

Abstract
Hyperactivity in striatum is associated with compulsive behaviors in obsessive-compulsive disorder (OCD) and related illnesses, but it is unclear whether this hyperactivity is due to intrinsic striatal dysfunction or abnormalities in corticostriatal inputs. Understanding the cellular and circuit properties underlying striatal hyperactivity could help inform the optimization of targeted stimulation treatments for compulsive behavior disorders. To investigate the cellular and synaptic abnormalities that may underlie corticostriatal dysfunction relevant to OCD, we used the Sapap3 knock-out (Sapap3-KO) mouse model of compulsive behaviors, which also exhibits hyperactivity in central striatum. Ex vivo electrophysiology in double-transgenic mice was used to assess intrinsic excitability and functional synaptic input in spiny projection neurons (SPNs) and fast-spiking interneurons (FSIs) in central striatum of Sapap3-KOs and wild-type (WT) littermates. While we found no differences in intrinsic excitability of SPNs or FSIs between Sapap3-KOs and WTs, excitatory drive to FSIs was significantly increased in KOs. Contrary to predictions, lateral orbitofrontal cortex-striatal synapses were not responsible for this increased drive; optogenetic stimulation revealed that lateral orbitofrontal cortex input to SPNs was reduced in KOs (∼3-fold) and unchanged in FSIs. However, secondary motor area (M2) postsynaptic responses in central striatum were significantly increased (∼6-fold) in strength and reliability in KOs relative to WTs. These results suggest that increased M2-striatal drive may contribute to both in vivo striatal hyperactivity and compulsive behaviors, and support a potential role for presupplementary/supplementary motor cortical regions in the pathology and treatment of compulsive behavior disorders.
SIGNIFICANCE STATEMENT These findings highlight an unexpected contribution of M2 projections to striatal dysfunction in the Sapap3-KO obsessive-compulsive disorder (OCD)-relevant mouse model, with M2 inputs strengthened by at least sixfold onto both spiny projection neurons and fast-spiking interneurons in central striatum. Because M2 is thought to be homologous to presupplementary/supplementary motor areas (pre-SMA/SMA) in humans, regions important for movement preparation and behavioral sequencing, these data are consistent with a model in which increased drive from M2 leads to excessive selection of sequenced motor patterns. Together with observations of hyperactivity in pre-SMA/SMA in both OCD and Tourette syndrome, and evidence that pre-SMA is a potential target for repetitive transcranial magnetic stimulation treatment in OCD, these results support further dissection of the role of M2 in compulsivity.
Keywords: central striatum, motor sequences, OCD, orbitofrontal cortex, SAPAP3, secondary motor cortex
Introduction
Although stereotyped and compulsive behaviors are prominent, disabling, and notoriously treatment-resistant, symptoms in multiple severe neuropsychiatric disorders, including Tourette syndrome (TS) (Leckman et al., 2010), grooming disorders (e.g., skin-picking, trichotillomania) (Chamberlain et al., 2009; Flessner et al., 2012), and obsessive-compulsive disorder (OCD) (Karno et al., 1988; Ayuso-Mateos, 2006; Menzies et al., 2008), little is known about their underlying neural mechanisms. Imaging studies in patients with OCD and other compulsivity-associated disorders have consistently identified both hyperactivity in the striatum (caudate head) and increased corticostriatal functional connectivity at baseline and when symptoms are expressed (Rauch et al., 1994, 1997; Saxena et al., 1998; Maia et al., 2008; Menzies et al., 2008; Chamberlain et al., 2009; Harrison et al., 2009; Leckman et al., 2010; Del Casale et al., 2011; Denys et al., 2013; Figee et al., 2013). However, the cellular and synaptic abnormalities that underlie this hyperactivity are unclear.
Determining whether striatal hyperactivity originates in striatum or in upstream cortical projections could help inform whether neuromodulatory treatments for OCD-related disorders should target cortical or subcortical regions. Although striatal deep brain stimulation has reported efficacy in OCD (Greenberg et al., 2010; Figee et al., 2013), studies demonstrating aberrant activity in corticostriatal circuits in OCD patients (Chamberlain et al., 2008; Menzies et al., 2008; Harrison et al., 2009; Del Casale et al., 2011; Figee et al., 2013; Nakamae et al., 2014) suggest that cortical regions may be useful targets for noninvasive neurostimulation via repetitive transcranial magnetic stimulation (rTMS), either through direct cortical effects or modulation of connected subcortical structures. Consistent with this idea, orbitofrontal cortex (OFC) and presupplementary motor area (pre-SMA), which are hyperactive in OCD patients (Maltby et al., 2005; Yücel et al., 2007; Leckman et al., 2010; Del Casale et al., 2011; de Wit et al., 2012; Grützmann et al., 2016), have been identified as promising targets for rTMS (Berlim et al., 2013). Determining how these hyperactive cortical regions interact with the striatum to generate both dysfunctional striatal activity and compulsive behaviors could help refine stimulation patterning for neuromodulatory treatments.
To begin to dissect the contributions of intrinsic striatal versus extrinsic cortical factors to abnormal OCD-relevant striatal activity, we used an animal model system that displays both striatal hyperactivity and compulsive behavior: Sapap3-KO mice (Welch et al., 2007; Burguière et al., 2013). Mutations in the Sapap3 gene, which encodes the synapse-associated protein 90/postsynaptic density-95-associated protein, have been associated with TS, pathological grooming disorders, and OCD in humans (Bienvenu et al., 2009; Züchner et al., 2009; Crane et al., 2011). Sapap3-KOs demonstrate ex vivo abnormalities in striatal spiny projection neurons (SPNs), including both increased NMDA-receptor mediated and reduced AMPA-receptor mediated transmission in dorsal striatum (Welch et al., 2007; Wan et al., 2011, 2014; Ade et al., 2016). Furthermore, consistent with human OCD literature, they also exhibit hyperactivity in central striatum at baseline and during compulsive grooming (Burguière et al., 2013). This hyperactivity is relieved by stimulation of lateral OFC (LOFC) inputs into central striatum, suggesting dysregulation of LOFC corticostriatal inputs to SPNs or fast-spiking interneurons (FSIs). However, the contribution of corticostriatal inputs from M2 to striatal hyperactivity in Sapap3-KOs has been unexplored, despite the fact that M2 is the mouse homolog of pre-SMA, a promising treatment target.
To investigate cellular and synaptic abnormalities that could contribute to pathologic striatal hyperactivity and resulting compulsive behaviors, we used acute slice electrophysiology to measure intrinsic excitability of SPNs and FSIs and characterize excitatory cortical inputs to central striatum in Sapap3-KOs and WT littermate controls. Although the intrinsic excitability of central striatal neurons was normal in Sapap3-KOs, we found substantial differences in functional cortical innervation. While LOFC input to SPNs was weakened in Sapap3-KOs, strength and reliability of M2 input to central striatum were substantially increased. These results are the first demonstration of upregulated M2 corticostriatal input in an OCD-relevant mouse model and highlight the potential role of pre-SMA/SMA in the pathology of compulsivity.
Materials and Methods
Animals.
Male and female Sapap3-KOs and WT littermates were maintained on C57BL/6 background and were derived from a colony initially established at Massachusetts Institute of Technology by Dr. Guoping Feng. For identification of FSIs, Sapap3-heterozygous (Sapap3-het) mice were bred with Sapap3-het::parvalbumin (PV)-cre mice to generate Sapap3-KO and WT littermates that were PV-cre hemizygous (see Fig. 1). PV-cre mice were derived from a mouse knockin of Cre recombinase directed by the PV promotor/enhancer (Pvalbtm1(cre)Arbr, The Jackson Laboratory; RRID:IMSR_JAX:017320). Mice were group housed with 2–5 mice per cage and ad libitum access to food and water. Mice underwent stereotaxic surgeries at postnatal days 35–39 or days 46–50 (p35-p39 or p46-p50). All experiments were approved by the Institutional Animal Use and Care Committee at the University of Pittsburgh in compliance with National Institutes of Health guidelines for the care and use of laboratory animals.
Figure 1.

Central striatum cell types were interrogated using ex vivo electrophysiology in double-transgenic Sapap3-KO//PV-cre mice. A, Double-transgenic mice were generated to investigate functional properties of FSIs. Sapap3-KO mice were bred with PV-cre mice to generate mice with one Sapap3-KO allele and one PV-cre allele (Sapap3-het//PV-Cre). Sapap3-het//PV-Cre mice were then used as breeders to generate Sapap3-KO//PV-cre and Sapap3-WT//PV-cre progeny. B, Top, AAV5-DIO-mCherry was injected in central striatum; resulting infection of cre-positive PV cells led to fluorescent labeling for targeted slice electrophysiology recordings. Scale bars: Inset top, 200 μm; Inset bottom, 10 μm. Bottom, Examples of evoked spiking traces in KO SPNs and FSIs (right). Calibration: 50 ms, 10 mV. C, No differences were observed between SPN I-O curves in Sapap3-KOs (4 animals, 25 cells) and WTs (4 animals, 14 cells). D, No differences were observed between FSI I-O curves in Sapap3-KOs (3 animals, 23 cells) and WTs (5 animals, 18 cells). E, Tissue sections from Sapap3-KO (N = 5) and WT (N = 8) mice were immunohistochemically stained for PV to perform FSI cell counts in central striatum (box). Scale bar, 500 μm. Inset, There was no difference observed in the number of FSIs in WTs versus KOs (rank sum = 44, p = 0.21, WRST).
Grooming assessments were conducted in a separate cohort of animals that had not undergone stereotaxic surgery, by placing Sapap3-KO and WT littermates individually into a 10 inch square Plexiglas chamber and video-recording behavior for 20 min. Grooming behavior was manually scored offline.
Stereotaxic surgeries.
Stereotaxic surgeries were performed under isoflurane anesthesia (2%). Burr holes were drilled over the target location, and virus was injected using either a Nanoject (Drummond Scientific) and glass pulled pipette or a syringe pump (Harvard Scientific) fitted with a syringe (Hamilton) connected to PE10 tubing and a 30 gauge cannula.
