

Abstract
Key points
Spasticity is a disorder of muscle tone that is associated with lesions of the motor system. This condition involves an overactive spinal reflex loop that resists the passive lengthening of muscles.
Previously, we established that application of anodal trans‐spinal direct current stimulation (a‐tsDCS) for short periods of time to anaesthetized mice sustaining a spinal cord injury leads to an instantaneous reduction of spasticity. However, the long‐term effects of repeated a‐tsDCS and its mechanism of action remained unknown.
In the present study, a‐tsDCS was performed for 7 days and this was found to cause long‐term reduction in spasticity, increased rate‐dependent depression in spinal reflexes, and improved ground and skill locomotion.
Pharmacological, molecular and cellular evidence further suggest that a novel mechanism involving Na‐K‐Cl cotransporter isoform 1 mediates the observed long‐term effects of repeated a‐tsDCS.
Abstract
Spasticity can cause pain, fatigue and sleep disturbances; restrict daily activities such as walking, sitting and bathing; and complicate rehabilitation efforts. Thus, spasticity negatively influences an individual's quality of life and novel therapeutic interventions are needed. We previously demonstrated in anaesthetized mice that a short period of trans‐spinal subthreshold direct current stimulation (tsDCS) reduces spasticity. In the present study, the long‐term effects of repeated tsDCS to attenuate abnormal muscle tone in awake female mice with spinal cord injuries were investigated. A motorized system was used to test velocity‐dependent ankle resistance and associated electromyographical activity. Analysis of ground and skill locomotion was also performed, with electrophysiological, molecular and cellular studies being conducted to reveal a potential underlying mechanism of action. A 4 week reduction in spasticity was associated with an increase in rate‐dependent depression of spinal reflexes, and ground and skill locomotion were improved following 7 days of anodal‐tsDCS (a‐tsDCS). Secondary molecular, cellular and pharmacological experiments further demonstrated that the expression of K‐Cl co‐transporter isoform 2 (KCC2) was not changed in animals with spasticity. However, Na‐K‐Cl cotransporter isoform 1 (NKCC1) was significantly up‐regulated in mice that exhibited spasticity. When mice were treated with a‐tsDCS, down regulation of NKCC1 was detected, and this level did not significantly differ from that in the non‐injured control mice. Thus, long lasting reduction of spasticity by a‐tsDCS via downregulation of NKCC1 may constitute a novel therapy for spasticity following spinal cord injury.
Keywords: spinal cord injury, spinal direct current stimulation, Spasticity
Key points
Spasticity is a disorder of muscle tone that is associated with lesions of the motor system. This condition involves an overactive spinal reflex loop that resists the passive lengthening of muscles.
Previously, we established that application of anodal trans‐spinal direct current stimulation (a‐tsDCS) for short periods of time to anaesthetized mice sustaining a spinal cord injury leads to an instantaneous reduction of spasticity. However, the long‐term effects of repeated a‐tsDCS and its mechanism of action remained unknown.
In the present study, a‐tsDCS was performed for 7 days and this was found to cause long‐term reduction in spasticity, increased rate‐dependent depression in spinal reflexes, and improved ground and skill locomotion.
Pharmacological, molecular and cellular evidence further suggest that a novel mechanism involving Na‐K‐Cl cotransporter isoform 1 mediates the observed long‐term effects of repeated a‐tsDCS.
Introduction
Spinal hyperexcitability is highly prevalent following spinal cord injury (SCI) and is problematic in at least one‐third of patients (Holtz et al. 2017). Spasticity is a form of involuntary contraction of muscles following sensory input induced by muscle stretch and it has been characterized as a velocity‐dependent phenomenon (Lance, 1990) as a result of the primary endings (1a) that are highly sensitive to the velocity of stretch (Brown & Matthews, 1966). Velocity‐dependent spasticity also develops in rats (Bennett et al. 1999; Bose et al. 2002; Marsala et al. 2005) and mice (Ahmed, 2014) following SCI. Because of its characteristics, spasticity negatively interferes with functional movements (Knutsson & Richards, 1979; Lamontagne et al. 2001) and, for patients, it can negatively affect their quality of life.
In a previous study, the short‐term effects of direct current flowing from the spinal cord to sciatic nerve on muscle tone of the triceps surae muscle in mice were examined (Ahmed, 2014). In the present study, spinal‐to‐sciatic trans‐spinal subthreshold direct current stimulation (tsDCS) is termed anodal‐tsDCS (a‐tsDCS) and sciatic‐to‐spinal tsDCS is termed cathodal‐tsDCS (c‐tsDCS). Spinal stimulation resulted in significant modulatory effects on muscle tone, with c‐tsDCS increasing muscle tone and a‐tsDCS reducing muscle tone (Ahmed, 2014). However, these were acute experiments performed in anaesthetized animals. Therefore, critical questions remained. How long can a‐tsDCS‐induced reset of spinal cord excitability be maintained? How long will this procedure influence muscle tone? How will a‐tsDCS influence the recovery of unskilled and skilled locomotion following SCI? The present study is based on the hypothesis that a‐tsDCS will cause long‐term normalization of spinal excitability, thereby promoting the maintenance of reduced muscle tone and improved recovery of unskilled and skilled locomotion in animals with SCI.
K‐Cl co‐transporter isoform 2 (KCC2) and Na‐K‐Cl co‐transporter isoform 1 (NKCC1) are responsible for establishing chloride (Cl) concentrations across nerve cell membranes (Misgeld et al. 1986). Because of the importance of the electrochemical Cl gradient in determining the strength of inhibition mediated by GABAA and glycine receptors, an imbalance in protein levels or the activities of KCC2 and NKCC1 has been predicted to lead to hyperexcitability and muscle dysfunction, particularly spasticity (Boulenguez et al. 2010; Modol et al. 2014). The mechanism of action underlying the long‐term effects of direct current stimulation on spinal or brain excitability is largely unknown. It has been proposed that tsDCS can cause long‐term changes in the excitability of spinal cord circuits (Bolzoni & Jankowska, 2015; Samaddar et al. 2017; Song et al. 2016; Wieraszko & Ahmed, 2016; Ahmed, 2013, 2017). Therefore, we propose that the effects of tsDCS on muscle tone are mediated by changes in protein expression of KCC2 and/or NKCC1 in stimulated spinal tissue.
To test this hypothesis, a combination of electrophysiology, a motorized system to test spasticity and locomotor analysis were used to reveal the long‐term influences of tsDCS on spasticity following SCI in awake mice. Pharmacological studies were performed to confirm causal relationship between a‐tsDCS, NKCC1 and reduction of spasticity. In these studies, we used bumetanide to block NKCC1, as well as tumour necrosis factor (TNF)‐α and MG‐132 to offset the effect of a‐tsDCS NKCC1 expression. In addition, quantitative real‐time PCR (qPCR), western blotting, and immunohistochemistry were performed to quantify relevant NKCC1 expression, aiming to identify a possible mechanism of action by which tsDCS influences muscle tone.
Methods
Ethical approval
All experiments were performed in accordance with the City University of New York review Board. The College of Staten Island (CSI) Institutional Animal Care and Use Committee, via Designated Member Review, approved this study in accordance with the Guide for the Care and Use of Laboratory Animals and CSI's Office of Laboratory Animal Welfare approved Public Health Service Animal Welfare Assurance, # A3718‐01. All procedures were completed in accordance with the guidelines and requirements of the Journal of Physiology.
Animals
Adult female CD‐1 mice (n = 156 weighing 27–37 g) were used for all of the animal studies performed. In total, 51 animals were used for the long‐term stimulation studies that included four groups. Animals with SCI were randomly divided into four groups: (i) a‐tsDCS‐treated group (n = 16); (ii) c‐tsDCS‐treated group (n = 15); (iii) sham‐treated group (n = 10); and (iv) a non‐injured control group (n = 10). Subgroups of these animals were also used to study long‐term changes in expression for KCC2, pKCC2, NKCC1 and pNKCC1. Short‐term effects of tsDCS on the expression of NKCC1 and pNKCC1 were studied in non‐injured control animals (n = 10). Pharmacological studies involving bumetanide (NKCC1 blocker) were conducted with SCI animals (n = 9) and non‐injured control animals (n = 3). Blocking degradation was conducted with respect to non‐injured (n = 2), injured sham‐treated (n = 3), injured and injected with MG‐132 (n = 2), and injured and injected with TNF‐α (n = 3). Meanwhile, qPCR studies were conducted with non‐injured control animals (n = 41). Non‐injured control animals were used to investigate the heat shock protein 70 (HSP70) response to a‐tsDCS (sham‐treated, n = 7; a‐tsDCS, n = 11). Dorsal root potential studies were performed on non‐injured animals (n = 14). Information about animal housing condition is provided elsewhere (Ahmed 2017). At the end of experiments and prior to tissue harvesting, animals were deeply anaesthetized using ketamine/xylazine (90/10 mg kg−1 i.p.).
Spinal cord contusion injury
Following administration of a ketamine/xylazine cocktail (90/10 mg kg−1 i.p.) to achieve deep anaesthesia, a laminectomy at thoracic T10 vertebra was performed to expose the T13 spinal segment. An infinite horizon (IH) spinal cord impactor (IH‐0400; Precision Systems & Instrumentation, Fairfax, VA, USA) was used to induce a contusive SCI force. The impact force level was 60 kdyn and was selected based on data collected from exploratory experiments that were conducted with this impactor. In these preliminary studies, contusion injuries were induced with increasing impact forces to optimize the SCI contusion force needed to produce spastic mice. Mice impacted at 60 kdyn produced incomplete spinal cord injury with ideal spastic behaviour at ∼2–3 weeks post‐injury. An animal with ideal spastic behaviour, as defined in the present study, is the level of spasticity that allows the animal to walk at score 3 or 4 (occasional planter stepping) of the BMS scale (Basso et al. 2006) and when animals show fanning or fisting of the paws digits and frequent dragging or crossing of the hindlimb during locomotion. Impact was applied by using a standard mouse tip (diameter 1.3 mm). Following the contusion procedure, overlying muscles and skin were sutured and the surgical site was covered with a layer of gentamicin sulphate ointment to prevent infection. Each mouse subsequently received a number and then was placed on a warming pad (30°C) for recovery. Following recovery from anaesthesia, each animal was placed in a separate cage. The recommended dose of Acetaminophen (1.64 mg ml−1) was orally administered once the animals retain consciousness. Daily examinations of the mice were performed to assess parameters such as wound healing and appetite and to ensure that a sterile environment is maintained. Sterile saline was injected subcutaneously as needed for hydration. DietGel (ClearH2O, Westbrook, ME, USA) and softened crushed Purina Mouse Chow (LabDiet, St Louis, MO, USA) were added to the cages to assist feeding. The bladder was emptied manually twice per day or as needed until automatic bladder function recovered.
Stretch apparatus
A computer‐controlled stretch device was assembled in our laboratory from a stepper motor (DMX‐UMD‐23m; ARCUS Technology Inc., Livermore, CA, USA), a foot presser mounted on a force‐displacement transducer (FT03; GRASS Technologies/NATUS, San Carlos, CA, USA) connected to a bridge amplifier (ADInstruments, Colorado Springs, CO, USA) and a mouse holder (Fig. 1). The transducer was permanently fixed to a linear rail‐guided carriage (SGB15NUU; ZNT Automatic Technology Co, Zhejiang, China). The rotational movement of the stepper motor is converted into linear displacement at the carriage by using a set of jointed flat‐aluminum rods that are connected with swivel hub bearings.
Figure 1. Set‐up and procedure for testing spasticity in mice.
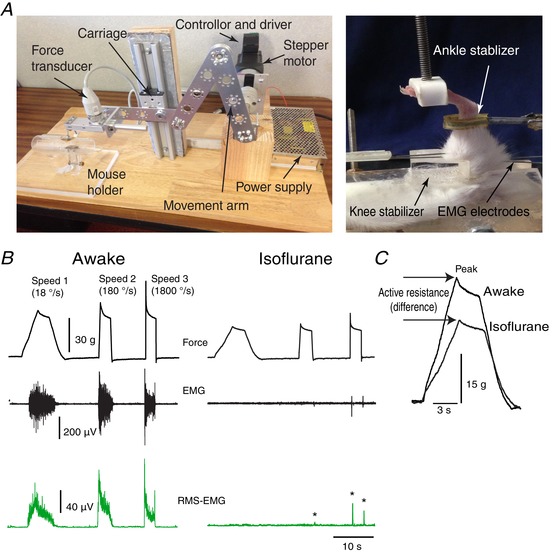
A, left: a stretch apparatus was constructed from a stepper motor, movement arm, isometric force transducer and mouse restrainer. The system was controlled by computer software. The image on the right shows how a limb would be positioned in the mouse holder. B, top: representative examples of the muscle resistance traces recorded at: speed 1 = 18° s−1; speed 2 = 180° s−1; and speed 3 = 1800° s−1. The trace on the left is from an animal that was awake, whereas the trace on the right was obtained with an animal that was asleep as a result of isoflurane administration. The bottom two traces (shown in green) represent raw and root squared EMG. *EMG during deep anaesthesia. C, force traces recorded for awake and asleep mice at speed 1 are superimposed to show the effect of isoflurane on measured active resistance, rising slope and peak. [Color figure can be viewed at wileyonlinelibrary.com]
Two different software programs were utilized to control the stepper motor and to perform the online (during) stretch and electromyogram (EMG) recordings. First, a stepper motor control software (DMX‐UMD GUI; ARCUS Technology Inc.) was re‐written to permit the execution of three consecutively timed stretches, with each stretch separated by two 10 s recovery intervals. The stepper motor was also programmed to provide three different stretch speeds: 18, 180 and 1800° s–1, which correspond to 3, 30 and 300 r.p.m., respectively. The second software package used was LabChart‐7, which was bundled with the PowerLab data acquisition system software (ADInstruments) that was used to gather and analyse data.
