

Abstract
Tissue ischemia, such as transient myocardial ischemia, leads to release of cellular RNA including microRNA into the circulation and extracellular space, but the biological function of the extracellular (ex-) RNA is poorly understood. We recently reported that cardiac RNA of both human and rodent origins induced cytokine production and immune cell activation. However, the identity of ex-RNA responsible for the pro-inflammatory effect remains unclear. In the current study, using a miRNA array, we profiled the plasma miRNAs four hours after transient myocardial ischemia (45 min) or sham procedure. Among 38 plasma miRNAs that were elevated following ischemia, eight were tested for their ability to induce cytokine response in macrophages and cardiomyocytes. We found that six miRNA mimics (-34a, -122, -133a, -142, -146a, -208a) induced cytokine production in a dose-dependent manner. The effects of miRNAs (-133a, -146a, -208a) were diminished by uridine→adenosine mutation and by RNase pretreatment. The miRNA-induced cytokine (MIP2, TNFα, IL-6) production was abolished in cells deficient of TLR7 or MyD88 or by a TLR7 antagonist, but remained the same in TLR3- or Trif-deficient cells. In vivo, mice i.p. injected with miR-133a or miR-146a had marked peritoneal neutrophil and monocyte migration, which was significantly attenuated in TLR7-/- mice. Moreover, locked nucleic acid (LNA) anti-miRNA inhibitors of these six miRNAs markedly reduced cardiac RNA-induced cytokine production. Taken together, these data demonstrate that ex-miRNA mimics (-34a, -122, -133a, -142, -146a, -208a) are potent innate immune activators and that the miRNAs most likely induce cytokine production and leukocyte migration through TLR7 signaling.
Introduction
We have previously demonstrated that within a few hours after transient myocardial ischemia (30-45 min), there is an increase in the plasma cell-free RNA concentration (1). To determine the potential role of these cellular RNA in inflammation and tissue injury, we and others have demonstrated that administration of RNase attenuates necrotic cell-induced cytokine production in cardiomyocytes and immune cells in vitro (1) and reduces myocardial infarction following transient ischemia in vivo (1, 2). These data suggest that extracellular (ex-) RNA may mediate necrosis-induced inflammation and contributes to myocardial ischemic injury. Moreover, cellular RNA, either purified from rodent and human hearts, or released from hypoxia-injured cardiomyocytes, induces multiple cytokine production in both cardiomyocytes and immune cells (3). We further show that ex-RNA-induced cytokine effect is significantly attenuated by TLR7 inhibitor or in TLR7-deficient cells, and was completely abolished by MyD88 deficiency (3). However, the nature and identity of the ex-RNA responsible for cytokine production is unclear.
miRNA are short, highly evolutionarily conserved, single-stranded (17-25 nucleotides) non-coding RNA (4-6). miRNA bind to the 3′ untranslated region of target mRNA and regulate gene expression either by inhibiting mRNA translation or inducing its degradation. A variety of cardiac miRNAs are modulated following acute myocardial infarction (7, 8) and some are released from ischemic myocardium into circulation (9-11). More than 200 miRNAs exist in the heart (12). Some are reportedly expressed in a tissue-specific manner (13), such as miR-208a in the heart (14). In addition, other muscle-enriched miRNAs, such as miR-1, miR-133 and miR-499, are highly expressed in cardiomyocytes and skeletal muscle cells (13). Several studies indicate that the circulating levels of miR-208a, miR-499, miR-1 and miR-133 are markedly elevated following acute myocardial infarction in patients and animals (15, 16) and may serve as sensitive biomarkers (11, 13). However, the specific biological function of these circulating miRNAs following cardiac ischemia and whether they play any particular role in myocardial inflammation/injury remains unclear.
In the current study, we hypothesize that multiple miRNAs are released into circulation following transient myocardial ischemia and that certain ex-miRNAs are capable of activating innate immunity and inducing cellular and tissue inflammation via specific TLR signaling. To test this, we profiled the circulating miRNAs in the plasma following a short period of coronary artery ligation and tested a set of eight miRNAs for their abilities to induce inflammation in macrophages/cardiomyocytes and in a mouse model of peritonitis. Moreover, using specific TLR antagonist and knockout mice, we tested the role of TLRs in mediating the miRNA-mediated effects.
Materials and Methods
Materials
miRNAs with phosphorothioate linkages were synthesized by Integrated DNA Technologies (Coralville, IA). The miRNA sequences were provided by miRBase 21 (http://www.mirbase.org/) (Table 1). All miRNA mutant derivatives were generated by replacing uridines with adenosines. LNA miRNA inhibitors and the control oligonucleotide (Negative Control A) were purchased from Exiqon (Vedbaek, Denmark). Collagenase 2 and RNase A of bovine pancreas were from Sigma-Aldrich (St. Louis, MO). DNase was from Thermo Scientific Inc. (Waltham, MA). Imiquimod (R837, TLR7 ligand) was provided by Invivogen (San Diego, CA). Pam3Cys (P3C) was purchased from Enzo Life (Plymouth Meeting, PA). Specific immunoregulatory DNA sequences (IRSs) with phosphorothioate linkages were synthesized by Integrated DNA Technologies (Coralville, IA) as previously described (17). The following sequences were tested: IRS661 (TLR7 inhibitor: 5′-TGCTTGCAAGCTTGCAAGCA-3′), IRS869 (TLR9 inhibitor: 5′-TCCTGGAGGGGTTGT-3′), and the control oligonucleotide (Con: 5′-TCCTGGCGGAAAAGT-3′). Antibodies were purchased from Cell Signaling Tech. (Danvers, MA).
Table 1.
Sequences of microRNAs and anti-miRNAs.
| miRNA | Sequences | Total nt | Number of U | |
|---|---|---|---|---|
| 1 | mmu-miR-133a-3p | UUUGGUCCCCUUCAACCAGCUG | 22 | 7 |
| 2 | mmu-miR-208a-3p | AUAAGACGAGCAAAAAGCUUGU | 22 | 4 |
| 3 | mmu-miR-146a-5p | UGAGAACUGAAUUCCAUGGGUU | 22 | 7 |
| 4 | mmu-miR-142-3p | UGUAGUGUUUCCUACUUUAUGGA | 23 | 11 |
| 5 | mmu-miR-122-5p | UGGAGUGUGACAAUGGUGUUUG | 22 | 8 |
| 6 | mmu-miR-34a-5p | UGGCAGUGUCUUAGCUGGUUGU | 22 | 9 |
| 7 | mmu-miR-192-5p | CUGACCUAUGAAUUGACAGCC | 21 | 5 |
| 8 | mmu-miR-210-3p | CUGUGCGUGUGACAGCGGCUGA | 22 | 5 |
|
|
||||
| anti-miRNA | Sequences | Total nt | ||
|
|
||||
| 1 | negative control A | TAACACGTCTATACGCCCA | 19 | |
| 2 | anti-miR-133a-3p | AGCTGGTTGAAGGGGACCA | 19 | |
| 3 | anti-miR-208a-3p | AAGCTTTTTGCTCGTCTTA | 19 | |
| 4 | anti-miR-146a-5p | AACCCATGGAATTCAGTTCTC | 21 | |
| 5 | anti-miR-142-3p | ATAAAGTAGGAAACACTAC | 19 | |
| 6 | anti-miR-122-5p | AAACACCATTGTCACACTC | 19 | |
| 7 | anti-miR-34a-5p | ACAACCAGCTAAGACACTGC | 20 | |
Animals
Wild-type (WT, C57BL/6), TLR3-/-, and TLR7-/- mice were purchased from The Jackson Laboratory (Bar Harbor, ME). MyD88-/- mice were generated by Kawai and colleagues (18). Trif-/- mice were generated by Yamamoto, et al. (19). Mice were 8-12 week-old, weighed between 20-30 g, and gender and age matched. Mice were fed the same bacteria-free diet (Prolab Isopro RMH 3000, LabDiet, Brentwood, MO) and water. The animal protocols were approved by the Subcommittee on Research Animal Care of Massachusetts General Hospital (Boston, MA). The experiments were performed in compliance with the guideline of the National Institutes of Health (Bethesda, MD). Simple randomization method was used to assign animals to various experimental conditions.