Viral injections were performed at p35-p39 and allowed to incubate for 3 weeks for optogenetic slice electrophysiology. Channelrhodopsin 2 (AAV2-hsyn-ChR2-eYFP or AAV2-hsyn-ChR2-mCherry, University of North Carolina Vector Core Facility, virus titer 3.1 × 1012) was injected unilaterally into either LOFC (anteroposterior 2.78, mediolateral 1.54, dorsoventral 1.65 mm; all coordinates from bregma and brain surface) or M2 (anteroposterior 2.74, mediolateral 1.54, dorsoventral 0.75 mm). Sapap3::PV-cre mice were injected with cre-dependent AAV5-DIO-mCherry into central striatum (anteroposterior 0.65, mediolateral 1.90, dorsoventral 3.00 mm) to target PV-positive interneurons.
Fluoro-Gold (2% in cacodylate buffer; Fluorochrome) iontophoresis was performed in central striatum (anteroposterior 0.65, mediolateral 1.90, dorsoventral 2.85 mm) at p46-p50 for retrograde anatomical tracing. Iontophoretic injections were conducted with 5 mA of current (7 s on, 7 s off) for 5–8 min. Animals were transcardially perfused 10 d after surgery.
Slice electrophysiology.
Coronal slices containing striatum (300 μm) were prepared using a VT1000S vibratome (Leica Microsystems) from brains of 8-week-old mice that had received ChR2 viral injections 3 weeks prior. Brains were sliced for recording with experimenter blind to genotype. Slices were cut in carbogenated HEPES ACSF containing the following (in mm): 20 HEPES, 92 NaCl, 1.2 NaHCO3, 2.5 KCl, 10 MgSO4, 0.5 CaCl2, 30 NaH2PO4, 25 glucose, 5 sodium ascorbate, 2 thiourea, and 3 sodium pyruvate, pH 7.25. Slices were allowed to recover for 15 min at 33°C in a chamber filled with N-methyl-D-glucamine-HEPES recovery solution (in mm): 93 N-methyl-D-glucamine, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 10 MgSO4, 0.5 CaCl2, 5 sodium ascorbate, 2 thiourea, and 3 sodium pyruvate. Slices were then held at room temperature for at least 1 h before recording in a holding solution that was similar to the HEPES cutting solution but with 1 mm MgSO4 and 2 mm CaCl2.
Recordings were conducted at 33°C in carbogenated ACSF (in mm) as follows: 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 12.5 glucose, 1 MgSO4, and 2 CaCl2. Picrotoxin (50 μm) was included in the ACSF to block all striatal local inhibitory activity, and DNQX (5 μm) was included in the ACSF for intrinsic firing rate data (see Fig. 1) and NMDA-mediated current recordings (see Fig. 6). For asynchronous release experiments, 2 mm CaCl2 was replaced with 2 mm SrCl2.
Figure 6.

Increased M2 input in central striatum is accompanied by increased NMDA currents. A, B, After recording M2 light-evoked AMPA-mediated currents at Vhold = −80 mV, cells were held at Vhold = 40 mV to record NMDA-mediated currents. Amplitude of NMDA-mediated current was measured 60 ms after the peak of the response, at which time the AMPA-mediated portion of the current has decayed. Gray represents WT. Pink represents KO. C, AMPA/NMDA ratios in SPNs were significantly lower in KOs relative to WTs (WT 4.09 ± 4.36, KO 1.94, IQR = 2.22; Z = 3.29, p = 9.89 × 10−4, WRST). D, AMPA/NMDA ratios were not significantly different between genotypes in FSIs (WT 16.67, IQR = 14.28, KO 11.79, IQR = 14.14; Z = 0.86, p = 0.39, WRST). E, F, Examples of isolated NMDA responses with pharmacological block of AMPA receptors with 5 μm DNQX. Gray represents WT. Pink represents KO. G, NMDA-mediated currents were significantly greater in SPNs in KO mice (174.77, IQR = 268.43 pA, 3 mice, 18 cells) relative to WTs (95.99 pA, IQR = 111.69 pA, 5 mice, 19 cells; Z = −2.23, p = 0.02, WRST). H, NMDA-mediated currents were significantly greater in FSIs in KO mice (108.61 pA, IQR = 79.91 pA, 3 mice, 16 cells) relative to WT mice (27.56, IQR = 75.81 pA, 4 mice, 11 cells; Z = −2.25, p = 0.02, WRST).
Data were collected with a MultiClamp 700B amplifier (Molecular Devices) and ITC-18 analog-to-digital board (HEKA) using Igor Pro software (Wavemetrics, RRID:SCR_000325) and custom acquisition routines (Recording Artist; Richard C. Gerkin, Phoenix). Current-clamp recordings were filtered at 10 kHz and digitized at 40 kHz; voltage-clamp recordings were filtered at 2 kHz and digitized at 10 kHz. Electrodes were made from borosilicate glass (pipette resistance, 2–6 mΩ). The internal solution for voltage-clamp recordings consisted of the following (in mm): 120 CsMeSO3, 15 CsCl, 8 NaCl, 0.5 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, and 5 QX-314. The internal solution for current-clamp recordings consisted of the following (in mm): 130 KMeSO3, 10 NaCl, 2 MgCl2, 0.16 CaCl2, 0.5 EGTA, 10 HEPES, 2 Mg-ATP, and 0.3 NaGTP.
Electrically evoked synaptic responses were elicited with 0.1 ms current pulses (50 ms or 100 ms interpulse interval [IPI]) passed through a glass stimulating electrode placed in striatum. Optogenetically evoked synaptic responses were elicited with two 1 ms pulses of light (100 ms IPI) filtered at 470 nm, delivered through the 60× objective of the rig microscope. Maximum responses were obtained by turning the LED to maximum power (1 mW). Trials were conducted 20 s apart, except for asynchronous EPSC (aEPSC) trials, which were conducted 12 s apart.
Histology.
Counts of PV-positive interneurons (FSIs) were conducted by staining 35 μm tissue sections containing central striatum with rabbit anti-PV (1:3000, overnight 4°C incubation, Swant, RRID:AB_10000343) and AlexaFluor-488 anti-rabbit (1:500, 3 h room temperature incubation, Abcam). Sections were counterstained with Hoechst (1:1000) to confirm cell bodies. The number of PV-positive cells in a 600-μm-square region over central striatum was counted and summed across six sections for each animal. Central striatum region was chosen based on known location of LOFC/M2 projection field overlap (anteroposterior 0.02–0.98 mm, mediolateral 1.90 mm, dorsoventral 2.85 mm).
Retrograde tracing experiments were analyzed by quantifying the number of Fluoro-Gold-labeled cell bodies in regions of the PFC delineated by the Paxinos Brain Atlas (Paxinos and Franklin, 2004). Sections (35 μm) were taken 105 μm apart and underwent a nuclear stain before imaging (NeuroTrace Nissl 640/660, Thermo Fisher Scientific). Sections containing central striatum (0.85–0.25 mm anteroposterior) were examined to ensure appropriate targeting and spread of Fluoro-Gold before proceeding with cell counting. For each animal, images of PFC sections (4–6 sections per animal, ∼3.08–2.34 mm anteroposterior) were imaged using the same parameters (20× magnification, 600 ms exposure). To normalize differences in Fluoro-Gold brightness and background in different animals, images were then manually thresholded so that the signal:background ratios were similar across animals. Cells were then automatically detected and counted in each ROI (Olympus CellSens, RRID:SCR_016238). Proportions of labeled cells in each region were determined by dividing each regional sum by the sum of the total number of cells detected in each animal (ipsilateral and contralateral hemispheres). Proportions were compared between genotype and region using a two-way repeated-measures ANOVA and post hoc contrasts.
Experimental design and statistical analysis.
Acute slice electrophysiology experiments were designed to include approximately equal numbers of Sapap3-KOs and WTs, taking into account animal availability. Animal and cell numbers for each experiment are reported in the corresponding figure legends. Approximately equal numbers of male and female mice were used. Recordings were obtained from 2 to 4 striatal slices per animal. All data were analyzed with genotype blind to the experimenter.
Firing rate (see Fig. 1) was calculated by counting the number of action potentials evoked with a 500 ms current step, and multiplied by 2 to obtain the spikes/s. All synaptic response amplitudes were calculated by finding the average amplitude of evoked EPSCs in five consecutive trials. Strontium asynchronous release events were manually detected within 500 ms after the light presentation, and the peak of the event was calculated and averaged over 20–160 events per cell. NMDA-mediated currents were calculated in two ways. First, cells were held at 40 mV, and the amplitude was found at 60 ms after the peak response. The AMPA/NMDA ratio was then calculated by dividing the average AMPA amplitude by the average NMDA amplitude. Second, in DNQX experiments, the peak of the NMDA-mediated current at Vhold = 40 mV was calculated and averaged across 5 trials.
Because data were not normally distributed, statistical differences between two groups of values were determined using Wilcoxon Rank Sum Tests (WRST) (see Figs. 1–6). To statistically compare input–output (I-O) curves across multiple stimulation currents (see Fig. 2), a two-way repeated-measures ANOVA was used because an appropriate nonparametric test could not be found. All values reported are median ± interquartile range (IQR) unless otherwise noted.
Figure 2.
