

Abstract
Spontaneous mutagenesis of synthetic genetic constructs by mobile genetic elements frequently results in the rapid loss of engineered functions. Previous efforts to minimize such mutations required the exceedingly time-consuming manipulation of bacterial chromosomes and the complete removal of insertional sequences (ISes). To this aim, we developed a single plasmid-based system (pCRIS) that applies CRISPR-interference to inhibit the transposition of bacterial ISes. pCRIS expresses multiple guide RNAs to direct inactivated Cas9 (dCas9) to simultaneously silence IS1, IS3, IS5 and IS150 at up to 38 chromosomal loci in Escherichia coli, in vivo. As a result, the transposition rate of all four targeted ISes dropped to negligible levels at both chromosomal and episomal targets. Most notably, pCRIS, while requiring only a single plasmid delivery performed within a single day, provided a reduction of IS-mobility comparable to that seen in genome-scale chromosome engineering projects. The fitness cost of multiple IS-knockdown, detectable in flask-and-shaker systems was readily outweighed by the less frequent inactivation of the transgene, as observed in green fluorescent protein (GFP)-overexpression experiments. In addition, global transcriptomics analysis revealed only minute alterations in the expression of untargeted genes. Finally, the transposition-silencing effect of pCRIS was easily transferable across multiple E. coli strains. The plasticity and robustness of our IS-silencing system make it a promising tool to stabilize bacterial genomes for synthetic biology and industrial biotechnology applications.
Keywords: genome stability, CRISPR-mediated gene silencing, transposable element, repression of transposition, decreased mutation rate
1. Introduction
De novo constructed DNA elements propagated in living cells frequently impede cellular fitness. In turn, evolutionary processes that are inactivating these DNA elements often release cells from growth retardation and thus lead to the replacement of the engineered population. Therefore these evolutionary processes, while resolving collateral burden, are hindering the central aim of biotechnology and synthetic biology: the predictable engineering of biological systems via the introduction of de novo constructed DNA elements (1).
Several strategies are already available to deal with this issue (2, 3): One possibility is to engineer redundant genetic circuits that can withstand mutations without loss-of-function (4). Another solution is to design DNA sequences that are less prone to mutation-acquisition, mainly by relying on experimental observations and computational designs (5, 6). Yet another way to limit the evolution of genes of interest is to link their expression to that of an essential gene (6, 7). Finally, the ultimate strategy to mitigate this issue is the reduction of the mutation rate of the engineered host. However, this latter solution until now remained limited to certain mutation types and a few bacterial models. For example, decreasing the rate of point mutations in bacterial cells has been achieved through the overexpression of individual genes [mutL (8) or nudG (9)], by removing the genes of error-prone DNA polymerases (polB, dinB and umuDC) (10), or by mutating the gene encoding RNase E (11). Also, the removal of the recA recombinase (12) or the dam methylase (13) have been shown to reduce the frequency of deletions, especially those arising by the homologous recombination of repetitive segments.
Strikingly, a recent study highlighted that insertional mutagenesis, caused primarily by insertion sequences (ISes) constitutes the main cause of instability of synthetic DNA elements in Escherichia coli (14). Bacterial ISes are most probably the simplest autonomous mobile genetic elements found in nature: they consist of a transposase gene (often split into two open reading frames) flanked by terminal inverted repeats (15). ISes have been known to make a measurable contribution to the spontaneous mutation rate of bacterial genomes, accounting for 3.9–98% of genetic changes (16–22). Therefore, genetic stabilization of bacterial hosts must include the complete elimination of their mobility. Based on this concept, the deletion of all transposable elements from the genomes of various E. coli strains (23–25) Corynebacterium glutamicum (26) and Acinetobacter baylyi (27) has already proven to be a successful strategy to increase the stability of chromosomal or plasmid-based synthetic constructs. One of these endeavors, which removed all mobile DNA (prophages, cryptic prophages, ISes) from the chromosome of E. coli MG1655 by subsequently executing 42 precise deletions, took several years to accomplish in our laboratory (23). Later, the availability of programmable CRISPR/Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins) (28, 29) endonucleases revolutionized the genome-wide inactivation of mobile DNA both in bacteria (24) and in mammalian cells (30, 31). Furthermore, an in vivo targeted base-editing technique has been recently developed that could potentially inactivate mobile DNA elements in E. coli using a CRISPR/Cas-targeted cytidine deaminase (32).
Recently, the discovery of transcriptional control by CRISPR/Cas interference (CRISPRi) has introduced a novel set of tools for the transcriptional reprogramming of living cells (33–35). Essentially, this system directs a catalytically inactive ‘dead’ Cas nuclease (dCas9) in vivo to virtually any target DNA specified by a short complementary RNA molecule (crRNA) and thereby achieves transcriptional-suppression of the target. Due to its robustness and portability, CRISPRi presents a versatile solution for genome-scale control of cellular phenotypes. By building on these advantageous properties, we present a system of plasmid-based CRISPR-interference termed pCRIS to simultaneously inhibit the major sources of insertional mutagenesis in various strains of E. coli. We demonstrate the ability of pCRIS to repress multiple bacterial transposases and consequently increase the genetic stability measured at both chromosomal and plasmid-borne loci.
2. Materials and methods
2.1 Strains, chemicals and media
Plasmids were constructed in E. coli strain MDS42 (23). Chromosomal mutation rates were measured in E. coli strains MG1655 (36) and BL21(DE3) (37). Plasmid stability was monitored in E. coli DH5αZ1 (38) and JM107MA2 (39).
Bacteria were grown in Luria-Bertani (LB) medium (40) or in mineral salts (MS) minimal medium, supplemented with 0.2% glucose (17).
Antibiotics (Sigma-Aldrich, St. Louis, MO, USA) were used in the following end-concentrations: chloramphenicol (Cm): 25 μg/ml, ampicillin (Ap): 100 μg/ml, kanamycin (Km): 25 μg/ml, cycloserine (Cyc): 0.04 mM. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was applied in 1 mM end concentration. Standard protocols were followed in all DNA manipulation and cloning processes unless otherwise specified (40).
2.2 CRISPR spacer design
CRISPR spacers were designed to bind the -10 box of targeted promoters. To define the -10 box of the transposase promoters, we analyzed the hypothetical promoter-region of each IS element using the Neural Network Promoter Prediction interface (41). The -10 box of IS5 has been experimentally determined before (42). For IS150, we targeted the -10 box of Promoter 1, published earlier (43). The promoter sequences were used as inputs at the Cas Online Designer (http://cas9.wicp.net) to obtain potential spacer sequences. The output CRISPR spacers were manually selected for complete overlap with the predicted -10 box. The Cas Online Designer was used to verify that the chosen spacers targeted all copies of an IS type within the E. coli genome, with no additional targets. Each spacer sequence was designed following a previous guideline (35). A dsDNA segment, containing a triple CRISPR array targeting IS1, IS5 and IS3 was synthesized by GenScript (Piscataway, NJ, USA). The two complementary strands of the IS150-specific spacer/repeat tandem were synthesized as oligonucleotides. Both the dsDNA and the oligonucleotides were supplemented with appropriate extensions to allow their cloning into the BsaI site of the pCRISPathBrick plasmid or its derivatives (see Supplementary Table S1).