Mouse holder
A mouse restraining system was fabricated in our laboratory (Fig. 1) from three components. (i) A clear Plexiglas [poly(methyl methacrylate)] acrylic tube served as the mouse holding chamber. (ii) An internally adjustable support system was made of a clear acrylic concave stabilization plate. The location of the plate was able to be adjusted linearly along the length of the chamber via a handle that protruded through a cut‐out that ran the entire length of the tube. In addition, the concave surface of the plate was designed to contour to the back of the animal, thereby securing it dorsally. This surface could also be adjusted externally to accommodate various sizes of mice. (iii) Four over‐skin stimulating electrodes (constructed in our laboratory) with each composed of a wick‐covered 1 × 1.5 cm stainless‐steel plate. The wick was soaked with 3% saline before every use. One of the electrodes was permanently fixed to the floor of the holding chamber (for the abdominal reference electrode), whereas a second electrode was permanently fixed to the middle of the stabilization plate (for the dorsal active spinal electrode) and the location of this plate could be adjusted to cover the thoracolumbar spinal column area that corresponds to lumbosacral spinal cord segments. The remaining two electrodes could be adjusted linearly by sliding them along cutouts present on the long sides of the stabilization plate, and these served as left and right sciatic nerve conductors, respectively. The electrodes were placed over the skin of the posterior side of upper thigh. The abdominal surface of the holding chamber had two openings, one for each of the animal's hindlimbs. Each opening was equipped with a knee stabilizer pad to ensure full knee extension during testing. Prior to testing, a cap with an opening in the centre was placed on the anterior end of the holding chamber for breathing and for isoflurane administration. To limit hindlimb movement to the ankle joint during stretching, another acrylic stand was created to secure the distal leg. The stand had an adjustable stainless steel ankle stabilizer clamp that could be moved in the x, y and z axes to achieve proper alignment of the foot under the presser, and to adjust the hip angle of the animal before stretching.
Circuit and power source
Passing current to peripheral nerves is required to attenuate muscle tone (Ahmed, 2014). Moreover, the direction and distribution of this current can be regulated (Ahmed, 2017). The tsDCS protocols of the present study required a modification of the trans‐spinal circuit that was originally designed in our laboratory to be used with anaesthetized animals (Ahmed, 2014). Briefly, the circuit was modified to non‐invasively pass direct current (DC) to the spinal cord and the sciatic nerve of the most affected limb by using over‐skin electrodes. To prevent evoking nerve activity, the current passing into the sciatic nerve electrode was attenuated by dividing the reference source into two branches: the first branch connected directly to the abdominal electrode and carried un‐attenuated current, whereas the second branch passed through a 300 KΩ resistor to attenuate current to the sciatic nerve. Current was supplied by a GRASS stimulator with a dedicated DC unit (S88; GRASS Technologies/NATUS). By switching the polarity of the current source, this circuit design could provide instant reversal of the current direction from anodal to cathodal, and vice versa. The circuit design was also effective in attenuating sciatic nerve current. For example, for each 1.5 mA that passed through the spinal‐abdominal electrodes, 300 μA passed through the sciatic nerve circuit. Monitoring of current parameters and verification of DC attenuation was performed at the beginning, during and upon completion of each experiment by use of a bench‐top digital multimeter (34401A; Agilent/Keysight Technologies, Santa Clara, CA, USA). Stimulation at this current density is considered subthreshold and causes no pain.
Repeated‐session tsDCS (long‐term) procedure
One week before treatment, all of the animals underwent initial baseline testing (Pre: the evaluation performed prior to the start of tsDCS or sham treatments) and the following data were recorded: the peak resistances at three different stretch speeds, concurrent EMG activity, and skill and ground locomotion testing. To apply stimulation, nine mouse holders were fabricated to provide tsDCS to multiple animals simultaneously.
Animals in the anodal and cathodal groups received 20‐min tsDCS (1.5 mA) treatments daily for 7 days. No sign of pain/distress (limb kicking, vocalization, partially closed eyelids, increased vibrissae movements or body shaking) was observed during tsDCS stimulation. Animals in the sham‐treated group were treated in parallel, although they did not receive tsDCS intervention. Meanwhile, the non‐injured group was only exposed to the testing procedure. Animals were re‐evaluated immediately after the 7 day tsDCS treatment period (E1), then also 2 weeks (E2) and 4 weeks post‐treatment (E3). Non‐injured control animals were evaluated twice, E1 and E2, and did not receive any treatment. After the last evaluation, E3 for all groups and E2 for non‐injured control, animals were tested for RDD of the Hoffman reflex. EMG data were collected by using the PowerLab system (ADInstruments), recorded at a sample rate of 10 KHz, and filtered at a low pass of 2 kHz and a high pass of 100 Hz.
Ground locomotion testing
The DigiGait system (Mouse Specifics Inc, Framingham, MA, USA) was used to quantify locomotor recovery following SCI and tsDCS treatment. All of the groups were tested four times with the DigiGait system: before, immediately after, and 2 and 3 weeks after tsDCS intervention. The non‐injured control group was tested twice, with a 2‐week interval between the tests. During testing, a minimum of 20 steps were collected at two speeds: 10 and 20 cm s−1. Three parameters were considered to be sensitive to spasticity in the triceps surae muscle during locomotion: peak area and rising and falling slopes of the paw area. Peak paw area was calculated based on the maximum contact area of the foot of the mouse with the DigiGait treadmill belt. Rising slope was considered an indicator of the speed of transition during the first half of the stance phase of the mouse locomotion cycle, whereas falling slope was considered an indicator of the speed of transition during the second half of the stance phase of the mouse locomotor cycle. The stance phase is the segment of the locomotion cycle when a mouse's paw is in contact with the treadmill belt.
Skill‐locomotion testing
A ladder‐wheel was assembled in our laboratory from a stepper motor, a driver and a controller (DMX‐UMD‐23; ARCUS Technology Inc.). The speed and direction of the wheel was controlled with custom‐written software. The wheel was made from Plexiglas to allow side visualization and the spaces between the rungs were variable to prevent learning. A camera was placed on the underside of each animal to record foot faults. Animals were tested prior to stimulation and once a week for 3 weeks after the stimulation ended. For each recorded session, a continuous 90 s video was analysed frame‐by‐frame according to a previously described foot fault scoring system (Metz & Whishaw, 2002, 2009; Farr et al. 2006) (Table 1). A score for each of the animal's hindlimbs was calculated for each criteria by multiplying the total number of foot faults by the corresponding score for each criterion according to Table 1. Scores for all of the criteria were subsequently added to obtain a total score for each of the hindlimbs.
Table 1.
Ladder wheel error scores: scoring system used to evaluate skilled walking (Metz and Whishaw 2009)
| Foot fault | Score | Description |
|---|---|---|
| Total miss | 0 | Assigned when a limb completely misses a rung and a fall occurs. A fall was defined as a limb deeply falling in‐between rungs with body posture and balance disturbed |
| Deep slip | 1 | Assigned when a limb is initially placed on a rung, then it slips off upon weight bearing and this causes a fall |
| Slight slip | 2 | The limb is placed on a rung and then slips off upon weight bearing. However, the slip does not result in a fall and the animal is able to maintain balance and a co‐ordinated gait |
| Replacement | 3 | The limb was placed on another rung but before it is weight, bearing it is quickly lifted and placed on another rung |
| Correction | 4 | A limb was placed on a rung and was quickly repositioned while remaining on the same rung |
| Partial placement | 5 | The limb is placed on a rung with either the wrist or digits of the forelimb, or with the heel or toes of the hindlimb |
| Correct placement | 6 | The mid‐portion of the palm of a limb is placed on a rung and can bear the animal's full weight |
RDD of the Hoffman reflex
After the last evaluation (E3), the mice were anaesthetized with a mixture of ketamine and xylazine (90 mg kg−1). The animals were then shaved at the hindlimb and pelvic area and two sets of recording electrodes were applied to record EMG, heart rate and breathing activity. Stimulation of the tibial nerve was achieved by using a concentric needle‐stimulating electrode. After positioning the animal on the recording station, heart rate (HR) and respiratory rate (RR) were monitored online. When an animal was deeply anaesthetized, both their HR and RR were generally faster, whereas their respiration was more shallow. At this level of anaesthesia, reflexes were difficult to evoke. Moreover, if reflexes were evoked, the RDD was very high, even in the animals that expressed severe spasticity. Therefore, in the exploratory experiments that were performed, the procedure was standardized by monitoring the relationship between both HR and RR and the levels of awareness of the animal and RDD. For example, as the animals awaken, their HR becomes regular at 2 Hz and then it becomes more irregular between 2 Hz and 4 Hz, whereas their RR becomes deeper and slower. When an irregular HR and deeper RR phase were first observed, the RDD protocol was started. This consisted of five trains of five pulses. The frequencies of the trains were: 0.1, 0.5, 1, 2 and 5 Hz. The waiting intervals between the trains were 1 min long. Testing was performed on both hindlimbs. Stimulation intensity was gradually increased until maximum H‐wave relative to M‐wave was observed, followed by the RDD protocol. Data were collected using the PowerLab system (ADInstruments), recorded at a sample rate of 10 KHz, and filtered at a low pass of 2 kHz and a high pass of 100 Hz. The data were averaged using the root square mean function with a 1 ms window by using a built‐in function of LabChart software (ADInstruments). Peaks of the averaged H‐wave were measured and percentage change was calculated relative to baseline values. RDD was calculated as the percentage reduction based on the difference calculated between the H‐wave amplitude induced by the fifth pulse and the first pulse in a train divided by the amplitude of the H‐wave induced by the first pulse, multiplied by 100, then multiplied by –1 to inverse the values.
In vivo dorsal root potential (DRP) recording
Animals were anaesthetized with ketamine/xylazine (100 and 10 mg kg−1, respectively). The lumbosacral spinal cord was exposed by laminectomy. The head and the spinal column of the animal were fixed using custom‐made clamping systems. The dura was opened and a dorsal root was exposed and teased into several filaments and cut distally. Two of these adjacent filaments were suctioned into glass pipettes of suction electrodes (A‐M System, Sequim, WA, USA) filled with 0.9% saline. One dorsal root was chosen for stimulation and the other for recording. a‐tsDCS electrode (constructed in our laboratory) was placed on the dorsum of the spinal cord, and the reference electrode was connected to the abdominal skin. a‐tsDCS electrodes were made from stainless steel sheets (0.5 × 1 cm) covered with conductive co‐polymer adhesive hydrogel tape (BTHG‐250X Hydrogel; Breachers Tape, IL, USA). The a‐tsDCS was delivered at 0.8 mA. Dorsal root stimulation was performed at supramaximal intensity. Ten dorsal root responses were recorded at 0.1 Hz before, during and 1 h after a‐tsDCS (n = 5). All recordings were passed through a headstage and amplified using modified model 1700 differential AC Amplifier (A‐M System) and digitized at 10 kHz. Recordings were also made before and 10 min after applying two blockers: 2–5 μL of 0.3% bumetanide dissolved in saline and 0.25% NaOH (NKCC1 blocker, n = 5), or 2–5 μL of 10 μm picrotoxin solution (GABAA receptors antagonist, n = 4). Blockers were applied directly on the exposed spinal cord.
Lumbar intrathecal injection of TNF‐α and MG‐132
We used a procedure that was described in details previously (Njoo et al. 2014), Briefly, animals were anaesthetized with 3% isoflurane. The lumbosacral area of the animal was shaved. Following the localization of the spinous process of L6, the needle (30 gauge) was carefully inserted between the groove of L5 and L6. A volume of 10 μL was injected using a 25 μL Hamilton syringe. The animals were moved back to the cage and allowed to recover for thirty minutes. Injections were made of either 200 ng of Recombinant Rat tumor necrosis factor alpha (TNF‐α; Peprotech, Rocky Hill, NJ, USA) dissolved in 10 μL of 0.9% sterile saline or 375 ng of the proteasome inhibitor MG‐132 (Santa Cruz Biotechnology, Dallas, TX, USA) dissolved in sterile DMSO (Sigma, St Louis, MO, USA) and diluted in 10 μL sterile saline.
