

Abstract
Intestinal fibrosis is a severe complication in patients with Crohn’s disease (CD). Unfortunately, the trigger leading to the development of intestinal fibrosis in the context of CD remains elusive. Here, we show that colonization by a CD-associated pathobiont adherent-invasive Escherichia coli (AIEC) promotes the development of intestinal fibrosis. Exogenously inoculated AIEC strain LF82 and commensal E. coli HS were gradually eradicated from the intestine in healthy mice. In Salmonella- or dextran sodium sulfate-induced colitis models, AIEC exploited inflammation and stably colonize the gut. Consequently, persistent colonization by AIEC LF82 led to substantial fibrosis. In contrast, commensal E. coli HS was unable to derive a growth advantage from inflammation, thereby failing to colonize the inflamed intestine or promote intestinal fibrosis. AIEC colonization potentiated the expression of the IL-33 receptor ST2 in the intestinal epithelium, which is crucial for the development of intestinal fibrosis. The induction of ST2 by AIEC LF82 was mediated by flagellin, as the FliC mutant failed to induce ST2. These observations provide novel insights into pathobiont-driven intestinal fibrosis and can lead to the development of novel therapeutic approaches for the treatment of intestinal fibrosis in the context of CD that target AIEC and/or its downstream IL-33-ST2 signaling.
Keywords: Crohn’s disease, fibrosis, gut microbiota, interleukin-33, flagellin
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the gastrointestinal tract, which comprises two major disease types, Crohn’s disease (CD) and ulcerative colitis (UC) 1. Although the precise etiology of IBD remains unknown, genetic predispositions and environmental factors, including the gut microbiota, diet, stress, smoking, cooperatively promote abnormal immune responses as well as the impairment of intestinal barrier function, thus leading to disease 2. Chronic inflammation often results in tissue fibrosis, especially in CD, frequently causing extensive local intestinal fibrosis and mechanical obstruction 3. Approximately 40% of CD patients with the ileal disease develop clinically apparent strictures 3. The strictures are clinically classified as fibrotic, inflammatory, or mixed types. Standard anti-inflammatory therapies, including anti-tumor necrotic factor-alpha (TNF-α) antibody treatment, can improve inflammatory strictures through the reduction of inflammation-mediated edema. On the other hand, conventional medical treatment is largely ineffective for fibrotic strictures. Hence, surgical resection and endoscopic dilation are utilized as treatment options for fibrotic strictures. Surgical resection is an option for more than 80% of stenosis patients 3. However, the post-surgery recurrence rate is close to 50% at 10 to 15 years following ileocecal resection. Likewise, the cumulative redilation-free rate after initial balloon dilation was 64% at 2 years and 47% at 3 years 4. Thus, existing strategies for stricture management in the context of CD are insufficient and new treatment options are desperately needed.
Accumulating evidence indicates that the gut microbiota plays a pivotal role in the development and progression of IBD. The gut microbial communities in IBD patients are known to be perturbed - a condition called gut dysbiosis 5. Gut dysbiosis results in the enrichment of potentially pathogenic members of commensal bacteria (pathobionts) and/or the reduction of protective bacteria, thereby rendering the microbiota pathogenic 6. Indeed, colonization by the dysbiotic gut microbiota isolated from IBD patients is sufficient to elicit abnormal immune activation in the intestinal mucosa 7. A pathotype of Escherichia coli, namely adherent-invasive E. coli (AIEC), has been identified as a potential pathobiont associated with IBD, particularly CD 8. Although colonization by AIEC only results in mild inflammation in normal mice 9, AIEC causes massive intestinal inflammation in genetically susceptible hosts. Moreover, AIEC aggravates dextran sulfate sodium (DSS)-induced or enteric pathogen-induced colitis 10, 11. Thus, AIEC is involved in the augmentation of inflammation in the context of IBD. However, the potential role of AIEC in the development of intestinal fibrosis remains incompletely understood. In CD patients, AIEC is isolated most frequently from the terminal ileum 8. Given that terminal ileum is the most common site for the formation of fibrotic stricture in CD 3, it is conceivable that AIEC strains target this section of the gut and contribute to the promotion of fibrosis.
Although an experimental model that would fully replicate the pathogenesis of intestinal fibrosis seen in IBD patients currently does not exist, various animal models have been used to study the mechanism of intestinal fibrosis associated with IBD 12. Chemically-induced colitis models (e.g, dextran sodium sulfate (DSS), trinitrobenzene sulfonic acid) are widely used to model fibrogenesis in IBD. However, these substances used to induce colitis and fibrosis are toxic to the intestinal epithelium and, therefore, these models might not mimic the natural course of fibrogenesis in IBD. In addition to the chemically-induced colitis models, infection with pathogenic bacteria can be used to induce intestinal fibrosis. It has been reported that Salmonella enterica serovar Typhimurium (S. Typhimurium) infection results in the development of severe fibrosis 13. S. Typhimurium infection recapitulates various pathogenic features of intestinal fibrosis seen in CD patients, such as Th1/Th17-skewed immune responses and induction of pro-fibrotic and extracellular matrix proteins. S. Typhimurium infection is therefore the optimal model for the investigation of immune pathways associated with fibrogenesis in CD 13. However, infection with this pathogen has not been implicated in the pathogenesis of CD. Hence, in this study, we have modified the Salmonella-induced intestinal fibrosis model and established an intestinal fibrosis model in which fibrosis is induced by the colonization of a CD-associated pathobiont AIEC. An attenuated strain of S. Typhimurium can cause Th1/Th17-skewed CD-like colitis, however it is insufficient for the induction of fibrosis 13. Here, we show that co-colonization with a CD-associated pathobiont AIEC promotes the development of intestinal fibrosis in mice infected with the attenuated strain of S. Typhimurium. AIEC colonization likewise promotes intestinal fibrosis in the DSS model of colitis, implying that the impact of AIEC colonization on the development of fibrosis is not model specific. AIEC colonization potentiates IL-33-ST2 signaling in the intestinal epithelium via production of flagellin. The AIEC-induced up-regulation of ST2 is critical for the induction of fibrosis. These observations are novel and identify new avenues for the treatment of intestinal fibrosis in IBD that focus on AIEC and AIEC-mediated IL-33-ST2 signaling.
Results
AIEC colonization persists in the inflamed intestine.
Infection with a murine non-typhoidal Salmonella is known to model various pathogenic features of intestinal fibrosis seen in CD patients and is hence widely used to study the immune pathways that are associated with fibrogenesis in CD 13. In this study, in order to address the extent to which colonization by CD-associated pathobionts contributes to intestinal fibrosis, we utilized a modified Salmonella-induced colitis and fibrosis model. A S. Typhimurium mutant strain that is deficient in type 3 secretion system encoded by the Salmonella pathogenicity island (SPI) 2 (ΔspiA mutant) was used. This mutant strain replicates normally in the intestine but fails to disseminate systemically, thereby causing chronic colitis without killing the infected hosts 13. Importantly, SPI-2 mutant Salmonella is unable to induce the development of intestinal fibrosis 13. SPF C57BL/6 mice were pre-treated with 20mg of streptomycin (Strep) (Fig. 1a). After 8 days, mice were challenged with the S. Typhimurium ΔspiA mutant strain (ST) and Salmonella colonization was monitored until day 21 post infection. To address the effect of AIEC colonization, mice were colonized with either a CD patient-derived AIEC strain LF82 8 or a human commensal E. coli strain HS 14 7 days prior to Salmonella or mock infection (Fig. 1a). Salmonella colonization levels reached >107 CFU/g feces on day 2 post-infection and then gradually diminished in the gut (Fig. 1b). Colonization levels of Salmonella were not affected by the presence of E. coli strains (Fig. 1b). Next, we assessed the colonization potential of E. coli strains. In the absence of Salmonella (mock infection), both HS and LF82 did not stably colonize the intestine and were gradually eradicated. In contrast, in Salmonella-infected mice, only LF82 persisted on days 7, 14 and 21 post Salmonella infection (Fig. 1c). These results suggest that AIEC, unlike commensal E. coli, is able to exploit Salmonella-caused inflammation and stably colonize the inflamed gut.
Figure 1. AIEC colonization persists in the inflamed intestine.