2.3 Plasmids
Plasmid pdCas9 (Addgene #46569) was a kind gift of Prof. Luciano Marraffini, obtained via Addgene (34). Plasmid pCRISPathBrick was constructed by modifying pdCas9 as described earlier (35). pCRISP_IS was constructed by cloning the triple CRISPR array targeting IS1, IS5 and IS3 into the BsaI site of pCRISPathBrick, following the protocol of Cress et al. (35). To obtain pCRIS, the two complementary oligonucleotides encoding the IS150-specific CRISPR spacer/repeat tandem were annealed and cloned into the BsaI site of pCRISP_IS. Plasmid pBDP_RFP_GFP, harboring a red- and a green-fluorescent protein (GFP) gene, driven by the bidirectional promoter BDP01 was a kind gift of Prof. Herbert M. Sauro (7). Plasmid pBDP_Km_GFP5 was constructed by replacing the rfp gene of pBDP_RFP_GFP with the kanamycin resistance (km) gene of plasmid pSG76-K (GenBank acc. no. Y09894.1), as follows: The pBDP_RFP_GFP plasmid, less the rfp gene was polymerase chain reaction (PCR)-amplified using primers pBDPfw and pBDPrev using Dream Taq DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA), while the km gene of pSG76-K was amplified with primers Km5 and Km3. The two PCR products were gel-purified with the Viogene Gel/PCR DNA Isolation Kit, (Viogene-Biotek Corporation, New Taipei, Taiwan R.O.C.) and fused using the NEBuilder kit (New England Biolabs, Ipswitch, MA, USA), according to the manufacturers’ instruction. The obtained plasmid, pBDP_Km_GFP5 constitutively expresses Ap-resistance and has an IPTG-inducible bidirectional promoter controlling the expression of Km-resistance and GFP. All plasmids were purified using the Zippy Plasmid Mini Prep Kit (Zymo Research Ltd., Orange County, CA, USA).
2.4 Measurement of growth rates
To measure bacterial growth rates in a flask-and-culture system, overnight starter cultures were diluted 100-fold into 100 ml LB+Cm medium and shaken in 500 ml flasks at 37°C in a water-bath with a rate of 225 rpm. Samples were obtained every 20 min and the optical density (OD) was measured at 550 nm. OD values above 0.5 were diluted 5-fold before measurement, and the obtained value was multiplied by 5. To infer the growth rate, the natural logarithm of the OD readings were plotted against time, and the linear segment of the obtained curve was used to add a linear trend-line using the respective function of Microsoft Excel. The slope of the obtained line corresponded to the growth rate. Doubling times were calculated with the formula DT = ln2/k, where DT = doubling time, k = growth rate. Final OD values of each culture were read after 24 h of incubation. Growth rates and final OD values were analyzed using unpaired, two-tailed t-tests.
Bacterial growth rates were also calculated for the cultures grown in microtiter plates during the adaptive laboratory evolution experiments measuring plasmid stability (see below). The OD600 values recorded on the second day of such experiments were fed as input into the ‘GrowthRates’ software (44). Output values were filtered by removing data originating from wells displaying an R-value <0.95 or negative lag time, followed by statistical analysis using unpaired, two-tailed t-tests.
2.5 Enumeration of IS copy numbers
The copy numbers of IS elements relevant to this study were enumerated in various E. coli strains relying on the whole genome sequences available at the NCBI Genome database. Manual screens were carried out using the ‘Search’ function of the graphical genome display using oligonucleotide sequences representing each IS element type. For IS1 and IS5, searches using multiple primer sequences were required to cover all divergent subtypes. The oligonucleotides used in each search process are indicated in the footnote of Table 1, their sequences are displayed in Supplementary Table S1.
Table 1.
The identified copy numbers of IS elements targeted in this study in various E. coli strains
| E. coli strain | GenBank acc. no. | IS1A relateda | IS1Fb | IS3c | IS5 relatedd | IS5Ye | IS150f | Total |
|---|---|---|---|---|---|---|---|---|
| K12 MG1655 | NC_000913.3 | 7 | 1 | 5 | 11 | 1 | 1 | 26 |
| BL21(DE3) | NC_012947.1 | 28 | 1 | 4 | 0 | 0 | 5 | 38 |
| B Rel606 | NC_012967.1 | 27 | 1 | 6 | 0 | 0 | 5 | 39 |
| DH10B | NC_010473.1 | 10 | 1 | 5 | 14 | 1 | 3 | 34 |
| DH5α | NZ_CP026085.1 | 0 | 0 | 4 | 1 | 0 | 2 | 7 |
| W3110 | NC_007779.1 | 6 | 1 | 5 | 17 | 1 | 1 | 31 |
Includes IS1A, B, D, G and R; counted using primer IS1A2.
Counted using primer IS1F1.
Counted using primer IS3ki1.
Includes IS5, IS5B and D; counted using primer IS5ki1.
Counted using primer IS5Y1.
Counted using primer IS150ki1.
2.6 RNA preparation and sequencing
Three parallel cultures of E. coli strain MG1655 carrying the control pCRISPathBrick plasmid and three cultures of MG1655 carrying the pCRIS plasmid were grown at 37°C in MS medium supplemented with 0.2% glucose and Cm. When reaching an OD540 value of 1, each culture was treated with the RNAprotect Bacteria Reagent (Qiagen, Leipzig, Germany), and cellular RNA was prepared using the E.Z.N.A. Bacteria RNA Kit (Omega bio-tek, Norcross, GA, USA) following instructions of the manufacturers.
Whole transcriptome sequencing was performed using TrueSeq RNA Library Preparation Kit v2 (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions with slight modifications. In brief, RNA quality and quantity measurements were performed using RNA ScreenTape and Reagents on TapeStation (all from Agilent, Santa Clara, CA, USA) and Qubit (Thermo Fisher Scientific, Waltham, MA, USA); only high-quality (RIN >8.0) total RNA samples were processed. Next, 1 µg of RNA was DNaseI (Thermo Fisher Scientific, Waltham, MA, USA) treated, the ribosomal RNA depleted using RiboZero Magnetic Kit for Gram-negative bacteria (Epicentre, Madison, WI, USA) and the leftover was ethanol precipitated. The success of rRNA removal was determined by measurement on TapeStation using high-sense RNA ScreenTape and Reagents (all from Agilent, Santa Clara, CA, USA). Next, RNA was purified and fragmented; first strand cDNA synthesis was performed using SuperScript II (Thermo Fisher Scientific, Waltham, MA, USA) followed by second strand cDNA synthesis, end repair, 3ʹ-end adenylation, adapter ligation and PCR amplification. All of the purification steps were performed using AmPureXP Beads (Beckman Coulter, Indianapolis, IN, USA). Final libraries were quality checked using D1000 ScreenTape and Reagents on TapeStation (all from Agilent, Santa Clara, CA, USA). The concentration of each library was determined using the KAPA Library Quantification Kit for Illumina (KAPA Biosystems, Wilmington, MA, USA). Sequencing was performed on an Illumina NextSeq instrument using the NextSeq 500/550 High Output Kit v2 (300 cycles; Illumina, San Diego, CA, USA) generating ∼10 million clusters for each sample.
2.7 Bioinformatic analysis of RNA-sequencing data
Following sequencing, raw paired-end Illumina reads were quality trimmed in CLC Genomics Workbench Tool (v.11.0, Qiagen bioinformatics, Aarhus, Denmark) using an error probability threshold of 0.01. No ambiguous nucleotide was allowed in trimmed reads. For filtering, reads were mapped on CRISPR spacer sequences using CLC with a length fraction of 0.9 and a sequence identity threshold of 0.95. Only those read pairs were kept for the subsequent RNA-Seq analysis that displayed no mapping against the CRISPR spacer construct. RNA-Seq analysis package from CLC was then used to map filtered reads on a custom-masked E. coli K12 MG1655 genome version (based on U00096.3, retaining only one unmasked copy from each relevant IS group: IS1A, IS1F, IS3, IS5, IS5Y and IS150). Only those reads were considered that displayed an alignment longer than 80% of the read length while showing at least 95% sequence identity against the reference genome. Next ‘Total gene read’ RNA-Seq count data were imported from CLC into R 3.3.2 for data normalization and differential gene expression analysis. Function ‘calcNormFactors’ from package ‘edgeR’ v.3.12.1 was used to perform data normalization based on the ‘trimmed mean of M-values’ (TMM) method (45). Log transformation, linear modeling, empirical Bayes moderation as well as the calculation of differentially expressed genes were carried out using ‘limma’ v. 3.26.9 (46). Genes showing at least 2-fold gene expression change with an FDR (false discovery rate) value below 0.05 were considered as significant.