Western blotting
Animals were deeply anaesthetized with ketamine and xylazine (100 and 10 mg kg−1, respectively) and spinal columns of the target area (below injury segments) were incised and placed in Petri dish, and then spinal cords were quickly pulled out and immediately placed in dry ice. The whole procedure was carried out in ∼20 s. Animals were immediately killed by abdominal aorta transection. The samples were subsequently homogenized in RIPA buffer (catalogue no. BP‐115; Boston Bio Products, Ashland, MA, USA) containing a Protease Inhibitor Cocktail (SC‐29131; Santa Cruz Biotechnology), Phosphatase Inhibitors Cocktail 11 (catalogue no. BP‐480; Boston Bio Products), 100 mm phenylmethane sulphonyl fluoride (catalogue no. BP‐481, Boston Bio Products) and 500 mm EDTA. Following the addition of RIPA cocktail to each sample (100 mg mL−1), the lysates were incubated on ice for 15 min before being sonicated for 0.5–1 min to achieve complete homogenization using Sonic Dismembrator (Fisher Scientific Springfield Township, NJ, USA). The samples were then centrifuged at 12,000g in a Sorvall Legend Micro 21R Centrifuge for 30 min to collect the supernatant fraction of each sample. Total protein concentrations were determined by using an iMark™ Microplate Absorbance Reader (Bio‐Rad, Hercules, CA, USA). Twenty micrograms of each sample were mixed with an equal volume of 2× sample buffer and electrophoresed on 10% SDS polyacrylamide gels. After the separated proteins were transferred to polyvinylidene fluoride membranes (Bio‐Rad), the membranes were blocked in 5% skim milk buffer for 2 h, and then were incubated at 4°C with the appropriate primary antibodies overnight. The primary antibodies used included: rabbit polyclonal HSP70/HSC‐70 (H‐300) antibody (dilution 1:1000; SC‐33575; Santa Cruz Biotechnology); anti‐KCC2 antibody (dilution 1:1000; ab49917; Abcam Inc., Cambridge, MA, USA); rabbit polyclonal Anti‐phosoho‐Ser940 potassium chloride cotransporter (KCC2) antibody (dilution 1:1000; p1551‐940; PhosphoSolutions, Aurora, CO, USA); mouse monoclonal NKCC1 (A‐6) antibody (dilution 1:1000; SC‐514774; Santa Cruz Biotechnology); and rabbit polyclonal anti‐phospho NKCC1 Thr212/Thr217 antibody (dilution 1:1000; ABS 1004; EMD Millipore, Burlington, MA, USA). The membranes were subsequently washed three times with 1× Tween 20‐Tris‐buffered saline (TTBS) and incubated with appropriate secondary antibodies in blocking buffer. The secondary antibodies included: goat anti‐rabbit immunoglobulin G‐horseradish peroxidase (HRP) antibody (dilution 1: 5000; SC‐2004; Santa Cruz Biotechnology) and goat anti‐mouse IgG‐HRP antibody (dilution 1:5000; SC‐2005; Santa Cruz Biotechnology). After 1 h at room temperature, the membranes were washed three times with 1× TTBS. Bound antibodies were visualized with Luminol/oxidizing solution, an HRP‐based Chemiluminescent Substrate (Boston Bio Products) and quantified with ImageJ (National Institutes of Health, Bethesda, MD, USA). The blots were subsequently incubated with 1× stripping buffer, 1× phosphate‐buffered saline (PBS) and 1× TTBS for 30 min, then were incubated in blocking buffer for 1 h. After an additional incubation with mouse monoclonal IgG β‐actin (C4) HRP antibody (dilution 1:2500; SC‐47778; Santa Cruz Biotechnology) in blocking buffer for 1 h, the blots were washed three times with 1× TTBS and then were imaged with Luminal/Oxidizing solution (Boston Bio Products). Bands were quantified with ImageJ.
Immunohistochemistry
HSP70 immunohistochemistry
To examine HSP70 expression in motor neurons following anodal and cathodal stimulation, animals were anaesthetized with a ketamine/xylazine solution and then perfused with PBS at room temperature, followed by 4% paraformaldehyde. Dissected spinal cord segments were post‐fixed overnight in the same fixative and then were transferred to 30% sucrose for cryoprotection. The spinal cord segments were frozen with dry ice, sectioned, and collected in PBS. After three washes in Tris A buffer, the sections were washed 1× with Tris B for 15 min and then were blocked in 10% normal goat serum diluted in Tris B. After 1 h, primary mouse monoclonal HSP70 (F‐3) antibody was added (dilution 1:500, sc‐373867; Santa Cruz Biotechnology) and the sections were incubated overnight on a shaker at 4°C. The next day, the sections were washed 3x and then were incubated with a biotinylated goat anti‐mouse antibody (dilution 1:500; BA‐9200; Vector Laboratories Burlingame, CA, USA). After 1 h, DyLight 488 (dilution 1:2000; SA‐5488; Vector Laboratories) was added. After another 30 min, the sections were washed twice with Tris A before the slices were mounted with medium containing 4',6‐diamidino‐2‐phenylindole (DAPI) (H‐1200; Vector Laboratories).
Choline acetyltransferase (ChAT) and NKCC1 immunohistochemistry
Spinal cord sections were washed three times in PBS. After washing, they were incubated in 10% rabbit serum in 0.1% Triton X‐100 in 0.1 m PBS for 1 h at room temperature. The sections were then incubated with anti‐ChAT primary antibody (dilution 1:500; AB144P; EMD Millipore Corp., Burlington, MA, USA) in the same 0.1% Triton X‐100 in 0.1 m PBS solution for 48–72 h at 4°C. Sections were the thoroughly washed to remove any excess antibody three times in PBS and then incubated with rabbit anti‐goat 568 nm (dilution 1:500) in the same 0.1% Triton X‐100 in 0.1 m PBS solution for 1 h at room temperature. Sections were again washed three times in 0.1 m PBS, blocked with 10% goat serum in 0.2% TritonX‐100 in 0.1 m PBS for 1 h at room temperature, followed by overnight incubation with NKCC1 primary antibody (dilution 1:400; sc: 514774, Santa Cruz Biotechnology) in 2% goat serum/0.2% Triton X‐100 in 0.1 m PBS solution at 4°C. The sections were washed three times with 0.1 m PBS and then incubated with goat anti‐mouse 488 (dilution 1:500) in 2% goat serum/ 0.2% Triton X‐100 in 0.1 m PBS solution for 1 h at room temperature. Tissue slices were washed at least three times with 0.1 m PBS and then mounted on glass slides and covered with glass cover slips using mounting medium with DAPI (H‐1200; Vector Laboratories Inc).
qPCR
RNA was isolated from spinal cord samples by using a Trizol‐based method (Rio et al. 2010), followed by extraction with chloroform and isopropyl alcohol. Briefly, each RNA sample was dissolved in diethyl dicarbonate water and then purified with a Qiagen RNAeasy kit (Qiagen, Hilden, Germany) in accordance with the manufacturer's instructions. RNA concentrations were measured with a Nanodrop 2000c instrument (Thermofisher Scientific, Waltham, MA, USA). Total RNA was converted to complementary DNA (cDNA) with the iScript™ Reverse Transcription Supermix (Bio‐Rad). For each reaction, 2 μg of total RNA was combined with gene specific primers (PrimePCR Assay Slc12a2). Samples that were obtained from stimulated and unstimulated animals were always assayed on the same plate. For each sample, amplified product differences for each transcript were measured from three replicates by using SYBR Green chemistry‐based detection. Beta‐actin (ACTB), TATA box binding protein (TBP) and hypoxanthine phosphoribosyltransferase 1 (HPRT1) were used as endogenous reference genes, and the primers used for amplification of these genes were PrimePCR Assay ACTB, PrimePCR Assay TBP and PrimePCR Assay HPRT1, respectively. The resulting three transcripts were selected for analysis based on a previous demonstration of their stable expression in the central nervous system (Valente et al. 2014; Walder et al. 2014). The total reaction volume was 10 μL and this included 5 μL of the supermix, primers, cDNA template and nuclease‐free water. The qPCR reactions were carried out in 384‐well plates with the CFX384 Real Time System (Bio‐Rad) and SsoADVANCED Universal SYBR Green Supermix (Bio‐Rad). CFX manager software (Bio‐Rad) was used with automatic baseline and threshold detection options selected. The resulting data were exported to Excel (Microsoft Corp., Redmond, WA, USA) and relative normalized expression was calculated for each sample using the geometric mean of the triplicates against the endogenous reference genes as a normalization factor.
Experimental design and statistical analysis
Based on the exploratory experiments that were performed, the ankle was stretched from an initial angle of 109° plantarflexion to 81° dorsiflexion (stretched by 28°) to induce a stretch reflex without damaging the muscle. The corresponding knee joint angle was at full extension and this was consistently maintained across all of the measurements. For each stretch parameter that was tested using the apparatus, two sets of data were collected: one set of data were collected when the animal was awake, and a second set of data were collected when the animal was under full isoflurane anaesthesia. The depth of anaesthesia was monitored in three ways: (i) pinching the animal's paw showing no reflex; (ii) observing an animal's breathing to be shallower; and (iii) complete disappearance of background EMG activity from the TS muscle. Data were collected when the animal was anaesthetized to represent the passive resistive forces of muscles and connective tissues. To isolate the neuronal‐produced component of stretch resistance from that caused by passive tissues, the value of the tested parameter collected during anaesthesia was subtracted from the value collected when the animal was awake. This method ensured that only the neuronal contribution of the stretch response would be analysed. Subtraction of the tail‐pinch and pedal reflexes were used to standardize the anaesthesia level at which the stretches were performed. However, isoflurane anaesthesia did not completely remove the reflex response during the higher speed stretches. The following stretch parameters were collected: active resistance peak amplitude and active slope and root mean square EMG (RMS EMG) area and amplitude (Fig. 1). Active peak resistance was calculated as the height of the resistance trace relative to baseline (before the stretch). Active slope was calculated based on the first derivative of the rising phase of the muscle resistance in response to stretching. RMS EMG amplitude was calculated based on the height of the EMG trace relative to baseline. RMS EMG area was calculated based on area under the curve.
Four investigators transferred the stretch apparatus data collected in LabChart software to Excel sheets and calculated the scores for each animal using embedded Excel sheet formulas. One investigator analysed the DigiGait (locomotion) and ladder wheel (skilled locomotion) videos, whereas another investigator analysed the data with embedded Excel sheet formulas to calculate animal scores. All six investigators were blinded to the study hypothesis. RMS equations were embedded in LabChart with a window of 0.5 ms to generate the RMS for EMG.
To investigate the effects of repeated tsDCS on muscle tone, a mixed factorial repeated measures experimental design was used in which treatment (cathode, anode, or sham) was the between‐subjects independent variable, and time and speed were the within‐subjects independent variables. Mixed factorial ANOVA (split‐plot ANOVA) was used to assess differences in peak resistance, slopes, EMG amplitude and area among the groups. Bonferroni confidence interval adjustment was also applied. Mixed factorial ANOVA was used to analyse the RDD data. A between groups comparison was performed using Tukey's post hoc test. One‐way repeated measures ANOVA (RM ANOVA) was used to analyse paw area and the rising and falling slopes in ground locomotion data. All assumptions for use of repeated‐measures ANOVA were fulfilled. Holm–Sidak post hoc correction was used to test differences between speeds for the stretch responses and skill locomotion. A two‐tailed paired t test was used to analyse the difference in skilled locomotion between the two evaluations of the non‐injured control groups.
SPSS, version 23 (IBM Corp., Armonk, NY, USA) was used to perform all of the statistical tests. To test for sphericity, Mauchly's test in SPSS was applied (P > 0.05). P < 0.05 was considered statistically significance. Data are presented as the mean ± SEM.
Results
Long‐term effects of DCS on spasticity outcome measures
Differences in the amplitudes of active muscle resistance were assessed by using mixed factorial ANOVA, with time (four levels: pre, E1, E2 and E3) and speed (three levels: speeds 1, 2 and 3) as within‐subjects variables, and treatment (three levels: anode, cathode and sham) as between‐subjects variable. No significant interaction between treatment and speed was observed (F = 0.4, P = 0.81). By contrast, a significant interaction between treatment and time of evaluation was observed (F = 2.36, P = 0.03). After adjustment for multiple comparisons (Bonferroni), a significant marginal mean difference between the anode‐ and cathode‐treated animals was identified (P < 0.001), as well as between the anode‐ and sham‐treated groups (P < 0.001). However, there was no significant difference between the cathode‐ and sham‐treated groups (P = 0.13) during E2, E3 and E4. There was also no significance difference between the anode‐, cathode‐ and sham‐treated animals at E1 (P > 0.05).