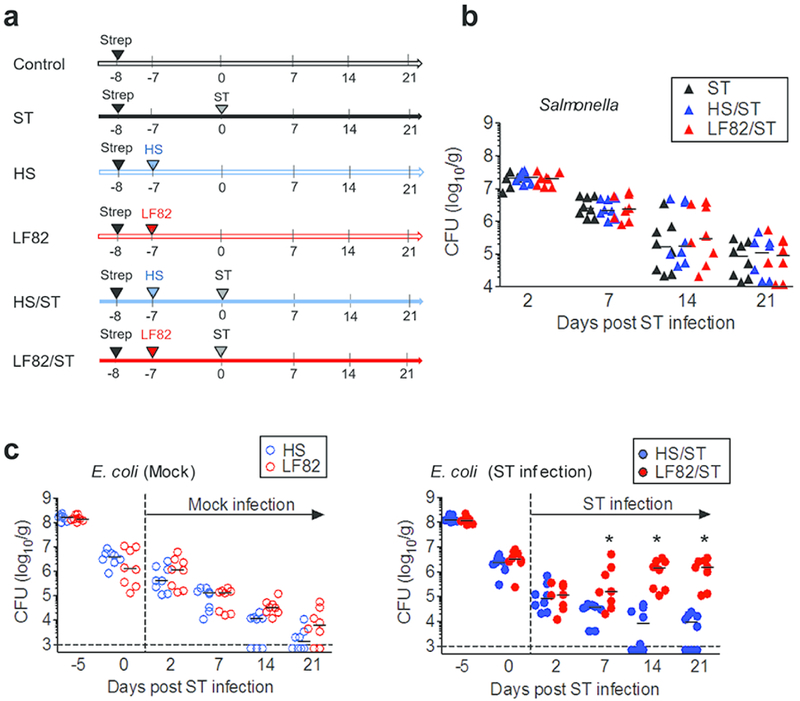
(a) SPF C57BL/6 mice were pre-treated with 20mg of streptomycin (Strep) 1 day prior to E. coli inoculation. Strep-treated mice were then inoculated with human commensal E. coli strain HS or human AIEC strain LF82 (1 × 109 CFU). The control and ST groups only received PBS. Seven days following E. coli colonization, mice were infected with 2×107 CFU of S. Typhimurium ΔspiA (ST). (b, c) Intestinal colonization (feces) of S. Typhimurium ΔspiA (b) and pre-colonized E. coli strains on indicated days post ST infection. Dots represent individual mice. Bars represent median. *; P < 0.05 by 2-Way ANOVA with Bonferroni post-hoc test.
AIEC colonization promotes colonic fibrosis.
Next, we assessed the effects of persistent AIEC colonization on the development of intestinal inflammation and fibrosis. In the acute phase (day 2), Salmonella-induced colitis (the expression of Tnf, Il6, Ifng) was not affected by the presence of E. coli strains (HS or LF82) (Fig. 2a). However, in the chronic phase (day 21), the presence of LF82 amplified the expression of pro-inflammatory cytokines induced by Salmonella infection in the colonic mucosa (Fig. 2b). In contrast, E. coli HS did not influence the severity of colitis caused by Salmonella, even in the chronic phase (Fig. 2b). Next, we examined colonic fibrosis to further assess the impact of AIEC colonization on Salmonella-induced chronic inflammation. Fibrosis was evaluated by assessing the expression of fibrosis-related factors Tgfb1 and Col1a2 in the colonic mucosa, Picro-sirius red staining and quantifying the amount of collagen deposited in the colonic tissues. Consistent with a previous report 13, infection with the mutant S. Typhimurium deficient in SPI-2 (ΔspiA) did not upregulate fibrosis-related genes or increase collagen deposition (Fig. 2c-e). Notably, upregulation of fibrosis genes and a massive deposition of collagen in the extracellular matrix (ECM), especially in the submucosal layer, were observed in the colonic mucosa of Salmonella/AIEC co-infected group (ST/LF82) (Fig. 2c-e). In contrast, HS colonization or Salmonella/HS co-colonization did not promote colonic fibrosis (Fig. 2c-e). These results indicate that persistent AIEC colonization results in chronic inflammation and fibrosis in the colon.
Figure 2. AIEC colonization promotes intestinal fibrosis in Salmonella-infected mice.
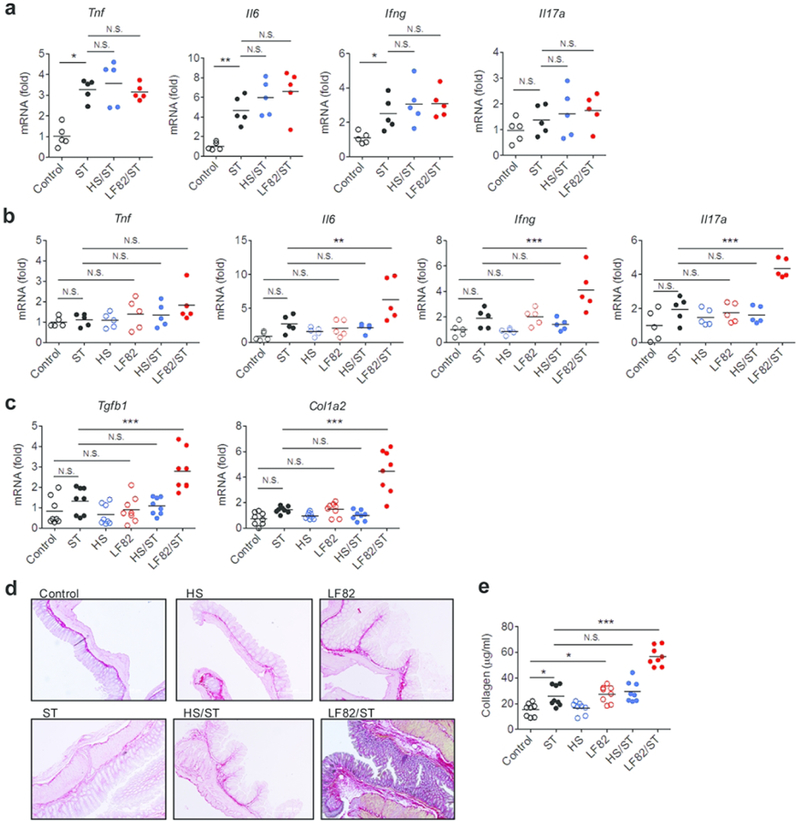
S. Typhimurium ΔspiA (ST) and E. coli (HS or LF82) infected mice were sacrificed on day 2 (a) and on day 21 post ST infection (b–e). Colonic tissues were harvested on day 2 (a) and on day 21 (b, c) post ST infection and mRNA expression of indicated cytokines was quantified by qPCR. The accumulation of collagen was examined by Picrosirius red staining on day 21 post ST infection (d) and collagen assay (e). Dots represent individual mice. Bars represent mean. N.S.; not significant, *; P < 0.05, **; P < 0.01, ***; P < 0.001 by 1-Way ANOVA with Bonferroni post-hoc test.
AIEC colonization promotes intestinal fibrosis in DSS-induced colitis.
Next, we examined the impact of AIEC colonization on the development of intestinal fibrosis in another IBD model. SPF C57BL/6 mice were pre-treated with 20mg of streptomycin 1 day prior to AIEC LF82 or commensal E. coli HS colonization. E. coli-colonized mice were then treated with 3.0% DSS for 5 days followed by regular water until day 21 (Fig. 3a). Commensal E. coli HS colonization did not affect body weight loss in DSS-treated mice (Fig. 3a). In contrast, mice colonized by AIEC LF82 lost significantly more body weight on day 14 compared to control mice not colonized by exogenous E. coli (none) (Fig. 3a). Consistent with the Salmonella-induced colitis model, LF82 colonized more efficiently than HS in DSS-treated mice during inflammation (Fig. 3b). Moreover, the colon tissue isolated from DSS-treated mice colonized by AIEC was significantly thicker than that isolated from DSS-treated mice colonized by HS or control DSS-treated mice not colonized by exogenous E. coli (Fig. 3c). However, colonic inflammation was not significantly enhanced by AIEC colonization (Fig. 3d). This evidence suggests that colon thickening in AIEC-colonized mice may not result from increased levels of inflammation. Rather, it is conceivable that AIEC colonization promoted colonic fibrosis in this model. Consistent with this notion, massive deposits of collagen were observed in the colonic tissue of DSS-treated mice colonized by LF82 compared to those colonized by HS or without E. coli colonization (Fig. 3e and 3f). These results suggest that, in addition to the colitis induced by a microbial pathogen, persistent AIEC colonization leads to the development of fibrosis in the colon in a chemically-induced colitis model.