2.8 Measurement of chromosomal mutation rate
To infer the rate of IS transposition to chromosomal targets, we used two established methods. The first method detects mutations of the cycA gene causing D-cycloserine resistance and has been described earlier (16). In brief, 20 parallel cultures of the tested strain were fully grown at 37°C in MS+Cm, and a fraction of each was plated on D-cycloserine-containing MS+Cm plates. The appropriate dilution of three random cultures was also plated on LB+Cm plates to infer the mean total cell number. The Ma-Sandri-Sarkar Maximum Likelihood method of fluctuation analysis was used to calculate the mutation per culture (m) based on the numbers of cycloserine-resistant colonies (47). The m-values of strains assayed in parallel were compared using unpaired, two-tailed t-tests, as described previously (48). To obtain and display a biologically more relevant measure of mutations, m was divided by the total cell number to yield the mutation rate (mutation/gene/generation) of the cycA gene. The m-values were not accepted to be significantly different for two strains if their total cell numbers were also significantly different, or if the mutation rates and the m-values displayed an opposite pattern. PCR amplification of the cycA locus using primers cycA1 and cycA2 allowed the classification of mutants as point mutants, deletion mutants or insertion mutants based on amplicon length. Further PCR-assay of the insertion mutants using IS-specific primers (Supplementary Table S1) yielded information on the contribution of each IS type to the disruption of the cycA gene.
The second method to monitor IS transposition measures the spontaneous activation of the cryptic bgl operon in cells starving on salicin-minimal plates (23). Cells to be assayed were grown to saturation in 20 parallel 1-ml cultures at 37°C in MS+Cm medium. Cells of each tube were pelleted, and plated onto MS+Cm plates containing 0.4% salicin (Alpha Aesar, Ward Hill, MA, USA) as the sole carbon source, and incubated at 37°C. The appearing colonies were counted and logged daily for 10 days. Total cell counts were obtained by plating appropriate dilutions onto LB+Cm plates. Statistical comparison of parallel lines was carried out in two ways: (i) Mutation frequencies were calculated by dividing the mean colony numbers observed on day 10 by the total cell count. Mutation frequencies were compared using Mann–Whitney U-tests. (ii) Total cell-normalized mean daily increments of mutant numbers were calculated for control and IS-silenced strains and compared using unpaired, two-tailed t-tests.
Classification of mutants as insertion mutants or ‘other’ types of mutants was based on the amplicon length of the PCR-amplified bgl region using primers bglR1 and bglR2 (Supplementary Table S1). Insertion mutants were called when PCR fragments were longer than amplicons obtained from wild-type cells. PCR analysis of insertion mutants using IS-specific and bgl-specific primers allowed the identification of the IS type integrating into the bgl regulatory region (bglR).
2.9 Adaptive laboratory evolution to measure plasmid stability
E scherichia coli DH5αZ1 or JM107MA2 cells carrying the pBDP_Km_GFP5 plasmid were transformed either with pCRISPathBrick (serving as the control) or pCRIS. A single colony from each transformation was inoculated and was fully grown in LB+Ap+Cm medium as a starter culture, diluted 1000-fold in fresh LB+Ap+Cm+IPTG medium and divided each into 48 parallel cultures of 200 μl. These cultures were shaken in 96-well microplates (Greiner Bio-One International, Kremsmünster, Austria) in a SynergyHT microplate reader (BioTek, Winooski, VT, USA) at 37°C, and supplemented with Km after 2 h. After overnight shaking and growth, the cultures were diluted 400-fold into fresh LB+Km+Cm+IPTG during their transfer to a new microplate. Such growth and 400-fold dilution was repeated three to seven more times. During growth, OD600 and GFP-fluorescence readings (excitation filter: 485/20, emission filter: 528/20) were recorded at 15-min intervals. The experiments were evaluated two ways: (i) The mean fluorescence of both cell lines was calculated every day from the daily peak GFP fluorescence values detected in each corresponding well, and was compared for the two lines using unpaired, two-tailed t-tests. (ii) Every day, each well was classified as ‘active’ or ‘inactive’ depending on whether the peak green fluorescence intensity reached a certain threshold value (3000 AU) or not, respectively, and the number of ‘inactive’ wells was compared for the two strains using χ2 tests.
To assess the type of mutations that were present after the adaptive evolution, we performed a PCR-based assay. PCR amplification of the VF2-VR segment (comprising the km and gfp genes) directly from the cultures allowed the identification of insertion mutants based on increased product length. Then, a second round of PCR was carried out on insertion mutants using primer VF2 and various IS-specific primers (Supplementary Table S1) to permit the identification of the exact IS types residing in the expression cassette.
2.10 Protein overexpression using a flask-and-shaker system
E scherichia coli DH5αZ1 carrying the pBDP_Km_GFP5 plasmid was transformed either with pCRISPathBrick (serving as the control) or pCRIS. Six colonies from each transformation were grown as starter cultures in 1 ml of LB+Cm+Ap medium in a shaker set to 37°C. After overnight growth, each culture was diluted 5000-fold into 5 ml cultures of LB+Cm+Ap+IPTG and was grown further for 2 h. Next, Km was added to each flask and shaking at 37°C continued for 40 h. At the end of growth, 100 μl of each culture was transferred to a 96-well microplate (Greiner Bio-One International, Kremsmünster, Austria) and the OD (at 600 nm) and the green fluorescence (excitation filter: 485/20, emission filter: 528/20) was measured in a SynergyHT microplate reader (BioTek, Winooski, VT, USA).
2.11 Plasmid availability
pCRISP_IS and pCRIS will be made available at Addgene https://www.addgene.org/. DNA sequences of pCRIS and pCRISP_IS have been deposited to GenBank (accession numbers: pCRIS: MK214496; pCRISP_IS: MK214497).
3. Results and discussion
3.1 Simultaneous silencing of up to 38 mobile genetic elements in E. coli
To develop a portable method that can downregulate insertional mutagenesis genome-wide, we established a plasmid-encoded CRISPRi system that targets selected transposases. Specifically, we targeted an extrachromosomally expressed, catalytically inactive SpCas9 (dCas9) to the promoter sequence in the left inverted repeat of selected mobile genetic elements in E. coli. We chose IS1, IS3, IS5, and IS150 to be targeted by our construct due to their main contribution to insertional mutagenesis in E. coli, as demonstrated by two previous analyses (16, 23).
We hypothesized that our system allows the inhibition of transposition by (i) transcriptional silencing of the transposase gene, and (ii) limiting the access of the transposase enzymes to the left inverted repeat of the mobile element and thereby inhibiting DNA cleavage and subsequent transposition. We implemented the well-established and robust pCRISPathBrick plasmid set that (i) constitutively expresses the dCas9, as well as one or more crRNAs of choice, (ii) permits CRISPR array assembly with relative ease and (iii) has been applied previously for multitarget silencing in vivo (35).