As shown in Fig. 2, a within‐groups analysis showed that animals treated with a‐tsDCS exhibited significant reductions in the amplitude of active muscle resistance. The mean scores at the different time intervals vs. the pre‐treatment score according to speed were significant: speed 1 (E1, P < 0.001; E2, P = 0.003; E3, P = 0.002), speed 2 (E1, P < 0.001; E2, P = 0.038; E3, P = 0.027) and speed 3 (E1, P < 0.001; E2, P = 0.004; E3, P = 0.046), each according to the Holm–Sidak method.
Figure 2. Repeated treatment with a‐tsDCS results in long‐term reduction of spasticity in mice with SCI.
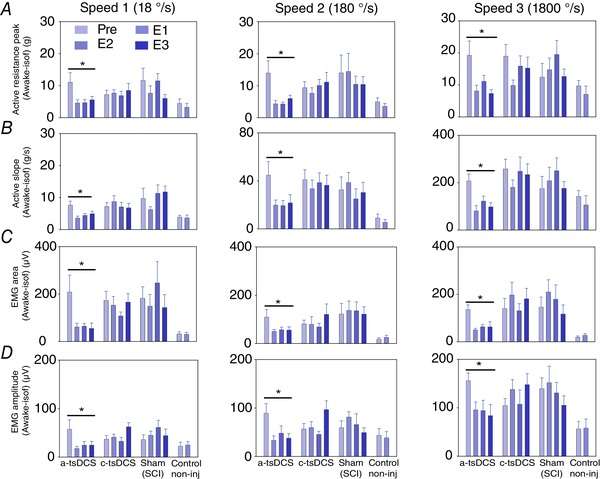
Animals were divided into four groups: an a‐tsDCS group (n = 16); a cathodal‐tsDCS group (n = 15); a sham‐treated group (n = 10); and a non‐injured control group (n = 8). A–C, a‐tsDCS caused a significant reduction in peak muscle resistance (A), rising slope (B), EMG area (C) and EMG amplitude (D). B–D, cathodal and sham treatments did not significantly differ. * P < 0.05, Holm–Sidak method. Pre, pre‐evaluation performed prior to the start of tsDCS or sham treatment; E1, first evaluation immediately performed after the end of 7 day tsDCS or sham treatment; E2, second evaluation performed 2 weeks post tsDCS or sham treatment; E3, third evaluation performed 4 weeks post tsDCS or sham treatment. Note time is relative to tsDCS and not to SCI. E1 and E2 for non‐injured control animals are the first and second evaluations, no treatment. Data are presented as the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Differences in active slope were assessed by using mixed factorial ANOVA (Fig. 2 B). The analysis showed significant interactions between time and treatments (F = 3.59, P = 0.03), yet no interaction between speed and treatments (F = 1.02, P = 0.41). Between‐group analysis further showed significant differences between the anode‐ and cathode‐treated groups (P = 0.015), as well as between the anode‐treated and sham‐treated groups (P = 0.04), during E2, E3 and E4. However, there was no difference between the sham‐treated and cathode‐treated animals (P = 0.829). A within‐subjects analysis also revealed that the anode‐treated group underwent a decrease in active slope over time (P < 0.01) and this was not observed for the other groups (P > 0.05).
The difference in EMG area was assessed by using mixed factorial ANOVA (Fig. 2 C). There was a significant interaction between time and treatments (F = 4.4, P = 0.0002), yet no interactions between speed and treatment (F = 0.6, P = 0.8). There were also no interactions between time, speed and treatments (F = 1.1, P = 0.34). A post hoc analysis identified a significant difference between the anode‐treated and cathode‐treated groups (P = 0.001) and between the anode‐treated and sham‐treated groups (P = 0.0001). By contrast, there was no difference between the cathode‐treated and sham‐treated groups (P = 0.34). A within‐subjects analysis further revealed that the anode‐treated group exhibited a decrease in EMG area over time (P < 0.01) and this was not observed for the other groups (P > 0.05).
The difference in EMG amplitude (awake minus isoflurane) was assessed by using mixed factorial ANOVA (Fig. 2 D). A significant interaction was observed between time and treatments (F = 3.63, P = 0.007). Meanwhile, there was no interaction between speed and treatments (F = 0.61, P = 0.66), nor between speed, time and treatments (F = 0.19, P = 0.9). A post hoc analysis identified a significant difference between the anode‐treated and cathode‐treated groups (P < 0.001) and between the anode‐treated and sham‐treated animals (P = 0.001). By contrast, there was no difference between the cathode‐treated and sham‐treated animals (P = 0.96). A within‐subjects analysis further identified that the anode‐treated group exhibited a decrease in EMG amplitude over time (P < 0.01) and this was not observed for the other groups (P > 0.05).
Repeated treatment of a‐tsDCS changes ground locomotion patterns
Locomotor characteristics were investigated by using the DigiGait system (Fig. 3). Animals were assessed at two speeds, 10 and 20 cm s−1, prior to tsDCS (pre), and then at three intervals following treatment: immediately following treatment (E1) and at 2 weeks (E2) and 4 weeks (E3) following treatment. Three relevant parameters were analysed at each of these time points: rising slope, peak paw area and falling slope.
Figure 3. Repeated treatment with a‐tsDCS leads to changes in locomotion patterns.
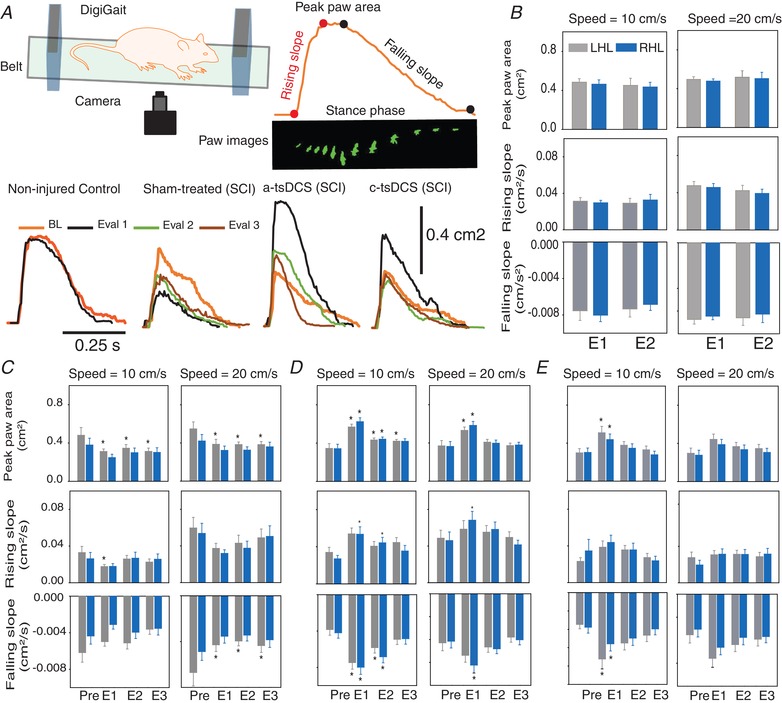
A, general set‐up of the DigiGait system. Locomotion was tested at two speeds: 10 cm s−1 and 20 cm s−1. A typical recording is shown at the top right, with corresponding rising slope, peak paw area and falling slope data labelled. The bottom recordings represent the stance phases from representative animals from each group. B, non‐injured control animals were evaluated twice with a 2 week interval between testing. The grey bars represent left hindlimb (LHL) data and the blue bars represent right hindlimb (RHL) data. E1 and E2: see Methods and Figure 1. The non‐injured control animals exhibited no difference in paw area, rising slope or falling slope between E1 and E2 for either limb. C, sham‐treated animals with SCI exhibited significant change in paw area. LHL paw area significantly increased during E1–E3 at slow speed. At the faster speed, paw area for the LHL significantly decreased during E1–E3. Rising slope significantly decreased during E1 at the slow speed for the LHL. At the faster speed, falling slope significantly decreased during E1–E3 for the LHL. D, a‐tsDCS‐treated animals exhibited significant changes in paw area. Paw area significantly increased during E1–E3 at the slow speed for the LHL, whereas this only occurred during E1 and E2 for the RHL. Paw area significantly increased during E1 at the faster speed for both the LHL and the RHL. Rising slope significantly increased at the slow speed during E1 and E2 for the RHL. Rising slope significantly increased during E1 at the faster speed only for the RHL. Falling slope significantly increased during E1 and E2 at the slow speed for the LHL and the RHL. Falling slope significantly increased during E1 at the faster speed only for the RHL. E, c‐tsDCS‐treated animals exhibited significant changes in paw area. Paw area significantly increased during E1 at the slow speed compared to baseline. Falling slope significantly increased during E1 and E2 for the LHL, and only during E1 for the RHL. * P < 0.05, Holm–Sidak method. Data are presented as the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
The non‐injured control animals showed no difference in paw area, rising slope or falling slope between the two evaluations that were performed for each of the two hind limbs (P > 0.05; two‐tailed paired t test). By contrast, the sham‐treated animals exhibited significant decreases in paw area for the left hindleg (LHL), yet not for the right hindleg (RHL), at the slower speed during the E1–E3 evaluations (RM ANOVA, F = 4.75, P = 0.009). At the faster speed, paw area significantly decreased at all of the evaluations (E1–E3) for the LHL (RM ANOVA, F = 5.54, P = 0.005), whereas paw area did not significantly change for the RHL. Rising slope significantly decreased during E1 at the slow speed for the LHL (RM ANOVA, F = 3.20, P = 0.039) and not for the RHL. By contrast, rising slope did not change significantly at the faster speed for both hindlimbs. Similarly, falling slope did not significantly decrease for either hindlimb at the slow speed, yet it significantly decreased during all of the evaluation points at the faster speed for the LHL (RM ANOVA, F = 4.47, P = 0.012) but not for the RHL (P < 0.05, Holm–Sidak method). The a‐tsDCS‐treated animals exhibited a significant increase in paw area for the LHL during all of the evaluations at slow speed (E1–E3) and only during E1 and E2 for the RHL (RM ANOVA for LHL: F = 12.70, P < 0.001; RM ANOVA for RHL: F = 17.32, P < 0.001). Paw area also significantly increased during E1 at the faster speed for both the LHL (RM ANOVA, F = 7.04, P < 0.001) and the RHL (RM ANOVA, F = 13.32, P < 0.001). Rising slope significantly increased at the slow speed during E1 and E2 for the RHL (RM ANOVA, F = 6.06, P = 0.002) but not for the LHL. Meanwhile, at the faster speed, rising slope only significantly increased during E1 for the RHL (RM ANOVA, F = 4.19, P = 0.011). Falling slope significantly increased during E1 and E2 at the slow speed for both the LHL (RM ANOVA, F = 10.03, P < 0.001) and the RHL (RM ANOVA, F = 13.48, P < 0.001). Meanwhile, falling slope significantly increased during E1 at the faster speed only for the RHL (RM ANOVA, F = 9.06, P < 0.001) (P < 0.05, Holm–Sidak method). In the c‐tsDCS‐treated animals, a significant increase in paw area was observed during E1 at the slow speed compared to baseline (RM ANOVA for LHL: F = 7.92, P < 0.001; RM ANOVA for RHL: F = 8.46, P < 0.001), whereas no change in paw area was observed during E2 or E3. There was also no significant change in paw area at any of the evaluation points at the faster speed, while rising slope remained unchanged at both the slow speed and the faster speed. By contrast, falling slope significantly increased during E1 and E2 for the LHL (RM ANOVA for slow speed: F = 6.72, P < 0.001) and only during E1 for the RHL (RM ANOVA for slow speed: F = 3.47, P = 0.025) (P < 0.05, Holm–Sidak method). At the faster speed, there was no significant change in falling slope.
a‐tsDCS treatment enhances recovery of skill locomotion
A computer‐controlled ladder wheel (Fig. 4 A) was used to investigate the effect of repeated tsDCS on skill locomotion. Mixed two‐way repeated measures ANOVA (variables: time and limb side) was applied to assess improvements in skill locomotion. Significant differences in skill locomotion were observed across the four time points examined in the anode‐treated group (F = 6.49, P = 0.002). The RHL of the anode‐treated group had a significantly higher mean score during E2 compared to E1 (P = 0.001, Bonferroni), whereas the LHL had a significantly higher score during E2 and E3 compared to E1 (P < 0.01, Bonferroni) (Fig. 4 A). However, significant differences were not observed for any of the other groups across the four time points (P > 0.05, Bonferroni).
Figure 4. Enhancement of motor control recovery following repeated anode treatment.