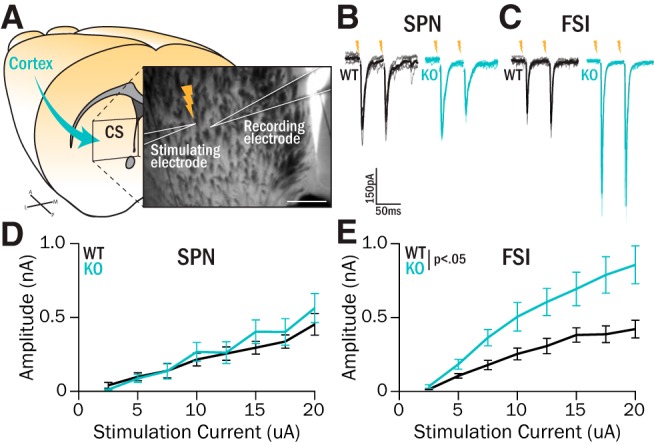
Excitatory drive to central striatum FSIs is increased in Sapap3-KOs. A, Nonspecific excitatory inputs from cortex were activated using intrastriatal stimulation while recording nearby cells in central striatum. Scale bar, 200 μm. B, C, Synaptic responses were evoked by passing current pulses (0.1 ms) through the stimulating electrode during whole-cell recordings of SPNs (B) or FSIs (C) in Sapap3-KOs and WT littermates. D, Evoked response I-O curves in SPNs were not different in KOs versus WTs (WTs: 8 animals, 14 cells; KOs: 6 animals, 12 cells; F(1) = 0.016, p = 0.90, ANOVA). At maximum stimulation intensity (20 μA), EPSC sizes were not different between WTs and KOs (Z = −0.75, p = 0.45, WRST). E, Evoked responses in FSIs were significantly greater in KOs relative to WTs (WTs: 6 animals, 13 cells; KOs: 6 animals, 12 cells; genotype, F(1) = 8.11, p = 0.014; interaction, F(7) = 4.05, p = 0.023, ANOVA). At maximum stimulation intensity (20 μA), KOs displayed significantly larger EPSCs (rank sum = 57, p = 0.0057, WRST).
Results
Intrinsic excitability of central striatal neurons is not different in Sapap3-KOs and WTs
Hyperactivity in central striatum SPNs has been implicated in the compulsive grooming phenotype observed in Sapap3-KOs (Burguière et al., 2013). Increased SPN activity could result from dysfunction at the level of striatum, including increased SPN intrinsic excitability or reduced FSI inhibitory activity. Because FSIs make up only 1% of the striatal cell population (Luk and Sadikot, 2001; Berke, 2011), we used a transgenic fluorescent strategy to target these neurons using acute slice electrophysiology. PV-Cre transgenic mice were bred with Sapap3-KOs to generate Sapap3-KO//PV-Cre and Sapap3-WT//PV-Cre offspring (Fig. 1A). Striatal FSIs were then labeled by injecting the PV-Cre transgenic mice with a cre-dependent fluorescent mCherry virus (Fig. 1B). Viral injections were conducted when mice were postnatal day 35–39 (p35-p39), and recordings were conducted at p56-p60. At this age, mice exhibit the overgrooming phenotype that is characteristic of the Sapap3-KO model (WT mean = 64.17, SEM = 9.01 s; KO mean = 204.35, SEM = 23.22 s; t(34) = −5.23, p = 8.53 × 10−6).
We first found that there were no genotype differences in SPN intrinsic excitability, as assessed by firing rate I-O curves (Fig. 1C). The slopes of the linear portion of the I-O curve were not significantly different between WTs and KOs (Table 1). Additionally, there were no significant differences in other SPN intrinsic properties, such as input resistance and resting potential (Table 1). This indicates that increased SPN intrinsic excitability does not contribute to in vivo hyperactivity in SPNs.
Table 1.
Intrinsic properties of FSIs and SPNs in Sapap3-KOs and WT littermatesa
| Median, IQR | FSIs |
SPNs |
||||
|---|---|---|---|---|---|---|
| WT | KO | p | WT | KO | p | |
| N (animals, cells) | 5, 18 | 3, 25 | 4, 14 | 4, 25 | ||
| Resting potential (mV) | −76.03, 9.03 | −81.17, 9.33 | 0.08 | −82.98, 13.67 | −86.18, 7.58 | 0.33 |
| Rheobase (pA) | 250, 150 | 300, 150 | 0.79 | 325, 150 | 225, 100 | 0.14 |
| Maximum firing rate (Hz) | 209, 124 | 250, 96 | 0.82 | 50, 9.50 | 44, 22 | 0.39 |
| I-O curve linear slope | 0.64, 0.64 | 0.56, 0.64 | 0.87 | 0.17, 0.11 | 0.22, 0.18 | 0.31 |
| Input resistance (mΩ) | 159.12,70.72 | 184.61, 108.91 | 0.12 | 299.80, 162.38 | 298.28, 120.76 | 0.70 |
aWT and KO groups were compared using WRST. No significant differences were observed.
Hyperactivity in SPNs could also be caused by reduced activity of FSIs, which could stem from either decreased FSI intrinsic excitability or fewer FSIs. However, there were no detectable genotype differences in intrinsic excitability of FSIs as assessed by firing rate I-O curves (Fig. 1D), I-O curve linear slope, or any other intrinsic properties (Table 1). To investigate whether Sapap3-KO mice have fewer FSIs in central striatum, we counted PV-immunoreactive cells in tissue sections from Sapap3-KO mice and WT littermates. Summing across six sections for each animal, the number of FSIs was not significantly different in Sapap3-KOs and WT littermates (p = 0.21, WRST; Fig. 1E, inset), suggesting that a reduction in the number of FSIs does not contribute to central striatal dysfunction.
Excitatory drive to FSIs in central striatum is increased in Sapap3-KOs
Next, we investigated whether the observed hyperactivity of SPNs in Sapap3-KO mice (Burguière et al., 2013) reflected differences in excitatory inputs to central striatum, which could augment SPN activity directly or via decreasing feedforward inhibition mediated by FSIs (Parthasarathy and Graybiel, 1997; Mallet et al., 2005; Gittis et al., 2010, 2011; Berke, 2011). To assess excitatory drive onto both FSIs and SPNs, we performed intrastriatal electrical stimulation in acute slices (Fig. 2A) and recorded EPSC responses (Fig. 2B,C). Electrical stimulation elicited robust EPSCs whose amplitudes increased with stimulus intensity in both SPNs and FSIs (Fig. 2D,E). In SPNs, EPSC amplitudes increased with similar slopes in WT and KO mice as a function of stimulation intensity (p = 0.90, ANOVA; Fig. 2D); at a maximum stimulation intensity of 20 μA, the average EPSC amplitude was 453 pA (SEM 74 pA) in WTs and 564 pA (SEM 99 pA) in KOs (p = 0.45, WRST). By contrast, evoked EPSCs in FSIs were consistently larger in KOs compared with WTs (genotype: p < 0.05; interaction: p < 0.05, ANOVA; Fig. 2E); at a maximum stimulus intensity of 20 μA, average EPSC amplitude was 422 pA (SEM 59 pA) in WTs and 857 pA (SEM 128 pA) in KOs (p < 0.01, WRST). No differences in paired-pulse ratio (PPR) or EPSC decay kinetics were observed in either cell type (Table 2).
Table 2.
PPR and decay constant obtained from all stimulation experiments in Sapap3-KOs and WT littermatesa
| Median, IQR | FSI |
SPN |
||||
|---|---|---|---|---|---|---|
| WT | KO | p | WT | KO | p | |
| Electrical stimulation | ||||||
| PPR 50 ms | 1.24, 0.32 | 1.10, 0.17 | 0.83 | 0.77, 0.16 | 0.86, 0.24 | 0.53 |
| N (animals, cells) | 3, 8 | 3, 3 | 5, 7 | 3, 6 | ||
| PPR 100 ms | 0.99, 0.25 | 1.15, 0.07 | 0.19 | 0.84, 0.38 | 0.86, 0.18 | 0.95 |
| N (animals, cells) | 3, 5 | 3, 9 | 5, 7 | 3, 6 | ||
| Decay constant (ms) | 3.77, 1.34 | 3.18, 0.94 | 0.07 | 6.49, 1.50 | 6.19, 2.14 | 0.25 |
| N (animals, cells) | 5, 12 | 5, 12 | 8, 13 | 6, 12 | ||
| LOFC stimulation | ||||||
| PPR | 0.36, 0.23 | 0.31, 0.19 | 0.39 | 0.27, 0.21 | 0.29, 0.11 | 0.90 |
| N (animals, cells) | 10, 21 | 5, 10 | 12, 33 | 5, 15 | ||
| Decay constant (ms) | 4.17, 1.71 | 4.63, 1.61 | 0.29 | 9.33, 5.70 | 7.10, 1.81 | 0.12 |
| N (animals, cells) | 10, 19 | 5, 9 | 12, 32 | 5, 10 | ||
| M2 stimulation | ||||||
| PPR | 0.40, 0.37 | 0.43, 0.28 | 0.37 | 0.32, 0.11 | 0.33, 0.27 | 0.71 |
| N (animals, cells) | 13, 30 | 7, 23 | 10, 21 | 7, 22 | ||
| Decay constant (ms) | 4.08, 1.72 | 4.02, 2.26 | 0.72 | 6.70, 2.84 | 6.89, 2.71 | 0.95 |
| N (animals, cells) | 11, 16 | 7, 22 | 8, 14 | 7, 17 | ||
aMedians and IQR reported for each value. WT and KO groups were compared using WRST. p values are reported for each comparison. No significant differences were observed.
M2 and LOFC are the main sources of cortical input to central striatum
Our synaptic data indicating that FSIs receive more excitatory drive in Sapap3-KO mice were surprising based on a previous in vivo study demonstrating overactive SPNs in KOs (Burguière et al., 2013). This motivated closer examination of the possible cortical sources of excitatory drive to the central striatum. We therefore iontophoretically infused the retrograde tracer Fluoro-Gold (Fluorochrome) into central striatum of Sapap3-KOs and WTs (Fig. 3A). To ensure consistency with slice electrophysiology data, mice were killed at p56-p60 after a 10 d incubation period. Only animals with confirmed injection sites in central striatum were used for analysis (Fig. 3B). Retrogradely labeled cell bodies were observed in the ipsilateral frontal cortices (Fig. 3C) and, to a lesser extent, in the contralateral frontal cortices (data not shown). Although we did not observe a strong laminar pattern, labeled cells were present in layer V, as anticipated based on past work (Levesque et al., 1996). To quantify the origins of these cortical-central striatum projections, the number of Fluoro-Gold-labeled cells was counted in the five cortical regions where labeling was observed, and the regional proportion of total labeled cells was calculated (Fig. 3C,D). In both WT and KO mice, many neurons projecting to central striatum were located in LOFC. However, unexpectedly, the highest proportion of labeled cells was located in the region directly dorsal to LOFC, M2 (Fig. 3C). Smaller numbers of neurons were also observed in medial orbitofrontal cortex (MOFC), prelimbic cortex (PL), and dorsolateral orbitofrontal cortex (DLOFC). There were no genotype differences in proportions of retrogradely labeled cells in any of these cortical regions (p = 0.10, ANOVA, Fig. 3D).