Spacers targeting these IS elements were designed, synthesized and cloned into the pCRISPathBrick plasmid to construct pCRIS (see Section 2). In this way, a total of 26 IS elements were targeted simultaneously by CRISPRi in E. coli K-12 MG1655, and 38 copies in BL21(DE3), as listed in Table 1.
3.2 Genome-wide simultaneous suppression of transposases from pCRIS
First, we investigated whether the expression of CRISPRi from a single episomal vector (pCRIS) is capable of suppressing transposases at multiple chromosomal loci. To this aim, the effect of the pCRIS plasmid on the host transcriptome was compared with that of a control plasmid using a standard Illumina RNAseq assay (see Section 2) and the expression of each gene was quantified as the frequency of Illumina sequencing reads that map to the given target (Supplementary Figures S1–S3).
Considering changes in transposase expression, the presence of pCRIS resulted in a more than 4-fold silencing of IS1A (t-test: P < 8 × 10−8), compared with a nonsuppressed control (Figure 1A). Moreover, the first open reading frame of IS1F (gene b4294) showed an even stronger, 11-fold suppression (t-test: P < 2 × 10−4) (Figure 1B). Dramatic knockdown was observed for the transposases of both the IS3 and the canonical IS5 elements, displaying more than 360- and 26-fold reduction of sequencing-read frequencies, respectively (P < 2 × 10−5, P < 2 × 10−14, respectively) (Figure 1C and D). Interestingly, no transcriptional knockdown was observed for IS150 (Supplementary Figure S4). The lack of changes in IS150 mRNA levels may indicate the necessity to re-define the active promoter of this element. This issue, along with other features of the transcriptomic dataset are discussed in Supplementary Note 1.
Figure 1.

Transcriptional changes of IS elements in E. coli K-12 MG1655 caused by the propagation of the pCRIS plasmid. The figure displays the frequency of Illumina sequencing reads mapping to IS1 (A), IS1F (B), IS3 (C) and IS5 (D). Star denotes significance (P < 2 × 10−4) based on t-test. Error bars represent SD, n = 3. CPM, counts per million reads.
3.3 pCRIS efficiently suppresses chromosomal insertional mutagenesis
Next, we assessed the effect of CRISPRi on the spontaneous chromosomal mutational landscape of E. coli. We sought to precisely evaluate insertional mutagenesis caused by endogenous ISes, primarily IS1, IS5, IS3 and IS150. For this aim, we applied two complementary mutation-detection systems. The first experimental setup detects deleterious mutations of the cycA gene. CycA, which is responsible for D-cycloserine susceptibility, can be inactivated by various loss-of-function mutations, including IS transposition (16, 23). Based on the numbers of D-cycloserine-resistant colonies observed, we carried out fluctuation analyses to compare the rate of mutations inactivating cycA in the presence and the absence of pCRIS-based transposase-suppression (see Section 2). In our tests, the mutation/culture values (m) were significantly lower in E. coli K-12 MG1655 in the presence of pCRIS compared with a nontargeting plasmid, pCRISPathBrick (Figure 2A). Notably, the observed effect of pCRIS-based IS-silencing on the mutation rate was comparable to that caused by the complete genomic deletion of ISes (23) which suggests the near-complete elimination of insertional mutagenesis.
Figure 2.
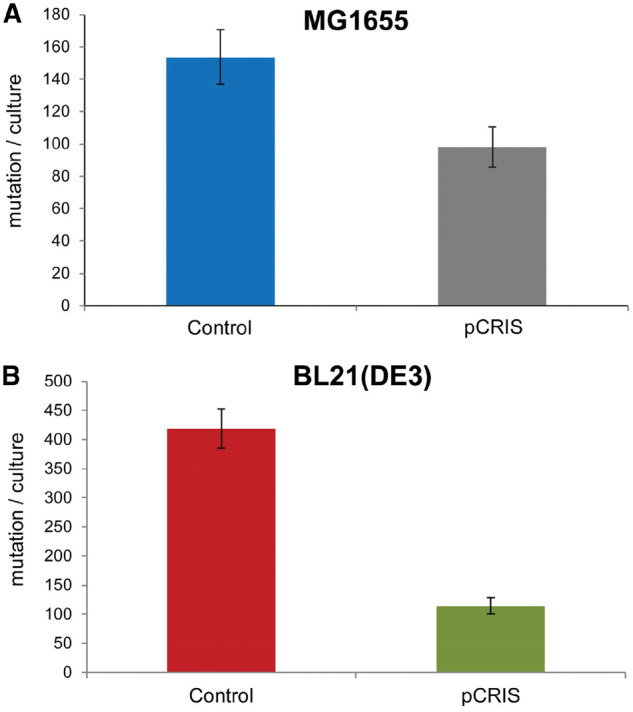
Effect of pCRIS-based IS-knockdown on the rate of cycA mutations in multiple E. coli strains: (A) E. coli K-12 MG1655 and (B) E. coli BL21(DE3). Error bars represent SD based on n = 3 independent measurements. *Significance (P < 0.05) based on t-test.
Strikingly, the reduction of insertional mutagenesis caused by pCRIS was even more remarkable in the industrial E. coli host, BL21(DE3). Applying the same assay, a 73% decrease in m was observed as compared with the control (Figure 2B). We attributed this large effect to the high fraction of IS150 insertions among cycA mutants in BL21(DE3), seen earlier (24). Confirming our hypothesis, PCR analysis of 100 E. coli BL21(DE3) cycA mutants indicated that in the nonsuppressed, parental strain, 78% and 7% of the mutations are caused by the transposition of IS150 and IS1, respectively (Figure 3A). However, in pCRIS-harboring E. coli BL21(DE3) cells only 1% of the resistant variants displayed IS integration with the rest being point mutants. Moreover, no IS1 transposition could be detected among the 100 tested colonies (Figure 3B). Therefore, the large decrease in mutation events seen in E. coli BL21(DE3) in the presence of pCRIS (Figure 2B) can be attributed to the near-complete elimination of IS transposition into cycA (Figure 3). The changes observed in the composition of mutants caused by the presence of pCRIS also underline the fact that IS150 mobility was dramatically downregulated despite the lack of transcriptional silencing.
Figure 3.
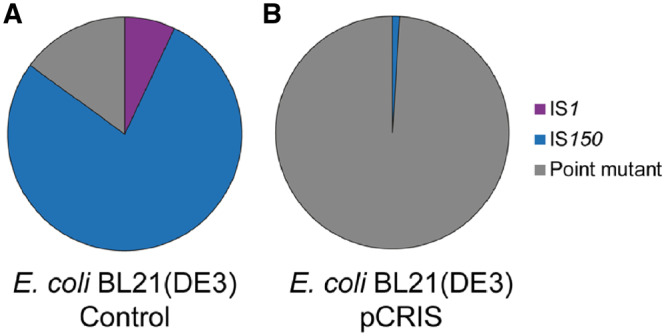
Effect of IS-knockdown on the spectrum of mutations inactivating cycA in E. coli BL21(DE3). (A) Effect of a nontargeting control plasmid (pCRISPathBrick); (B) effect of pCRIS-based transposase-silencing. χ2 test: P < 10−5.
The second mutagenesis assay that we applied tests the activation of the cryptic bgl operon in E. coli that leads to the ability of mutants to grow on salicin-minimal medium. This assay was chosen for three noteworthy features: First, it investigates insertional mutagenesis at a locus distinct from cycA. Second, it is highly specific for IS transposition (17): in case of wild-type E. coli K-12 MG1655, more than 90% of salicin-assimilating colonies contain an IS element (primarily IS1 and IS5) at the bglR locus. Finally, besides detecting mutations in growing cultures, it is also capable of quantifying mutation events in nongrowing living cells, starving on salicin-minimal medium.