Figure 3. AIEC colonization promotes intestinal fibrosis in DSS-induced colitis mice.
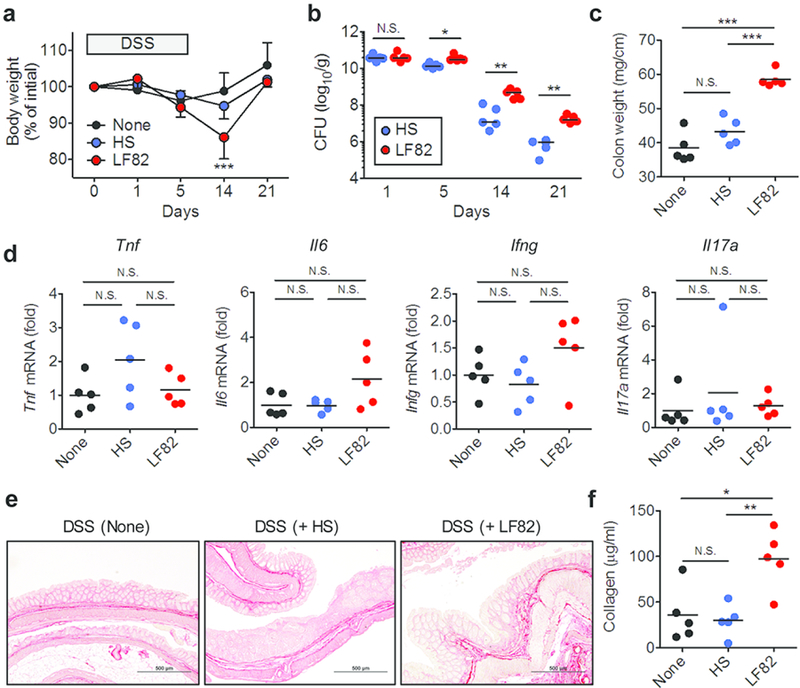
SPF C57BL/6 mice were pre-treated with 20mg of streptomycin (Strep) 1 day prior to E. coli inoculation. Strep-treated mice were then inoculated with human commensal E. coli strain HS or human AIEC strain LF82 (1 × 109 CFU each). E. coli-colonized mice were then treated with 3.0% DSS for 5 days followed by regular water. Mice were sacrificed 21 days post DSS administration. (a) Body weight change. Data shown are mean ± SD (N=5). ***; P < 0.001 by 2-Way ANOVA with Bonferroni post-hoc test (vs None). (b) Intestinal colonization (feces) of E. coli strains. Dots represent individual mice. Bars represent median. N.S.; not significant, *; P < 0.05, **; P < 0.01 by Man-Whitney U test. (c) Colon weight (per length) at day 21. (d) The mRNA expression of pro-inflammatory cytokines in the colonic mucosa. (e, f,) The accumulation of collagen was examined by Picrosirius red staining on day 21 (e) and collagen assay (f). (c–f) Dots represent individual mice (mean ± SD). N.S.; not significant, *; P < 0.05, **; P < 0.01, ***; P < 0.001 by 1-Way ANOVA with Bonferroni post-hoc test.
AIEC does not directly promote collagen deposition in fibroblasts.
To unravel how AIEC colonization promotes the development of fibrosis, we first tested whether AIEC colonization directly initiates collagen deposition in fibroblasts, which play a central role in organ fibrogenesis. We stimulated human primary colonic fibroblasts with LF82 or HS, and then measured the expression of fibrosis-related factors TGFB1 and COL1A2. As shown in Fig. 4, stimulation with neither LF82 nor HS elicited the expression of TGFB1 and COL1A2 in human fibroblasts. In contrast, stimulation with E. coli significantly upregulated the expression of pro-inflammatory cytokine TNF (Fig. 4). These results suggest that AIEC does not directly induce fibrotic programs in fibroblasts, although these cells can sense the bacterium and elicit inflammatory responses.
Figure 4. AIEC does not directly promote collagen deposition in colonic fibroblasts.
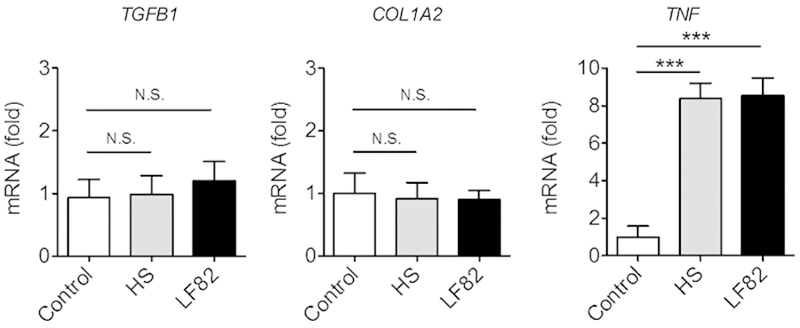
Human primary colonic fibroblasts (1 × 104 cells/500 µl/well; 24 well plate) were stimulated with E. coli HS or LF82 at MOI=10 for 3 hr followed by 15 hrs of additional culture in the presence of gentamycin (100 µg/ml) to prevent bacterial overgrowth. The expression of fibrosis-related mediators and a pro-inflammatory cytokine was quantified by qPCR. Data shown are mean ± SD (N=6). *; P < 0.05 by 1-Way ANOVA with Bonferroni post-hoc test.
AIEC colonization promotes intestinal fibrosis via the activation of IL-33-ST2 signaling.
IL-33, an IL-1 family cytokine, is implicated in the pathogenesis of fibrotic diseases in various organs 15. IL-33 binds to its receptor ST2, leading to the activation of nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs) that cooperatively drive the production of pro-inflammatory and type 2 helper T cell (Th2)-associated cytokines 15. Th2 cytokines, especially IL-13, promote TGF-β production and collagen deposition in the ECM 16. In our model, mono-colonization with Salmonella or E. coli strains (commensal HS or AIEC LF82) did not induce the expression of Il33 and Il1rl1 (gene encodes ST2) (Fig. 5a). In contrast, co-colonization with Salmonella and AIEC (group ST/LF82) resulted in a marked upregulation of Il33 and Il1rl1 in the colon, while HS colonization did not augment the expression of these genes in Salmonella-infected animals (HS/ST group) (Fig. 5a). Moreover, AIEC LF82 colonization, unlike commensal E. coli HS, induced the expression of Il1rl1 in the DSS model of colitis (Fig. 5b). Next, we validated these results in human CD patients. Using a publically available dataset (GSE75214) 17, we discovered that the expression of IL-33 (IL33) and ST2 (IL1RL1) are significantly upregulated in the ileal mucosa of active CD patients compared to that of normal controls or inactive CD patients (Fig. 5c). To assess the role of IL-33-ST2 signaling in AIEC-elicited intestinal fibrosis, we attempted to block this pathway during AIEC and Salmonella co-infection. Streptomycin-treated mice were colonized with LF82 for 7 days, followed by Salmonella infection for 21 days. The anti-ST2 blocking antibody or an isotype control IgG was injected intraperitoneally 3 times a week starting from day 7 post Salmonella infection (Fig. 5d). Salmonella and E. coli (LF82) colonization was not influenced by blockade of IL-33-ST2 signaling (Fig. 5e). Likewise, blockade of IL-33-ST2 signaling did not affect the expression of pro-inflammatory cytokines, such as TNF-α, IL-6, IFN-γ and IL-17A (Fig. 5f). In contrast, IL-33-ST2 signaling blockade suppressed AIEC colonization-induced up-regulation of fibrosis-related genes in the colonic mucosa (Fig. 5g). Consistently, diminished IL-33-ST2 signaling significantly attenuated the accumulation of collagen in the colon (Fig. 5h. i). These data suggest that AIEC colonization promotes intestinal fibrosis via the activation of IL-33-ST2 signaling.
Figure 5. AIEC colonization promotes intestinal fibrosis via the activation of IL-33-ST2 signaling.