Mouse myocardial ischemia-reperfusion (I/R) model
The surgical procedures of myocardial ischemic injury were performed as previously described (1, 20, 21). In brief, male mice were anesthetized by intra-peritoneal injection of ketamine (120 mg/kg) and xylazine (4 mg/kg), intubated, and ventilated. Body temperature was maintained at 36.5 to 37.5°C on a heating pad. A left intercostal thoracotomy was performed to expose the heart. The left anterior descending coronary artery was visualized and ligated with 7-0 silk sutures under a surgical microscope. Myocardial ischemia was confirmed by myocardial blanching and ECG change on Lead II. Forty-five min after LAD occlusion, the ligature was released and reperfusion was visually confirmed. Sham-operated mice underwent the same procedure with a suture passed under the LAD but without ligation. Four hours after reperfusion, the blood was collected from the heart in an EDTA-containing tube and the plasma samples were subsequently used for microRNA measurements.
RNA extraction and quantitation
RNA in the plasma was extracted using Trizol LS and quantified using Quant-iT™ RNA assay kit (Life Technologies, Grand Island, NY) as reported previously (1, 3).
Circulating microRNA array
EDTA-anticoagulated serum samples were prepared for the miRNA array assay (Firefly™ microRNA Assay, http://www.fireflybio.com/technology). The assay system was based on encoded hydrogel particles and used a unique post-hybridization ligation-based scheme to fluorescently label bound microRNA targets. After hybridizing the microRNA targets to the target-specific probes attached to the hydrogel Firefly™ Particles, a universal biotinylated adapter was ligated to the captured targets. A fluorescent reporter binds to the universal adapter and is ultimately used for microRNA detection in a flow cytometer. Since RNA extraction was no longer needed to detect miRNA with this technology, we were able to detect miRNA array in a tiny volume of plasma (100 μl) without risk of losing RNA. This is particularly advantageous for mouse study.
Circulating MicroRNA Detection by qRT-PCR
Total RNA was isolated from 0.20 ml mouse plasma using Trizol LS (Life Technologies, Grand Island, NY) extraction procedure as described above. The C. elegans miRNA (cel-miR-39) was added before RNA extraction following the instruction of miRNeasy Serum/Plasma Spike-in Control (Qiagen, Valencia, CA). RNA Pellets were suspended in 14 μl of RNase-free water and 12 μl samples were used for Reverse transcription (RT) according to the protocol of miScript II RT kit (Qiagen, Valencia, CA). Mouse miRNAs and cel-miR-39 were quantified by using miScript SYBR Green PCR kit and corresponding primers following the protocol of the manufacturer (Qiagen, Valencia, CA). Relative expression was calculated using the comparative Ct method (2-ΔΔCt).
Cell isolation and culture
Bone marrow-derived macrophage (BMDM) culture
Bone marrow cells were harvested from the tibias and femurs of mice, cultured, and differentiated into macrophages in the presence of 10 ng/ml macrophage colony-stimulating factor as described previously with minor modifications (21). Briefly, cells were re-suspended in RPMI 1640 culture medium and seeded in a 96-well plate (2 × 105 cells/well) in CO2 incubator at 37 °C. Three days later, culture media were changed and the macrophages were ready for experiments at day 5.
Rat neonatal cardiomyocyte (CM) culture
Rat neonatal CMs were prepared as described previously with minor modifications (22). Briefly, the hearts were isolated, dissected from major vessels, and cut into small pieces. The heart tissues were then incubated in ADS buffer (pH 7.35, 116 mM NaCl, 20 mM HEPES, 0.8 mM Na2HPO4, 5.6 mM glucose, 5.4 mM KCl, and 0.8 mM MgSO4) containing 0.4 mg/ml collagenase 2 and 0.6 mg/ml pancreatin (Worthington, Lakewood, NJ) at 37 °C for 8 min in a shaker. Cell suspension was slowly removed, and the remaining myocardial tissues were further incubated with the enzyme buffer for 9 more times. Cell suspensions were collected, spun, and re-suspended in DMEM containing 20% FBS and 4.5% D-glucose. Fibroblasts were removed by plating cells on 10-cm dishes for 70 min. Neonatal CMs were then plated in a 96-well plate, pre-coated with 5 μg/ml fibronectin and 0.2 mg/ml gelatin (Sigma-Aldrich, St. Louis, MO), (0.8 × 105 cells/well) and incubated in CO2 incubator at 37 °C for 36 h before experiments.
Treatments of cell cultures
Cell culture medium was changed to pre-warmed serum-free RPMI 1640 medium containing 0.05% BSA for 1 h before treatment. miRNA mimics were mixed with lipofectamine 3000 (Life Technologies, Grand Island, NY) following the manufacturer's instruction and incubated for 5 min at room temperature before applied to cell cultures. For nucleases digestion experiments, microRNA (50 nM in BMDM and 150 nM in rCM) was incubated with RNase (10 μg) or DNase (1 U) at room temperature for 30 min before packed with lipofectamine 3000. For the IRSs inhibition experiments, CMs were treated with 300 nM IRSs or control sequence that had been packed with lipofectamine 3000, for 1 h before microRNA treatment.
Cytokines
Media were prepared at 4 °C and stored at -80 °C and subsequently measured for MIP-2, TNFα, and IL-6 using ELISA kits (R&D systems, Minneapolis, MN). Final cytokine concentrations were calculated based on a standard curve constructed in each experiment.
Western Blotting
Cells were lysed in NP-40 buffer and cell lysates were centrifuged at 12,000 g at 4°C for 30 min. Proteins in supernatants were separated in 4-12% gradient Bis-Tris SDS-PAGE and immunoblotted with antibodies (1:1000) against phospho-JNK, JNK, phospho-p38, p38, phospho-ERK, ERK, I-κBα, and GAPDH (all from Cell Signaling Tech, Danvers, MA) as reported previously (21).
In vivo microRNA administration
After shaving the furs and sterilizing the skin with 70% ethanol, mice were administered with lipofectamine-packed miR-133a-3p (20 μg/mouse) or lipofectamine alone through intra-peritoneal injection (i.p.) using 31 G insulin syringe. The injection site was subsequently covered by an adhesive 3M Tegaderm film to prevent infection. Twenty hours later, the peritoneal lavage was harvested as described previously (21). In brief, 2 ml of normal saline was injected into the peritoneal space and mixed thoroughly by gentle massage of the abdomen. About 1.2 ml of the peritoneal lavage was collected and centrifuged. The cell pellets were suspended. Total peritoneal cells were manually counted. Eight × 105 cells were incubated with 1:100 diluted specific anti-mouse Ly-6G (BD Biosciences, San Jose, CA) and anti-mouse F4/80 (eBiosciences, San Diego, CA) at 4°C for 30 min. After washing, the neutrophils, resident macrophages, and recruited monocytes were determined by flow cytometry gating as Ly-6G+, F4/80high, or F4/80low, respectively.
miRNA inhibition
BMDMs or rCMs were treated with 10 μg/ml RNA purified from the heart. Six miRNA inhibitors (specifically for miRNAs: miR-133a, -208a, -146, -142, -122, -134a) were mixed together at the indicated concentration. The anti-miR combo (final concentration = 100 nM) was added to the culture media before the RNA treatment. In a separate group, a control oligonucleotide was used instead.
The Uptake of miRNA
BMDMs isolated from WT or TLR7 KO mice were treated with fluorescent 6-FAM-conjugated miR-133a-3p (50 nM, packed with lipofectamine). Four hours later, fluorescent images were taken under a microscope. The cells were detached by trypsin digestion and the intracellular fluorescence was determined by flow cytometry.