When comparing pCRIS-containing E. coli K-12 MG1655 cells to controls carrying a nontargeting plasmid, this assay revealed a 4.4-fold decrease in overall mutation frequency in stationary phase (Figure 4). PCR analysis of the salicin-utilizing colonies after 10 days uncovered that the frequency of insertion mutants decreased nearly 11-fold, with the near-complete elimination of IS1-transposition into bglR (Figure 4). The daily rate of the emergence of salicin-assimilating cells during the 10-day stationary phase also displayed a 5-fold reduction in the presence of pCRIS (P < 10−3) (Supplementary Figure S5). Based on these two assays detecting mutation events at two distinct chromosomal loci, IS-knockdown from a single plasmid was found to efficiently downregulate insertional mutagenesis in two different strains of E. coli.
Figure 4.
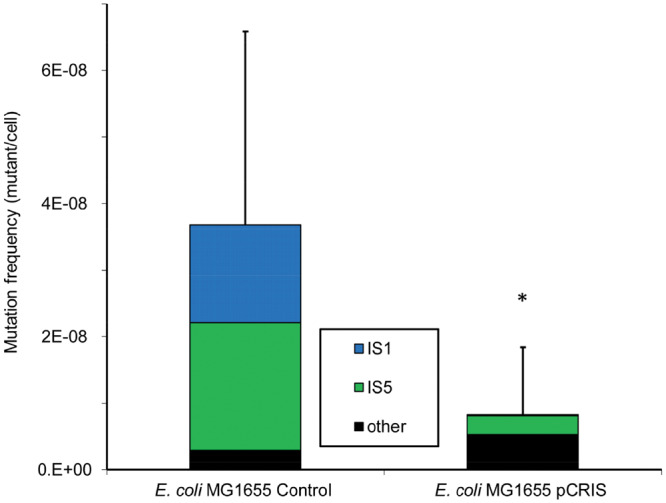
Effect of IS-knockdown on the frequency and spectrum of mutations activating the bgl operon of E. coli MG1655. The frequency and composition of salicin-assimilating mutants observed on day 10 are shown. The control strain carried the nontargeting pCRISPathBrick plasmid, pCRIS represents active IS-silencing. *P < 10−4 (Mann–Whitney U-test).
3.4 Transposase suppression increases the genetic stability of plasmid-borne synthetic DNA
Finally, we sought to investigate the effects of transposase-suppression on the genetic stability of plasmid-borne synthetic DNA elements during their long-term propagation in E. coli. To estimate the genetic stability of a synthetic DNA construct under mock-industrial conditions, we carried out adaptive evolution experiments utilizing a previously established plasmid-based system that is reportedly susceptible to insertional mutagenesis (7).
Specifically, our test plasmid pBDP_Km_GFP5 carries a gfp gene and an oppositely oriented kanamycin-resistance gene, driven by a bidirectional LacI-repressible promoter (7), called the km-gfp cassette. In its induced state kanamycin-selection transcriptionally links GFP expression to the host cell’s survival and thereby prevents mutations that disrupt the bidirectional promoter. As a result, the burden of GFP production is most often relieved by the IS-mediated interruption of its gene, instead of the otherwise commonly seen promoter mutations. In our evolutionary experiments, continuously induced pBDP_Km_GFP5 was propagated in the LacI-overexpressing DH5αZ1 host in 48 parallel cultures for 60–65 generations in the presence of kanamycin. Notably, the timeframe and the number of cell divisions in these experiments were industrially relevant as cell-growth from a seeding starter to a fermentation volume of 1–200 m3 requires approximately 60 generations. The effect of IS-knockdown was assessed by comparing pCRIS-carrying strains with control lines harboring the nontargeting pCRISPathBrick plasmid. To evaluate mutational dynamics in the course of the adaptation, we continuously monitored GFP expression and the OD600 value of each culture and thereby assessed the integrity of the synthetic construct (Section 2).
The means of the maximal intensities were calculated each day for each strain to follow the day-to-day changes in the average heterologous protein production (Supplementary Figure S6). By the end of laboratory evolution, the median fluorescence of the bacterial populations dropped by 16% when pCRIS was present in cells, in contrast to the 35% decrease without controlling insertional mutagenesis (P < 2 × 10−3) (Figure 5). Similar results were obtained when repeating the experiment in E. coli JM107MA2 (Supplementary Figures. S7 and S8).
Figure 5.

The effect of IS-silencing on plasmid stability in E. coli DH5αZ1. The figure shows the T0-normalized, relative GFP signal (AU) of 48 parallelly passaged cultures of E. coli DH5αZ1 that are either carrying the nontargeting, control plasmid (pCRISPathBrick) or the IS-knockdown plasmid, pCRIS. Fluorescence intensities were compared on initiation (T0) and after 60 generations (T60) in the course of adaptive laboratory evolution. Center lines show the medians; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, outliers are represented by dots; crosses represent sample means. n = 48 sample points. *Marks the significant difference of the means (P < 2 × 10−3) assessed by a Mann–Whitney U-test.
To investigate the mutational processes and dynamics behind synthetic construct inactivation, we monitored the transfer-to-transfer changes observable in each culture in the course of our laboratory adaptive evolution experiment. Each well was classified as ‘inactive’ or ‘active’ based on its maximal GFP fluorescence intensity, and the number of ‘inactive’ cultures was compared for the two strains. After 7 days and 60 generations of serial passage (T60), only one ‘inactive’ culture was found for pCRIS while the nonsuppressed control inactivated 11 cultures, thus indicating a significant decrease in the frequency of inactivating mutations (χ2 test: P < 0.005) (Figure 6A).
Figure 6.

Effect of IS-knockdown on fluorescence of individual cultures. (A) Maximum fluorescence intensities (AU) of the individual cultures in the presence of either an IS-silencing plasmid (pCRIS) or a nontargeting control plasmid (pCRISPathBrick), after 7 days (60 generations) of continuous propagation (T60). (B) Analysis of the mutagenic spectrum within the km-gfp cassette of pBDP_Km_GFP5 from individual cell-populations (n = 48) at T60. Arrows mark PCR products displaying an increased length due to IS transposition into km-gfp.
Next, we sought to investigate the causative mutations behind inactivation. PCR amplification of the km-gfp cassette demonstrated that the dominant genotype in each ‘inactive’ culture corresponded to an elongated PCR product, thereby indicating that each inactivating mutation was caused by IS transposition into pBDP_Km_GFP5 (Figure 6B). Further PCR-based analysis of these insertion mutants in the nonsuppressed control strain revealed that 10 out of 11 was caused by IS2 transposition, with IS10 being responsible for the remaining one. The single insertion seen in the IS-silenced strain was caused by IS10. The repetition of our adaptive laboratory evolution in a further strain of E. coli (JM107MA2) revealed a smaller, but significant stabilizing effect for pCRIS (Supplementary Figures S7 and S8). The significant suppression of IS2 transposition, albeit not its complete elimination was observed in JM107MA2, as well (Supplementary Table S4). Since our CRISPR array did not target IS2, this observation was surprising. The possible background of this intriguing phenomenon is discussed in Supplementary Note 2.