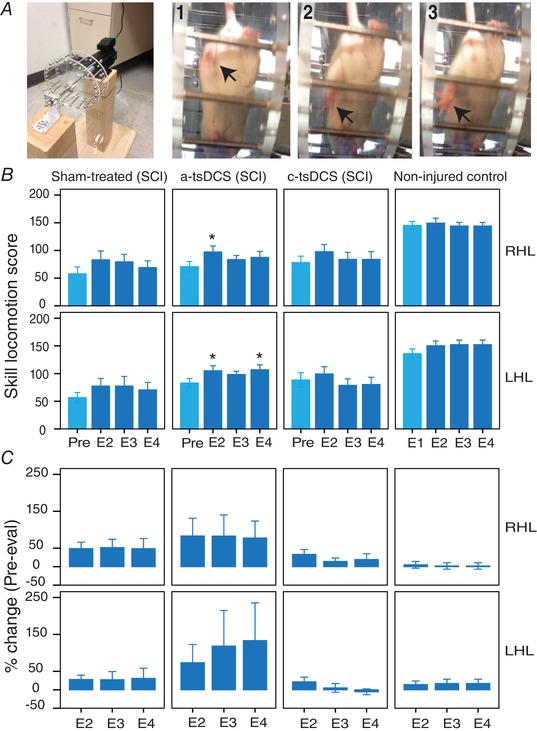
A, left: image of the ladder‐wheel; right: consecutive frames (1‐3) showing a total miss by a left hind limb. B, mean scores of skilled locomotion for the four time points evaluated (see Methods). Sham, SCI group that was sham‐treated; a‐tsDCS, SCI group that was treated daily for 7 days with a‐tsDCS; c‐tsDCS, SCI group that was treated daily for 7 days with c‐tsDCS; non‐injured control, no injury/no treatment;. C, percentage change from pre‐treatment evaluations or first evaluations for the healthy controls (Pre‐eval) to E2‐E4. Data are presented as the mean ± SEM. * P < 0.05 compared to Pre‐evaluation. [Color figure can be viewed at wileyonlinelibrary.com]
Next, we compared percentage change (from pre‐evaluation) between the groups (Fig. 4 B). Mixed two‐way repeated measures ANOVA detected significant main effects among the groups (F = 5.97, P = 0.016). For example, the anode‐treated group exhibited a significantly higher percentage change in their scores compared to the cathode‐treated group (P = 0.027, Bonferroni) and the healthy group (P = 0.048, Bonferroni). For the anode‐treated group, a higher percentage change was observed compared with the sham group, although the change was not significant (P > 0.05). There were no significant differences among the other groups (P > 0.05). Overall, these findings reveal that enhanced recovery of motor control of skill locomotion occurred in the animals that were treated with a‐tsDCS.
RDD of H‐reflex following tsDCS treatment
Mixed two‐way repeated measures ANOVA was also conducted to assess changes in RDD. The treatment conditions (e.g. independent factor, four levels: non‐injured control, sham, anode and cathode) resulted in significant differences in the main effect among the different levels of treatment (F = 3.0, P = 0.038). In addition, frequency was identified as a significant main effect (within‐subjects repeated factor, five levels: 0.1, 0.5, 1, 2 and 5 Hz) (F = 13.8, P < 0.001). However, there was no statistically significant interaction observed between frequency and treatment condition (F = 1.35, P = 0.192). Thus, the effects from different levels of treatment do not depend on what level of frequency is present.
A multiple comparison test (Tukey) was applied to identify which group(s) differ from the others. As shown in Fig. 5, the mean scores of RDD at frequencies of 1, 2 and 5 Hz were significantly higher compared to the RDD score at 0.1 Hz (5 Hz vs. 1 Hz, P < 0.001; 4 Hz vs. 1 Hz, P = 0.006; 3 Hz vs. 1 Hz, P = 0.024). Within the sham group, the mean scores of RDD at all of the frequencies did not significantly differ compared to the RDD score at 0.1 Hz (P > 0.05). Within the anode‐treated group, the mean scores of RDD at frequencies of 2 and 5 Hz were significantly higher compared to the RDD score at 0.1 Hz (5 Hz vs. 1 Hz, P < 0.001; 4 Hz vs. 1 Hz, P < 0.001), whereas the mean RDD score at 1 Hz within the cathode‐treated group was significantly higher compared to the mean RDD at 0.1 Hz (P = 0.017).
Figure 5. RDD of the Hoffman reflex after repeated sessions of tsDCS stimulation.
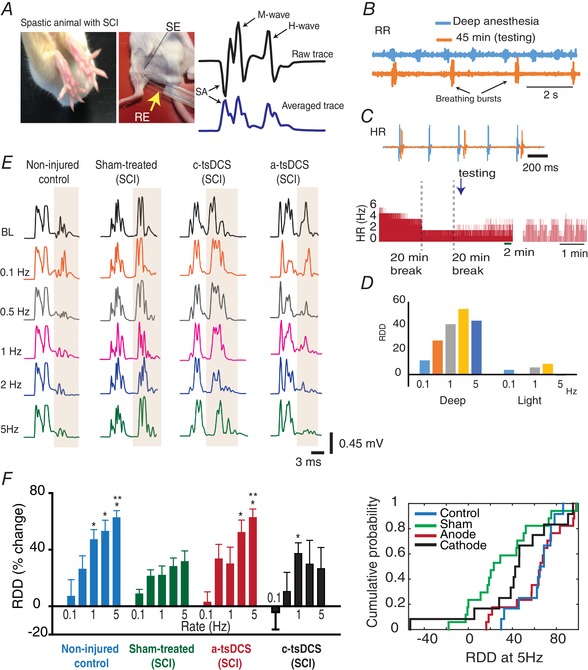
A, left: observable characteristics of spasticity (fanning of toes) in an animal with SCI; right: recording set‐up: SE, stimulating electrode; RE, recording electrode; SA, stimulus artefact. The two recording traces shown at the far right represent a raw trace (top) and the root‐mean square calculated trace (bottom). B, respiratory rate (RR) during deep anaesthesia (blue) and 45 min after an anaesthetic injection (orange) are shown. C, top: heart rate (HR) during deep anaesthesia (blue) and 45 min after an anaesthetic injection (orange) are shown; bottom: full recording, with breaks, showing the HR at which RDD testing took place. D, changes that occurred when RDD was tested during deep anaesthesia (Deep) vs. light anaesthesia (Light). E, left: representative recordings from the groups indicated. F, left: average RDD for the groups indicated. RDD was significantly higher in the control group (n = 12) for rates 1–5 Hz compared to rate 0.1 Hz (P < 0.001); right: cumulative probability distribution of the groups indicated for RDD at 5 Hz. *Significant difference from RDD at 0.1 Hz in the same group; **Significant compared to respective RDD in the sham group. Data are presented as the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Between‐group comparisons (Tukey) were performed and significant differences in RDD scores at 5 Hz were observed. For example, the RDD score at 5 Hz was significantly higher in the anode‐treated group compared to the scores for the sham‐ and cathode‐treated groups (P < 0.05). Meanwhile, the non‐injured control group had a significantly higher RDD score at 5 Hz compared to the cathode‐treated group (P = 0.016). Overall, these findings provide support for a physiological mechanism to underlie the observed reduction in spasticity that was observed following repeated anode stimulation.
Expression of KCC2, p‐KCC2, NKCC1 and p‐NKCC1 following repeated tsDCS treatment
Changes in KCC2 protein levels were investigated in the lumbosacral region of the spinal cord below the site of injury 4 weeks after the animals received 7 days of repeated tsDCS treatment. Reductions in KCC2 and pKCC2 protein levels were observed following cathodal and a‐tsDCS treatments compared to the non‐injured control group (Fig. 6 A). However, there were no significant differences between the protein levels among the groups (P > 0.05). The level of the co‐transporter, NKCC1, was also examined (Fig. 6 B). Kruskal–Wallis one‐way ANOVA detected significant differences between the median values among the treatment groups for NKCC1 (H = 31.4, P < 0.001). In the non‐injured control healthy animals, low levels of NKKC1 expression were detected in their lumbosacral spinal cord regions. By contrast, the levels of NKCC1 protein in the sham‐treated animals with SCI and spasticity and in the cathodal‐treated groups were significantly higher compared to the control healthy group (P < 0.05, Dunn's method). In the anodal‐treated group, a lower level of NKCC1 protein was detected, and the difference between this level and that of the non‐injured control group was not significant (P > 0.05, Dunn's method).
Figure 6. Changes in expression detected for KCC2, p‐KCC2, NKCC1 and p‐NKCC1 following repeated tsDCS.
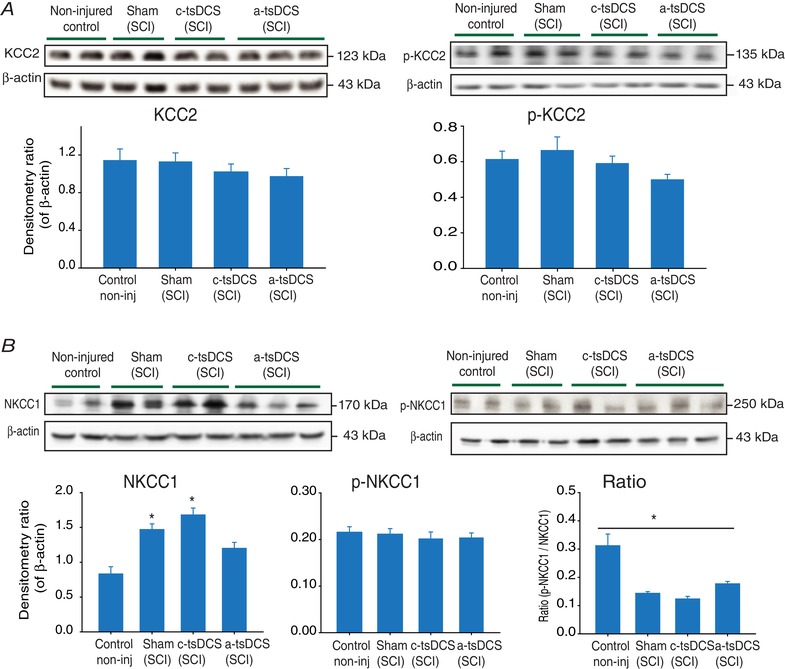
Representative animals from the experimental groups were randomly selected to investigate these molecular changes. A, left: KCC2 expression was unchanged in the sham‐treated group and in the anode‐ and cathode‐treated groups compared to the control group; right: pKCC2 expression was unchanged in the sham‐treated group and in the cathode‐treated and anode‐treated groups compared to the control group. B, NKCC1 expression was significantly increased in the sham‐treated and cathode‐treated groups, whereas the level in the anode‐treated group did not differ from that of the control group. P‐NKCC1 expression was not changed between groups. Detection of β‐actin was included as a loading control. * P < 0.05. Data are presented as the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Next, we investigated levels of phosphorylated NKCC1 (p‐NKCC1). Among the groups examined, there were no differences in p‐NKCC1 levels. However, when the ratio of p‐NKCC1 to total NKCC1 was calculated, one‐way ANOVA detected a statistically significant difference among the treatment groups (F = 16.4, P < 0.001), with the anodal‐treated group (0.17 ± 0.01, P < 0.001), the cathodal‐treated group (0.12 ± 0.01, P < 0.001) and the sham‐treated group (0.14 ± 0.01, P < 0.001) exhibiting significantly lower ratios compared to the non‐injured control group (0.31 ± 0.04) (Holm–Sidak method).
To establish a temporal association between stimulation and expression of NKCC1, the immediate effect of stimulation on NKCC1 expression was investigated in intact animals (Fig. 7). Briefly, three groups of animals received either anodal, cathodal or sham tsDCS treatments and then spinal cord samples from the stimulated sites were collected 2.5 h later. The stimulation‐induced changes in NKCC1 expression were similar to those observed in the animals that received repeated treatment as described above (Fig. 7). Moreover, the absolute value of p‐NKCC1 was slightly increased in the cathodal‐treated group, and was slightly reduced in the anodal‐treated group, compared to the sham‐treated group. When the ratio of p‐NKCC1 to total NKCC1 was calculated, one‐way ANOVA detected a statistically significant difference among the treatment groups (F = 5.96, P = 0.006). For example, the ratios for the anodal‐treated and cathodal‐treated groups were 0.77 ± 0.18 (P = 0.01) and 0.28 ± 0.5 (P = 0.8), respectively. In comparison, the ratio for the sham‐treated group was 0.31 ± 3.6 (Holm–Sidak method). The above data were confirmed using immunohistochemistry staining of spinal motor neurons (Fig. 7 A). The data shows a significant reduction in NKCC1 signal in motor neurons after a‐tsDCS, and an increase after cathodal‐tsDCS compared to sham condition. Taken together, these data indicate a possible mechanism by which a‐tsDCS may cause long‐term changes in spinal cord excitability.
Figure 7. NKCC1 and p‐NKCC1 expression following one stimulation session in intact mice.