(a, b) The mRNA expression of IL-33 (Il33) and its receptor ST2 (Il1rl1) in the colonic mucosa Salmonella/E.coli co-colonization (day 21) (a) and DSS + E. coli (day 21) (b) models. (c) mRNA expression of ST2 (IL1RL1) and IL-33 (IL33) genes in the ileal tissue from control subjects (N=11), patients with inactive CD (N=16) and active CD (N=51). Data were derived from Gene Expression Omnibus (GEO) dataset GSE75214. Bars represent median. N.S.; not significant, *; P < 0.05, **; P < 0.01, ***; P < 0.001 by Kruskal-Wallis test. (d) Mice pre-colonized with AIEC strain LF82 were infected with S. Typhimurium ΔspiA (ST). Anti-ST2 blocking antibody and isotype control IgG (50µg/mouse) were injected intra-peritoneally 3 times per week starting on day 7 post Salmonella infection (6 times total). Mice were sacrificed 21 days following ST infection. (e) Intestinal colonization (feces) of S. Typhimurium ΔspiA (left) and pre-colonized E. coli LF82 (right) on indicated days following ST infection. (f) mRNA expression of indicated cytokines 21 days post ST infection. (g) Expression of fibrosis-related genes in the colonic mucosa (day 21). (h, i) Accumulation of collagen was quantified by Picro-sirius red staining (h) and collagen assay (i). Dots represent individual mice. Bars represent median (e) or mean (f, g, i). N.S.; not significant, *; P < 0.05, **; P < 0.01, ***; P < 0.001 by 1-Way ANOVA with Bonferroni post-hoc test.
CD-associated E. coli strains induce ST2 expression in IECs.
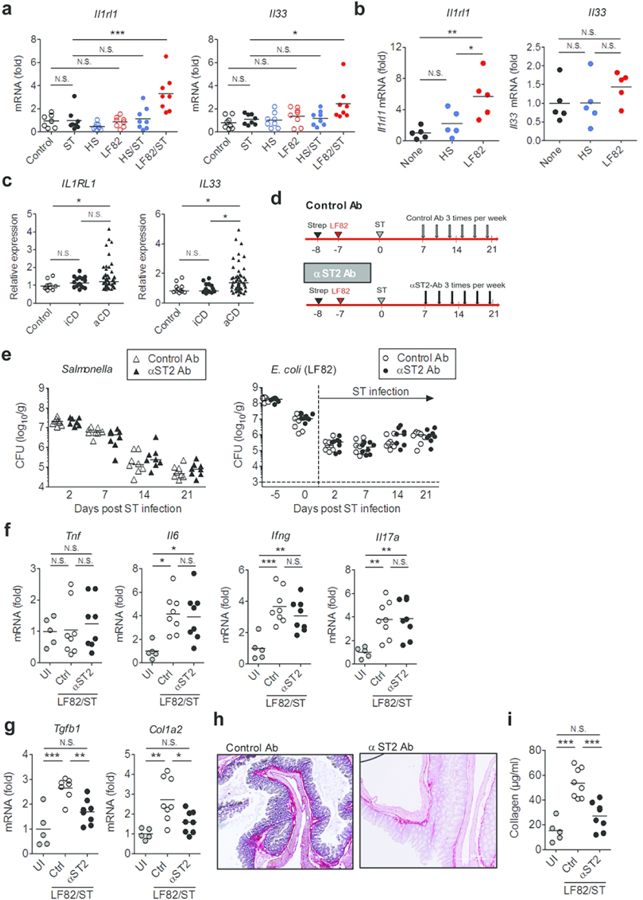
The IL-33 receptor ST2 is known to be expressed in various immune and non-immune cells. In the gastrointestinal tract, it is reported that intestinal epithelial cells (IECs) express high levels of ST2 18. Likewise, IECs are major producers of its ligand IL-33 19. Hence, we asked whether AIEC colonization can elicit the expression of ST2 and/or IL-33 in IECs. To this end, T84 cells, a human colonocyte cell line, were stimulated with E. coli HS or LF82 and the expression of IL-33 and ST2 was assessed by qPCR. As a control, human primary colonic fibroblasts were also stimulated with E. coli strains. As expected, only stimulation with AIEC LF82 elicited a marked upregulation of IL33 (IL-33) and IL1RL1 (ST2) expression (Fig. 6a). Neither E. coli strains induced the expression of these genes in fibroblasts (Fig. 6a). Next, we confirmed this result in vivo. To this end, we examined the expression of ST2 in the colonic mucosa of Salmonella-infected and Salmonella/E. coli co-infected mice (Fig. 6b). Consistent with the in vitro result, co-infection with Salmonella and AIEC (LF82/ST) resulted in the expression of ST2 protein in the colonic epithelium (Fig. 6b), whereas mono-infection with Salmonella (ST) or co-infection with Salmonella and E. coli HS (HS/ST) did not cause ST2 expression in the colonic mucosa (Fig. 6b). These results suggest that the CD-associated E. coli strain induces ST2 expression in the colonic epithelium.
Figure 6. CD-associated E. coli strains induce ST2 expression in IECs.
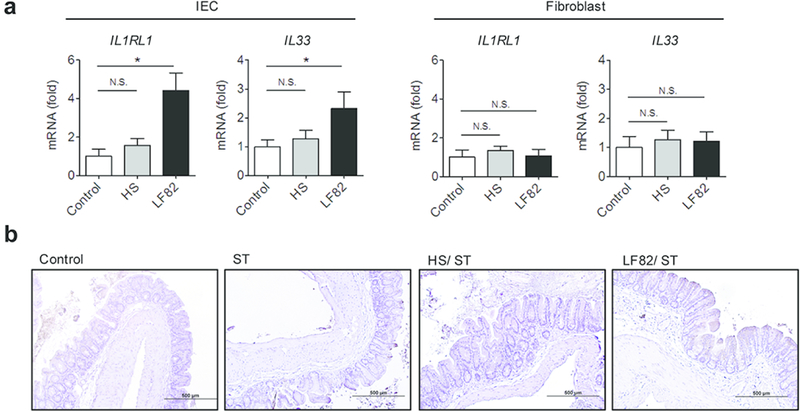
(a) Human IEC T84 cells and primary colonic fibroblasts (1 × 105 cells/500 µl/well; 24 well plate) were stimulated with E. coli HS or LF82 at MOI=10 for 3 hr followed by 15 hrs of additional culture in the presence of gentamycin (100 µg/ml) to prevent bacterial overgrowth. mRNA expression of IL-33 (IL33) and ST2 (IL1RL1) was quantified by qPCR. Data shown are mean ± SD (N=6). N.S.; not significant, *; P < 0.05 by 1-Way ANOVA with Bonferroni post-hoc test. (b) Strep-treated mice were then inoculated with human commensal E. coli strain HS or human AIEC strain LF82 (1 × 109 CFU). The control and ST groups only received PBS. Seven days following E. coli colonization, mice were infected with 2×107 CFU of S. Typhimurium ΔspiA (ST). At day 21 post ST infection, the colonic tissues were harvested and ST2 was strained. Representative images of ST2 immunohistochemistry are shown.
Flagellin mediates the induction of ST2
Since one of AIEC’s pathogenic features is its ability to adhere to and invade IECs 8, we next assessed whether epithelial adhesion/invasion trigger the expression of these genes. To this end, we utilized a panel of AIEC and non-AIEC strains isolated from CD patients (except HS) and used them to stimulate IECs. Consistent with the results obtained from LF82, all tested AIEC strains (strain LF16, LF31, LF73, and LF82) elicited the expression of IL1RL1 in IECs (Fig. 7a). The non-AIEC strains, with the exception of LF1, were not able to induce IL1RL1 (Fig. 7a). Although two AIEC strains (LF82 and LF73) were able to induce the expression of IL33, all the other AIEC and non-AIEC strains failed to up-regulate IL33 expression (Fig. 7a). These results suggest that the ability of E. coli to adhere/invade to IECs is not required for the induction of ST2. Notably, IL-33 induction was not induced by most of CD-associated E. coli strains, indicating that the induction of ST2, rather than its ligand IL-33, might be a key fibrosis-initiating event resulting from pathobiont colonization in the context of CD. Next, we sought to identify the microbial factor(s), present in the CD-associated pathobiont LF82, involved in the induction of ST2. We employed the isogenic mutant strains of LF82 that lack FimH (adhesin) 20, FliC (flagellin synthesis) 21, OmpA or OmpC (outer membrane proteins) 22. T84 cells were stimulated with WT LF82 or the mutant strains, and the expression of IL1RL1 and IL33 was assessed by qPCR. The LF82 ΔfimH mutant induced IL1RL1 at levels similar to WT LF82, indicating that epithelial adhesion is not required for this process (Fig. 7b). Similarly, the ΔompA and ΔompC mutants were able to induce the expression of IL1RL1. Notably, the LF82 ΔfliC mutant was significantly impaired in its ability to induce IL1RL1, suggesting that flagellin is a key bacterial factor driving the expression of ST2 in IECs. Interestingly, all tested mutant strains, including the LF82 ΔfliC mutant, induced IL-33 with equal efficiency as their parental strain WT LF82.