Figure 3.
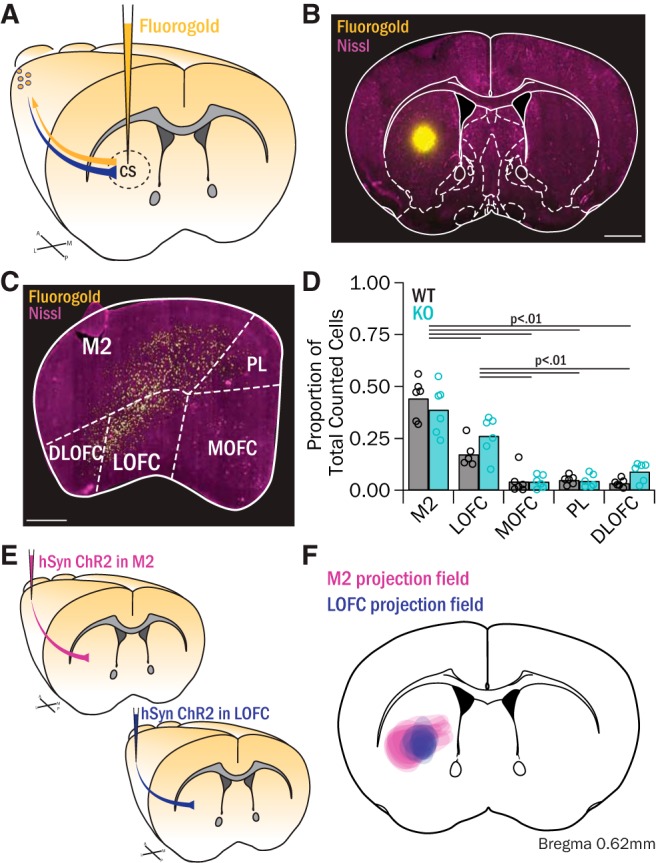
Retrograde tracing shows that M2 and LOFC project to central striatum. A, Fluoro-Gold was injected into central striatum of Sapap3-KO (n = 6) and WT (n = 6) mice to retrogradely label cortical projection cells. B, Example of a Fluoro-Gold injection into central striatum. Scale bar, 1 mm. C, Labeled cortical cells were present throughout frontal cortex in M2, LOFC, MOFC, PL, and DLOFC. Scale bar, 500 μm. D, No genotype difference in counted cells was observed (F(1) = 3.22, p = 0.10, ANOVA). However, there were differences in the proportions of labeled cells counted across regions (F(1) = 60.19, p = 0.000, ANOVA). Ipsilateral M2 showed the highest proportion of labeled cells (WT: 44.3 ± 3.9%; KO: 38.9 ± 4.8%), which was significantly greater than the proportions in LOFC (p < 0.05), MOFC (p < 0.001), PL (p < 0.001), and DLOFC (p < 0.001, post hoc contrasts). LOFC contained a significantly higher proportion of labeled cells (WT: 17.3 ± 2.4%; KO: 26.3 ± 3.9%, p < 0.001) than MOFC (WT: 4.2 ± 2.4%; KO: 4.1 ± 1.3%, p < 0.001), PL (WT: 5.0 ± 0.8%; KO: 4.5 ± 1.3%, p < 0.001), and DLOFC (WT: 3.5 ± 0.8%; KO: 9.0 ± 1.8%; p < 0.001, post hoc contrasts). Data are reported as mean ± SEM. E, AAV2-hSyn-ChR2-EYFP was injected into M2 (5 WTs, 2 KOs) or LOFC (8 WTs, 3 KOs) to map anterograde projection fields in striatum. F, The fluorescence territories of terminals from M2 or LOFC were mapped onto a representative atlas image, demonstrating that M2 and LOFC have partially overlapping inputs in central striatum.
To further characterize the projections of LOFC and M2 to central striatum, the anterograde virus AAV2-hSyn-ChR2-EYFP was injected into either LOFC or M2 of Sapap3-KOs and WTs (Fig. 3E). The projection fields in central striatum were qualitatively assessed, and their coverage was mapped onto a representative atlas image (Fig. 3F, magenta: M2 projection fields; violet: LOFC projection fields). This assessment highlights that both M2 and LOFC send projections to central striatum, and thus may contribute to corticostriatal dysfunction underlying striatal hyperactivity in Sapap3-KOs.
LOFC input to central striatum is reduced in Sapap3-KO SPNs but unchanged in FSIs
Based on our anatomical results, we integrated ChR2 into our slice electrophysiology experiments to study region-specific inputs to central striatum. We first focused on LOFC projections, as this region has been implicated in compulsive behavior (Saxena et al., 1998; Maltby et al., 2005; Chamberlain et al., 2008; Burguière et al., 2013) and related behavioral constructs, including action selection, cognitive flexibility, and reversal learning (Schoenbaum et al., 2002; Takahashi et al., 2009; Sul et al., 2010; Dalton et al., 2016; Gremel et al., 2016). To selectively measure the strength of LOFC inputs onto FSIs and SPNs in KOs versus WTs, a pan-neuronal virus (AAV2-hSyn-ChR2-EYFP) was injected into the LOFC of Sapap3-KO/PV-Cre or WT/PV-Cre mice, and recordings were performed in central striatum after 3 weeks of virus incubation (p56-p60) (Fig. 4A,B). LOFC axon terminals in central striatum were stimulated with two brief pulses of light, and evoked EPSCs recorded in FSIs and SPNs were compared between Sapap3-KOs and WT littermates (Fig. 4C,D). Activation of LOFC terminals evoked large EPSCs in SPNs in WT mice (445 pA; IQR = 637 pA), but these responses were reduced ∼3.2-fold in KOs (138; IQR = 110 pA; p < 0.001, WRST; Fig. 4F). To address whether the decreased EPSC amplitudes in KO SPNs were due to a decrease in presynaptic release probability, we calculated PPR; no significant differences were observed (Table 2), suggesting a postsynaptic effect.
Figure 4.
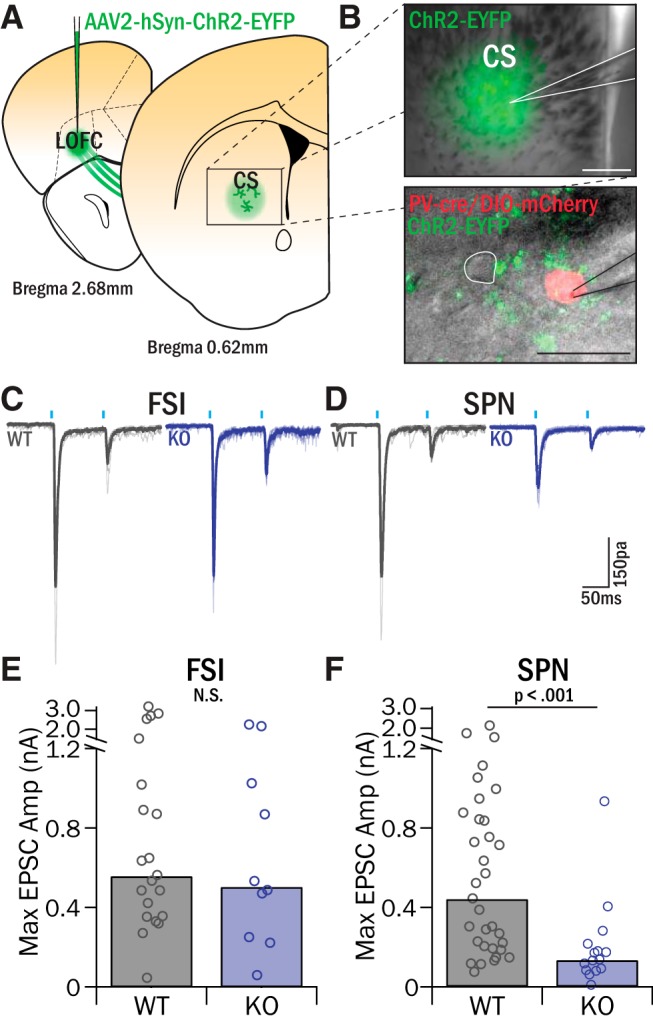
LOFC inputs to SPNs have reduced amplitude in Sapap3-KOs, whereas inputs to FSIs are unchanged. A, AAV2-hSyn-ChR2-EYFP was injected into LOFC to selectively identify LOFC projections in central striatum. B, Top, Recordings were targeted to the EYFP-labeled LOFC projection zone in central striatum. Scale bar, 200 μm. Bottom, Whole-cell patch voltage-clamp recordings (Vhold = −80 mV) were conducted in FSIs (identified by the presence of mCherry) and nearby SPNs (white outline). Scale bar, 25 μm. C, D, Brief pulses of light (1 mW, 10 ms pulse, 100 ms IPI) filtered at 470 nm and delivered through the 60× microscope objective-evoked synaptic release from ChR2-infected LOFC terminals, and the resulting EPSCs were recorded in FSIs (C) and SPNs (D) and compared between Sapap3-KOs and WT littermates. E, LOFC-evoked EPSCs in FSIs were no different in KOs and WTs (WT: 563.38 pA, IQR = 661.69 pA, 5 animals, 10 cells; KO: 509.81 pA, IQR = 680.86 pA, 10 animals, 21 cells; Z = 0.57, p = 0.57, WRST). F, SPNs in central striatum had significantly smaller LOFC-evoked EPSCs in KOs relative to WTs (WT: 444.84 pA, IQR = 637.43 pA, 12 animals, 33 cells; KO: 137.92 pA, IQR = 110.20 pA, 3 animals, 15 cells; Z = 3.43, p = 6.14 × 10−4, WRST).