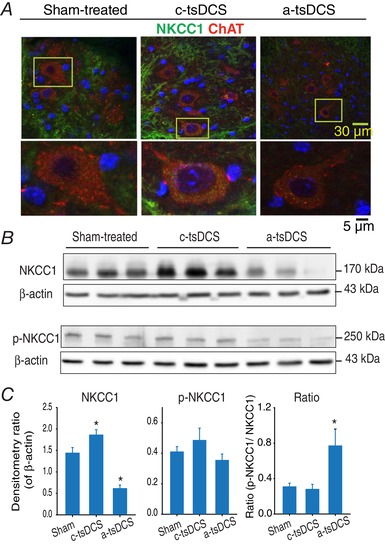
A, confocal images of spinal motor neurons stained for NKCC1 (green) and ChAT (red). Motor neurons showed an increase and decrease in NKCC1 after cathodal and a‐tsDCS respectively. B, western blots performed for samples collected 2.5 h after stimulation to detect NKCC1 and p‐NKCC1. Higher levels and lower levels of NKCC1 were detected after cathodal vs. anodal stimulation, respectively, compared to the Sham samples. Detection of β‐actin was included as a loading control. C, band density data from (A), including calculations of p‐NKCC1/total NKCC1 ratios. * P < 0.05. Data are presented as the mean ± SEM. [Color figure can be viewed at wileyonlinelibrary.com]
Detection of NKCC1 mRNA
The effect of tsDCS on mRNA levels of NKCC1 was also examined in the spinal cord tissues that were collected 2.5 h after stimulation. According to one‐way ANOVA, the mean values among the treatment groups differed significantly (F = 3.8, P = 0.03). It was observed that a‐tsDCS did not cause a significant change in NKCC1 mRNA levels (0.85 ± 0.37, P = 0.45, n = 14), whereas c‐tsDCS led to a significant increase in NKCC1 mRNA levels (1.14 ± 0.49, P = 0.03, n = 15) compared to the sham‐treated group (0.74 ± 0.22; n = 12). These results are consistent with the protein expression data described above.
a‐tsDCS induces an HSP70 response
As described above, a‐tsDCS reduced expression of NKCC1 protein, yet did not affect the level of NKCCC1 mRNA. These results suggested involvement of a degradation process, and HSP70 has been shown to enhance proteasomal and lysosomal pathways of protein degradation (Reeg et al. 2016). Therefore, expression of HSP70 was investigated following a‐tsDCS to strengthen and explain the qPCR and western blot data for NKCC1 levels. For these experiments, intact anaesthetized animals received one session of a‐tsDCS and target tissues were collected 2 h later. A robust and significant increase in HSP70 expression was induced in the anodal‐treated group compared to the sham‐treated group (P = 0.03, t test) (Fig. 8). Immunofluorescence staining of motor neurons further demonstrated that an overall increase in HSP70 expression occurred following anodal treatment. These stainings showed localization of HSP70 in and around nuclei, in the cytoplasm, and in dendrites. In addition, staining with ChAT was performed to identify spinal motor neurons and DAPI staining identified nuclei (data not shown) (Fig. 7).
Figure 8. a‐tsDCS‐induced HSP70 response.
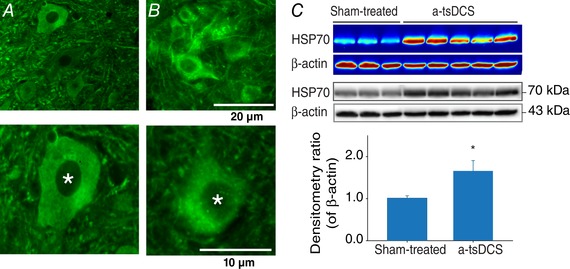
Because a‐tsDCS led to a reduction in NKCC1 protein levels and not NKCC1 mRNA levels, the potential for HSP70 to mediate the degradation of NKCC1 was investigated. A, example of spinal cord sections from sham‐treated non‐injured animals. B, example of spinal cord sections from non‐injured anaesthetized animals received one session of a‐tsDCS. Samples were stained for HSP70 (green). A and B, top: motor neuron nuclei; bottom: enlarged images of motor neurons. Asterisks indicate cell nuclei. Scale bars are indicated. C, western blots showed a significant and consistent increase in HSP70 expression in the a‐tsDCS‐treated animal samples vs. the sham‐treated samples (* P = 0.03). Detection of β‐actin was performed as a loading control. Both colour maps and the original blots are shown. Bottom: mean band densities for HSP70 relative to β‐actin in the sham‐treated and a‐tsDCS‐treated samples. [Color figure can be viewed at wileyonlinelibrary.com]
Bumetanide, a NKCC1 blocker, reduced spasticity in animals with SCI
As described above, NKCC1 was found to be overexpressed in animals with spasticity, and levels of NKCC1 normalized after repeated a‐tsDCS. Therefore, the effect of bumetanide, a NKCC1‐specific blocker, on animals with spasticity following SCI was investigated. Bumetanide was tested at two concentrations: 30 and 60 mg kg−1. One hour after bumetanide was administered at 30 mg kg−1 (n = 5), moderate reductions in spasticity were observed (data not shown). Following the administration of 60 mg kg−1 bumetanide, peak muscle resistance and EMG were measured. One‐way RM ANOVA detected significant differences among the various time points assayed (e.g. 5, 20, 30 and 45 min after the injection) (F = 83.8, P < 0.001). A multiple comparison test (Holm–Sidak method) also showed a significant reduction in peak resistance (34%) and corresponding EMG at all of the time points assayed compared to the baseline measurements (P < 0.001) (Fig. 9). Thirty minutes after the bumetanide injection, slow stretches were also observed to reduce background activity and resting muscle tone, whereas these aspects were increased in the spastic animals (Fig. 9 A and C). Taken together, these findings establish a strong link between high levels of NKCC1 expression and spasticity. Furthermore, when non‐injured control animals were injected with 60 mg kg−1 bumetanide, 21% of the animals exhibited a reduced reflexive stretch response. Thus, NKCC1 may also have functional consequences for spinal cords in normal mice.
Figure 9. The NKCC1 blocker, bumetanide, reduced spasticity in animals with SCI.
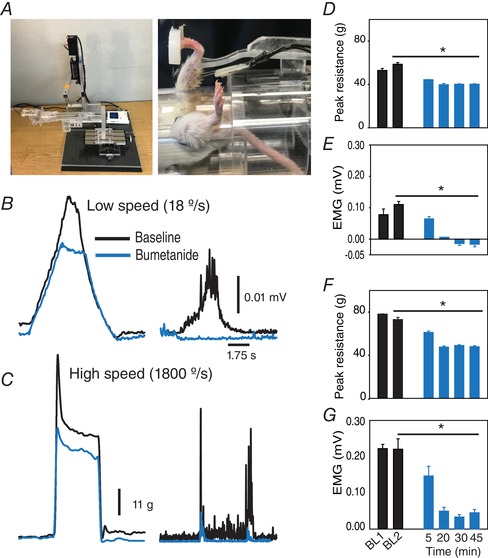
A, photographs of a stretch apparatus and mouse holder used to perform these experiments. Note that this apparatus is a modified version from the device in Figure 1. B and C, representative traces of muscle resistance (left) and concurrent EMG (right) at low speed (B) and high speed (C). D–G, decreases in muscle resistance and concurrent EMG at low speed and high speed, respectively, in each case, following bumetanide injection (n = 9 mice). The negative values of EMG indicate stretches caused depression of EMG relative to background activity prior to stretching. Data are presented as mean ± SEM. * P < 0.05 compared to baseline 2 [BL2 was measured 5 min following the first baseline (BL1)]. [Color figure can be viewed at wileyonlinelibrary.com]
Direct linkage between NKCC1 expression, spinal pre‐synaptic inhibition and a‐tsDCS‐induced reduction of spasticity
To provide direct causal relationship between a‐tsDCS‐induced reduction in spasticity and NKCC1 expression, we performed experiments in which we used TNF‐α or MG‐132 to block the degradation. Both TNF‐α and MG‐132 were shown to prevent the degradation of NKCC1 (Ding et al. 2014; Huang et al. 2014; Pozdeev et al. 2017); therefore, we injected mice intrathecally with TNF‐α and MG‐132 30 min before a‐tsDCS application. The peak reflex stretch force was tested before a‐tsDCS and immediately after and 10, 30, 60 and 120 min after the cessation of stimulation. As shown in Fig. 10, in animals injected with TNF‐α or MG‐132, the peak resistance was returned to baseline 2 h after the cessation of a‐tsDCS (Fig. 10 C) compared to a‐tsDCS only (Fig. 10 D). Note that Fig. 10 D compares responses from the three groups to the fastest speed only. These findings make direct connection between a‐tsDCS induced reduction of NKCC1 expression and a reduction of spasticity by a‐tsDCS. Animals were then killed and spinal tissues were collected for further analysis using western blotting to quantify the level of NKCC1. As shown in Fig. 10 A and B, both MG‐132 and TNF‐α prevented the effect of a‐tsDCS on NKCC1 expression, as seen in Figs 6 and 7. Expression of NKCC1 in SCI animals injected with MG‐132 or TNF‐α combined with a‐tsDCS was 272.7% of non‐injured control. Sham‐treated (SCI) group expression of NKCC1 was 285.9% of non‐injured control.
Figure 10. Mechanism of action of a‐tsDCS.
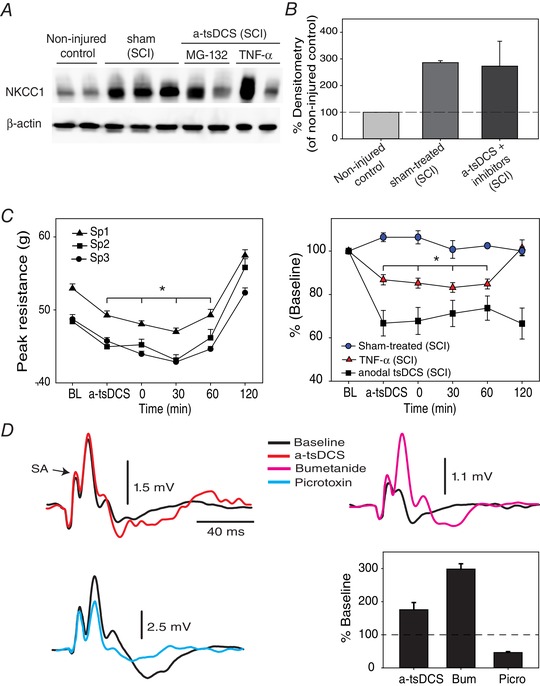
A, intrathecal injection of MG‐132 and TNF‐α blocked a‐tsDCS induced‐reduction of NKCC1 expression. Control, non‐injured and non‐stimulated mice; sham, injured but sham‐stimulated; a‐tsDCS, injured and stimulated. SCI indicates animals group with spinal cord injury. B, summary graph showing averages from different groups. C, blocking degradation of NKCC1 prevents a‐tsDCS induced reduction of spasticity. Animals with spasticity received intrathecal injection of TNF‐α or MG‐132 (n = 5) 30 min before applying a‐tsDCS and were then tested for spasticity. Left: anodal caused reduction of peak resistance that was significantly lower than baseline; however, peak resistance in response to three speeds of stretch returned exceeding baseline 2 h following the end of 20 min stimulation. Right: comparison between three groups with SCI and spasticity (speed 3 only): sham‐treated was not injected or stimulated; TNF‐α was injected and stimulated; and stimulated but not injected with TNF‐α or MG‐132. Note that these experiments were performed using the stretch device in Fig. 9 A. D, modulation of DRP by a‐tsDCS and bumetanide. DRP traces are shown on the left and top right. Bottom right: DRP was increased by a‐tsDCS and by bumetanide but reduced by picrotoxin. Bum, bumetanide; Picro, picrotoxin. [Color figure can be viewed at wileyonlinelibrary.com]
Next, we performed electrophysiological experiments to investigate the effect of a‐tsDCS and bumetanide on spinal presynaptic inhibition. As shown in Fig. 10 D, DRP were recorded before and 1 h following a‐tsDCS. a‐tsDCS significantly increased DRP (baseline median, 3.37 vs. after a‐tsDCS median, 1.74 mV; P = 0.008; Mann–Whitney rank sum test). Moreover, local application of bumetanide on the exposed spinal cord significantly increased DRP (main baseline, 0.62 ± 0.2 mV vs. main after bumetanide, 2.08 ± 0.2 mV, P < 0.001, t test). To provide evidence that DRP are GABAergic in nature, we applied picrotoxin (GABAA antagonist), which caused significant reduction in DRP amplitude (baseline median, 5.01 mV vs. after picrotoxin median 2.1 mV; P < 0.001; Mann–Whitney rank sum test). Taken together, the results provide direct evidence linking a‐tsDCS action to its downregulating effect on NKCC1 consequently enhancing spinal inhibition.