Co-localization of miRNA and TLR7
BMDMs or rCMs were treated with lipofectamine-packed 6-FAM-conjugated miR-133a-3p (50 or 100 nM, respectively). Four hours later, the cells were fixed and co-stained with specific antibodies to TLR7 (Novus, Littleton, CO) and EEA1 (Santa Cruz, Dallas, Texas). The fluorescent imaging was analyzed by confocal microscopy (Nikon). The nuclei were stained with DAPI.
Statistical Analysis
Statistical analysis was performed using Graphpad Prism 5 software (Graphpad, La Jolla, CA). Unless stated otherwise, the distributions of the continuous variables were expressed as the mean ± SD. For those cytokine levels below detection limit, values input at the detection limit were used. The statistical significance of the difference between groups was measured by two-way ANOVA with post hoc analysis. The null hypothesis was rejected for P<0.05 with the two-tailed test.
Results
Transient myocardial ischemia leads to an increase in circulating miRNAs
Four hours after transient myocardial ischemia (45 min of coronary artery ligation) or sham procedures, mouse plasma RNA was extracted and quantified. As shown in Fig. 1A, there was a significant increase in the plasma RNA concentration in the mice subjected to ischemic injury (I/R) as compared with sham mice (465±164 vs. 248±62, P<0.05). The RNA measurement with a RNA fluorescent dye was validated as the purified RNA was completely degraded by RNase, but not DNase, treatment (Fig. 1A).
Figure 1. Multiple circulating miRNAs were increased following myocardial ischemia/reperfusion.

Mice were subjected to sham or myocardial ischemia/reperfusion (I/R) procedures. I/R mice had a left anterior descending coronary artery (LAD) ligation for 45 min followed by 4 h reperfusion, whereas sham mice only underwent left thoracotomy without coronary artery ligation. In separate experiments, EDTA-treated plasma or serum was harvested for RNA quantitation, miRNA profiling by miRNA array, and qRT-PCR (as detailed in Experimental Procedures). A, Blood RNA quantitation following myocardial I/R. Each error bar represents mean ± SD. * P < 0.05, *** P < 0.001. n = 4 in each group. B, Heatmap of circulating miRNA array. C, Quantitative data of increased circulating miRNA following myocardial I/R compared with Sham group. Each error bar represents mean ± SE. n = 6 in Sham group, n = 9 in I/R group. D, Mean fluorescent intensity of selected eight circulating miRNAs in Sham and I/R mice. Each error bar represents mean ± SE. n = 6 in Sham group, n = 9 in I/R group. E, The increase of multiple circulating miRNAs validated by qRT-PCR. Each error bar represents mean ± SD. * P < 0.05, *** P < 0.001. n = 10 in Sham group and n = 17 in MI/R group.
We profiled the circulating plasma miRNAs following transient myocardial ischemia protocol noted above. Among the panel of 68 miRNAs reportedly related to cardiac diseases (Fig. 1B) (http://www.abcam.com/multiplex-mirna-assay-cardiology-panel-circulating-ab204063-references.html), 38 were significantly elevated in the ischemia group (I/R) compared to those in sham group (Fig. 1C). The complete miRNA array data (accession number GSE74951) is available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74951. There are 31 miRNAs that exhibited>2-fold increase in the plasma of the I/R mice compared with that of the sham mice.
To test the function of the miRNAs, we selected eight miRNAs with the I/R/sham ratio > 2 and with fluorescence intensity > 100 as indicated by the arrows in Fig. 1B-C. These were miR-146a, miR-133a, miR-122, miR-34a, miR-208a, miR-192, and miR-210. Fig. 1D illustrates the mean fluorescent intensity of these eight miRNAs in both sham and I/R groups. Moreover, we validated, using qRT-PCR, the change of these eight miRNAs in the plasma samples from a separate set of sham and I/R animals. As shown in Fig. 1E, with the exception of miR-142a, the other 7 miRNAs, including miR-146a, miR-133a, miR-122, miR-34a, miR-208a, miR-192, and miR-210, were all significantly increased in the plasma samples isolated from I/R mice compared to those from sham mice.
miRNA mimics induce a dose-dependent cytokine production in macrophages and cardiomyocytes
We tested the eight miRNA mimics (Table 1) for their ability to induce cytokine production. As shown in Fig. 2A-B, of the eight miRNA mimics, six miRNAs, i.e., miR-146a, miR-133a, miR-142a, miR-122, miR-34a, and miR-208a, induced a dose-dependent MIP-2 response in both macrophages (BMDM) and cardiomyocytes (rCM). miR-142, miR-146a, and miR-133 seemed the most potent while miR-34a and miR-122 were the weakest. In contrast, miR-192 and miR-210 induced no MIP-2 production at the same concentrations. When tested in a combination, the six miRNAs (miR-146a, -133a, -142a, -122, -34a, and -208a) mixture induced a dose-dependent MIP-2 production with EC50 of 18 nM and 44 nM in macrophages and cardiomyocytes, respectively (Fig. 2C-D).
Figure 2. Multiple synthetic miRNA mimics induced cytokine production in both macrophages and cardiomyocytes.

Bone marrow-derived macrophages (BMDM, A, C, E, and G) or neonatal rat cardiomyocytes (rCM, B, D, F, and H) were treated with selected 8 miRNA mimics (A and B), mixture of six miRNAs (miR-133a, -146a, -142a, -122, -34a, -208a) (C and D), miRNA U→A mutants (miR-133a, -146a, -208a) ((E and F), or in the presence of RNase or DNase (G and H). Sixteen hours later, the culture media were collected and cytokine expression in the media was measured by MIP-2 ELISA. Each error bar represents mean ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001. n = 3 in each group. Con: control.
U→A miRNA mutation and RNase diminish miRNA-induced cytokine response
To test the specificity of miRNA-induced cytokine production, we mutated miR-133a, miR-208a, and miR-146a by replacing uridine (U) with adenosine (A). As shown in Fig. 2E-F, the miRNAU→A mutants failed to induce MIP-2 production in both BMDM and cardiomyocytes. Moreover, RNase, but not DNase, pretreatment of the miRNA mimics prior to lipofectamine packaging completely abolished cytokine response-induced by miRNAs in BMDM and cardiomyocytes, respectively (Fig. 2G-H). These data confirmed the specificity of the miRNA-induced MIP-2 response.
miRNA mimics induce cytokine response in a TLR7/MyD88-dependent manner
Early studies have established TLR3 and TLR7 as the sensors for viral RNA and TLR9 for DNA (23-26). Thus, we hypothesize that miRNA mimics induce cytokine production through the intracellular RNA sensors, TLR3 or TLR7 and their signaling molecule Trif or MyD88. We isolated BMDM from WT, TLR7-/-, MyD88-/-, TLR3-/-, and Trif-/- mice and treated them with specific TLR3 or TLR7 ligand or various miRNA mimics. As shown in Fig. 3A, the six miRNA mimics, R837 (TLR7 ligand) and Pam3cys (TLR2 ligand) induced various degrees of MIP2 response in macrophages. TLR7 deletion completely blocked the R837-induced MIP-2 response, but had no effect on the Pam3cys (P3C)-induced response. Importantly, TLR7 deficiency blocked all 6 miRNA-induced MIP-2 production (Fig. 3A). Moreover, genetic deletion of MyD88, the key adaptor of several TLRs including TLR7, blocked the MIP-2 response induced by the miRNAs and the TLR7/TLR2 ligands (Fig. 3A). In contrast, TLR3 or Trif deficiency had no impact on the miRNA-induced cytokine response. We also measured both TNFα and IL-6 production in response to the miRNA treatments in BMDM. As shown in Fig. 3, similar to MIP2 response, miR-146a and -142 induced strong TNFα and IL-6 responses, which were completely abolished in TLR7 and MyD88-deficient cells (Fig. 3B-C).