In contrast to our findings in SPNs, WTs and KOs had similar EPSC amplitudes in FSIs following LOFC terminal stimulation. Maximally evoked EPSCs were 563 pA (IQR = 662 pA) in WT and 510 pA (IQR = 681 pA) in KOs (p = 0.57, WRST; Fig. 4E). These data reveal that LOFC input to central striatum is reduced onto SPNs, but not FSIs, in Sapap3-KOs. Given our findings of increased excitatory drive to FSIs using electrical stimulation, as well as no difference in overall excitatory drive to SPNs, these findings in LOFC suggested an alternative source of increased excitatory input to central striatum.
M2 input to central striatum is increased in Sapap3-KOs
Surprisingly, our anatomical tracing study demonstrated that the largest source of neurons projecting to central striatum was M2, rather than its neighboring cortical area, LOFC. Because M2 is associated with planning of sequenced movements (Sul et al., 2011; Cao et al., 2015; Li et al., 2015) and is thought to be homologous to pre-SMA (which is implicated in OCD) (Mantovani et al., 2010; de Wit et al., 2012), we investigated whether M2 was a source of upregulated excitatory drive to central striatum in Sapap3-KOs (Fig. 5A). Using optogenetics and acute slice physiology in WTs, we found that optical stimulation of M2 terminals in central striatum evoked EPSCs in FSIs and SPNs that were much smaller than those evoked by LOFC stimulation (FSIs: 84 pA, IQR = 158 pA; SPNs: 43 pA, IQR = 136 pA; both p < 0.0001, WRST; Fig. 5B–E). Furthermore, in a number of cells, EPSCs were either not reliably evoked by each laser pulse or were not evoked at all. To quantify this, each recorded cell was categorized as having reliability between 100% (5 of 5 responses) and 0% (0 of 5 responses). In WT mice, SPNs and FSIs had an average response reliability of 65.4% and 85.9%, respectively (Fig. 5F).
Figure 5.
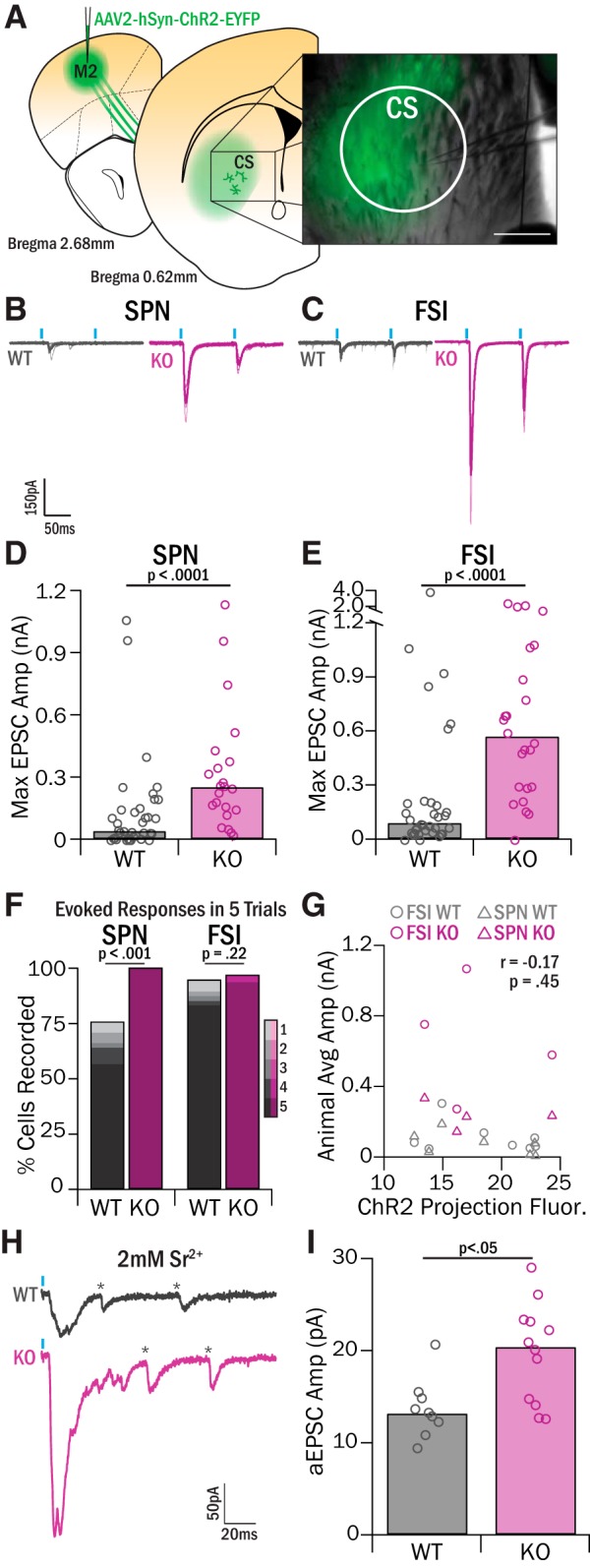
M2 input to CS is increased in Sapap3-KOs. A, AAV2-hSyn-ChR2-EYFP was injected into M2 to selectively label M2 projections in central striatum. Recordings were targeted to the central striatum region, which included the medial portion of the EYFP-labeled M2 projection zone. Scale bar, 200 μm. B, C, Brief pulses of 470 nm light (1 mW, 10 ms pulse, 100 ms IPI) evoked synaptic release from ChR2-infected M2 terminals, and the resulting EPSCs were recorded in SPNs (B) and FSIs (C) in Sapap3-KOs and WT littermates. D, SPNs in the central striatum of KOs had significantly greater EPSC amplitudes than WTs (WT: 42.58 pA, IQR = 136.36 pA, 15 animals, 37 cells; KO: 254.28 pA, IQR = 221.90 pA, 7 animals, 22 cells; Z = −3.87, p = 1.11 × 10−4, WRST). E, FSIs in central striatum of KOs had significantly greater EPSC amplitudes than WTs (WT: 85.81 pA, IQR = 158.33 pA, 15 animals, 34 cells; KO: 564.65 pA, IQR = 640.84 pA, 8 animals, 24 cells; Z = −4.22, p = 2.44 × 10−5, WRST). F, Response reliability for each cell was assessed by quantifying the number of optically evoked responses in five trials of stimulation. SPN mean response reliability was significantly increased in KOs relative to WTs (WT 65.41%, SEM = 7.17%; KO 100%; Z = −2.93, p = 0.0034, WRST). FSI mean response reliability was greater in KOs, but not significantly (WT 85.88%, SEM = 5.31%; KO 95, SEM = 4.21%; Z = −0.93, p = 0.22, WRST). G, Average fluorescence intensity of ChR2-infected M2 projections was calculated and correlated with the overall average amplitude of EPSCs recorded in FSIs (10 WTs, 4 KOs) and SPNs (9 WTs, 4 KOs) in a given animal. There was not a significant correlation (r = −0.17, p = 0.45, Pearson's R). H, Example traces of strontium aEPSC events in WT and KO SPNs. *Detected aEPSC events. I, SPNs in KO mice showed significantly greater aEPSCs relative to WT mice (WT = 13.25 pA, IQR = 2.27 pA, 2 mice, 9 cells; KO = 20.48 pA, IQR = 8.59 pA, 3 mice, 12 cells; Z = −2.38, p = 0.02, WRST).
In contrast, we found a dramatic increase in the strength of synaptic input from M2 onto both FSIs and SPNs in Sapap3-KOs. EPSC amplitudes were increased ∼6-fold in SPNs (254 pA, IQR = 222 pA, p < 0.001) and ∼6.6-fold in FSIs (565 pA, IQR = 641 pA, p < 0.0001, WRST; Fig. 5B–E). Sapap3-KOs also exhibited increased response reliability relative to WTs in SPNs (100%, p < 0.01, WRST) and FSIs (95.0%, p = 0.35, WRST; Fig. 5F). This general increase in synaptic strength was not due to a difference in viral expression levels because terminal fluorescence in central striatum was similar across genotypes, and we saw no correlation between EPSC amplitude and fluorescence intensity (Fig. 5G). Thus, unlike LOFC projections, which exhibited an SPN-specific decrease in Sapap3-KOs, M2 projections to central striatum were broadly increased in amplitude and reliability in KOs relative to WTs.
Increases in M2 input to central striatum are driven by postsynaptic changes
Upregulation in M2 drive of central striatum responses could reflect abnormalities in M2 cortical presynaptic terminals, or postsynaptic changes in striatal neurons. To evaluate whether presynaptic changes in M2-central striatum (CS) inputs were present, we calculated PPR. No genotype differences in PPR were observed in either FSIs or SPNs (Table 2), suggesting a postsynaptic alteration in glutamate receptor number or composition. The decay kinetics of EPSCs were also similar in WTs and KOs (Table 2), suggesting no change in subunit composition of AMPA receptors.
To investigate whether strengthened M2-CS synapses were a consequence of a postsynaptic alteration in the number of AMPA receptors, M2-evoked aEPSCs were recorded in SPNs in the presence of 2 mm strontium chloride and 0 mm calcium chloride (Fig. 5H) to measure the strength of quantal release events. aEPSC amplitudes were significantly greater in Sapap3-KOs (20.48 pA, IQR = 8.59 pA) relative to WTs (13.25 pA, IQR = 2.27 pA, p < 0.05, WRST; Fig. 5I), indicating that M2 synapses are strengthened in part by an upregulation in postsynaptic AMPA receptors.