3.5 The fitness effect of IS-knockdown during flask-scale growth is outweighed by higher transgene stability
A potential disadvantage of using dCas-mediated IS-knockdown could be its adverse effect on cellular growth parameters, with special emphasis on the biomass yield (final OD) of the bacterial culture. To test the effect of pCRIS on bacterial fitness, we recorded the growth curves of 100 ml cultures of E. coli BL21(DE3) in a flask-and-shaker system. The presence of pCRIS caused a detectable decrease in both the growth rate and the final OD compared with that of the nontargeting plasmid pCRISPathBrick or the empty vector pACYC184 (Supplementary Figure S12). Since pCRISPathBrick did not cause any significant growth retardation compared with pACYC184, it is tempting to assume that the attachment of dCas to the chromosome at 38 loci and not the overexpression of dCas impairs growth. Interestingly, in a microplate-based growth assay, pCRIS did not affect the growth rate or final OD, as displayed by the growth parameters extracted from the plasmid-stability experiments described above (Supplementary Figure S13).
An important question when using IS-knockdown to stabilize the expression of heterologous proteins is whether the gain acquired from the increased half-life of protein overexpression can put up with, or outweigh the adverse effect of the decreased biomass. To test this, we expressed GFP from the pBDP_Km_GFP5 plasmid in E. coli DH5α in the presence of either pCRIS or the nontargeting pCRISPathBrick plasmid, by simultaneously growing populations in twelve 5-ml flask cultures. IPTG induction was applied for 12 generations and full growth of the cultures was obtained after 42 h. As summarized on Figure 7A, the presence of pCRIS decreased the mean final OD by 12.7% compared with the control (P < 10−3). The mean GFP fluorescence of the cultures, that models heterologous protein production, however, was not impaired by the presence of pCRIS but displayed a slight improvement compared with the control (Figure 7B). This difference is explained by the deterioration of GFP expression observed in two of the six control flasks (Figure 7C), decreasing the control’s mean GFP signal and increasing its SD (Figure 7B). The OD-normalized fluorescence, often used as a proxy for mean cellular protein expression, is nevertheless increased by 31.7% in the presence of pCRIS compared with that of the control plasmid (P < 0.05) (Figure 7D). Thus, this experiment stands in good agreement with those results obtained in our microplate-based assay (Figure 6), which used smaller volumes and more parallels.
Figure 7.

Effect of pCRIS on growth parameters and GFP expression of flask-scale cultures. Six parallels each of E. coli DH5αZ1 carrying plasmid pBDP_Km_GFP5 and either plasmid pCRIS (blue bars) or the nontargeting control plasmid pCRISPathBrick (red bars) was induced with IPTG for 42 h, followed by the endpoint-measurement of OD600 and GFP fluorescence values. (A) Mean OD600 values; (B) Mean GFP fluorescence values; (C) GFP fluorescence measured in each individual flask culture at the end of induction; (D) OD600-normalized fluorescence mean values. Error bars indicate SD (n = 6). Asterisks indicate the results of unpaired, two-tailed t-tests: *P < 0.05, **P < 10−3.
In sum, these results demonstrated that continuous IS-knockdown is capable of extending the half-life of episomally propagated synthetic genetic constructs under conditions that are similar to industrial fermentations. The ability of pCRIS to downregulate IS transposition in the absence of growth (i.e. in cells starving on salicin-minimal medium) may indicate its potential applicability in production systems where the bacterial culture has ceased replication. We expect that pCRIS can offer a portable alternative to time-consuming, genome-wide elimination of ISes (23, 24) where increased genetic stability is required. However, follow-up studies will be needed to evaluate the effects of pCRIS on industrially applied biosynthetic pathways and under fermentation conditions.
One of the greatest challenges limiting synthetic biology is the fact that biological systems constantly evolve, and the direction of evolution seldom matches that desired by bioengineers (1). Decelerating evolution by limiting mutational processes provides a promising strategy to increase genetic stability, however the genome-scale elimination of transposition has, to date lacked a simple and generally applicable solution.
Our work addresses this problem by introducing a portable system, termed pCRIS that reduces insertional mutagenesis within multiple E. coli strains and circumvents the time-consuming nature of previous genome-engineering endeavors. pCRIS displays several unique features, it is: (i) portable, demonstrated by its application in distinct E. coli strains; (ii) instantaneous, requiring only a simple transformation into the strain-of-interest; (iii) reversible, a plasmid curing is sufficient to regain wild-type mutation rate in the host; and (iv) highly specific for insertional mutagenesis.
Several examples are available in the literature to limit the rate of single-nucleotide exchanges or homologous recombination-driven rearrangements in bacterial hosts by inactivating a handful of genes (8–13). Such a simple and rapid solution to counteract transposable elements has been lacking, despite increasing evidence concerning their role in transgene-inactivation. For example, Rugbjerg et al. (14), investigated the spectrum of mutations inactivating industrially applicable mevalonate-producing pathways. Their results showed a striking dominance of insertions with IS transposition being responsible for 69.5–99.9% of mutations in E. coli DH10B. Moreover, a significant number of case studies from basic- and applied-research have also reported the interruption of cloned DNA segments by IS elements (39, 49–54).
In this work, we demonstrate the CRISPRi-based knockdown of IS-activities in multiple E. coli strains, including the biotechnologically useful production host, E. coli BL21(DE3). The system, termed pCRIS, efficiently suppressed the mobility of four types of IS elements (IS1, IS3, IS5 and IS150), that notably distinguishing it from other endeavors (55). By expressing a single array of targeting constructs that directs a catalytically inactive SpCas9 to the transposases-of-interest, IS transposition to chromosomal and episomal targets was demonstrated to be significantly downregulated in various experimental setups. Importantly, the expression of less than 0.3% of all E. coli genes differed upon IS-knockdown, which demonstrates the unique specificity of plasmid-based IS suppression (Supplementary Figure S3). Albeit multiplex gene silencing has been established before using the same approach (35), to the best of our knowledge, this is the first time multiple crRNAs each targeting multiple loci have been used effectively to control transcription. We demonstrate this ‘multi-multi’ strategy to successfully control the mobility of up to 38 copies of IS elements in E. coli BL21(DE3). Most notably, pCRIS, while requiring only a single plasmid delivery performed within a single day, provided a reduction of IS-mobility comparable to that seen in genome-scale chromosome engineering projects (23, 24). We demonstrate that although multiple IS-silencing may cause a fitness defect in flask-and-shaker systems, the less frequent inactivation of the overexpressed gene can counteract, and even outweigh this issue. In our tests, this beneficial effect of pCRIS to avoid mutations inactivating protein overexpression was readily apparent even in a single growth session in shake-flask cultures, within as few as 12 generations (Figure 7C). It may thereby provide rapid strain stabilization for a wide range of applications in basic- and applied-research where IS transposition is the primary source of deleterious mutations while circumventing the time-consuming nature of genome-scale engineering.
However, our system also has some limitations, as the current design of pCRIS may not necessarily be the optimal choice for all projects that require IS suppression. First, the prevalence of IS types found in inactivated mutants shows dependence on the target sequence, the host strain and possibly other factors as well, which may require modification of the plasmid. The user-friendly cloning system of pCRISPathBrick, however, allows rapid cloning of repeat-spacer tandems targeting further mobile elements (35). Second, pCRIS is based on a p15A origin of replication that can be incompatible with several other production-plasmids and may be restricted to certain hosts. The use of broad-host replication origins, combined with modular plasmid assembly strategies can provide solutions for such issues for a wide range of applications (56, 57). Third, the constitutive overexpression of dCas shown here can itself cause a negative fitness effect on the host, which may prove to be deleterious when combined with the overexpression of a toxic gene of interest. Such cases may necessitate the fine-tuning of dCas9 using inducible promoters, or require the chromosomal integration of the CRISPR/dCas machinery. Attempts to reduce dCas toxicity, albeit for different applications than ours, have also been published (58). Finally, certain plasmid-based expression systems are not interrupted by insertions at all but display other modes of inactivation, such as deletions caused by replication slippage (7). Further solutions, discussed in Section 1 must therefore always be kept in mind.