Figure 3. miRNA mimics elicited cytokine production through TLR7/MyD88 signaling.

A, The deficiency of TLR7 and MyD88, but not TLR3 and Trif, completely abolished miRNA mimic-induced MIP2 production in BMDM. Bone marrow-derived macrophages (BMDM) were isolated from wild-type and genetically modified mice and treated with various lipofectamine-complexed miRNA mimics (50 nM), R837 (TLR7 ligand, 0.25 μg/ml), or Pam3Cys (P3C, TLR2 ligand, 10 ng/ml). B-C, The deficiency of TLR7 and MyD88, but not TLR3 and Trif, completely abolished miRNA mimic-induced TNFα and IL-6 production in BMDM, respectively. D, Specific TLR7 antagonist (IRS661) attenuated miRNA mixture-induced cytokine response in rCM. Neonatal cardiomyocytes were cultured and treated with 300 nM lipofectamine-complexed IRSs or a control oligo for 1 h before treatments of miRNA (150 nM of mixture of 6 miRNA mimics: miR-133a, -146a, -142a, -122, -34a, -208a), R837 (0.5 μg/ml), or Pam3cys (P3C, 30 ng/ml). Sixteen h after treatment, the culture media were collected and cytokine expression in the media was measured by MIP-2 ELISA. Each error bar represents mean ± SD. * P < 0.05, ** P< 0.01, *** P < 0.001. n = 3 in each group. Con: control; IRS: immunoregulatory sequence.
Next, we demonstrated the critical role of TLR7 in the miRNA-induced MIP2 production in rat cardiomyocytes. As shown in Fig. 3D, compared to the control oligonucleotide (Con.) or the TLR9 antagonist (IRS869), IRS661, a specific TLR7 inhibitory oligonucleotide, markedly decreased R837 (TLR7 ligand)-induced MIP-2 production (50% decrease), but had no impact on Pam3cys-induced effect. Most importantly, IRS661, but not IRS869 (17), significantly attenuated the 6 miRNA mixture (miR-133a, -146a, -142a, -122, -34a, -208a)-induced MIP-2 response (88.1% reduction). Collectively, these data suggest that TLR7-MyD88 signaling likely mediates the miRNA-elicited cytokine production.
miRNA mimics activate intracellular MAPKs and NF-κB pathways
To determine the intracellular signaling pathways of these miRNA mimics, we tested the phosphorylation of the three MAP kinases, JNK, ERK, and p38, at 30, 60, and 90 min following the miRNA mixture treatment. As shown in Fig. 4A-C, phosphorylation of all three MAP kinases was time-dependent and significantly increased at 90 min after the miRNA treatment. To measure the activation of NF-κB signaling, we detected the total amount of IκB, the degradation of which leads to the nuclear translocation and subsequently activation of NF-κB. The IκB level was decreased by 70% at 90 min following the miRNA treatment (Fig. 4D). Together, these data suggest that miRNA mimics are capable of activating the intracellular MAPKs and NF-κB pathways.
Figure 4. miRNA mimic activates MAPKs and NF-κB in BMDM.

Mouse bone marrow-derived macrophages (BMDM) were treated with the mixture of the six miRNAs (50 nM) for 0, 30, 60, or 90 min. The cell lysates were harvested and tested for phosphorylation of JNK (A), ERK (B), p38 (C), and degradation of I-κBα (D) using immunoblotting blot. The data were quantitated as the fold change of the phospho-JNK/JNK, phospho-p38/p38, phospho-ERK/ERK, and I-κBα/GAPDH ratio as compared to that of the time 0. Each error bar represents mean ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. time 0 group. n = 3 in each group.
miRNA-133a-3p promotes peritoneal leukocyte migration in vivo
To examine whether or not miRNA induces inflammation in vivo, we administered miR-133a-3p mimic into the mouse peritoneal space (20 μg/mouse) and harvested the peritoneal lavage 20 h later. As shown in Fig. 5A, the total peritoneal cells were 5.8±0.5 × 106 in the WT mice injected with lipofectamine (Lipo control). Flow cytometry analysis revealed that among the gated peritoneal cells, 0.6% was Ly-6G+ neutrophils (NE), 8.9% F4/80low recruited monocytes (MO), and 16.6% F4/80high resident macrophages (Mϕ) (Fig. 5B-C). The remaining cells (Ly-6G-/F4/80-) were most likely B and T lymphocytes, and other immune cells (NK and dendritic cells) (27, 28). In WT mice injected with miR-133a-3p, there were significantly more peritoneal cells (9.2±0.6 × 106 vs. 5.8±0.5 × 106, P < 0.01), with a significant increase in neutrophils (15.0±3.1%), F4/80low infiltrated monocytes (19.8±3.5%), but decreased F4/80high resident macrophages (4.1±1.4%, P < 0.01) (Fig. 5A-C) compared to the lipofectamine control group. Compared with WT mice, TLR7-/- mice had reduced number of the peritoneal cells (6.7±0.5 × 106 vs. 9.2±0.6 × 106), neutrophils (4.6±1.3%, P < 0.05), infiltrated monocytes (11.9±2.4%), but sustained F4/80high resident macrophages (11.9±2.0%) (Fig. 5A-C) following miR-133a administration. MyD88 deficiency completely abolished miR-133a-induced peritoneal leukocytes migration (Fig. 5A-C). Surprisingly, Trif-/- mice had even more total peritoneal leukocytes than WT mice after miR-133a injection (14.5±1.2 × 106, P < 0.01). However, there was no significant difference in the percentage of leukocyte population between miRNA-treated WT and Trif-/- mice (Fig. 5A-C). Of note, the absolute numbers of the three cell types are summarized in Table 2. Taken together, these data suggest that miR-133a-elicited peritoneal leukocyte migration, i.e., the influx of neutrophils/monocytes and the efflux of resident macrophages) in vivo is through TLR7→MyD88 signaling.
Figure 5. TLR7/MyD88 signaling mediates miR-133a-3p mimic-induced leukocyte migration in vivo.

Wild-type (WT), TLR7-/-, MyD88-/-, or Trif-/- male mice were administered i.p. with 20 μg of miR-133a-3p mimic or lipofectamine alone (Lipo.). Twenty hours later, the peritoneal lavage was harvested and the peritoneal cells were spun down. The total peritoneal cells were counted and the neutrophils (NE), resident macrophages (Mϕ), and recruited monocytes (MO) in the peritoneal cavity were determined by flow cytometry as detailed in Experimental Procedures. A, The total peritoneal cells in each group. B, The percentage of NE (Ly-6G+), Mϕ (F4/80high), and MO (F4/80low) among total peritoneal cells. Each error bar represents mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001. n = 6 mice in MyD88-/--Lipo Group, n = 4 mice in other Lipo Group, n = 6 mice in all miRNA group. C, Representative flow cytometry pictures gated for Ly-6G+, F4/80high, and F4/80low in each group. Lipo: lipofectamine; miR: miR-133a-3p.
Table 2.
The peritoneal leukocyte numbers following lipofectamine or miR-133a-3p mimic i.p. injection in WT and KO mice.
| Neutrophils | Monocytes | Macrophages | ||
|---|---|---|---|---|
| (×105) | (×105) | (×105) | ||
| WT | Lipo | 0.4±0.2 | 5.3±1.2 | 9.9±2.3 |
| miR-133a | 14.1±3.2*** | 19.0±4.1** | 3.6±1.0 | |
|
| ||||
| TLR7-/- | Lipo | 0.1±0.0 | 3.4±1.2 | 12.4±3.9 |
| miR-133a | 3.2±1.1††† | 7.8±1.5†† | 7.6±1.1 | |
|
| ||||
| MyD88-/- | Lipo | 0.0±0.0 | 1.4±0.6 | 5.0±2.2 |
| miR-133a | 0.0±0.0††† | 2.9±0.8††† | 10.8±3.2 | |
|
| ||||
| Trif-/- | Lipo | 0.0±0.0 | 3.3±1.3 | 8.5±1.8 |
| miR-133a | 11.9±1.3*** | 21.3±3.6*** | 4.1±0.9 | |
Lipo: lipofectamine
P<0.05,
P<0.01,
P<0.001 vs. individual Lipo group.