Increases in NMDA-mediated currents are also present at M2 synapses
Sapap3-KO mice have been shown to have alterations in NMDA signaling that can impact synaptic strength, including differential expression of NMDA receptor subunits, increased NMDA-mediated currents in dorsolateral striatum, and reduced AMPA/NMDA ratios in dorsolateral striatum (Welch et al., 2007; Wan et al., 2011). We therefore also assessed AMPA/NMDA ratios at M2 synapses in central striatum. AMPA-mediated currents were measured at a holding potential of −80 mV; neurons were then voltage-clamped to 40 mV to remove the Mg2+ block from NMDA channels, and EPSCs were measured again (Fig. 6A,B). NMDA currents were estimated by taking the peak EPSC amplitude measured 60 ms after onset, a time when the majority of AMPA receptors have closed. In SPNs, AMPA/NMDA ratios were significantly lower in KOs (1.95, IQR = 2.22) relative to WTs (4.09, IQR = 4.34, p < 0.001, WRST; Fig. 6C). In contrast, there were no detectable differences in the FSI AMPA/NMDA ratio in KOs (11.79, IQR = 14.14) and WTs (16.67, IQR = 14.28, p = 0.39, WRST; Fig. 6D).
The fact that AMPA/NMDA ratios were decreased in KO SPNs despite the strong increase in AMPA currents (Fig. 5) suggested that NMDA currents are increased even more than AMPA currents. To better isolate NMDA currents by eliminating the possibility of AMPA current contamination, light-evoked currents were measured at Vhold = 40 mV in the presence of DNQX in a separate experiment (Fig. 6E,F). Increased NMDA-mediated currents in SPNs were observed in Sapap3-KOs (174.77 pA, IQR = 268.43 pA) relative to WTs (95.99, IQR = 111.69 pA, p < 0.05, WRST; Fig. 6G). These data are consistent with the idea that SPN NMDA currents are increased proportionately more than AMPA currents in KOs. We observed similar findings in FSIs: KOs had significantly greater NMDA-mediated currents (108.61, IQR = 79.91 pA) relative to WTs (27.56, IQR = 75.81 pA, p < 0.05, WRST; Fig. 6H). To account for the lack of change in FSI AMPA/NMDA ratio, this suggests that, in contrast to SPN findings, AMPA- and NMDA-mediated currents in FSIs are increased similarly in Sapap3-KOs. Together, these data suggest that strengthened M2 inputs in central striatum of Sapap3-KOs are caused by an increase in both AMPA and NMDA receptor-mediated currents at these synapses.
Discussion
Our data reveal an unexpected contribution of M2 projections to corticostriatal dysfunction in a transgenic mouse model that exhibits OCD-relevant compulsive behavior, Sapap3-KOs. We found no abnormalities in intrinsic striatal properties in KOs but found substantial alteration in cortical inputs to central striatum. First, we found a reduction of LOFC input specifically onto SPNs, in contrast to predictions that LOFC-SPN input would be upregulated. Even more strikingly, we found that M2 inputs are strengthened by at least ∼6-fold onto both SPNs and FSIs in central striatum (Fig. 7). These results suggest a model in which increased responsiveness of central striatum to input from M2 contributes to the generation of OCD-relevant striatal hyperactivity.
Figure 7.
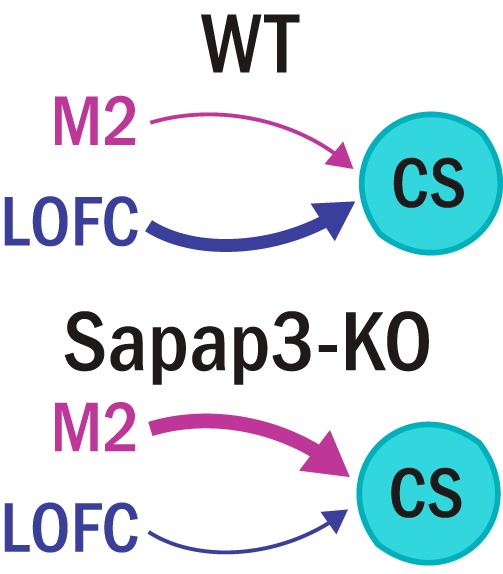
Ex vivo synaptic physiology suggests a model of imbalanced cortical input to central striatum. In WT mice, the prominent functional input to central striatal cells is LOFC, and cells receive weak input from M2. In Sapap3-KO mice, the normally strong input from LOFC is reduced, and M2 functional input is substantially increased.
These findings are the first demonstration of increased strength of M2-striatal circuits in an OCD-relevant mouse model. Similar to SMA/pre-SMA in humans and primates (Isoda and Hikosaka, 2007), M2 in mice is involved in behavioral planning and movement preparation during behavioral selection (Gremel and Costa, 2013; Guo et al., 2014; Cao et al., 2015; Li et al., 2015; Rothwell et al., 2015; Barthas and Kwan, 2017). Of particular interest, M2-striatal projections have been shown to be critical for initiation of behavioral sequences (Rothwell et al., 2015). Our data indicate that increased strength of M2-central striatum projections in Sapap3-KOs includes both increased amplitude and increased reliability of central striatal SPN responses to evoked M2 input. This heightened functional connectivity and increased response reliability could result in striatal neurons that are primed to respond to M2 activity, which in turn could increase the likelihood of initiating specific sequenced behaviors. Previous work has also shown that artificially increasing M2-dorsal striatum activity via ChR2 after a behavioral sequence was initiated led to decreased completion of the sequence (Rothwell et al., 2015). This evidence supports the hypothesis that abnormally strengthened M2 input to central striatum leads to more initiations and fewer completions of grooming behavior sequences in Sapap3-KOs.
The upregulation of M2 input to central striatum results from increased AMPA and NMDA currents, consistent with other ex vivo observations in striatum of Sapap3-KOs (Welch et al., 2007; Wan et al., 2011). Increased NMDA activity at M2 striatal synapses may lead to abnormal corticostriatal plasticity in an already strengthened projection. For instance, it has been shown that KOs have higher levels of the NR2B subunit in the striatum relative to WTs (Welch et al., 2007), and increased NR2B expression in hippocampus has been associated with enhanced LTP and heightened learning (Cao et al., 2007; Xu et al., 2009). This suggests that pathologically enhanced plasticity mechanisms may be present at M2 corticostriatal synapses, which could contribute to abnormal learning phenotypes (Burguière et al., 2013) and persistence of compulsive grooming behavior (Welch et al., 2007) observed in Sapap3-KOs.
Coincident with increased strength and reliability of M2-striatal input in Sapap3-KOs, we observed weakened LOFC input to central striatal SPNs that may also play a role in the generation of OCD-relevant compulsive behaviors. Generally, LOFC is known to support functions essential for flexible selection of behaviors, including reversal learning (Schoenbaum et al., 2002; Bohn et al., 2003; Sul et al., 2010). Consistent with this, Gremel et al. (2016) reported that activity in LOFC corticostriatal projections is essential for updating action-value associations (Gremel et al., 2016). These findings suggest that our observed downregulation of LOFC corticostriatal inputs could further bias Sapap3-KOs toward decreased flexible action selection and increased compulsivity and/or habit formation.
Together, this evidence supports a model in which normally balanced activity of M2 and LOFC is necessary to properly select goal-directed and habitual actions, and strengthened M2 drive and/or reduced LOFC input to central striatum could play a role in generating compulsive grooming behavior in Sapap3-KO mice. These findings highlight central striatum as a potential critical node for behavioral selection. We propose that mouse central striatum corresponds to the human caudate head because this region receives input from both LOFC and anterior pre-SMA in humans, but in-depth studies by neuroanatomists and in vivo functional assessments in rodents are needed to accurately define the human homolog of central striatum in rodents, including tracing studies in mice, rats, and nonhuman primates. In Sapap3-KO mice, the shift toward M2 motor control and away from LOFC associative control of central striatum may particularly interfere with flexible switching between habitual and goal-directed behaviors, which has been implicated in OCD symptomatology (Gillan and Robbins, 2014). For instance, reduced activity in LOFC corticostriatal terminals is necessary for habit expression, and habit expression is dependent on endocannabinoid-mediated LTD (Gremel et al., 2016); because both phenomena are enhanced in Sapap3-KOs (Chen et al., 2011), KOs could display enhanced habit expression.
Whereas the synaptic data presented here describe an intriguing shift to M2-cortical influence over central striatum in Sapap3-KOs, our ex vivo data do not directly explain baseline SPN hyperactivity in vivo. However, there are several possibilities for how the described circuit abnormalities may lead to increased SPN firing rates in vivo. While we observed no difference in overall presynaptic excitatory drive to SPNs in KOs versus WTs, we did observe a substantial increase in M2 reliability. This increased reliability may cause increased drive of SPNs that we cannot capture with ex vivo physiology, especially if M2 itself is hyperactive or hypersynchronous in vivo. In addition, SPN hyperactivity has been described during compulsive grooming as well as at baseline (Burguière et al., 2013). If M2-central striatum circuits are specifically engaged during sequenced behaviors, such as grooming, this could lead to behavior-specific generation of larger evoked postsynaptic responses, resulting in striatal hyperactivity. Increased drive to FSIs could also play a role in SPN hyperactivity. While FSIs are canonically thought to strongly inhibit SPN firing (Owen et al., 2018), other reports show that FSI firing can potentiate activity in a subset of SPNs (O'Hare et al., 2017). It has also been suggested that FSIs can tune SPN firing to task-relevant events (Lee et al., 2017; O'Hare et al., 2017; Owen et al., 2018). Increased FSI drive could therefore contribute to enhanced neural activity specifically during performance of sequenced motor behaviors, such as grooming in Sapap3-KOs. It is important to note that we did not measure FSI-SPN synapses in our study; changes in FSI-SPN synaptic strength in KOs could affect interpretation of our findings. Future studies investigating the in vivo activity of M2 and central striatal SPNs are needed to understand how our observed ex vivo dysfunction leads to compulsive behaviors.