Discussion
In the present study, the long‐term effects of administering tsDCS for 20 min day–1 for 7 days were investigated in a mouse model of spasticity induced by SCI. Mice treated with a‐tsDCS exhibited significant reductions in spasticity compared to the other groups (Fig. 2). Ground and skill locomotion also significantly improved in the a‐tsDCS‐treated animals following treatment. Furthermore, these functional improvements occurred on a similar time scale increased walking parameters indicative of reduced spasticity, and reduced NKCC1 protein expression. Additionally, electrophysiological measurements following E3 revealed significant increase in RDD in a‐tsDCS treat animals, suggesting restoration of spinal inhibition. These results, in combination with the observations that spasticity was reduced following bumetanide treatment and NKCC1 levels were reduced after one session of anodal tsDCS, suggest that NKCC1 has an important role in the ability of a‐tsDCS to reduce spasticity.
During the gait cycle, when muscles are being rapidly lengthening, spasticity results in the untimely activation of muscles (Lamontagne et al. 2001) causing gait abnormalities (Krawetz & Nance, 1996). In the present study, to assess the effects of tsDCS treatment on locomotion‐related spasticity (Fig. 3), three parameters of the gait‐cycle were investigated: peak paw area, rising slope and falling slope. These parameters were examined based on their sensitivities to changes in the stance phase of the gait cycle. The rising slope corresponds to the progressive lowering of the foot to the ground as the animal shifts its body weight to that foot. The shifting continues until midstance, which is the point at which the middle of the stance phase is achieved, and the foot is flat on the ground. Midstance is also when the second parameter, peak paw area, is measured. Falling slope then follows the end of midstand, and it begins as the heel starts to elevate (heel off) and continues as the foot gradually prepares for push off to propel the animal forward. During most of the stance phase, the TS muscles undergo progressive lengthening as the ankle flexes and the body moves forward (Lamontagne et al. 2001). In spastic animals, this lengthening triggers untimely locomotion‐interfering reflexive contractions as a result of impaired reflex modulation that is associated with spasticity (Faist et al. 1999). Therefore, the significant increases in the falling and rising slopes that were observed following anodal treatment (Fig. 3 D, middle and lower) indicate that an enhanced transition and a decreased transition time occurred. A reduction in the peak paw area in the DigiGait locomotion test is an indication of abnormality in the paw placement, which can be caused by locomotion related spasticity as it occurs in human with spasticity (Lamontagne et al. 2001). The peak paw area for a subset of these animals increased during midstance at both of the speeds tested (Fig. 3 D, top) compared to the sham‐treated animals (Fig. 3 C, top). In the injured sham‐treated group (Fig. 3 C), peak area of the LHL was progressively reduced, which is an indication of developing locomotion‐related spasticity. This conclusion is supported by the parallel reduction of rising and falling slopes. The progressive reduction in peak area and falling slope in injured sham‐treat animals (Fig. 3 C) implies that a‐tsDCS not only reduced locomotion‐related spasticity, but also stopped its development in animals treated with a‐tsDCS. The discrepancy in the results with respect to peak paw area and rising and falling slopes between RHL and LHL (Fig. 3 C) could be because of differences in the initial status (Pre) between the two limbs.
Skill locomotion was tested with a ladder wheel and it was significantly improved in the animals treated with a‐tsDCS (Fig. 4). By contrast, the skill locomotion score for the non‐injured control animals did not improve over time, thereby indicating that learning was not possible in this task. Thus, the significant increases in the scores of the a‐tsDCS‐treated animals reflect true recovery of skill locomotion and this was training independent. Furthermore, given that skill locomotion in mice involves the brain (Farr et al. 2006), the results of the preaent study suggest that a‐tsDCS treatment may potentially uncover and/or strengthen certain corticospinal connections (Ahmed, 2011; Ahmed & Wieraszko, 2012; Zareen et al. 2017).
RDD involves an attenuation of H‐reflex amplitude following repetitive stimulations (Lloyd & Wilson, 1957; Ishikawa et al. 1966; Meinck, 1976) and it is reduced in spastic humans (Nielsen et al. 1993; Aymard et al. 2000) and animals (Lee et al. 2014b; Hedegaard et al. 2015). It is hypothesized that the mechanisms that underlie post‐activation depression involve a decrease in the probability that neurotransmitter quantal release from Ia afferent will occur as a result of its prior activation (Kuno, 1964a,b; Hirst et al. 1981; Lev‐Tov & Pinco, 1992). Post‐activation depression is functionally beneficial during voluntary movement because it lowers synaptic efficacy of Ia afferent, thereby preventing clonus and oscillations (Hultborn & Nielsen, 1998). Significant increases in RDD were observed in the anode group compared to the sham group following short‐term tsDCS (data not shown) and long‐term tsDCS (Fig. 6). The short‐term effect is consistent with functional modulation of DC current at the synaptic level (Eccles et al. 1962). The long‐term results observed in the present study may be explained by a persistent modification of synaptic strength following a‐tsDCS, and this could be achieved with restoration of spinal inhibition by reducing expression of NKCC1.
Following SCI, expression levels of KCC2 and NKCC1 were found to be altered (Cramer et al. 2008; Hasbargen et al. 2010; Lee et al. 2014a). Furthermore, an imbalance between these cotransporters can lead to spinal hyperexcitability, and hence, spasticity, spasms and/or pain (Hasbargen et al. 2010; D'Amico et al. 2014). In rats, SCI causes downregulation of KCC2, and this has been shown to correlate with spasticity (Boulenguez et al. 2010). In the present study, an insignificant reduction in the expression of KCC2 or p‐KCC2 was observed in the sham‐treated injured animals. Because this result is inconsistent with previously published data (Boulenguez et al. 2010), western blots were performed twice with all of the samples. In general, our KCC2 results are in agreement with previously reported results, showing no changes in the expression of KCC2 after SCI (Modol et al. 2014). It should also be noted that an anti‐phospho‐serine 940 KCC2 antibody was used in the present study. Phosphorylation at this site is mediated by protein kinase C and it enhances the stability of KCC2 at the cell surface and increases ion transport (Lee et al. 2007). Meanwhile, expression of NKCC1 was markedly elevated in our SCI animals without a significant change in the absolute level of p‐NKCC1.These results indicate that, in CD‐1 mice, an elevated level of NKCC1 expression is probably a crucial factor in modulating spinal excitability following SCI. This conclusion is supported by the observation that spasticity was reduced following treatment with the NKCC1‐specific blocker, bumetanide (Fig. 9).
To demonstrate a contemporaneous effect of tsDCS on NKCC1 expression, short‐term experiments were performed 2.5 h after a single session of stimulation was applied to anaesthetized non‐injured control animals. A robust polarity‐dependent effect of tsDCS was observed (Fig. 7) and neither injury, nor wakefulness was critical to this effect. Furthermore, a significant reduction in the level of NKCC1 was detected after anodal stimulation, and this was not accompanied by any significant change in the level of NKCC1 mRNA. These results suggest that a mechanism involving protein degradation should be considered, and this may be a Ca+2‐dependent process (Reynolds et al. 2007). Moreover, in our previous study, Ca+2 accumulation was found to significantly increase in nervous tissue following anodal stimulation (Wieraszko & Ahmed, 2016), and this may be a result of increased glutamate release in spinal cord tissue (Ahmed & Wieraszko, 2012). In the present study, no evidence of a direct link between calcium accumulation and NKCC1 degradation was investigated, although the simultaneous occurrence of the two events following anodal stimulation suggests a strong association. Previously, calcium‐dependent degradation of NKCC1 was observed (Reynolds et al. 2007), as well as a degradation of proteins following glutamate treatment (Guo & Wang, 2007). To strengthen this association, we investigated HSP70, a protein, which is known to enhance the flow of substrates through proteasomal or lysosomal pathways for degradation. In the present study, a‐tsDCS was found to induce a significant HSP70 response (Fig. 8). Thus, because a‐tsDCS increases glutamate release, increases calcium accumulation, induces an HSP70 response and downregulates NKCC1 protein, yet not its mRNA, this strongly suggests that a‐tsDCS‐induces protein degradation processes. Moreover, the results shown in Fig. 10, indicating that the proteasome inhibitor MG‐132 blocks the action of a‐tsDCS on spasticity by preventing anodal‐induced reduction of NKCC1, further strengthen our notion of a‐tsDCS‐induced protein degradation processes. However, further studies are needed to investigate the details of the signal transduction mechanisms potentially involved in mediating the actions of a‐tsDCS. Meanwhile, the observations that c‐tsDCS increased levels of NKCC1 protein and mRNA indicate that c‐tsDCS mediates a distinct mechanism involving de novo protein synthesis.
p‐NKCC1 is a dimer of the mature form of NKCC1 that has a molecular weight of ∼250 kDa. Because this form is distinct from NKCC1 based on molecular weight (MW, 135/170 kDa), p‐NKCC1 is not part of NKCC1 band. Currently, the role of phosphorylation in relation to the activity of NKCC1 in nervous tissue remains unknown. It has been demonstrated that upregulation of NKCC1 in brain tissue contributes to ischaemic damage in the absence of elevated NKCC1 activity by phosphorylation (Yan et al. 2003), thereby indicating that NKCC1 is an active form. Correspondingly, the upregulation of NKCC1 that was detected in the present study may be implicated in spinal disinhibition (Laird et al. 2004). Moreover, the absolute level of p‐NKCC1 remained unchanged between the groups examined, and a 34% reduction in reflexive muscle resistance was observed in the animals with SCI vs. a 21% reduction in non‐injured controls after bumetanide injection. These results support the hypothesis that NKCC1 is active in the absence of phosphorylation.
Pre‐synaptic inhibition is an important factor in regulating the monosynaptic reflexes (Eccles et al. 1963; Levy, 1977; Hackman et al. 1997). In vivo measurements of DRP have been used to estimate spinal presynaptic inhibition (Grunewald & Geis, 2014). In the present study, we found electrophysiological evidence that presynaptic inhibition was increased by a‐tsDCS, in agreement with the findings by Kaczmarek et al. (2017), and by bumetanide (NKCC1 blocker) (Fig. 10 E). Additionally, a‐tsDCS, and bumetanide reduce spasticity (Figs 2 and 9). Together with the results shown in Figure 10 A–D, it is demonstrated that downregulation of NKCC1 expression by a‐tsDCS is required for its action on spasticity.
In summary, the results of the present study demonstrate that a‐tsDCS can cause long‐term reduction in spasticity in CD‐1 mice with SCI. In addition, ground and skill locomotion were improved without additional training. We also show that increased expression of NKCC1 was associated with spasticity. A specific role for NKCC1 was further demonstrated via pharmacological inhibition of NKCC1 and establishing a direct relationship between downregulation of NKCC1 by a‐tsDCS and reduction of spasticity. Taken together, the results of the present study provide strong evidence that downregulation of NKCC1 expression has an important role in mediating the action of a‐tsDCS.
Additional information
Competing interests
ZA holds several patents related to tsDCS and is the scientific founder of PathMaker Neurosystems Inc., a company developing spinal stimulation systems in direct relationship with the present work. No other author has any conflicting interests relating to the work reported in this article.
Author contributions
Experiments were performed in the laboratory of ZA. ZA conceived and designed the study with inputs of repeated stimulation experiments from WM. WM, SB, SS, MH, PT, MA, AG, MM, MA and ZA contributed to data acquisition. ZA analysed the data, prepared the figures and wrote the original manuscript, and edited the final version of the manuscript. All authors read and approved the final version of the manuscript submitted for publication. All authors agree to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was funded by NIH grant, R21 NS088957‐01A1 to ZA.
Acknowledgements
We thank the staff at the Advanced Imaging facility. We also thank all staff of the Animal Facility at College of Staten Island, especially Joanne Niekrash and Anamaria Rodriguez.
Biography
Wagdy Mekhael received a clinical doctorate in physical therapy in 2009 and a PhD in neuroscience in 2017. He performed the current work as part of his PhD thesis in the laboratory of Zaghloul Ahmed. His research interests include identifying mechanisms by which direct current modulates neuronal plasticity. His current work aims to delineate the effects of combining direct current stimulation with other therapeutic interventions to treat central nervous system injuries. Wagdy is currently a doctoral lecturer at the College of Staten Island of the City University of New York.

Edited by: Ole Paulsen & Dario Farina
Linked articles This article is highlighted in Perspectives article by Gómez‐Soriano et al. To read this article, visit https://doi.org/10.1113/JP277740.
This is an Editor's Choice article from the 15 April 2019 issue.