P<0.05,
P<0.01,
P<0.001 vs. WT/miR-133a group.
miR-146a-5p induces peritoneal leukocytes migration in vivo
To further test the ability of ex-miRNAs to induce acute inflammation in vivo, we injected another miRNA mimic, miR-146a-5p, into WT and TLR7-/- mice. For these experiments, we used the miR-146a (uridine→adenosine) mutant as the control. As shown in Fig. 6, compared to the mice without any injection, the miR-146aU→A mutant-injected WT mice only had mild neutrophil recruitment (2.8±0.7% vs. none). The total peritoneal cell numbers and the populations of monocytes and macrophages were the same (Fig. 6A-E). Similar to miR-133a, miR-146a-5p i.p. injection led to a marked increase in total peritoneal cells (9.1±3.1 × 106 vs. 3.2±1.2 × 106, P < 0.001), neutrophils (15.8±7.6% vs. 2.8±0.7%, P < 0.01), monocytes (31.1±9.9% vs. 9.2±1.6%, P < 0.001), and significant reduction in the peritoneal resident macrophages (2.8±1.2% vs. 12.02±6.1%, P < 0.05). Of note, the absolute numbers of the three cell types are summarized in Table 3. In comparison, systemic deletion of TLR7 nearly completely abolished miR-146a-induced peritoneal neutrophil and monocyte recruitment and macrophage efflux (Fig. 6A-E, Table 3). These data further demonstrate that certain miRNA mimics are capable of inducing acute inflammation by promoting leukocytes migration in vivo through a local TLR7-sensing mechanism.
Figure 6. miR-146a-5p mimic, but not its U→A mutant, elicited peritoneal leukocyte migration in vivo via TLR7.

Wild-type (WT), and TLR7-/- male mice were administered i.p. with 20 μg of synthetic miR-146a-5p or mutant (Mut). Twenty hours later, the peritoneal lavage was harvested and spun down. The total peritoneal cells were counted and the neutrophils (NE), resident macrophages (Mϕ), and recruited monocytes (MO) in the peritoneal cavity were determined by flow cytometry as detailed in Experimental Procedures. A, The total peritoneal cells in each group. B-D, The percentage of NE (Ly-6G+, B), Mϕ (F4/80high, C), and MO (F4/80low, D) among total peritoneal cells. Each error bar represents mean ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001. n = 4 in each Group. E, Representative flow cytometry pictures gated for Ly-6G+, F4/80high, and F4/80low in each group. None: without injection; miR: miR-146a-5p.
Table 3.
The peritoneal leukocyte numbers following i.p. injection of lipofectamine, miR-146aU→A mutant, or miR-146a in WT and KO mice.
| Neutrophils | Monocytes | Macrophages | ||
|---|---|---|---|---|
| (×105) | (×105) | (×105) | ||
| WT | Lipo | 0.0±0.0 | 1.9±0.4 | 5.0±1.0 |
| Mut | 0.9±0.2 | 3.0±0.7 | 3.6±0.9 | |
| miR-146a | 16.0±6.3**†† | 30.3±9.0***††† | 2.6±0.7 | |
|
| ||||
| TLR7-/- | Lipo | 0.1±0.0 | 4.3±0.4 | 11.7±2.1 |
| Mut | 4.2±1.9 | 4.8±1.1 | 9.2±2.5 | |
| miR-146a | 0.8±0.3‡‡ | 4.5±0.9‡‡‡ | 14.6±4.5‡‡ | |
Lipo: lipofectamine, Mut: miR-146aU→A mutant
P<0.05,
P<0.01,
P<0.001 vs. individual Lipo group.
P<0.05,
P<0.01,
P<0.001 vs. individual Mut group.
P<0.05,
P<0.01,
P<0.001 vs. WT/miR-146a group.
Endogenous miRNAs play a role in cardiac RNA-induced cytokine response
To test the role of endogenous miRNAs in cytokine production, we employed anti-miR combo containing LNA anti-miRNA inhibitors against the selected six miRNAs (miR-133, -208, -146, -142, -122, -34) or a negative control nucleotide (Table 1), aiming to block the endogenous miRNAs in purified cellular RNA. We first determined the efficacy of the anti-miR inhibitor combo in blocking the six miRNA mixture-induced cytokine production in both BMDM and cardiomyocyte cultures. The anti-miR combo, at concentration of 100 nM and added to cell cultures one hour prior to the miRNA mixture, almost completely abolished MIP-2 and TNFα production (Fig. 7A-B). Importantly, cardiac RNA-elicited MIP-2 production (3) was also markedly inhibited by the anti-miR inhibitor combo with 82.4% or 44.6% reduction in MIP-2 production in BMDM or cardiomyocytes, respectively, and 76.6% reduction in TNFα production in BMDM. Similar results were obtained in IL-6 and TNFα production. These data suggest that miRNAs contribute significantly to the endogenous cardiac RNA-induced cytokine production.
Figure 7. miRNA inhibitor combo attenuated cardiac RNA-induced cytokine response.

Bone marrow-derived macrophages (BMDM, A-B) or cardiomyocytes (rCM, C) were cultured and pretreated with the mixture of six miRNA inhibitors (100 nM) or Control oligonucleotide as noted in the Materials and Methods followed by the treatment with cardiac RNA (10 μg/ml) or miRNA mixture (100 nM). Sixteen h later, the culture media were collected and MIP-2 or TNFα expression in the media was measured by ELISA. Each error bar represents mean ± SD. *** P < 0.001. n = 3 in each group. Con: control; miRs: the mixture of six miRNA mimics.
miRNA is co-localized with endosomes and TLR7 in macrophages and cardiomyocytes
To determine whether or not miRNA mimics are taken up by cells and reside with endosomes, we first incubated macrophages (BMDM) with fluorescence-labeled miR-133a (6-carboxyfluorescein (6-FAM)-miR-133a) for 4 hours and imaged cell-associated 6-FAM-miR-133a by flow cytometry and fluorescent microscopy. As indicated in Fig. 8A-B, there was a strong fluorescence intensity associated with WT BMDMs but no significant difference between TLR7-/- and WT BMDM in the fluorescence intensity as determined by flow cytometry. Microscopic imaging showed cytosolic localization of fluorescent FAM-133a in both WT and TLR7-/- BMDM (Fig. 8C). Using fluorescent confocal microscopy, we imaged endosome, TLR7, and miR-133 presented in the same BMDM or cardiomyocytes (Fig. 8D-E, upper panels). The two individual images were then merged together (Fig. 8D-E, lower panels). We found that the early endosome antigen 1 (EEA1) (red in BMDM and purple in cardiomyocytes, respectively) was largely co-stained with TLR7 (purple in BMDM, Fig. 8D and red in cardiomyocytes, Fig. 8E) in both macrophages and cardiomyocytes. The green fluorescence-labeled miRNA-133 was taken up into the endosome and partially co-localized with TLR7. These data suggest that miRNA could reach TLR7 within the endosome after a period of incubation with macrophages and cardiomyocytes.
Figure 8. The co-localization of miRNA and TLR7 within endosome.