Figure 7. The induction of ST2 by AIEC is mediated by flagellin.
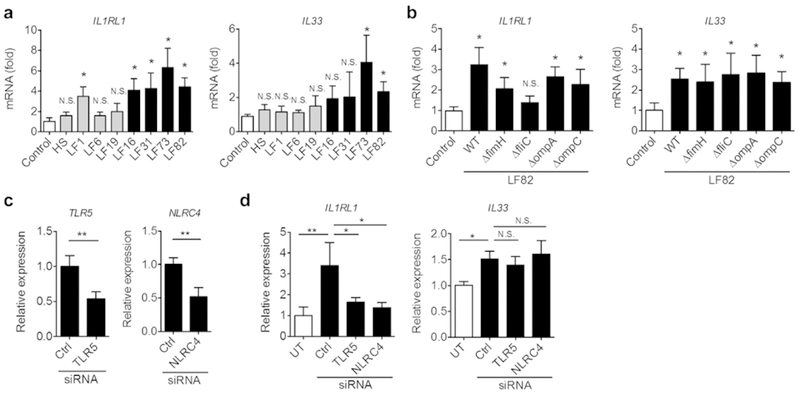
(a) T84 cells were stimulated with human commensal E. coli strain HS or E. coli strains isolated from CD patients. Gray bars indicate non-AIEC strains and black bars indicate AIEC strains based on adhesion/invasion assay. (b) T84 cells were stimulated with AIEC LF82 or various mutants of LF82 at MOI=10 for 3 hr followed by 15 hrs of additional culture in the presence of gentamycin (100 µg/ml) to prevent bacterial overgrowth. mRNA expression of IL-33 (IL33) and ST2 (IL1RL1) was quantified by qPCR. Data shown are mean ± SD (N=6). N.S.; not significant, *; P < 0.05 by Dunnett test (vs control). (c) TLR5 and NLRC4 mRNA were knocked-down in T84 cells using siRNA specific to human TLR5 or NLRC4. Relative expression to Ctrl Non-targeting pool siRNA are shown. Data shown are mean ± SD (N=3). **; P < 0.01 by Student-t test. (d) TLR5 and NLRC4 mRNA were knocked-down in T84 cells using siRNA. Cells were then stimulated with AIEC LF82 at MOI=10 or medium (untreated; UT) for 3 hr followed by 15 hrs of additional culture in the presence of gentamycin (100 mg/ml) to prevent bacterial overgrowth. mRNA expression of ST2 (IL1RL1) and IL-33 (IL33) was quantified by qPCR. Data shown are mean ± SD (N=3). N.S.; not significant, *; P < 0.05, **; P < 0.01 by 1-Way ANOVA with Bonferroni post-hoc test.
To identify the receptor involved in AIEC-mediated ST2 expression in IECs, we next knocked-down the known flagellin receptors TLR5 and NLRC4 in IECs. The introduction of siRNA significantly reduced the expression of these receptors in T84 cells (Fig. 7c). Our results showed that both TLR5 and NLRC4 are involved in the induction of ST2 by AIEC (Fig. 7d). To validate the role of flagellin in the induction of intestinal fibrosis in the attenuated Salmonella/AIEC co-infection model, we next co-infected streptomycin-treated mice with the SPI-2 mutant of Salmonella and WT or the ΔfliC mutant of LF82 (Fig. 8a). Flagellin production by AIEC did not affect the ability of Salmonella to colonize the intestine and Salmonella was eliminated from the mice in both WT LF82/ST and ΔfliC LF82/ST groups with the same efficiency (Fig. 8b). Likewise, the WT and ΔfliC LF82 strains colonized equally well in the inflamed gut, indicating that flagellin is not required for adaptation to the inflammatory microenvironment (Fig. 8b). The expression of pro-inflammatory cytokines in the colonic mucosa was not attenuated in the ΔfliC LF82/ST group compared to the WT LF82/ST group (Fig. 8c), suggesting that LF82 is able to augment Salmonella-induced inflammation in the absence of flagellin secretion. However, the ΔfliC mutant LF82 did not induce the expression of ST2 in the colonic mucosa (Fig. 8d, e). As a result of impaired ST2 induction, the expression of fibrosis-related genes was significantly reduced in the ΔfliC LF82/ST group compared to the WT LF82/ST group (Fig. 8f), thereby leading to the attenuated accumulation of collagen in the colon (Fig. 8g, h). These data suggest that AIEC’s flagellin promotes intestinal fibrosis.
Figure 8. Flagellin is required to promote intestinal fibrosis by a CD-associated E. coli.
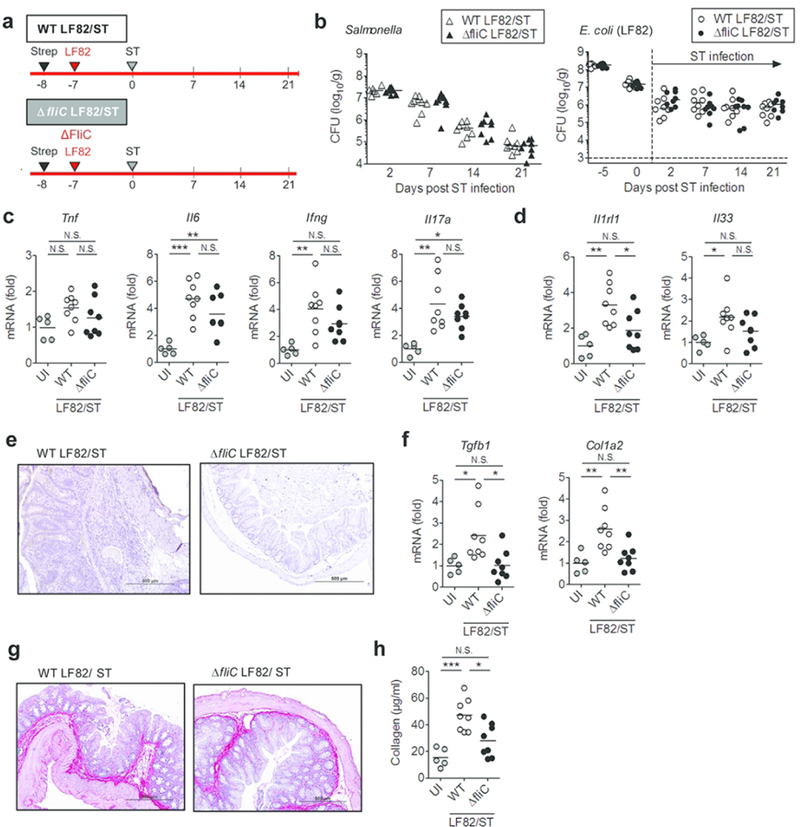
(a) SPF C57BL/6 mice were pre-treated with 20mg of streptomycin (Strep) 1 day prior to E. coli inoculation. A CD-associated AIEC strain LF82 (WT or ΔfliC mutant) strains (1 × 109 CFU) were then inoculated to Strep-treated mice. Seven days after AIEC colonization, mice were infected with 2×107 CFU of Salmonella typhimurium ΔspiA (ST). (b) Intestinal colonization (in feces) of ST and AIEC LF82 (WT or ΔfliC mutant) at indicated days post ST infection. Dots represent individual mice. Bars represent median. (c) Expression of inflammation-related genes in the colonic mucosa (day 21 ST). (d) The mRNA expression of IL-33 (Il33) and its receptor ST2 (Il1rl1) in the colonic mucosa. (e) Representative images of ST2 immunohistochemistry (day 21). (f) Expression of fibrosis-related genes in the colonic mucosa (day 21 ST). (g, h) Accumulation of collagen was examined by Sirius red staining (g) and collagen assay (h). Dots represent individual mice. Bars represent mean. N.S.; not significant, *; P < 0.05, **; P < 0.01, ***; P < 0.001 by 1-Way ANOVA with Bonferroni post-hoc test.