Our findings of increased reliability of M2-central striatum synapses are intriguing in light of functional imaging studies that have identified hyperactivity in pre-SMA in OCD subjects relative to healthy controls (Yücel et al., 2007; de Wit et al., 2012; Grützmann et al., 2016). In addition, pre-SMA activity has been associated with the “urge” to move in both TS patients and healthy subjects (Bohlhalter et al., 2006; Neuner et al., 2014). Together with our findings, these studies suggest that hyperactivity in pre-SMA-striatal circuits could promote compulsive behaviors through increased responsiveness of striatal SPNs, and that decreasing pre-SMA activity through low-frequency rTMS could be therapeutic. However, it is important to note evidence suggesting that pre-SMA hyperactivity in OCD patients is compensatory, with higher pre-SMA activity correlating with lower Yale-Brown Obsessive-Compulsive Scale scores (van Velzen et al., 2014). This would suggest that the appropriate therapeutic intervention would be to use high-frequency rTMS to enhance compensation in OCD subjects whose pre-SMA activity is in the lower range. These discrepancies highlight the fact that the ideal treatment intervention would involve closed-loop technology that could tune neural activity up or down depending on specific context, task requirements, and individuals' symptom levels. However, it is important to cautiously interpret our results with respect to the human OCD literature. While we believe it is critical to further explore the role of M2-striatal circuits in compulsivity using preclinical models, our findings should be translated with caution, and should not be used as the basis for treatment recommendations.
Our data highlight M2-striatal pathology in an OCD-relevant mouse model and support a novel conceptual model in which Sapap3-KO mice display a shift toward increased motor control and decreased associative control over central striatum. In vivo investigations of these circuits will be essential to determine whether the observed alterations in synaptic weights result in an imbalance of corticostriatal activity that leads to aberrant behavioral selection, with a bias toward repeating sequenced motor programs associated with compulsive grooming. In addition, these findings support the potential clinical importance of further investigations of pre-SMA/SMA function in OCD patients. Our data raise the intriguing possibility that pre-SMA is a more important treatment target for decreasing compulsive behaviors in OCD and related disorders than previously suspected.
Footnotes
This work was supported by National Institute of Mental Health F31MH110125 to V.L.C., National Science Foundation DMS 1516288 and National Institutes of Health R00 NS076524 to A.H.G., and BRAINS R01MH104255, McKnight Neuroscience Scholar Award, MQ Fellows Award, Burroughs Wellcome Fund CAMS Award, and Klingenstein Fellowship in the Neurosciences to S.E.A. These data were previously presented in abstract/poster form.
The authors declare no competing financial interests.
References
- Ade KK, Wan Y, Hamann HC, O'Hare JK, Guo W, Quian A, Kumar S, Bhagat S, Rodriguiz RM, Wetsel WC, Conn PJ, Dzirasa K, Huber KM, Calakos N (2016) Increased metabotropic glutamate receptor 5 signaling underlies obsessive-compulsive disorder-like behavioral and striatal circuit abnormalities in mice. Biol Psychiatry 80:522–533. 10.1016/j.biopsych.2016.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso-Mateos JL. (2006) Global burden of obsessive-compulsive disorder in the year 2000. Geneva: World Health Organization. [Google Scholar]
- Barthas F, Kwan AC (2017) Secondary motor cortex: where 'sensory' meets 'motor' in the rodent frontal cortex. Trends Neurosci 40:181–193. 10.1016/j.tins.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke JD. (2011) Functional properties of striatal fast-spiking interneurons. Front Syst Neurosci 5:45. 10.3389/fnsys.2011.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlim MT, Neufeld NH, Van den Eynde F (2013) Repetitive transcranial magnetic stimulation (rTMS) for obsessive-compulsive disorder (OCD): an exploratory meta-analysis of randomized and sham-controlled trials. J Psychiatr Res 47:999–1006. 10.1016/j.jpsychires.2013.03.022 [DOI] [PubMed] [Google Scholar]
- Bienvenu O, Wang Y, Shugart Y, Welch J, Grados M, Fyer A, Rauch S, McCracken J, Rasmussen S, Murphy D (2009) Sapap3 and pathological grooming in humans: results from the OCD collaborative genetics study. Am J Med Genet B Neuropsychiatr Genet 150:710–720. 10.1002/ajmg.b.30897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlhalter S, Goldfine A, Matteson S, Garraux G, Hanakawa T, Kansaku K, Wurzman R, Hallett M (2006) Neural correlates of tic generation in Tourette syndrome: an event-related functional MRI study. Brain 129:2029–2037. 10.1093/brain/awl050 [DOI] [PubMed] [Google Scholar]
- Bohn I, Giertler C, Hauber W (2003) Orbital prefrontal cortex and guidance of instrumental behaviour in rats under reversal conditions. Behav Brain Res 143:49–56. 10.1016/S0166-4328(03)00008-1 [DOI] [PubMed] [Google Scholar]
- Burguière E, Monteiro P, Feng G, Graybiel AM (2013) Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science 340:1243–1246. 10.1126/science.1232380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao VY, Ye Y, Mastwal S, Ren M, Coon M, Liu Q, Costa RM, Wang KH (2015) Motor learning consolidates arc-expressing neuronal ensembles in secondary motor cortex. Neuron 86:1385–1392. 10.1016/j.neuron.2015.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Cui Z, Feng R, Tang YP, Qin Z, Mei B, Tsien JZ (2007) Maintenance of superior learning and memory function in NR2B transgenic mice during ageing. Eur J Neurosci 25:1815–1822. 10.1111/j.1460-9568.2007.05431.x [DOI] [PubMed] [Google Scholar]
- Chamberlain SR, Menzies L, Hampshire A, Suckling J, Fineberg NA, del Campo N, Aitken M, Craig K, Owen AM, Bullmore ET, Robbins TW, Sahakian BJ (2008) Orbitofrontal dysfunction in patients with obsessive-compulsive disorder and their unaffected relatives. Science 321:421–422. 10.1126/science.1154433 [DOI] [PubMed] [Google Scholar]
- Chamberlain SR, Odlaug BL, Boulougouris V, Fineberg NA, Grant JE (2009) Trichotillomania: neurobiology and treatment. Neurosci Biobehav Rev 33:831–842. 10.1016/j.neubiorev.2009.02.002 [DOI] [PubMed] [Google Scholar]
- Chen M, Wan Y, Ade K, Ting J, Feng G, Calakos N (2011) Sapap3 deletion anomalously activates short-term endocannabinoid-mediated synaptic plasticity. J Neurosci 31:9563–9573. 10.1523/JNEUROSCI.1701-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane J, Fagerness J, Osiecki L, Gunnell B, Stewart SE, Pauls DL, Scharf JM (2011) Family-based genetic association study of DLGAP3 in Tourette syndrome. Am J Med Genet B Neuropsychiatr Genet 156:108–114. 10.1002/ajmg.b.31134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GL, Wang NY, Phillips AG, Floresco SB (2016) Multifaceted contributions by different regions of the orbitofrontal and medial prefrontal cortex to probabilistic reversal learning. J Neurosci 36:1996–2006. 10.1523/JNEUROSCI.3366-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Casale A, Kotzalidis GD, Rapinesi C, Serata D, Ambrosi E, Simonetti A, Pompili M, Ferracuti S, Tatarelli R, Girardi P (2011) Functional neuroimaging in obsessive-compulsive disorder. Neuropsychobiology 64:61–85. 10.1159/000325223 [DOI] [PubMed] [Google Scholar]
- Denys D, de Vries F, Cath D, Figee M, Vulink N, Veltman DJ, van der Doef TF, Boellaard R, Westenberg H, van Balkom A (2013) Dopaminergic activity in Tourette syndrome and obsessive-compulsive disorder. Eur neuropsychopharmacol 23:1423–1431. 10.1016/j.euroneuro.2013.05.012 [DOI] [PubMed] [Google Scholar]
- de Wit SJ, de Vries FE, van der Werf YD, Cath DC, Heslenfeld DJ, Veltman EM, van Balkom AJ, Veltman DJ, van den Heuvel OA (2012) Presupplementary motor area hyperactivity during response inhibition: a candidate endophenotype of obsessive-compulsive disorder. Am J Psychiatry 169:1100–1108. 10.1176/appi.ajp.2012.12010073 [DOI] [PubMed] [Google Scholar]
- Figee M, Luigjes J, Smolders R, Valencia-Alfonso CE, van Wingen G de Kwaasteniet B, Mantione M, Ooms P, de Koning P, Vulink N, Levar N, Droge L, van den Munckhof P, Schuurman PR, Nederveen A, van den Brink W, Mazaheri A, Vink M, Denys D (2013) Deep brain stimulation restores frontostriatal network activity in obsessive-compulsive disorder. Nat Neurosci 16:386–387. 10.1038/nn.3344 [DOI] [PubMed] [Google Scholar]
- Flessner CA, Knopik VS, McGeary J (2012) Hair pulling disorder (trichotillomania): genes, neurobiology, and a model for understanding impulsivity and compulsivity. Psychiatry Res 199:151–158. 10.1016/j.psychres.2012.03.039 [DOI] [PubMed] [Google Scholar]
- Gillan CM, Robbins TW (2014) Goal-directed learning and obsessive-compulsive disorder. Philos Trans R Soc Lond B Biol Sci 369:20130475. 10.1098/rstb.2013.0475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC (2010) Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci 30:2223–2234. 10.1523/JNEUROSCI.4870-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Leventhal DK, Fensterheim BA, Pettibone JR, Berke JD, Kreitzer AC (2011) Selective inhibition of striatal fast-spiking interneurons causes dyskinesias. J Neurosci 31:15727–15731. 10.1523/JNEUROSCI.3875-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg BD, Gabriels LA, Malone DA Jr, Rezai AR, Friehs GM, Okun MS, Shapira NA, Foote KD, Cosyns PR, Kubu CS, Malloy PF, Salloway SP, Giftakis JE, Rise MT, Machado AG, Baker KB, Stypulkowski PH, Goodman WK, Rasmussen SA, Nuttin BJ (2010) Deep brain stimulation of the ventral internal capsule/ventral striatum for obsessive-compulsive disorder: worldwide experience. Mol Psychiatry 15:64–79. 10.1038/mp.2008.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremel CM, Costa RM (2013) Premotor cortex is critical for goal-directed actions. Front Comput Neurosci 7:110. 10.3389/fncom.2013.00110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremel CM, Chancey JH, Atwood BK, Luo G, Neve R, Ramakrishnan C, Deisseroth K, Lovinger DM, Costa RM (2016) Endocannabinoid modulation of orbitostriatal circuits gates habit formation. Neuron 90:1312–1324. 10.1016/j.neuron.2016.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grützmann R, Endrass T, Kaufmann C, Allen E, Eichele T, Kathmann N (2016) Presupplementary motor area contributes to altered error monitoring in obsessive-compulsive disorder. Biol Psychiatry 80:562–571. 10.1016/j.biopsych.2014.12.010 [DOI] [PubMed] [Google Scholar]