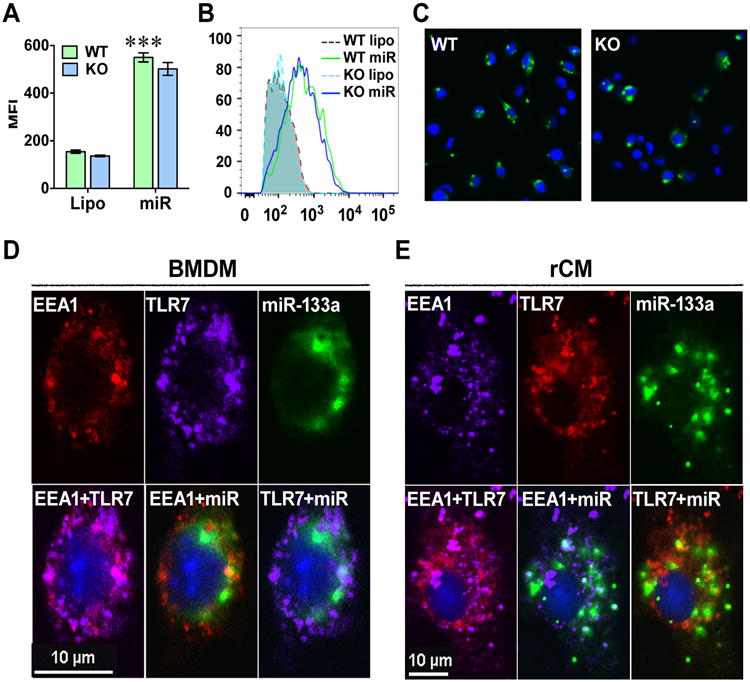
A-C, The cellular uptake of miRNA is independent of TLR7. Bone marrow-derived macrophages (BMDM) were isolated from wild-type (WT) and TLR7-/- (KO) mice and treated with 6-FAM conjugated miR-133a-3p (50 nM). Four hours later, the cells were imagined under fluorescent microscope and detached for intracellular fluorescent quantitation by flow cytometry. A. Quantitative data of flow cytometry. B. Representative data of flow cytometry. C. Representative fluorescent image. Each error bar represents mean ± SD. *** P < 0.001. n = 3 in each group. Lipo: lipofectamine; miR: 6-FAM-miR-133a-3p. D-E, miRNA can reach TLR7 within endosome in both BMDM (D) and cardiomyocytes (E). BMDM isolated from wild-type mice or rat neonatal cardiomyocytes were treated with 6-FAM-conjugated miR-133a-3p (50 nM). Four hours later, the cells were fixed for immunohistochemistry staining using antibodies against EEA1 (an endosome marker) or TLR7. Upper panels: Individual fluorescent imaging of EEA1, TLR7, and miR-133a in macrophages (BMDM) and rat cardiomyocytes (rCM). Lower panels: Merged fluorescent images as indicated. Blue represents the nucleistained by DAPI.
Discussion
In the current study, we tested the hypothesis that multiple cellular miRNAs are released into the circulation following myocardial I/R and that certain extracellular miRNAs are capable of inducing cellular and tissue inflammation via specific TLRs. First, we profiled circulating miRNAs in a mouse model of transient myocardial ischemic injury and found 38 out of 68 miRNAs were significantly increased following I/R compared with sham animals. Out of 38 miRNAs, eight single-stranded miRNAs were synthesized and tested for their abilities to induce inflammation. Six miRNA mimics, i.e., miR-34a, miR-122, miR-133a, miR-142, miR-146a, miR-208a, induced dose-dependent cytokine production in macrophages and cardiomyocytes. Of interest, each of these six miRNA mimics exhibits a different potency in inducing MIP-2 production with miR-142, miR-146a, and miR-133 being the most potent. The Second, the miRNA mimic-induced cytokine response appeared to be dependent on TLR7→MyD88 signaling as either TLR7 or MyD88 deficiency blocked the effect of the miRNA mimics. Third, the administration of miR-133a or miR-146a into the peritoneal space induced significant neutrophil and monocyte recruitment, which was also dependent of TLR7→MyD88 signaling. Fourth, we found that inhibition of endogenous miRNAs (miR-34a, miR-122, miR-133a, miR-142, miR-146a, miR-208a) markedly attenuated the cardiac RNA-induced cytokine response, demonstrating an important role of endogenous cardiac miRNAs in promoting cellular inflammation. Finally, the confocal microscope imaging data demonstrates that miR-133a-3p resides closely with TLR7 within the endosomes in macrophages and cardiomyocytes.
To identify the possible miRNA candidates that may play a critical role in myocardial I/R injury and inflammation, we profiled the circulating levels of 68 miRNAs that were reportedly related to cellular and tissue responses to various cardiovascular diseases, including myocardial ischemic infarction (29, 30), coronary artery diseases (31), heart failure (32), and peripheral artery diseases (33). We found that 38 plasma cell-free miRNAs were significantly increased following myocardial I/R. Of importance, some of these miRNAs, such as miR-208, -133, -1, and -499, are reportedly associated with patients with acute myocardial infarct and have been proposed as sensitive biomarkers for myocardial ischemic injury (11, 13). To test their pro-inflammatory effect, we selected eight miRNAs based on the following criteria: 1) the I/R/Sham ratio > 2, 2) fluorescence intensity > 100, seven of which were independently validated using qRT-PCR in a separate set of experiments. Moreover, these miRNAs are reportedly related to host immunological functions (i.e., miR-192-5p, -122-5p, -34a-5p, -210-3p, and -146a-5p) and expressed at relatively high levels in myocardial and skeletal muscles (i.e., miR-208a-3p and -133a-3p). Six miRNAs (miR-34a, miR-122, miR-133a, miR-142, miR-146a, miR-208a) exhibited a remarkable ability to induce cytokine production, e.g., MIP-2, TNFα, and IL-6. The MIP-2-inducing effect of these miRNA mimics was dose-dependent with an EC50 at nM range, sensitive to RNase (but not DNase) digestion, uridine-dependent as U→A mutation led to a complete loss of the activities of miR-133a, miR-208a, and miR-146a. The miRNA-induced MIP-2 response was also mediated via TLR7-MyD88 signaling, not TLR3-Trif, as both TLR7- and MyD88-deficient macrophages failed to respond to these miRNA mimics. This finding was further supported by the observation that the specific TLR7 antagonist, IRS661, blocked the effect of the miRNA mixtures in cardiomyocytes.
To determine whether or not endogenous miRNAs possess similar proinflammatory activities, we employed six anti-miRNAs to block endogenous target miRNAs and found that the cardiac RNA-elicited cytokine response was significantly decreased by the anti-miRNA treatment as compared with an oligonucleotide control. Of note, in our studies the control oligo exhibits non-specific inhibition on the miRNA-induced MIP-2 production. And this effect seems dose-dependent. At the concentration at or below 25 nM, the control oligo had minimal inhibition on the MIP-2 production induced by 50 nM of miRNA mixrure (data not shown). Nevertheless, this data supports the notion that extracellular miRNAs of cardiac origin may contribute to the RNA-induced inflammation. Since other yet to be identified cellular miRNAs or non-coding RNA may possess similar proinflammatory properties, it is not surprising that blocking of these six miRNAs only leads to partial attenuation of the RNA-induced cytokine production. It is worth noting that in vivo, most circulating miRNAs are enveloped in some types of carriers, such as extracellular vesicles (e.g., exosomes and microvesicles) or proteins (e.g., HDL or Ago-2). These carriers protect RNA from degradation induced by circulating RNase. Indeed, we found that while in the absence of exosome or lipofectamine, the “naked” miRNA-induced cytokine production is sensitive to RNase digestion as illustrated in Fig. 2, our recent pilot study suggests that exosome- and exosome-associated miRNA-induced cytokine production is resistant to RNase treatment (data not shown).
