interacts with the ER stress protein STING in response to both acute exposure to ethanol (13) and diet-induced obesity (22). Therefore, we hypothesized that the non-transcriptional functions of IRF3 could provide a critical link between induction of ER stress and hepatocellular injury. Induction of CYP2E1 (Figure 3A), associated with ethanol-induced ER stress (23), and accumulation of 4-hyroxynonenal adducts (Figure 3B), an indicator of oxidative stress, in response to Gao-binge were independent of genotype. Further, phosphorylation of eIF2, an up-stream mediator of the ER stress response (Figure 3C), as well as expression of the spliced form of XBP-1 (Figure 3D) and multiple mRNA for ER stress response proteins, including DR5, ero1α, Bak, GADD34, grp94, and grp78, were all increased in response to Gao-binge ethanol exposure in all genotypes (Figure 3D). Interestingly, in some measures of ER stress (e.g. CHOP protein, mRNA for grp78 and grp94), the baseline expression in the Irf3−/− , but not Irf3S1/S1, mice was moderately elevated even in pair-fed mice (Figure 3C/D) and even further elevated in response to Gao-binge. Taken together, these data indicate that multiple measures of ER stress were dissociated from other indicators of liver injury, including ALT/AST and cytokine production. Indeed, these data are more consistent with a potentially protective role of the ER stress response at this stage of hepatocellular injury.
Figure 3: Ethanol-induced oxidative and ER stress in liver in response to Gao-binge ethanol exposure was independent of Irf3 genotype.

C57BL/6, Irf3−/− and Irf3S1/S1 mice were exposed to the Gao-binge model as in Figure 1. A) Expression of CYP2E1 was measured by Western blot and normalized to HSC70. B) Formalin-fixed paraffin-embedded sections of liver were de-paraffinized and accumulation of 4-hydroxynonenal (4-HNE) adducts assessed by immunohistochemistry. Images were acquired at 10X. C) Phosphorylated-eIF2α, total eIF2α and CHOP10 protein in liver lysates was assessed by Western blot and normalized to HSC70. D) Expression of the spliced form of XBP1, as well as additional mRNA for ER stress markers, was measured in liver and normalized to 18S rRNA. Values represent means ± SEM, n=4–8 per group. Values with different superscripts are significantly different from each other, p<0.05, assessed by ANOVA..
The Gao-binge model of ethanol-induced liver injury is characterized by a robust infiltration of neutrophils that contributes to hepatocellular inflammation and injury (24). Despite evidence for reduced inflammation and hepatocellular injury in Irf3−/− (e.g. lower ALT/AST, lower expression of inflammatory cytokines), NIMPR14 staining of neutrophils in the liver increased in response to Gao-binge independently of genotype (Figure 4A/B). Further, while the percentage of CD45+ non-parenchymal cells (Figure 4C) was not affected by diet or genotype, the percentage of CD45+/Ly6G+ cells was actually higher in Irf3−/− in response to Gao-binge ethanol exposure, compared to C57BL/6 and Irf3S1/S1 mice (Figure 4D/E). Similarly, the expression of Ly6G mRNA (Figure 4G) was increased in response to Gao-binge ethanol exposure in all genotypes. Indicators of neutrophil activation, including the percentage of CD45+/CD11b+/Ly6G+ cells and the expression of ELANE, an indicator of neutrophil extravasation, were also increased in response to Gao-binge ethanol exposure in all genotypes (Figure 4F/H and Supplemental Figure 4). Taken together, these data indicate that neither the transcriptional or non-transcriptional functions of IRF3 contribute to the accumulation of activated neutrophils in the liver in response to Gao-binge ethanol exposure.
Figure 4: Neutrophil accumulation in liver in response to Gao-binge ethanol exposure was independent of Irf3 genotype.

C57BL/6, Irf3−/− and Irf3S1/S1 mice were exposed to the Gao-binge model as in Figure 1. A/B) NIMPR14 expression was visualized by immunohistochemistry after 6 h. A) Formalin-fixed paraffin-embedded sections of liver were de-paraffinized and expression of NIMPR14 assessed by immunohistochemistry. B) Images were acquired at 10X and the number of positive cells enumerated. C/D/E/F) Total non-parenchymal cells were isolated from the liver and analyzed by flow cytometry. The percentage of C) CD45+, D/E) CD45+/Ly6G+ and F) CD45+/ CD11b+/Ly6G+ was determined by flow cytometry. D) Representative flow diagrams for CD45+/Ly6G+ are illustrated. G/H) Expression of G) Ly6G and H) ELANE mRNA in whole liver was assessed by qRT-PCR and normalized to 18S rRNA. Values represent means ± SEM, n=4–8 per group. Values with different superscripts are significantly different from each other, p<0.05, assessed by ANOVA..