4. Summary and conclusion
In summary, we applied CRISPRi to repress the transposition of multiple IS types in multiple strains of E. coli. Constitutively expressing dCas9, tracrRNA and four IS-specific crRNAs from pCRIS was shown to increase genetic stability of both chromosomal- and plasmid-based genetic circuits in vivo. RNA sequencing analysis unveiled the highly specific nature of IS-knockdown, and also revealed that transcriptional silencing is not a necessary prerequisite of transposition-inhibition. Albeit the binding of dCas to multiple chromosomal loci had a detectable adverse effect on the growth of flask-scale cultures, the longer half-life of transgene expression was shown to outweigh the decrease in biomass. IS-silencing was also demonstrated in non-dividing bacterial cells, indicating the potential functionality of this system in stationary-phase production cultures. The portable nature of pCRIS makes it easily applicable in a number of further hosts. In addition, the robustness of CRISPRi will permit the system to be easily reprogrammed to target other IS types and thereby yield a full set of strain-specific IS-silencing plasmids. With such possibilities in mind, we expect that the rationale of pCRIS will be broadly applicable to decelerate transposition-driven evolutionary processes in a wide variety of laboratory model-strains and industrially applied bacterial hosts.
Supplementary Material
Acknowledgments
We thank Prof. Luciano Marraffini and Prof. Herbert M. Sauro for providing plasmids, Prof. Antal Kiss for providing strains and Prof. György Pósfai for helpful discussions.
Funding
This work was supported by the National Research, Development, and Innovation Office of Hungary (NKFIH) [Grant No. K119298 (to T.F.) and the GINOP-2.3.2-15-2016-00001 (to T.F.)] and by grants from the European Research Council H2020-ERC-2014-CoG 648364—Resistance Evolution (to C.P.), the Wellcome Trust (to C.P.) and GINOP (MolMedEx TUMORDNS) GINOP-2.3.2-15-2016-00020, GINOP (EVOMER) GINOP-2.3.2-15-2016-00014 (to C.P.); the ‘Lendület’ Program of the Hungarian Academy of Sciences (to C.P.); and a PhD fellowship from the Boehringer Ingelheim Fonds (to Á.N.).
Conflict of interest statement. None declared.
References
- 1. Endy D. (2005) Foundations for engineering biology. Nature, 438, 449–453. [DOI] [PubMed] [Google Scholar]
- 2. Renda B.A., Hammerling M.J., Barrick J.E. (2014) Engineering reduced evolutionary potential for synthetic biology. Mol. Biosyst., 10, 1668–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bull J.J., Barrick J.E. (2017) Arresting evolution. Trends Genet., 33, 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tyo K.E., Ajikumar P.K., Stephanopoulos G. (2009) Stabilized gene duplication enables long-term selection-free heterologous pathway expression. Nat. Biotechnol., 27, 760–765. [DOI] [PubMed] [Google Scholar]
- 5. Jack B.R., Leonard S.P., Mishler D.M., Renda B.A., Leon D., Suarez G.A., Barrick J.E. (2015) Predicting the genetic stability of engineered DNA sequences with the EFM calculator. ACS Synth. Biol., 4, 939–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sleight S.C., Bartley B.A., Lieviant J.A., Sauro H.M. (2010) Designing and engineering evolutionary robust genetic circuits. J. Biol. Eng., 4, 12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang S., Sleight S.C., Sauro H.M. (2013) Rationally designed bidirectional promoter improves the evolutionary stability of synthetic genetic circuits. Nucleic Acids Res., 41, e33.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harris R.S., Feng G., Ross K.J., Sidhu R., Thulin C., Longerich S., Szigety S.K., Winkler M.E., Rosenberg S.M. (1997) Mismatch repair protein MutL becomes limiting during stationary-phase mutation. Genes Dev., 11, 2426–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kamiya H., Iida E., Murata-Kamiya N., Yamamoto Y., Miki T., Harashima H. (2003) Suppression of spontaneous and hydrogen peroxide-induced mutations by a MutT-type nucleotide pool sanitization enzyme, the Escherichia coli Orf135 protein. Genes Cells, 8, 941–950. [DOI] [PubMed] [Google Scholar]
- 10. Csorgo B., Feher T., Timar E., Blattner F.R., Posfai G. and (2012) Low-mutation-rate, reduced-genome Escherichia coli: an improved host for faithful maintenance of engineered genetic constructs. Microb. Cell Fact., 11, 11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deatherage D.E., Leon D., Rodriguez A.E., Omar S.K., Barrick J.E. (2018) Directed evolution of Escherichia coli with lower-than-natural plasmid mutation rates. Nucleic Acids Res., 46, 9236–9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vapnek D., Alton N.K., Bassett C.L., Kushner S.R. (1976) Amplification in Escherichia coli of enzymes involved in genetic recombination: construction of hybrid ColE1 plasmids carrying the structural gene for exonuclease I. Proc. Natl. Acad. Sci. USA, 73, 3492–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Troester H., Bub S., Hunziker A., Trendelenburg M.F. (2000) Stability of DNA repeats in Escherichia coli dam mutant strains indicates a Dam methylation-dependent DNA deletion process. Gene, 258, 95–108. [DOI] [PubMed] [Google Scholar]
- 14. Rugbjerg P., Myling-Petersen N., Porse A., Sarup-Lytzen K., Sommer M.O.A. (2018) Diverse genetic error modes constrain large-scale bio-based production. Nat. Commun., 9, 787.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mahillon J., Chandler M. (1998) Insertion sequences. Microbiol. Mol. Biol. Rev., 62, 725–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feher T., Cseh B., Umenhoffer K., Karcagi I., Posfai G. (2006) Characterization of cycA mutants of Escherichia coli. An assay for measuring in vivo mutation rates. Mutat. Res., 595, 184–190. [DOI] [PubMed] [Google Scholar]
- 17. Hall B.G. (1998) Activation of the bgl operon by adaptive mutation. Mol. Biol. Evol., 15, 1–5. [DOI] [PubMed] [Google Scholar]
- 18. Halliday J.A., Glickman B.W. (1991) Mechanisms of spontaneous mutation in DNA repair-proficient Escherichia coli. Mutat. Res., 250, 55–71. [DOI] [PubMed] [Google Scholar]
- 19. Chao L., Vargas C., Spear B.B., Cox E.C. (1983) Transposable elements as mutator genes in evolution. Nature, 303, 633–635. [DOI] [PubMed] [Google Scholar]
- 20. Feher T., Bogos B., Mehi O., Fekete G., Csorgo B., Kovacs K., Posfai G., Papp B., Hurst L.D., Pal C. (2012) Competition between transposable elements and mutator genes in bacteria. Mol. Biol. Evol., 29, 3153–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Orgel L.E., Crick F.H. (1980) Selfish DNA: the ultimate parasite. Nature, 284, 604–607. [DOI] [PubMed] [Google Scholar]
- 22. Schneider D., Lenski R.E. (2004) Dynamics of insertion sequence elements during experimental evolution of bacteria. Res. Microbiol., 155, 319–327. [DOI] [PubMed] [Google Scholar]
- 23. Pósfai G., Plunkett G., Fehér T., Frisch D., Keil G.M., Umenhoffer K., Kolisnychenko V., Stahl B., Sharma S.S., de Arruda M.. et al. (2006) Emergent properties of reduced-genome Escherichia coli. Science, 312, 1044–1046. [DOI] [PubMed] [Google Scholar]