Out of eight miRNA mimics tested, miR-192 and miR-210 failed to induce cytokine response. We wondered whether there was any sequence consensus that determines the activity of the miRNAs. Several sequence motifs have been reported important for ssRNA effects. For example, it has been reported that GUUGUGU motif is important for the neuron activity of let-7b and miR-599 since let-7a, which contains GUUGUAU, failed to elicit inward current (34, 35). Others have reported the importance of GU-rich element (24, 36, 37) and the amount of uridine ribonucleotides (38) for ssRNA activities. In our study, we could not identify GUUGUGU motif among the six miRNAs that induce robust MIP-2 response in cardiomyocytes and macrophages. However, five of the six active miRNAs contain seven or more uridine ribonucleotides in their sequences, whereas the two miRNAs (miR-192 and -210) that failed to induce MIP-2 response have only five uridines (Table 1). These and the miRNA U→A mutant data demonstrate the specificity of the observed ex-miRNA effects and seem to suggest the importance of uridine nucleotide for the miRNA-induced innate immune responses including cytokine response and peritoneal leukocyte migration in vivo. This notion is further supported by a recent study where Zhang, et al. generated crystal structures of TLR7 complexes and identified two ligand binding sites (39). One of the ligand binding sites is specific for uridine moieties in ssRNA and the other site binds guanosine, and that TLR7 is synergistically activated by guanosine and uridine-containing ssRNA.
We found that miR-133a and miR-146a mimics were capable of inducingly-6G+ neutrophil and F4/80low monocyte infiltration when injected into the peritoneal space. In the case of miR-146a, the U→A mutant failed to induce the effect, a finding consistent with the in vitro cytokine data. Moreover, the in vivo effects of both miR-133a and -146a appeared through TLR7 signaling as they were significantly attenuated by the deletion of TLR7. Deletion of MyD88, but not Trif, completely blocked miR-133a-induced leukocyte recruitment. These data suggest that the miRNA-elicited leukocytes migration is in part dependent of TLR7 and completely dependent of MyD88. Since MyD88 is the key adaptor for all TLR signaling with exception of TLR3, we speculate that other TLRs, such as TLR8, may sense miRNAs and have contributed to miR133- and miR146a-induced leukocyte migration.
We found that both cardiac RNA (3) and synthetic miRNA significantly decreased the number of the peritoneal F4/80high macrophage after intraperitoneal administration. Consistent with this finding is the observed rapid disappearance of resident macrophages (F4/80high) following i.p. administration of bacterial antigens (40, 41), thioglycollate (27, 42) or LPS (27). Geoffrey, et al. report that during inflammation resolution, macrophages migrate rapidly into the draining lymphatics instead of dying locally (43). The macrophage efflux induced by diverse peritoneal inflammatory mediators may play an essential role in the antigen presentation and the development of the adaptive immunity (44-46). In this context, our data clearly demonstrate the ability of miRNAs to accelerate macrophage efflux via TLR7 signaling although the exact mechanism remains to be investigated.
We tested the possibility that TLR7 mediates miRNA transmembrane uptake in macrophages and that lack of cytokine response in TLR7-/- cells might be due to disrupted miRNA transport in these cells. Using flow cytometry, we found that TLR7-/- macrophages had similar level of miRNA uptake as those isolated from WT macrophages. Confocal imaging data demonstrate that miRNA is localized to the endosome inside the cells and co-localized with TLR7 within endosome, a necessary prerequisite to ligand binding to TLR7. Together, these data suggest that TLR7 deficiency does not interfere with the uptake of miRNA and the absence of cytokine production in TLR7-deficient BMDM is most likely due to the lack of miRNA sensing.
We did not test the efficacy of LNA anti-miRNA inhibitors to block circulating and myocardial target ex-miRNAs in vivo and their abilities to inhibit myocardial inflammation after I/R, which is considered a limitation of this study. Instead, we used the peritonitis as an intermediate model to establish the pro-inflammatory property of these miRNAs in vivo. We are currently in the process of testing the efficacy of various protocols of delivering LNA anti-miRNAs in vivo. We anticipate that demonstrating the efficacy of anti-miRNA inhibitors to neutralize circulating/myocardial target miRNAs would be essential to determine the critical role of endogenous miRNAs in the development of myocardial inflammation and injury following transient ischemia.
In summary, we found that multiple miRNAs were increased in the blood circulation four hours after transient myocardial ischemia. We demonstrated that a group of miRNAs, e.g., miR-34a, miR-122, miR-133a, miR-142, miR-146a, miR-208a, were capable of inducing a profound inflammatory response in innate immune cells as well as in cardiomyocytes. In vivo, miRNA133 and miR-146a induce neutrophil and monocyte peritoneal recruitment, and resident macrophage efflux after intraperitoneal administration. We further identified the pivotal role of TLR7→MyD88 signaling in mediating these pro-inflammatory effects. Together, our data suggest that certain miRNAs are a potent TLR7 ligand and may contribute to cellular/tissue innate immune response following release from damaged cells.
Acknowledgments
This work was supported in part by NIH R01-GM097259 and R01-GM117233 (WC).
Abbreviations
- BMDM
bone marrow-derived macrophage
- EEA1
early endosome antigen 1
- IRS
immunoregulatory DNA sequence
- R837
Imiquimod
- rCM
rat neonatal cardiomyocytes
- Trif
Toll/IL-1 receptor domain-containing adapter-inducing interferon β
Footnotes
Conflict of Interest: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Chen C, Feng Y, Zou L, Wang L, Chen HH, Cai JY, Xu JM, Sosnovik DE, Chao W. Role of Extracellular RNA and TLR3-Trif Signaling in Myocardial Ischemia-Reperfusion Injury. J Am Heart Assoc. 2014;3:e000683. doi: 10.1161/JAHA.113.000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cabrera-Fuentes HA, Ruiz-Meana M, Simsekyilmaz S, Kostin S, Inserte J, Saffarzadeh M, Galuska SP, Vijayan V, Barba I, Barreto G, Fischer S, Lochnit G, Ilinskaya ON, Baumgart-Vogt E, Boning A, Lecour S, Hausenloy DJ, Liehn EA, Garcia-Dorado D, Schluter KD, Preissner KT. RNase1 prevents the damaging interplay between extracellular RNA and tumour necrosis factor-alpha in cardiac ischaemia/reperfusion injury. Thromb Haemost. 2014;112:1110–1119. doi: 10.1160/TH14-08-0703. [DOI] [PubMed] [Google Scholar]
- 3.Feng Y, Chen H, Cai J, Zou L, Yan D, Xu G, Li D, Chao W. Cardiac RNA Induces Inflammatory Responses in Cardiomyocytes and Immune Cells via Toll-like Receptor 7 Signaling. J Biol Chem. 2015;290:26688–26698. doi: 10.1074/jbc.M115.661835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 7.Ye Y, Perez-Polo JR, Qian J, Birnbaum Y. The role of microRNA in modulating myocardial ischemia-reperfusion injury. Physiol Genomics. 2010;43:534–542. doi: 10.1152/physiolgenomics.00130.2010. [DOI] [PubMed] [Google Scholar]
- 8.D'Alessandra Y, Pompilio G, Capogrossi MC. MicroRNAs and myocardial infarction. Curr Opin Cardiol. 2012;27:228–235. doi: 10.1097/HCO.0b013e3283522052. [DOI] [PubMed] [Google Scholar]
- 9.Gupta SK, Bang C, Thum T. Circulating microRNAs as biomarkers and potential paracrine mediators of cardiovascular disease. Circ Cardiovasc Genet. 2010;3:484–488. doi: 10.1161/CIRCGENETICS.110.958363. [DOI] [PubMed] [Google Scholar]