Since neutrophil accumulation was independent of IRF3 genotype, we hypothesized that the nontranscriptional activity of IRF3 contributed to the pathogenesis of ethanol-induced liver injury via modulation of other innate immune cell populations in the liver. Therefore, we characterized the interactions between ethanol and Irf3 genotype on the percentage of resident macrophages and infiltrating monocytes in the liver by flow cytometry. F4/80+ Kupffer cell numbers were not affected by ethanol exposure or genotype (Figure 5A). Ly6C+ monocytes were predominantly of the Ly6Clow phenotype (Figure 5B) rather than Ly6Chigh cells (Figure 5C). The relative percentage of Ly6Clow monocytes increased and Ly6Chigh decreased in Irf3−/− mice compared to the other genotypes. This shift is consistent with a potential restorative/anti-inflammatory function of Ly6Clow monocytes in protecting mice from ethanol-induced injury (6). Sorted Ly6Chigh monocytes expressed more CCR2 compared to Ly6Clow cells (Supplemental Figure 5A) ,while expression of IFNβ and IFIT1 mRNA was higher in Ly6Clow monocytes (Supplemental Figure 5B/C).
Figure 5: Irf3 genotype influenced monocyte accumulation and phenotype in liver in response to Gao-binge ethanol exposure.

C57BL/6, Irf3−/− and Irf3S1/S1 mice were exposed to the Gao-binge model as in Figure 1. Total non-parenchymal cells were isolated from the liver and the percentage of A) CD45+/F4/80+ cells, B) CD45+/Ly6Clow and C) CD45+/Ly6Chigh cells was assessed by flow cytometry. D/E/F) Expression of cytokine and chemokine, as well as G/H/I) markers of monocyte/macrophage polarization was measured by qRT-PCR in isolated NPCs. Values represent means ± SEM, n=4–8 per group. Values with different superscripts are significantly different from each other, p<0.05, assessed by ANOVA..
Expression of pro-inflammatory cytokines in the total population of isolated non-parenchymal cells revealed a predominant pro-inflammatory milieu in C57BL/6 and Irf3S1/S1 mice, compared to Irf3−/− mice (Figure 5D/E/F), consistent with the higher proportion of Ly6Clow to Ly6Chigh in the Irf3−/− mice. Further, increased expression of Arg-1, CD36 and FABP4 were indicative of a restorative/anti-inflammatory repertoire in Irf3−/−, compared to C57BL/6 and Irf3S1/S1 mice (Figure 5G/H/I). Taken together, these data suggest that the non-transcriptional function of IRF3 regulated the proportions of specific populations of Ly6Clow/Ly6C high monocytes in the liver in ethanol-induced liver injury.
The differences in Ly6Clow and Ly6Chigh infiltrating monocytes between IRF3 genotypes was not due to a difference the numbers of Ly6Clow or Ly6Chigh monocytes in the bone marrow, circulation or spleen between genotypes (Supplemental Figure 6). Therefore, we hypothesized that if the non-transcriptional activity of IRF3 contributed to apoptosis of hepatic immune cells, the absence of this IRF3-mediated RIPA activity in the Irf3−/− could contribute to increases in specific populations of infiltrating monocytes in response to Gao-binge ethanol exposure. Isolated non-parenchymal cells were stained with FITC-Annexin V Apoptosis Detection Kit to detect apoptotic cells. Interestingly, Gao-binge ethanol exposure decreased apoptosis in Ly6G+ cells in C57BL/6 and Irf3S1/S1 mice, while Irf3−/− mice had low percent Annexin V positive neutrophils in both pair-fed and Gao-binge conditions (Figure 6A). In contrast, neither diet nor genotype affected the percentage of Annexin V+/F4/80+ cells (Figure 6B). Ethanol exposure increased Annexin V+/CD45+/ Ly6Clow monocytes from C57BL/6 and Irf3S1/S1, but not Irf3−/−, mice (Figure 6C), consistent with the higher accumulation of Ly6Clow monocytes in the Irf3−/− mice (Figure 5B). Finally, the percentage of Annexin V+/CD45+/Ly6Chigh monocytes was very low and independent of diet or genotype (Figure 6D). Taken together, these data suggest that the non-transcriptional function of IRF3 contributes to apoptosis in specific populations of immune cells in the liver, modulating the balance of immune cells active within the hepatic environment in ethanol-induced liver injury.