- Guo ZV, Li N, Huber D, Ophir E, Gutnisky D, Ting JT, Feng G, Svoboda K (2014) Flow of cortical activity underlying a tactile decision in mice. Neuron 81:179–194. 10.1016/j.neuron.2013.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison BJ, Soriano-Mas C, Pujol J, Ortiz H, López-Solà M, Hernández-Ribas R, Deus J, Alonso P, Yücel M, Pantelis C, Menchon JM, Cardoner N (2009) Altered corticostriatal functional connectivity in obsessive-compulsive disorder. Arch Gen Psychiatry 66:1189–1200. 10.1001/archgenpsychiatry.2009.152 [DOI] [PubMed] [Google Scholar]
- Isoda M, Hikosaka O (2007) Switching from automatic to controlled action by monkey medial frontal cortex. Nat Neurosci 10:240–248. 10.1038/nn1830 [DOI] [PubMed] [Google Scholar]
- Karno M, Golding JM, Sorenson SB, Burnam MA (1988) The epidemiology of obsessive-compulsive disorder in five US communities. Arch Gen Psychiatry 45:1094–1099. 10.1001/archpsyc.1988.01800360042006 [DOI] [PubMed] [Google Scholar]
- Leckman JF, Bloch MH, Smith ME, Larabi D, Hampson M (2010) Neurobiological substrates of Tourette's disorder. J Child Adolesc Psychopharmacol 20:237–247. 10.1089/cap.2009.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Holley SM, Shobe JL, Chong NC, Cepeda C, Levine MS, Masmanidis SC (2017) Parvalbumin interneurons modulate striatal output and enhance performance during associative learning. Neuron 93:1451–1463.e4. 10.1016/j.neuron.2017.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque M, Charara A, Gagnon S, Parent A, Deschenes M (1996) Corticostriatal projections from layer V cells in rat are collaterals of long-range corticofugal axons. Brain Res 709:311–315. 10.1016/0006-8993(95)01333-4 [DOI] [PubMed] [Google Scholar]
- Li N, Chen TW, Guo ZV, Gerfen CR, Svoboda K (2015) A motor cortex circuit for motor planning and movement. Nature 519:51–56. 10.1038/nature14178 [DOI] [PubMed] [Google Scholar]
- Luk KC, Sadikot AF (2001) GABA promotes survival but not proliferation of parvalbumin-immunoreactive interneurons in rodent neostriatum: an in vivo study with stereology. Neuroscience 104:93–103. 10.1016/S0306-4522(01)00038-0 [DOI] [PubMed] [Google Scholar]
- Maia TV, Cooney RE, Peterson BS (2008) The neural bases of obsessive-compulsive disorder in children and adults. Dev Psychopathol 20:1251–1283. 10.1017/S0954579408000606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Le Moine C, Charpier S, Gonon F (2005) Feedforward inhibition of projection neurons by fast-spiking GABA interneurons in the rat striatum in vivo. J Neurosci 25:3857–3869. 10.1523/JNEUROSCI.5027-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltby N, Tolin DF, Worhunsky P, O'Keefe TM, Kiehl KA (2005) Dysfunctional action monitoring hyperactivates frontal-striatal circuits in obsessive-compulsive disorder: an event-related fMRI study. Neuroimage 24:495–503. 10.1016/j.neuroimage.2004.08.041 [DOI] [PubMed] [Google Scholar]
- Mantovani A, Simpson HB, Fallon BA, Rossi S, Lisanby SH (2010) Randomized sham-controlled trial of repetitive transcranial magnetic stimulation in treatment-resistant obsessive-compulsive disorder. Int J Neuropsychopharmacol 13:217–227. 10.1017/S1461145709990435 [DOI] [PubMed] [Google Scholar]
- Menzies L, Chamberlain SR, Laird AR, Thelen SM, Sahakian BJ, Bullmore ET (2008) Integrating evidence from neuroimaging and neuropsychological studies of obsessive-compulsive disorder: the orbitofronto-striatal model revisited. Neurosci Biobehav Rev 32:525–549. 10.1016/j.neubiorev.2007.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamae T, Sakai Y, Abe Y, Nishida S, Fukui K, Yamada K, Kubota M, Denys D, Narumoto J (2014) Altered fronto-striatal fiber topography and connectivity in obsessive-compulsive disorder. PLoS One 9:e112075. 10.1371/journal.pone.0112075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner I, Werner CJ, Arrubla J, Stöcker T, Ehlen C, Wegener HP, Schneider F, Shah NJ (2014) Imaging the where and when of tic generation and resting state networks in adult Tourette patients. Front Hum Neurosci 8:362. 10.3389/fnhum.2014.00362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hare JK, Li H, Kim N, Gaidis E, Ade K, Beck J, Yin H, Calakos N (2017) Striatal fast-spiking interneurons selectively modulate circuit output and are required for habitual behavior. Elife 6:e26231. 10.7554/eLife.26231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen SF, Berke JD, Kreitzer AC (2018) Fast-spiking interneurons supply feedforward control of bursting, calcium, and plasticity for efficient learning. Cell 172:683–695.e15. 10.1016/j.cell.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy HB, Graybiel AM (1997) Cortically driven immediate-early gene expression reflects modular influence of sensorimotor cortex on identified striatal neurons in the squirrel monkey. J Neurosci 17:2477–2491. 10.1523/JNEUROSCI.17-07-02477.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB (2004) The mouse brain in stereotaxic coordinates. Houston, TX: Gulf Professional Publishing. [Google Scholar]
- Rauch SL, Jenike MA, Alpert NM, Baer L, Breiter HC, Savage CR, Fischman AJ (1994) Regional cerebral blood flow measured during symptom provocation in obsessive-compulsive disorder using oxygen 15-labeled carbon dioxide and positron emission tomography. Arch Gen Psychiatry 51:62–70. 10.1001/archpsyc.1994.03950010062008 [DOI] [PubMed] [Google Scholar]
- Rauch SL, Savage CR, Alpert NM, Dougherty D, Kendrick A, Curran T, Brown HD, Manzo P, Fischman AJ, Jenike MA (1997) Probing striatal function in obsessive-compulsive disorder: a PET study of implicit sequence learning. J Neuropsychiatry Clin Neurosci 9:568–573. 10.1176/jnp.9.4.568 [DOI] [PubMed] [Google Scholar]
- Rothwell PE, Hayton SJ, Sun GL, Fuccillo MV, Lim BK, Malenka RC (2015) Input- and output-specific regulation of serial order performance by corticostriatal circuits. Neuron 88:345–356. 10.1016/j.neuron.2015.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Brody AL, Schwartz JM, Baxter LR (1998) Neuroimaging and frontal-subcortical circuitry in obsessive-compulsive disorder. Br J Psychiatry Suppl 35:26–37. [PubMed] [Google Scholar]
- Schoenbaum G, Nugent SL, Saddoris MP, Setlow B (2002) Orbitofrontal lesions in rats impair reversal but not acquisition of go, no-go odor discriminations. Neuroreport 13:885–890. 10.1097/00001756-200205070-00030 [DOI] [PubMed] [Google Scholar]
- Sul JH, Kim H, Huh N, Lee D, Jung MW (2010) Distinct roles of rodent orbitofrontal and medial prefrontal cortex in decision making. Neuron 66:449–460. 10.1016/j.neuron.2010.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sul JH, Jo S, Lee D, Jung MW (2011) Role of rodent secondary motor cortex in value-based action selection. Nat Neurosci 14:1202–1208. 10.1038/nn.2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi YK, Roesch MR, Stalnaker TA, Haney RZ, Calu DJ, Taylor AR, Burke KA, Schoenbaum G (2009) The orbitofrontal cortex and ventral tegmental area are necessary for learning from unexpected outcomes. Neuron 62:269–280. 10.1016/j.neuron.2009.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Velzen LS, Vriend C, de Wit SJ, van den Heuvel OA (2014) Response inhibition and interference control in obsessive-compulsive spectrum disorders. Front Hum Neurosci 8:419. 10.3389/fnhum.2014.00419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Feng G, Calakos N (2011) Sapap3 deletion causes mGluR5-dependent silencing of AMPAR synapses. J Neurosci 31:16685–16691. 10.1523/JNEUROSCI.2533-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Ade KK, Caffall Z, Ilcim Ozlu M, Eroglu C, Feng G, Calakos N (2014) Circuit-selective striatal synaptic dysfunction in the Sapap3 knockout mouse model of obsessive-compulsive disorder. Biol Psychiatry 75:623–630. 10.1016/j.biopsych.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding JD, Feliciano C, Chen M, Adams JP, Luo J, Dudek SM, Weinberg RJ, Calakos N, Wetsel WC, Feng G (2007) Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 448:894–900. 10.1038/nature06104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Chen RQ, Gu QH, Yan JZ, Wang SH, Liu SY, Lu W (2009) Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J Neurosci 29:8764–8773. 10.1523/JNEUROSCI.1014-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yücel M, Harrison BJ, Wood SJ, Fornito A, Wellard RM, Pujol J, Clarke K, Phillips ML, Kyrios M, Velakoulis D, Pantelis C (2007) Functional and biochemical alterations of the medial frontal cortex in obsessive-compulsive disorder. Arch Gen Psychiatry 64:946–955. 10.1001/archpsyc.64.8.946 [DOI] [PubMed] [Google Scholar]
- Züchner S, Wendland JR, Ashley-Koch AE, Collins AL, Tran-Viet KN, Quinn K, Timpano KC, Cuccaro ML, Pericak-Vance MA, Steffens DC, Krishnan KR, Feng G, Murphy DL (2009) Multiple rare SAPAP3 missense variants in trichotillomania and OCD. Mol Psychiatry 14:6–9. 10.1038/mp.2008.83 [DOI] [PMC free article] [PubMed] [Google Scholar]