Discussion
It has become evident that the gut microbiota in IBD patients is significantly perturbed compared to non-IBD subjects 5. However, the impact of gut dysbiosis on disease outcomes remains poorly understood. A recent report has demonstrated that colonization of mice with dysbiotic microbiotas obtained from CD patients sufficiently recapitulates the pro-inflammatory responses seen in CD patients 7. Thus, dysbiosis in IBD might be accompanied by an abnormal accumulation of pathobionts whose colonization likely elicits intestinal inflammation. However, the identities of IBD-associated pathobionts remain largely unknown. AIEC is an E. coli pathotype found to be more prevalent in the gastrointestinal tract of patients with CD 23. As of now, the consequences of chronic colonization by AIEC in CD patients are incompletely understood.
In the present study, we have demonstrated that persistent gut colonization by the AIEC strain LF82 potentiated the development of intestinal fibrosis in Salmonella-induced colitis model in mice. Interestingly, colonization by a human commensal E. coli strain HS (a symbiont strain) did not affect intestinal fibrosis, indicating that induction of fibrosis is a specific feature of AIEC. Notably, neither pathobiont nor symbiont E. coli strains influenced intestinal inflammation caused by Salmonella infection, suggesting that promotion of fibrosis, resulting from AIEC colonization, cannot be explained by considering AIEC-triggered inflammation alone. One possible explanation is that AIEC adapts better to intestinal inflammation compared to the commensal E. coli strain and an increased abundance of E. coli might lead to intestinal fibrosis regardless of AIEC signatures. In this context, we have found that Salmonella-induced intestinal inflammation facilitated LF82 colonization, while not affecting HS. Neither E. coli strain was able to stably colonize the intestine in the absence of Salmonella. In IBD patients, it is well-documented that Enterobacteriaceae (phylum Proteobacteria) are over-represented in the gut microbiota 5. In general, a healthy gut microbiota consists of obligate anaerobes, such as Clostridiales and Bacteroidales, while bacteria belonging to the family Enterobacteriaceae, including E. coli, are only minor constitutes. On the other hand, intestinal inflammation tends to foster blooms of Enterobacteriaceae at the expense of obligate anaerobes. Intestinal inflammation elicits the infiltration of inflammatory immune cells in the mucosa, such as neutrophils. These immune cells express oxidative enzymes (e.g., iNOS, NOX1, DUOX2, PHOX, SOD, and MPO) capable of generating antimicrobial radicals, such as superoxide, peroxide, hypochloride, nitric oxide, and peroxynitrite. Further oxidation of these molecules yields nitrates, N-oxides, S-oxides and other compounds that serve as electron acceptors. Facultative anaerobes, including Enterobacteriaceae, rely on these products to utilize anaerobic respiration in the intestine 24, 25. Since both AIEC and commensal E. coli can utilize anaerobic respiration, it is conceivable that AIEC employs specific, as yet unknown, mechanisms that allow it to adapt to the inflammatory environment with greater efficiency. Further study is needed to unravel the unique metabolic pathways utilized by AIEC to adapt to the inflammatory environment.
In this study, we have demonstrated that AIEC colonization elicits expression of the IL-33 receptor ST2 in IECs, which in turn augments IL-33 signaling and promotes the development of intestinal fibrosis. IL-33 is a member of the IL-1 cytokine family that has been discovered relatively recently 15. IL-33 engages its receptor ST2 expressed on various immune and non-immune cells, and activates its downstream signaling 15. The IL-33-ST2 signaling pathway is considered to be crucial for type 2 immune responses since it induces the production of IL-5 and IL-13 in Th2 cells and/or granulocytes. IL-33 is a protective cytokine important for promotion of wound healing 15. However, excessive tissue regeneration mediated by IL-33 can lead to the development of fibrosis in various organs, such as liver cirrhosis, systemic sclerosis and pulmonary fibrosis 26–28. Thus, the role of IL-33-ST2 in IBD remains controversial. Experimental animal IBD models have been used to demonstrate that IL-33-ST2 signaling promotes intestinal inflammation via enhancing IL-4 29. On the other hand, mice deficient in ST2 develop more severe colitis than control mice in DSS-induced and TNBS-induced IBD models, indicating that IL-33-ST2 signaling has a protective role 18. Likewise, a recent study has also reported that IL-33 aids tissue healing in the gut by enhancing the differentiation of goblet cells and M2 type macrophages, thus protecting mice from DSS and TNBS-induced colitis 18. This discrepancy might in part be due to variability in the genetic backgrounds of experimental animals. The former study utilized Balb/c (Th2 skewed) animals whereas C57BL/6 (Th1 skewed) animals were used in the latter two studies. This may indicate that the IL-33-ST2 pathway plays different roles in UC and CD, as these two disease forms are associated with differently skewed adaptive immunity 1. In human IBD, IL-33 expression is reported to be elevated in the inflamed mucosa, mainly in UC and, to a lesser extent, in CD 30, 31. In contrast, blood levels of IL-33 are found to decrease only in UC patients as compared to healthy control subjects 32. Additionally, blood levels of soluble ST2 (sST2), which antagonizes the activity of IL-33, are elevated in both UC and CD 32. In this study, we analyzed the publically available GEO dataset to discover that the expression of both IL-33 and ST2 is up-regulated in the ileal mucosa of patients with active CD, although this analysis cannot discriminate between soluble and membrane-bound ST2, which play opposite roles with respect to IL-33 signaling.
Various cell types are known to express membrane-bound ST2. ST2 is involved in IL-33 signaling and is primarily expressed, under normal conditions, in IECs in both mice and humans 18. During intestinal inflammation, ST2-positive cells can also be found in subepithelial infiltrates in both murine colitis models as well as human IBD patients 18. Therefore, it is possible that other ST2-expressing cells also contribute to AIEC-induced intestinal fibrosis. Recent accumulating evidence suggests that group 2 innate lymphoid cells (ILC2), known to express high levels of ST2, play a crucial role in the development of fibrosis via activating Th2 immune responses, as demonstrated in various models of fibrosis, such as liver and lung 26, 33. However, a recent report has shown ILC2 to be dispensable for the development of intestinal fibrosis 34. Thus, ILC2 might not be involved in AIEC-induced intestinal fibrosis.
In this study, we have been able to demonstrate that AIEC colonization enhances ST2 expression in IECs, which in turn augments the sensing of IL-33 in the intestine. However, the identity of pathways downstream of IL-33-ST2 that leads to fibrosis remains elusive. Since the blocking of ST2 resulted in a decreased expression of TGF-β and collagen in the intestine, AIEC-mediated activation of the IL-33-ST2 pathway might eventually lead to the up-regulation of TGF-β, thereby promoting collagen deposition in fibroblasts. In the lung, pulmonary epithelial cells express TGF-β 35. Thus, it is possible that IECs produce TGF-β in response to IL-33 and promote collagen expression in fibroblasts through mutual interaction. Another possibility is that IL-33 signaling in IECs alters the function of IECs, which in turn induce the activation and/or differentiation of other TGF-β producing cells, such as macrophage/dendritic cells (DCs). For example, thymic stromal lymphoprotein (TSLP), which is induced by IL-33, is known to condition DCs and skew them toward the type 2 phenotype, including up-regulation of TGF-β expression 36. It follows, then, that TSLP conditioned-DCs might mediate pro-fibrotic responses. Further research is required to identify the downstream mediators that link IL-33 signaling and intestinal fibrosis.
Our present results indicate that flagellin, a principal component of bacterial flagella, is a key molecule produced by AIEC that activates the expression of ST2 in IECs and subsequently causes the development of fibrosis. The role of bacterial flagella in locomotion is well documented 37. A decrease in the expression of type 1 pili, required for cell adhesion, has recently been observed in an isogenic mutant of LF82 that lacks fliC, the gene that encodes flagellin 37. As a result, the fitness and virulence of the LF82 ΔfilC mutant were significantly impaired both in vitro and in vivo 37. In our study, the ST2 inducing capacity of AIEC LF82 remained intact when the fimH gene, which encodes adhesin and thus facilitates AIEC adhesion to IECs, was mutated. Consistently, a non-AIEC strain LF1, which cannot adhere/invade IECs, was also capable of inducing the expression of ST2 in IECs. It has been reported that TLR5 plays an important role in the sensing of AIEC 38. Consistently, we found that AIEC colonization induces ST2 expression in IECs through TLR5. In addition to TLR5, our result indicates that the activation of NLRC4 signaling by flagellin may also be used for the induction of ST2.