Figure 6: Non-transcriptional function of IRF3 contributed to increased apoptosis of liver immune cells in response to Gao-binge ethanol exposure.

C57BL/6, Irf3−/− and Irf3S1/S1 mice were exposed to the Gao-binge model as in Figure 1. Total non-parenchymal cells were isolated from the liver and the percentage of immune cells also positive for Annexin V was assessed by flow cytometry. Annexin V positive cells also A) CD45+/Ly6G+, B) CD45+/F4/80+ cells, C) CD45+/Ly6Clow and D) CD45+/Ly6Chigh. Values represent means ± SEM, n=4 per group. Values with different superscripts are significantly different from each other, p<0.05, assessed by ANOVA..
While the IRF3-RIPA pathway is known to result in apoptosis in response to viral infection, it is not yet known if this mechanism plays a role in metabolic diseases (25). Therefore, we investigated the ability of the TLR3 ligand Poly (I:C) and the TLR4 ligand LPS to activate the IRF3-RIPA pathway in primary cultures of rat hepatic macrophages and hepatic macrophages isolated from C57BL/6, Irf3−/− and Irf3S1/S1 mice. Hepatic macrophages were stimulated with 25 μg/ml Poly (I:C) or 100 ng/ml LPS and the co-localization of IRF3 with mitochondria assessed by confocal microscopy. Both ligands induced the translocation of IRF3 to mitochondria after 6 h, with a more robust response observed with Poly (I:C) (Figure 7A/B). Poly (I:C) also stimulated a co-localization of IRF3 with Bax (Figure 7C). If Poly (I:C) induced translocation of IRF3 to the mitochondria and co-localization with Bax resulted in IRF3-RIPA, then we would expect to detect cleavage products of caspase-9, caspase-3 and PARP (25). Indeed, these indicators of IRF3-RIPA were increased in hepatic macrophages after 6 h challenge with Poly (I:C) (Figure 7D). Finally, we tested whether Poly (I:C) stimulated translocation of IRF3 to mitochondria in macrophages required the non-transcriptional activity of IRF3. Indeed, stimulation with Poly (I:C) induced translocation of IRF3 to the mitochondria in isolated hepatic macrophages from C57BL/6 and Irf3S1/S1 mice; IRF3 was undetectable in Irf3−/− mice (Figure 7E).
Figure 7: Poly (I:C) stimulated co-localization of IRF3 with mitochondria and Bax, as well as cleavage of caspases, in primary cultures of hepatic macrophages.

Hepatic macrophages were isolated from rats, cultured overnight and then challenged with or without 25 μg/ml Poly (I:C) or 10 ng/ml LPS for 6 h. A/B) Co-localization of IRF3 with mitochondria (Mito-tracker) and C) Bax was assessed by confocal microscopy. Nuclei were stained with DAPI. Yellow arrows highlight areas of co-localization. Images are representative of at least 6 independent hepatic macrophage isolations. D) Cleavage of caspase-9, caspase-3 and PARP was assessed by Western blot, semi-quantified and normalized to HSC70. Values represent means ± SEM, n=4. Values with different superscripts are significantly different from each other, p<0.05, assessed by ANOVA.. E) Hepatic macrophages were isolated from C57BL/6, Irf3−/− and Irf3S1/S1 and cultured overnight prior to challenge with 25 μg/ml Poly (I:C). Co-localization of IRF3 with mitochondria (Mito-tracker) was assessed by confocal microscopy. Nuclei were stained with DAPI. Yellow arrows highlight areas of co-localization. Images are representative of at least 3 independent macrophage isolations per genotype.