- 24. Umenhoffer K., Draskovits G., Nyerges Á., Karcagi I., Bogos B., Tímár E., Csörgő B., Herczeg R., Nagy I., Fehér T.. et al. , (2017) Genome-wide abolishment of mobile genetic elements using genome shuffling and CRISPR/Cas-assisted MAGE allows the efficient stabilization of a bacterial chassis. ACS Synth. Biol., 6, 1471–1483. [DOI] [PubMed] [Google Scholar]
- 25. Park M.K., Lee S.H., Yang K.S., Jung S.C., Lee J.H., Kim S.C. (2014) Enhancing recombinant protein production with an Escherichia coli host strain lacking insertion sequences. Appl. Microbiol. Biotechnol., 98, 6701–6713. [DOI] [PubMed] [Google Scholar]
- 26. Choi J.W., Yim S.S., Kim M.J., Jeong K.J. (2015) Enhanced production of recombinant proteins with Corynebacterium glutamicum by deletion of insertion sequences (IS elements). Microb. Cell Fact., 14, 207.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suarez G.A., Renda B.A., Dasgupta A., Barrick J.E. (2017) Reduced mutation rate and increased transformability of transposon-free Acinetobacter baylyi ADP1-ISx. Appl. Environ. Microbiol, 83, e01025–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jansen R., Embden J.D., Gaastra W., Schouls L.M. (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol., 43, 1565–1575. [DOI] [PubMed] [Google Scholar]
- 29. Mojica F.J., Diez-Villasenor C., Soria E., Juez G. (2000) Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol., 36, 244–246. [DOI] [PubMed] [Google Scholar]
- 30. Niu D., Wei H.J., Lin L., George H., Wang T., Lee I.H., Zhao H.Y., Wang Y., Kan Y., Shrock E.. et al. (2017) Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science, 357, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang L., Guell M., Niu D., George H., Lesha E., Grishin D., Aach J., Shrock E., Xu W., Poci J.. et al. (2015) Genome-wide inactivation of porcine endogenous retroviruses (PERVs). Science, 350, 1101–1104. [DOI] [PubMed] [Google Scholar]
- 32. Banno S., Nishida K., Arazoe T., Mitsunobu H., Kondo A. (2018) Deaminase-mediated multiplex genome editing in Escherichia coli. Nat. Microbiol., 3, 423–429. [DOI] [PubMed] [Google Scholar]
- 33. Shapiro R.S., Chavez A., Collins J.J. (2018) CRISPR-based genomic tools for the manipulation of genetically intractable microorganisms. Nat. Rev. Microbiol., 16, 333–339. [DOI] [PubMed] [Google Scholar]
- 34. Bikard D., Jiang W., Samai P., Hochschild A., Zhang F., Marraffini L.A. (2013) Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res., 41, 7429–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cress B.F., Toparlak O.D., Guleria S., Lebovich M., Stieglitz J.T., Englaender J.A., Jones J.A., Linhardt R.J., Koffas M.A. (2015) CRISPathBrick: modular combinatorial assembly of type II-A CRISPR arrays for dCas9-mediated multiplex transcriptional repression in E. coli. ACS Synth. Biol., 4, 987–1000. [DOI] [PubMed] [Google Scholar]
- 36. Blattner F.R., Plunkett G., Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F.. et al. (1997) The complete genome sequence of Escherichia coli K-12. Science, 277, 1453–1462. [DOI] [PubMed] [Google Scholar]
- 37. Jeong H., Barbe V., Lee C.H., Vallenet D., Yu D.S., Choi S.H., Couloux A., Lee S.W., Yoon S.H., Cattolico L.. et al. (2009) Genome sequences of Escherichia coli B strains REL606 andx BL21(DE3). J. Mol. Biol., 394, 644–652. [DOI] [PubMed] [Google Scholar]
- 38. Lutz R., Bujard H. (1997) Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res., 25, 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blumenthal R.M., Gregory S.A., Cooperider J.S. (1985) Cloning of a restriction-modification system from Proteus vulgaris and its use in analyzing a methylase-sensitive phenotype in Escherichia coli. J. Bacteriol., 164, 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sambrook J., Fritch E.F., Maniatis T. (1987) Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 41. Reese M.G. (2001) Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem., 26, 51–56. [DOI] [PubMed] [Google Scholar]
- 42. Sawers R.G. (2005) Transcript analysis of Escherichia coli K-12 insertion element IS5. FEMS Microbiol. Lett., 244, 397–401. [DOI] [PubMed] [Google Scholar]
- 43. Schwartz E., Kroger M., Rak B. (1988) IS150: distribution, nucleotide sequence and phylogenetic relationships of a new E. coli insertion element. Nucleic Acids Res., 16, 6789–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hall B.G., Acar H., Nandipati A., Barlow M. (2014) Growth rates made easy. Mol. Biol. Evol., 31, 232–238. [DOI] [PubMed] [Google Scholar]
- 45. Robinson M.D., McCarthy D.J., Smyth G.K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 43, e47.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sarkar S., Ma W.T., Sandri G.H. (1992) On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica, 85, 173–179. [DOI] [PubMed] [Google Scholar]
- 48. Rosche W.A., Foster P.L. (2000) Determining mutation rates in bacterial populations. Methods, 20, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fernandez C., Larhammar D., Servenius B., Rask L., Peterson P.A. (1986) Spontaneous insertions into cosmid vector—a warning. Gene, 42, 215–219. [DOI] [PubMed] [Google Scholar]
- 50. Nakamura K., Inouye M. (1981) Inactivation of the Serratia marcescens gene for the lipoprotein in Escherichia coli by insertion sequences, IS1 and IS5; sequence analysis of junction points. Mol. Gen. Genet., 183, 107–114. [DOI] [PubMed] [Google Scholar]
- 51. Prather K.L., Edmonds M.C., Herod J.W. (2006) Identification and characterization of IS1 transposition in plasmid amplification mutants of E. coli clones producing DNA vaccines. Appl. Microbiol. Biotechnol., 73, 815–826. [DOI] [PubMed] [Google Scholar]
- 52. Rood J.I., Sneddon M.K., Morrison J.F. (1980) Instability in tyrR strains of plasmids carrying the tyrosine operon: isolation and characterization of plasmid derivatives with insertions or deletions. J. Bacteriol., 144, 552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Muller J., Reinert H., Malke H. (1989) Streptokinase mutations relieving Escherichia coli K-12 (prlA4) of detriments caused by the wild-type skc gene. J. Bacteriol., 171, 2202–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rawat P., Kumar S., Pental D., Burma P.K. (2009) Inactivation of a transgene due to transposition of insertion sequence (IS136) of Agrobacterium tumefaciens. J. Biosci., 34, 199–202. [DOI] [PubMed] [Google Scholar]
- 55. Geng P., Leonard S.P., Mishler D.M., Barrick J.E. (2018) Synthetic genome defenses against selfish DNA elements stabilize engineered bacteria against evolutionary failure. ACS Synth. Biol., doi: 10.1021/acssynbio.8b00426. [DOI] [PubMed]
- 56. Martinez-Garcia E., Aparicio T., Goni-Moreno A., Fraile S., de Lorenzo V. (2015) SEVA 2.0: an update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res., 43, D1183–D1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Silva-Rocha R., Martinez-Garcia E., Calles B., Chavarria M., Arce-Rodriguez A., de Las Heras A., Paez-Espino A.D., Durante-Rodriguez G., Kim J., Nikel P.I.. et al. (2013) The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res., 41, D666–D675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang S., Voigt C.A. (2018) Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res., 46, 11115–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.