Our study has demonstrated that flagellin, produced by AIEC, is critical for the induction of ST2 expression in IECs. However, it remains unclear why only AIEC induces ST2 and promotes intestinal fibrosis; after all, both AIEC and commensal E. coli HS have flagellin. In this regard, it has been recently shown that AIEC LF82, unlike commensal E. coli HS, produces flagellin when exposed to intestinal mucus 37. Thus, it is conceivable that AIEC produces more flagellin than commensal E. coli and this hypothesis would explain why only AIEC elicits flagellin-mediated pro-fibrotic signaling and subsequent development of intestinal fibrosis. Related to this notion, it also remains unclear why infection with the SPI-2 Salmonella mutant per se fails to induce fibrosis, since it also harbors flagellin. A simple explanation is that colonization of the mutant Salmonella peaked on day 2 post infection and was dramatically eradicated in the later phase of infection. On the other hand, AIEC colonization persisted longer and its abundance in the later phase of infection was higher than that of Salmonella. Thus, persistent colonization by a flagellated pathobiont resulted in intestinal fibrosis. Another possibility may be related to the capacity of the bacterium to survive intracellularly. It has been reported that SPI-2 is essential for the survival of the pathogen in phagocytic cells, such as macrophages 39. Since AIEC can survive within infected macrophages 8, it is possible that intracellular survival of pathobionts within intestinal macrophages contributes to pro-fibrotic events induced by IL-33-ST2 signaling. However, it is largely unclear how macrophage activation by AIEC affects ST2 induction in the colonic epithelium. Thus, further study is required to fully understand the mechanisms by which CD-associated pathobionts induce intestinal fibrosis.
Similarly, the extent to which other bacteria, in addition to AIEC, contribute to susceptibility to intestinal fibrosis in CD patients remains uncertain. In this regard, it has been repeatedly reported that flagellated bacteria are enriched and the luminal flagellin load is elevated in CD patients compared to normal controls 7, 40. Thus, dysbiotic microbiotas rich in flagellated bacteria are able to elicit intestinal fibrosis regardless of the abundance of AIEC. Consistently, colonization of germ-free mice with the gut microbiotas isolated from CD patients leads to ST2 expression in the colon 7. Thus, CD-associated pathobionts, including AIEC, contribute to the development of intestinal fibrosis via production of flagellin.
Taken together, we have unveiled a novel link between gut dysbiosis and the risk for intestinal fibrosis. Treatment of gut dysbiosis by antibiotics, probiotics, or fecal microbiota transplantation to eradicate AIEC and/or other flagellated pathobionts could be a novel therapeutic approach used to prevent intestinal fibrosis in CD. Alternatively, blockade of IL-33-ST2 signaling has the potential to be a treatment option for fibrosis in the context of CD patients that does not perturb the gut microbiota.
Materials and Methods
Animals
Eight to ten week-old specific pathogen-free (SPF) wild-type C57BL/6 mice were originally purchased from Jackson Laboratories. Mice were then bred and maintained in a specific pathogen-free (SPF) animal facility at the University of Michigan. All animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan.
Bacterial strains
Kanamycin- and streptomycin (Strep)-resistant Salmonella enterica serovar Typhimurium (S. Typhimurium) SL1344 ΔspiA 41 was used in mouse infection experiments. Escherichia coli strains used in this study were commensal E. coli strain HS 14, AIEC strains LF82, LF16, LF31 and LF73 isolated from CD patients 8, and non-AIEC-type strains LF1, LF6 and LF19 isolated from CD patients. Mutant LF82 strains ΔfimH, ΔfliC, ΔompA, and ΔompC were also used. All bacteria were grown in Luria-Bertani (LB) broth at 37°C with shaking (180 rpm).
Salmonella infection and E. coli colonization
Mice were pre-treated with 20 mg/100 βl/mouse of streptomycin (Strep) 1 day prior to E. coli inoculation. Strep-treated mice were then inoculated with human E. coli strain HS or LF82 (1 × 109 CFU). Seven days after E. coli colonization, mice were infected with 2×107 CFU of S. Typhimurium SL1344 ΔspiA. To determine bacterial numbers in the feces, fecal pellets were collected from individual mice, homogenized in cold PBS, and plated at serial dilutions on MacConkey agar plates containing 50 µg/ml Strep or ampicillin (Amp). CFU counts were determined after overnight incubation at 37°C. IL-33-ST2 signaling in vivo was blocked by the intraperitoneal administration of 50 µg of an anti-mouse ST2 antibody or control rat IgG (Genentech. Inc) 3 times per week. 21 days following Salmonella infection, colonic tissues were harvested and fixed in buffered 4% paraformaldehyde. Fixed tissues were then embedded in paraffin, sectioned, and stained with heamatoxylin and eosin or picro-sirius red (Picrosciences, Inc.).
Quantitative real-time PCR
RNA was extracted from colonic tissues using the E.Z.N.A.® Total RNA Kit I (Omega Bio-tek). Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative PCR was performed with SYBR Green qPCR Kits (Radiant molecular tolls). The primers used are listed in supplementary table 1. The cycling conditions were 95°C for 3 mins and then 39 cycles of 95°C for 15 sec, 60°C for 1 min. The relative mRNA expression of target gene was normalized against that of the β-actin gene in the same RNA preparation. The following primer sets were used for amplifications: Human hIL1RL1-Fw; 5’- CTG TCT GGC CCT GAA TTT GC −3’, Human IL1RL1-Rv; 5’- AGC AGA GTG GCC TCA ATC CA −3’, Human IL33-Fw; 5’- ATC CCA ACA GAA GGC CAA AG −3’, Human IL33-Rv; 5’- CCA AAG GCA AAG CAC TCC AC −3’, Human TGFB1-Fw; 5’- CTA ATG GTG GAA ACC CAC AAC G −3’, Human TGFB1-Rv; 5’- TAT CGC CAG GAA TTG TTG CTG −3’, Human COL1A2-Fw; 5’- TCT GGA TGG ATT GAA GGG ACA −3’, Human COL1A2-Rv; 5’- CCA ACA CGT CCT CTC TCA CC −3’, Human TNF-Fw; 5’- GCC TCT TCT CAT TCC TGC TT −3’, Human TNF-Rv; 5’- TGG GAA CTT CTC ATC CCT TTG −3’, Human Actb-Fw; 5’- CTC TTC CAG CCT TCC TTC CTG −3’, Human Actb-Rv; 5’- GAA GCA TTT GCG GTG GAC GAT −3’. Mouse Il1rl1-Fw; 5’- AGA CCT GTT ACC TGG GCA AG −3’, Mouse Il1rl1-Rv; 5’- CAC CTG TCT TCT GCT ATT CTG G −3’, Mouse Il33-Fw; 5’- TCC TTG CTT GGC AGT ATC CA −3’, Mouse Il33-Rv; 5’- TGC TCA ATG TGT CAA CAG ACG −3’, Mouse Tgfb1-Fw; 5’- CTC CCG TGG CTT CTA GTG C −3’, Mouse Tgfb1-Rv; 5’- GCC TTA GTT TGG ACA GGA TCT G −3’, Mouse Col1a2-Fw; 5’- TGG ATA CGC GGA CTC TGT TG −3’, Mouse Col1a2-Rv; 5’- CCC TTT CGT ACT GAT CCC GAT T −3’, Mouse Tnf-Fw; 5’- GCC TCC CTC TCA TCA GTT CT −3’, Mouse Tnf-Rv; 5’- CAC TTG GTG GTT TGC TAC GA −3’, Mouse Il6-Fw; 5’- GAG GAT ACC ACT CCC AAC AGA CC −3’, Mouse Il6-Rv; 5’- AAG TGC ATC ATC GTT GTT CAT ACA −3’, Mouse Ifng-Fw; 5’- ATC TGG AGG AAC TGG CAA AA −3’, Mouse Ifng-Rv; 5’- TTC AAG ACT TCA AAG AGT CTG AGG TA −3’, Mouse Il17a-Fw; 5’- CAG GGA GAG CTT CAT CTG TGT −3’, Mouse Il17a-Rv; 5’- GCT GAG CTT TGA GGG ATG AT −3’, Mouse Actb-Fw; 5’- AAG TGT GAC GTT GAC ATC CG −3’, Mouse Actb-Rv; 5’- GAT CCA CAT CTG CTG GAA GG −3’.