DISCUSSION
IRF3 is an important mediator of innate immune function, exhibiting both transcriptional and non-transcriptional activity. IRF3-mediated transcription requires activation via phosphorylation, leading to the induction of IFN-β and antiviral genes (9). In contrast, IRF3, independent of phosphorylation or transcription, can also trigger the pro-apoptotic RIPA pathway via interaction with Bax and translocation to mitochondria (10,11). The nontranscriptional RIPA activity of IRF3 contributes to its anti-viral immune function; however, the role of RIPA in the innate immune contributions to metabolic diseases, such as AH, has not been investigated. Irf3−/− mice are protected from chronic ethanol-induced liver injury, suggesting that IRF3 plays a critical role in the progression of ALD (12,13). Interestingly, in bone marrow transplant studies, Irf3-deficiency in hepatocytes actually exacerbated ethanol-induced injury, associated with impaired expression of Type I IFNs by hepatocytes (12). Making use of a novel knock-in mouse that only expresses the non-transcriptional activity of IRF3, we find that the non-transcriptional activity of IRF3-mediated RIPA also contributes to liver injury in the Gao-binge (acute on chronic) model of ethanol-induced liver injury. Our data delineate highly novel functions for the non-transcriptional functions of IRF3 in the progression of ethanol-induced liver injury. These data demonstrate that, in addition to the anti-viral function of RIPA (11), RIPA also plays an important function in maintaining innate immune homeostasis in the liver via regulation of apoptosis of infiltrating monocytes in the liver. Taken together with previous reports for a contribution of the transcriptional activity of IRF3 in hepatocytes (12), these data indicate that both transcriptional and non-transcriptional activity of IRF3 make cell-type specific contributions to the progression of ethanol-induced liver injury.
Gao-binge ethanol exposure increased multiple measures of hepatocellular injury, including increased ALT/AST, triglycerides and expression of inflammatory cytokines. Irf3−/−, but not Irf3S1/S1, mice were protected from these indicators of injury, indicating that the nontranscriptional activity of IRF3 was sufficient to drive IRF3-dependent injury in response to ethanol. One of the non-transcriptional functions of IRF3 was identified in a murine model of obesity-induced NASH wherein IRF3 restricts the movement of the p65 submit of NFκB to the nucleus via an interaction with IKKβ (14). However, in the Gao-binge model, induction of inflammatory cytokine and chemokine expression, known to be dependent on NFκB activity during ethanol exposure (2,3,6), was robustly increased in liver of both C57BL/6 and Irf3S1/S1 mice, suggesting that, in contrast to obesity-induced NASH (14), the non-transcriptional activity of IRF3 does not restrict the activity of hepatic NFκB within the context of ethanol-induced liver injury. The Gao-binge model of ethanol-induced liver injury is also characterized by accumulation of neutrophils in the liver and the development of ER stress (19,20,24). Interestingly, Gao-binge-induced neutrophil accumulation and ER stress were independent of Irf3 genotype. These data indicate that not only are neutrophil accumulation and ER stress independent of IRF3 activity, but these pathways of Gao-binge-induced injury are likely necessary, but not sufficient, to induce hepatocellular injury.
Instead, we find that the non-transcriptional activity of IRF3 modulated the accumulation of infiltrating monocytes in the liver in response to Gao-binge ethanol exposure. Infiltrating monocytes are known to contribute to both inflammation, typically characterized as Ly6Chigh, and restoration/repair, characterized as Ly6Clow, in a number of models of liver injury (26), including chronic ethanol-induced steatosis/early inflammation (27) and carbon tetrachloride-induced fibrosis (28). Depending on the stage of injury/repair, infiltrating monocytes can acquire multiple phenotypes, exhibiting pro-inflammatory, anti-inflammatory or pro-resolution/remodeling functional activity (6). At 6 h post-binge, Ly6Clow monocytes were the predominant infiltrating monocytes in the liver of wild-type mice, with no increase in numbers in response to Gao-binge (Figure 5). However, in Irf3−/− mice, there was a robust increase in Ly6Clow infiltrating monocytes and a reduction in Ly6Chigh infiltrating monocytes, favoring a reduction in the production of pro-inflammatory cytokines by hepatic non-parenchymal cells and an induction of mRNAs associated with restorative monocytes, including Arg-1, CD36 and FABP4. Importantly, this protective accumulation of Ly6Clow monocytes was abrogated in the C57BL/6 and Irf3S1/S1 mice, consistent with the increased expression of inflammatory mediators and hepatocyte injury in these mice (Figure 5).