References
- Ahmed Z (2011). Trans‐spinal direct current stimulation modulates motor cortex‐induced muscle contraction in mice. J Appl Physiol (1985) 110, 1414–1424. [DOI] [PubMed] [Google Scholar]
- Ahmed Z (2013). Effects of cathodal trans‐spinal direct current stimulation on mouse spinal network and complex multijoint movements. J Neurosci 33, 14949–14957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z (2014). Trans‐spinal direct current stimulation alters muscle tone in mice with and without spinal cord injury with spasticity. J Neurosci 34, 1701–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z (2017). Effects of cathodal trans‐spinal direct current stimulation on lower urinary tract function in normal and spinal cord injury mice with overactive bladder. J Neural Eng 14, 056002. [DOI] [PubMed] [Google Scholar]
- Ahmed Z & Wieraszko A (2012). Trans‐spinal direct current enhances corticospinal output and stimulation‐evoked release of glutamate analog, D‐2,3‐(3)H‐aspartic acid. J Appl Physiol (1985) 112, 1576–1592. [DOI] [PubMed] [Google Scholar]
- Aymard C, Katz R, Lafitte C, Lo E, Penicaud A, Pradat‐Diehl P & Raoul S (2000). Presynaptic inhibition and homosynaptic depression: a comparison between lower and upper limbs in normal human subjects and patients with hemiplegia. Brain 123, 1688–1702. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM & Popovich PG (2006). Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23, 635–659. [DOI] [PubMed] [Google Scholar]
- Bennett DJ, Gorassini M, Fouad K, Sanelli L, Han Y & Cheng J (1999). Spasticity in rats with sacral spinal cord injury. J Neurotrauma 16, 69–84. [DOI] [PubMed] [Google Scholar]
- Bolzoni F & Jankowska E (2015). Presynaptic and postsynaptic effects of local cathodal DC polarization within the spinal cord in anaesthetized animal preparations. J Physiol 593, 947–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose P, Parmer R & Thompson FJ (2002). Velocity‐dependent ankle torque in rats after contusion injury of the midthoracic spinal cord: time course. J Neurotrauma 19, 1231–1249. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean‐Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M & Vinay L (2010). Down‐regulation of the potassium‐chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med 16, 302–307. [DOI] [PubMed] [Google Scholar]
- Brown M & Matthews P (1966). On the subdivision of the efferent fibres to muscle spindles into static and dynamic fusimotor fibres In: Control and Innervation of Skeletal Muscle, ed. Dundee Andrew B., pp. 18–31. University of St Andrews, St Andrews. [Google Scholar]
- Cramer SW, Baggott C, Cain J, Tilghman J, Allcock B, Miranpuri G, Rajpal S, Sun D & Resnick D (2008). The role of cation‐dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain 4, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amico JM, Condliffe EG, Martins KJ, Bennett DJ & Gorassini MA (2014). Recovery of neuronal and network excitability after spinal cord injury and implications for spasticity. Front Integr Neurosci 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B, Frisina RD, Zhu X, Sakai Y, Sokolowski B & Walton JP (2014). Direct control of Na(+)‐K(+)‐2Cl(‐)‐cotransport protein (NKCC1) expression with aldosterone. Am J Physiol Cell Physiol 306, C66‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Kostyuk PG & Schmidt RF (1962). The effect of electric polarization of the spinal cord on central afferent fibres and on their excitatory synaptic action. J Physiol 162, 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Schmidt R & Willis WD (1963). Pharmacological Studies on Presynaptic Inhibition. J Physiol 168, 500–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faist M, Ertel M, Berger W & Dietz V (1999). Impaired modulation of quadriceps tendon jerk reflex during spastic gait: differences between spinal and cerebral lesions. Brain 122, 567–579. [DOI] [PubMed] [Google Scholar]
- Farr TD, Liu L, Colwell KL, Whishaw IQ & Metz GA (2006). Bilateral alteration in stepping pattern after unilateral motor cortex injury: a new test strategy for analysis of skilled limb movements in neurological mouse models. J Neurosci Methods 153, 104–113. [DOI] [PubMed] [Google Scholar]
- Grunewald B & Geis C (2014). Measuring spinal presynaptic inhibition in mice by dorsal root potential recording in vivo. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L & Wang Y (2007). Glutamate stimulates glutamate receptor interacting protein 1 degradation by ubiquitin‐proteasome system to regulate surface expression of GluR2. Neuroscience 145, 100–109. [DOI] [PubMed] [Google Scholar]
- Hackman JC, Holohean AM & Davidoff RA (1997). Role of metabotropic glutamate receptors in the depression of GABA‐mediated depolarization of frog primary afferent terminals. Neuroscience 81, 1079–1090. [DOI] [PubMed] [Google Scholar]
- Hasbargen T, Ahmed MM, Miranpuri G, Li L, Kahle KT, Resnick D & Sun D (2010). Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Ann N Y Acad Sci 1198, 168–172. [DOI] [PubMed] [Google Scholar]
- Hedegaard A, Lehnhoff J, Moldovan M, Grondahl L, Petersen NC & Meehan CF (2015). Postactivation depression of the Ia EPSP in motoneurons is reduced in both the G127X SOD1 model of amyotrophic lateral sclerosis and in aged mice. J Neurophysiol 114, 1196–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst GD, Redman SJ & Wong K (1981). Post‐tetanic potentiation and facilitation of synaptic potentials evoked in cat spinal motoneurones. J Physiol 321, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz KA, Lipson R, Noonan VK, Kwon BK & Mills PB (2017). Prevalence and effect of problematic spasticity after traumatic spinal cord injury. Arch Phys Med Rehabil 98, 1132–1138. [DOI] [PubMed] [Google Scholar]
- Huang LQ, Zhu GF, Deng YY, Jiang WQ, Fang M, Chen CB, Cao W, Wen MY, Han YL & Zeng HK (2014). Hypertonic saline alleviates cerebral edema by inhibiting microglia‐derived TNF‐alpha and IL‐1beta‐induced Na‐K‐Cl Cotransporter up‐regulation. J Neuroinflammation 11, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultborn H & Nielsen JB (1998). Modulation of transmitter release from Ia afferents by their preceding activity – a ‘postactivation depression’ In: Presynaptic Inhibition and Neural Control, ed. Rudomin P, Romo R. & Mendell L, pp. 178–191. Oxford University Press, New York, NY. [Google Scholar]
- Ishikawa K, Ott K, Porter RW & Stuart D (1966). Low frequency depression of the H wave in normal and spinal man. Exp Neurol 15, 140–156. [DOI] [PubMed] [Google Scholar]
- Kaczmarek D, Ristikankare J & Jankowska E (2017). Does trans‐spinal and local DC polarization affect presynaptic inhibition and post‐activation depression? J Physiol 595, 1743–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutsson E & Richards C (1979). Different types of disturbed motor control in gait of hemiparetic patients. Brain 102, 405–430. [DOI] [PubMed] [Google Scholar]
- Krawetz P & Nance P (1996). Gait analysis of spinal cord injured subjects: effects of injury level and spasticity. Arch Phys Med Rehabil 77, 635–638. [DOI] [PubMed] [Google Scholar]
- Kuno M (1964a). Mechansim of facilitation and depression of the excitatory synaptic potential in spinal motoneurones. J Physiol 175, 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno M (1964b). Quantal components of excitatory synaptic potentials in spinal motoneurones. J Physiol 175, 81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird JM, Garcia‐Nicas E, Delpire EJ & Cervero F (2004). Presynaptic inhibition and spinal pain processing in mice: a possible role of the NKCC1 cation‐chloride co‐transporter in hyperalgesia. Neurosci Lett 361, 200–203. [DOI] [PubMed] [Google Scholar]
- Lamontagne A, Malouin F & Richards CL (2001). Locomotor‐specific measure of spasticity of plantarflexor muscles after stroke. Arch Phys Med Rehabil 82, 1696–1704. [DOI] [PubMed] [Google Scholar]
- Lance JW (1990). What is spasticity? Lancet 335, 606. [DOI] [PubMed] [Google Scholar]
- Lee HH, Walker JA, Williams JR, Goodier RJ, Payne JA & Moss SJ (2007). Direct protein kinase C‐dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem 282, 29777–29784. [DOI] [PubMed] [Google Scholar]
- Lee HK, Ahmed MM, King KC, Miranpuri GS, Kahle KT, Resnick DK & Sun D (2014a). Persistent phosphorylation of NKCC1 and WNK1 in the epicenter of the spinal cord following contusion injury. Spine J 14, 777–781. [DOI] [PubMed] [Google Scholar]
- Lee S, Toda T, Kiyama H & Yamashita T (2014b). Weakened rate‐dependent depression of Hoffmann's reflex and increased motoneuron hyperactivity after motor cortical infarction in mice. Cell Death Dis 5, e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev‐Tov A & Pinco M (1992). In vitro studies of prolonged synaptic depression in the neonatal rat spinal cord. J Physiol 447, 149–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy RA (1977). The role of GABA in primary afferent depolarization. Prog Neurobiol 9, 211–267. [DOI] [PubMed] [Google Scholar]
- Lloyd DP & Wilson VJ (1957). Reflex depression in rhythmically active monosynaptic reflex pathways. J Gen Physiol 40, 409–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsala M, Hefferan MP, Kakinohana O, Nakamura S, Marsala J & Tomori Z (2005). Measurement of peripheral muscle resistance in rats with chronic ischemia‐induced paraplegia or morphine‐induced rigidity using a semi‐automated computer‐controlled muscle resistance meter. J Neurotrauma 22, 1348–1361. [DOI] [PubMed] [Google Scholar]
- Meinck HM (1976). Occurrence of the H reflex and the F wave in the rat. Electroencephalogr Clin Neurophysiol 41, 530–533. [DOI] [PubMed] [Google Scholar]
- Metz GA & Whishaw IQ (2002). Cortical and subcortical lesions impair skilled walking in the ladder rung walking test: a new task to evaluate fore‐ and hindlimb stepping, placing, and co‐ordination. J Neurosci Methods 115, 169–179. [DOI] [PubMed] [Google Scholar]
- Metz GA & Whishaw IQ (2009). The ladder rung walking task: a scoring system and its practical application. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld U, Deisz RA, Dodt HU & Lux HD (1986). The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science 232, 1413–1415. [DOI] [PubMed] [Google Scholar]
- Modol L, Mancuso R, Ale A, Francos‐Quijorna I & Navarro X (2014). Differential effects on KCC2 expression and spasticity of ALS and traumatic injuries to motoneurons. Front Cell Neurosci 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Petersen N, Ballegaard M, Biering‐Sorensen F & Kiehn O (1993). H‐reflexes are less depressed following muscle stretch in spastic spinal cord injured patients than in healthy subjects. Exp Brain Res 97, 173–176. [DOI] [PubMed] [Google Scholar]
- Njoo C, Heinl C & Kuner R (2014). In vivo SiRNA transfection and gene knockdown in spinal cord via rapid noninvasive lumbar intrathecal injections in mice. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozdeev VI, Lang E, Gorg B, Bidmon HJ, Shinde PV, Kircheis G, Herebian D, Pfeffer K, Lang F, Haussinger D, Lang KS & Lang PA (2017). TNFalpha induced up‐regulation of Na(+),K(+),2Cl(‐) cotransporter NKCC1 in hepatic ammonia clearance and cerebral ammonia toxicity. Sci Rep 7, 7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeg S, Jung T, Castro JP, Davies KJA, Henze A & Grune T (2016). The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radic Biol Med 99, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A, Parris A, Evans LA, Lindqvist S, Sharp P, Lewis M, Tighe R & Williams MR (2007). Dynamic and differential regulation of NKCC1 by calcium and cAMP in the native human colonic epithelium. J Physiol 582, 507–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio DC, Ares M Jr., Hannon GJ & Nilsen TW (2010). Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc 2010, pdb prot5439. [DOI] [PubMed] [Google Scholar]
- Samaddar S, Vazquez K, Ponika D, Toruno P, Sahbani K, Begum S, Abouelela A, Mekhael W & Ahmed Z (2017). Trans‐spinal direct current stimulation modulates migration and proliferation of adult newly‐born spinal cells in mice. J Appl Physiol (1985) 122, 339–353. [DOI] [PubMed] [Google Scholar]
- Song W, Amer A, Ryan D & Martin JH (2016). Combined motor cortex and spinal cord neuromodulation promotes corticospinal system functional and structural plasticity and motor function after injury. Exp Neurol 277, 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente V, Teixeira SA, Neder L, Okamoto OK, Oba‐Shinjo SM, Marie SK, Scrideli CA, Paco‐Larson ML & Carlotti CG Jr (2014). Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT‐PCR. Ann Neurosci 21, 62–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walder RY, Wattiez AS, White SR, Marquez de Prado B, Hamity MV & Hammond DL (2014). Validation of four reference genes for quantitative mRNA expression studies in a rat model of inflammatory injury. Mol Pain 10, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieraszko A & Ahmed Z (2016). Direct current‐induced calcium trafficking in different neuronal preparations. Neural Plast 2016, 2823735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Dempsey RJ, Flemmer A, Forbush B & Sun D (2003). Inhibition of Na(+)‐K(+)‐Cl(‐) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res 961, 22–31. [DOI] [PubMed] [Google Scholar]
- Zareen N, Shinozaki M, Ryan D, Alexander H, Amer A, Truong DQ, Khadka N, Sarkar A, Naeem S, Bikson M & Martin JH (2017). Motor cortex and spinal cord neuromodulation promote corticospinal tract axonal outgrowth and motor recovery after cervical contusion spinal cord injury. Exp Neurol 297, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]