Collagen assay
Colonic specimens were homogenized in 2.5M acetic acid containing 0.1 mg/ml pepsin and stored at −80°C until sufficient quantity was obtained. The samples were then dried overnight at 37ºC. Total soluble collagen was quantified by Soluble Collagen Assay (Cell Biolabs, Inc).
Cell culture
Human primary colonic fibroblasts were isolated from disease-free surgical margins found in surgically resected colonic specimens obtained from a diverticulitis patient. Human fibroblasts were then cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics (penicillin/streptomycin). Human colon carcinoma cells (T84) were grown in Ham’s F12 medium + DMEM (1:1) supplemented with 10% FBS and antibiotics. Cells (1 × 105 cells/500 µl /well; 24 well plate) were stimulated with E. coli at a MOI=10 for 3 hr followed by 15 hrs of additional culture in the presence of gentamycin (100 µg/ml) to prevent bacterial overgrowth.
Knock-down of flagellin receptors
SMARTpool siRNA for human TLR5 and NLRC4 were purchased from Dharmacon. The siRNAs or Non-targeting control siRNA (Dharmacon) were transfected to T84 cells using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturers’ instructions. Two days post-transfection, the cells were stimulated with E. coli (MOI=10) for 3 hr followed by 15 hrs of additional culture in the presence of gentamycin (100 µg/ml).
Immunohistochemistry
The colonic tissues were harvested, fixed with 4% paraformaldehyde and embedded in paraffin. The tissue samples were then cut into 4 µm thick sections. Rehydrated paraffin sections were boiled in a microwave oven for epitope retrieval using a sodium citrate buffer (pH 6, 10 min). Slides were stained with an anti-mouse ST2 antibody (Abcam, ab25877, 1:500 dilution) overnight at 4°C and then incubated with a rabbit HRP polyclonal antibody for 1 h at room temperature. HRP was then visualized with DAB (VECTOR).
Statistical Analyses
Statistical analyses were performed using GraphPad Prism software version 5.0 (GraphPad Software, Inc, San Diego, CA). Differences between 2 groups were evaluated using Student’s t test (parametric) or the Mann–Whitney U test (nonparametric). For comparisons of more than 3 groups, statistical analysis was performed using 1-way analysis of variance (parametric) or the Kruskal–Wallis test (nonparametric), followed by the Bonferroni correction for parametric samples, or the Dunn test for nonparametric samples as a post hoc test. P values less than 0.05 were considered significant.
Acknowledgments
The authors thank the University of Michigan Center for Gastrointestinal Research (UMCGR) (NIH 5P30DK034933) for technical assistance. This work was supported by Kenneth Rainin Foundation Innovator Award (N.K.), National Institute of Health DK110146 and DK108901 (N.K.), T32 DK094775 (T.L.M.), Crohn’s and Colitis Foundation of America (N. K and H. N.-K.), Mitsukoshi Health and Welfare Foundation (J.I.), JSPS Postdoctoral Fellowship for Research Abroad (S.K. and H. N.-K.), the Uehara Memorial Foundation Postdoctoral Fellowship Award (S.K. and K.S.), Clinical and Translational Science Awards (CTSA) Program (S.K.), and Prevent Cancer Foundation (S.K.). All data necessary to reproduce the results presented herein are available in the main text.
Footnotes
Disclosures: The authors declare no competing interests
References
- 1.Xavier RJ, Podolsky DK Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Ananthakrishnan AN Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol 12, 205–217 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Rieder F, Fiocchi C, Rogler G Mechanisms, Management, and Treatment of Fibrosis in Patients With Inflammatory Bowel Diseases. Gastroenterology 152, 340–350.e346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Latella G, Di Gregorio J, Flati V, Rieder F, Lawrance IC Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol 50, 53–65 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Sartor RB, Wu GD Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 152, 327–339.e324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamada N, Seo SU, Chen GY, Nunez G Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 13, 321–335 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Nagao-Kitamoto H et al. Functional characterization of inflammatory bowel disease-associated gut dydbiosis in gnotobiotic mice. Cellular and Molecular Gastroenterology and Hepatology 2, 468–481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darfeuille-Michaud A et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127, 412–421 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Small CL, Reid-Yu SA, McPhee JB, Coombes BK Persistent infection with Crohn’s disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat Commun 4, 1957 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carvalho FA, Barnich N, Sauvanet P, Darcha C, Gelot A, Darfeuille-Michaud A Crohn’s disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm Bowel Dis 14, 1051–1060 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Small CL, Xing L, McPhee JB, Law HT, Coombes BK Acute Infectious Gastroenteritis Potentiates a Crohn’s Disease Pathobiont to Fuel Ongoing Inflammation in the Post-Infectious Period. PLoS Pathog 12, e1005907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rieder F, Kessler S, Sans M, Fiocchi C Animal models of intestinal fibrosis: new tools for the understanding of pathogenesis and therapy of human disease. Am J Physiol Gastrointest Liver Physiol 303, G786–801 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grassl GA, Valdez Y, Bergstrom KS, Vallance BA, Finlay BB Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology 134, 768–780 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Rasko DA et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 190, 6881–6893 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmitz J et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Fichtner-Feigl S et al. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J Immunol 178, 5859–5870 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Vancamelbeke M et al. Genetic and Transcriptomic Bases of Intestinal Epithelial Barrier Dysfunction in Inflammatory Bowel Disease. Inflamm Bowel Dis 23, 1718–1729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sedhom MA et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 62, 1714–1723 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moussion C, Ortega N, Girard JP The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One 3, e3331 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dreux N et al. Point mutations in FimH adhesin of Crohn’s disease-associated adherent-invasive Escherichia coli enhance intestinal inflammatory response. PLoS Pathog 9, e1003141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnich N, Boudeau J, Claret L, Darfeuille-Michaud A Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn’s disease. Mol Microbiol 48, 781–794 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Rolhion N, Carvalho FA, Darfeuille-Michaud A OmpC and the sigma(E) regulatory pathway are involved in adhesion and invasion of the Crohn’s disease-associated Escherichia coli strain LF82. Mol Microbiol 63, 1684–1700 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Palmela C et al. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 67, 574–587 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Winter SE et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamada N, Chen GY, Inohara N, Nunez G Control of pathogens and pathobionts by the gut microbiota. Nat Immunol 14, 685–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McHedlidze T et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 39, 357–371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koca SS et al. The IL-33 gene is related to increased susceptibility to systemic sclerosis. Rheumatology international 36, 579–584 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Wu L et al. IL-33 Can Promote the Process of Pulmonary Fibrosis by Inducing the Imbalance Between MMP-9 and TIMP-1. Inflammation (2018). [DOI] [PubMed]
- 29.Pushparaj PN et al. Interleukin-33 exacerbates acute colitis via interleukin-4 in mice. Immunology 140, 70–77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beltran CJ et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis 16, 1097–1107 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Pastorelli L et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A 107, 8017–8022 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seo DH et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Scientific reports 7, 851 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mindt BC, Fritz JH, Duerr CU Group 2 Innate Lymphoid Cells in Pulmonary Immunity and Tissue Homeostasis. Frontiers in immunology 9, 840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo BC et al. The orphan nuclear receptor ROR alpha and group 3 innate lymphoid cells drive fibrosis in a mouse model of Crohn’s disease. Science immunology 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denney L et al. Pulmonary Epithelial Cell-Derived Cytokine TGF-beta1 Is a Critical Cofactor for Enhanced Innate Lymphoid Cell Function. Immunity 43, 945–958 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Son A et al. TWEAK/Fn14 pathway promotes a T helper 2-type chronic colitis with fibrosis in mice. Mucosal Immunol 6, 1131–1142 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Sevrin G et al. Adaptation of adherent-invasive E. coli to gut environment: Impact on flagellum expression and bacterial colonization ability. Gut Microbes 1–17 (2018). [DOI] [PMC free article] [PubMed]
- 38.Chassaing B, Koren O, Carvalho FA, Ley RE, Gewirtz AT AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 63, 1069–1080 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buckner MM, Croxen MA, Arena ET, Finlay BB A comprehensive study of the contribution of Salmonella enterica serovar Typhimurium SPI2 effectors to bacterial colonization, survival, and replication in typhoid fever, macrophage, and epithelial cell infection models. Virulence 2, 208–216 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duck LW et al. Isolation of flagellated bacteria implicated in Crohn’s disease. Inflamm Bowel Dis 13, 1191–1201 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW Simultaneous identification of bacterial virulence genes by negative selection. Science 269, 400–403 (1995). [DOI] [PubMed] [Google Scholar]