It is interesting to note that there is a predominant accumulation of Ly6Clow monocytes at 6 h after the Gao-binge at 20–30% of CD45+ cells, compared to only 2–3% of Ly6Chigh monocytes (Figure 5B/C). This contrasts to the predominant recruitment of Ly6Chigh monocytes early in response to CCl4, which then convert to a Ly6Clow restorative phenotype (28). Given the very low numbers of Ly6Chigh monocytes in the liver, it is unlikely that there was a strong conversion of Ly6Chigh to Ly6Clow monocytes within the 6 h time frame. In contrast, it is possible that the nature of the hepatocellular injury in Gao-binge recruits Ly6Clow monocytes, which patrol endothelial surfaces for injury (29), rather than Ly6Chigh monocytes, which are recruited to sites of inflammation (29).
The increased accumulation of Ly6Clow infiltrating monocytes in the Irf3−/− mice could be due to enhanced recruitment and/or decreased removal of cells. Since IRF3-mediated RIPA is identified as a non-transcriptional function of IRF3 and apoptosis of immune cells is an important mechanism for regulation of inflammatory responses, we investigated the interaction between Gao-binge-induced accumulation of infiltrating monocytes with Irf3 genotype. Interestingly, Gao-binge ethanol exposure had a differential impact on apoptosis of hepatic immune cells in wild-type mice, as assessed by Annexin V staining, with ethanol decreasing neutrophil apoptosis and increasing apoptosis in Ly6Clow monocytes, with no apparent apoptosis in resident Kupffer cells or Ly6Chigh monocytes (Figure 6). Further, the non-transcriptional activity of IRF3 also differentially contributed to apoptosis in specific immune cell populations, with no role in neutrophils, but increasing apoptosis in Ly6Clow monocytes. Taken together, these data suggest that apoptosis is ongoing during the acute response to ethanol binge, likely contributing to the dynamic remodeling of cell populations in the liver and that IRF3-mediated RIPA is one mechanism contributing to this dynamic regulation of immune cell populations in the liver in response to ethanol.
Supplementary Material
HIGHLIGHTS.
Interferon regulatory factor 3 (IRF3) has both transcriptional and non-transcriptional activity
Gao-binge ethanol exposure increases both the phosphorylation and ubiquitination of IRF3.
Irf3−/− are protected from Gao-binge ethanol-induced liver injury but mice expressing only non-transcriptional IRF3 activity are not protected.
The non-transcriptional activity of IRF3 modulates the innate immune environment of the liver in the Gao-binge model of acute on chronic ethanol-induced liver injury by increasing the apoptotic cell death of immune cells that promote the resolution of injury.
Financial support:
This work was supported in part by NIH grants; P50 AA024333, U01AA021890 and RO1AA023722 (LEN); R01 AA023722 (LEN and XL); RO1AI073303 (GS); R21AA026017 (SC); F32 AA024595 and K99AA026648 (KLP) and F31 AA024017 (DB). This work was also supported in part by the Case Western Reserve University/Cleveland Clinic CTSA UL1RR024989 and utilized the Leica SP5 confocal/multi-photon microscope that was purchased with partial funding from National Institutes of Health SIG grant 1S10RR026820.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors have declared that no conflict of interest exists.