- 10.Zhu H, Fan GC. Extracellular/circulating microRNAs and their potential role in cardiovascular disease. Am J Cardiovasc Dis. 2011;1:138–149. [PMC free article] [PubMed] [Google Scholar]
- 11.Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. 2012;110:483–495. doi: 10.1161/CIRCRESAHA.111.247452. [DOI] [PubMed] [Google Scholar]
- 12.Li C, Pei F, Zhu X, Duan DD, Zeng C. Circulating microRNAs as novel and sensitive biomarkers of acute myocardial Infarction. Clin Biochem. 2012;45:727–732. doi: 10.1016/j.clinbiochem.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fichtlscherer S, Zeiher AM, Dimmeler S. Circulating microRNAs: biomarkers or mediators of cardiovascular diseases? Arterioscler Thromb Vasc Biol. 2011;31:2383–2390. doi: 10.1161/ATVBAHA.111.226696. [DOI] [PubMed] [Google Scholar]
- 14.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 15.D'Alessandra Y, Devanna P, Limana F, Straino S, Di Carlo A, Brambilla PG, Rubino M, Carena MC, Spazzafumo L, De Simone M, Micheli B, Biglioli P, Achilli F, Martelli F, Maggiolini S, Marenzi G, Pompilio G, Capogrossi MC. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur Heart J. 2010;31:2765–2773. doi: 10.1093/eurheartj/ehq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang GK, Zhu JQ, Zhang JT, Li Q, Li Y, He J, Qin YW, Jing Q. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J. 2010;31:659–666. doi: 10.1093/eurheartj/ehq013. [DOI] [PubMed] [Google Scholar]
- 17.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–1139. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 20.Feng Y, Zhao H, Xu X, Buys ES, Raher MJ, Bopassa JC, Thibault H, Scherrer-Crosbie M, Schmidt U, Chao W. Innate immune adaptor MyD88 mediates neutrophil recruitment and myocardial injury after ischemia-reperfusion in mice. Am J Physiol Heart Circ Physiol. 2008;295:H1311–H1318. doi: 10.1152/ajpheart.00119.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Y, Zou L, Si R, Nagasaka Y, Chao W. Bone marrow MyD88 signaling modulates neutrophil function and ischemic myocardial injury. Am J Physiol Cell Physiol. 2010;299:C760–769. doi: 10.1152/ajpcell.00155.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Si R, Feng Y, Chen HH, Zou L, Wang E, Zhang M, Warren HS, Sosnovik DE, Chao W. Myocardial ischemia activates an injurious innate immune signaling via cardiac heat shock protein 60 and Toll-like receptor 4. J Biol Chem. 2011;286:31308–31319. doi: 10.1074/jbc.M111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 24.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 25.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 26.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 27.Ghosn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, Bortoluci KR, Almeida SR, Herzenberg LA. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci U S A. 2010;107:2568–2573. doi: 10.1073/pnas.0915000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray A, Dittel BN. Isolation of mouse peritoneal cavity cells. J Vis Exp. 2010 doi: 10.3791/1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Recchioni R, Marcheselli F, Olivieri F, Ricci S, Procopio AD, Antonicelli R. Conventional and novel diagnostic biomarkers of acute myocardial infarction: a promising role for circulating microRNAs. Biomarkers. 2013;18:547–558. doi: 10.3109/1354750X.2013.833294. [DOI] [PubMed] [Google Scholar]
- 30.Wang R, Li N, Zhang Y, Ran Y, Pu J. Circulating microRNAs are promising novel biomarkers of acute myocardial infarction. Intern Med. 2011;50:1789–1795. doi: 10.2169/internalmedicine.50.5129. [DOI] [PubMed] [Google Scholar]
- 31.Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Roxe T, Muller-Ardogan M, Bonauer A, Zeiher AM, Dimmeler S. Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010;107:677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- 32.Ellis KL, Cameron VA, Troughton RW, Frampton CM, Ellmers LJ, Richards AM. Circulating microRNAs as candidate markers to distinguish heart failure in breathless patients. Eur J Heart Fail. 2013;15:1138–1147. doi: 10.1093/eurjhf/hft078. [DOI] [PubMed] [Google Scholar]
- 33.Stather PW, Sylvius N, Wild JB, Choke E, Sayers RD, Bown MJ. Differential microRNA expression profiles in peripheral arterial disease. Circulation. 2013;6:490–497. doi: 10.1161/circgenetics.111.000053. [DOI] [PubMed] [Google Scholar]
- 34.Park CK, Xu ZZ, Berta T, Han Q, Chen G, Liu XJ, Ji RR. Extracellular microRNAs activate nociceptor neurons to elicit pain via TLR7 and TRPA1. Neuron. 2014;82:47–54. doi: 10.1016/j.neuron.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gantier MP, Tong S, Behlke MA, Xu D, Phipps S, Foster PS, Williams BR. TLR7 is involved in sequence-specific sensing of single-stranded RNAs in human macrophages. J Immunol. 2008;180:2117–2124. doi: 10.4049/jimmunol.180.4.2117. [DOI] [PubMed] [Google Scholar]
- 36.Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ, Zanesi N, Crawford M, Ozer GH, Wernicke D, Alder H, Caligiuri MA, Nana-Sinkam P, Perrotti D, Croce CM. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A. 2012;109:E2110–2116. doi: 10.1073/pnas.1209414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D, Habbel P, Kalin R, Franzoni E, Rybak A, Nguyen D, Veh R, Ninnemann O, Peters O, Nitsch R, Heppner FL, Golenbock D, Schott E, Ploegh HL, Wulczyn FG, Lehnardt S. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci. 2012;15:827–835. doi: 10.1038/nn.3113. [DOI] [PubMed] [Google Scholar]
- 38.Diebold SS, Massacrier C, Akira S, Paturel C, Morel Y, Reis e Sousa C. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur J Immunol. 2006;36:3256–3267. doi: 10.1002/eji.200636617. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Ohto U, Shibata T, Krayukhina E, Taoka M, Yamauchi Y, Tanji H, Isobe T, Uchiyama S, Miyake K, Shimizu T. Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity. 2016;45:737–748. doi: 10.1016/j.immuni.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Nelson DS, Boyden SV. The loss of macrophages from peritoneal exudates following the injection of antigens into guinea-pigs with delayed-type hypersensitivity. Immunology. 1963;6:264–275. [PMC free article] [PubMed] [Google Scholar]
- 41.Bultmann B, Sodomann CP, Heymer B, Haferkamp O, Schmidt WC. Loss of macrophages from peritoneal exudates of sensitized rats after injection of streptococcal antigens. Int Arch Allergy Appl Immunol. 1971;41:491–500. doi: 10.1159/000230544. [DOI] [PubMed] [Google Scholar]
- 42.Melnicoff MJ, Horan PK, Morahan PS. Kinetics of changes in peritoneal cell populations following acute inflammation. Cell Immunol. 1989;118:178–191. doi: 10.1016/0008-8749(89)90367-5. [DOI] [PubMed] [Google Scholar]
- 43.Bellingan GJ, Caldwell H, Howie SE, Dransfield I, Haslett C. In vivo fate of the inflammatory macrophage during the resolution of inflammation: inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J Immunol. 1996;157:2577–2585. [PubMed] [Google Scholar]
- 44.Bellingan GJ, Xu P, Cooksley H, Cauldwell H, Shock A, Bottoms S, Haslett C, Mutsaers SE, Laurent GJ. Adhesion molecule-dependent mechanisms regulate the rate of macrophage clearance during the resolution of peritoneal inflammation. J Exp Med. 2002;196:1515–1521. doi: 10.1084/jem.20011794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao C, Lawrence DA, Strickland DK, Zhang L. A specific role of integrin Mac-1 in accelerated macrophage efflux to the lymphatics. Blood. 2005;106:3234–3241. doi: 10.1182/blood-2005-03-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomez IG, Tang J, Wilson CL, Yan W, Heinecke JW, Harlan JM, Raines EW. Metalloproteinase-mediated Shedding of Integrin beta2 promotes macrophage efflux from inflammatory sites. J Biol Chem. 2012;287:4581–4589. doi: 10.1074/jbc.M111.321182. [DOI] [PMC free article] [PubMed] [Google Scholar]