Reference List
- 1.Guirguis J, Chhatwal J, Dasarathy J, Rivas J, McMichael D, Nagy LE, McCullough AJ, and Dasarathy S. Clinical impact of alcohol-related cirrhosis in the next decade: estimates based on current epidemiological trends in the United States. Alcohol Clin Exp Res 2015; 39: 2085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao B and Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011; 141: 1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagy LE, Ding WX, Cresci G, Saikia P, and Shah VH. Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes. Gastroenterology 2016; 150: 1756–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeluru A, Cuthbert JA, Casey L, and Mitchell MC. Alcoholic Hepatitis: Risk Factors, Pathogenesis, and Approach to Treatment. Alcohol Clin Exp Res 2016; 40: 246–55. [DOI] [PubMed] [Google Scholar]
- 5.McCullough RL, McMullen MR, Sheehan MM, Poulsen KL, Roychowdhury S, Chiang DJ, Pritchard MT, Caballeria J, and Nagy LE. Complement Factor D protects mice from ethanol-induced inflammation and liver injury. Am J Physiol Gastrointest Liver Physiol 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ju C and Mandrekar P. Macrophages and Alcohol-Related Liver Inflammation. Alcohol Res 2015; 37: 251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Endotoxemia Rao R. and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009; 50: 638–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parihar A, Eubank TD, and Doseff AI. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J Innate Immun 2010; 2: 204–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chattopadhyay S and Sen GC. dsRNA-activation of TLR3 and RLR signaling: gene induction-dependent and independent effects. J Interferon Cytokine Res 2014; 34: 427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel JL, and Sen GC. Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity 2016; 44: 1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chattopadhyay S, Yamashita M, Zhang Y, and Sen GC. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol 2011; 85: 3708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petrasek J, Dolganiuc A, Csak T, Nath B, Hritz I, Kodys K, Catalano D, Kurt-Jones E, Mandrekar P, and Szabo G. Interferon regulatory factor 3 and type I interferons are protective in alcoholic liver injury in mice by way of crosstalk of parenchymal and myeloid cells. Hepatology 2011; 53: 649–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, Fitzgerald KA, and Szabo G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U S A 2013; 110: 16544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang XA, Zhang R, She ZG, Zhang XF, Jiang DS, Wang T, Gao L, Deng W, Zhang SM, Zhu LH, Guo S, Chen K, Zhang XD, Liu DP, and Li H. Interferon regulatory factor 3 constrains IKKbeta/NF-kappaB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 2014; 59: 870–85. [DOI] [PubMed] [Google Scholar]
- 15.Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, Tenen D, Roh HC, Kong X, Kazak L, Ahmad R, and Rosen ED. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest 2016; 126: 2839–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iracheta-Vellve A, Petrasek J, Gyongyosi B, Satishchandran A, Lowe P, Kodys K, Catalano D, Calenda CD, Kurt-Jones EA, Fitzgerald KA, and Szabo G. Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. J Biol Chem 2016; 291: 26794–26805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen JI, Roychowdhury S, DiBello PM, Jacobsen DW, and Nagy LE. Exogenous thioredoxin prevents ethanol-induced oxidative damage and apoptosis in mouse liver. Hepatology 2009; 49: 1710–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, Llorente C, Pan SQ, Yang Q, Li Y, Lazaro R, Ansong C, Smith RD, Bataller R, Morgan T, Schnabl B, and Tsukamoto H. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology 2018; 67: 1737–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai Y, Xu MJ, Koritzinsky EH, Zhou Z, Wang W, Cao H, Yuen PS, Ross RA, Star RA, Liangpunsakul S, and Gao B. Mitochondrial DNA-enriched microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. JCI Insight 2017; 2: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Ni HM, Dorko K, Kumer SC, Schmitt TM, Nawabi A, Komatsu M, Huang H, and Ding WX. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget 2016; 7: 17681–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cresci GA, Glueck B, McMullen MR, Xin W, Allende D, and Nagy LE. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J Gastroenterol Hepatol 2017; 32: 1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mao Y, Luo W, Zhang L, Wu W, Yuan L, Xu H, Song J, Fujiwara K, Abe JI, LeMaire SA, Wang XL, and Shen YH. STING-IRF3 Triggers Endothelial Inflammation in Response to Free Fatty Acid-Induced Mitochondrial Damage in Diet-Induced Obesity. Arterioscler Thromb Vasc Biol 2017; 37: 920–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ji C, Mehrian-Shai R, Chan C, Hsu YH, and Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res 2005; 29: 1496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertola A, Mathews S, Ki SH, Wang H, and Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013; 8: 627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chattopadhyay S, Yamashita M, Zhang Y, and Sen GC. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol 2011; 85: 3708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brempelis KJ and Crispe IN. Infiltrating monocytes in liver injury and repair. Clin Transl Immunology 2016; 5: e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang M, Frasch SC, Li G, Feng D, Gao B, Xu L, Ir D, Frank DN, Bratton DL, and Ju C. Role of gp91(phox) in hepatic macrophage programming and alcoholic liver disease. Hepatol Commun 2017; 1: 765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon-Walker TT, Williams MJ, Dunbar DR, Manning JR, van Rooijen N, Fallowfield JA, Forbes SJ, and Iredale JP. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A 2012; 109: E3186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ginhoux F and Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 2014; 14: 392–404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.