

Abstract
Key points
The lateral superior olive (LSO), a brainstem hub involved in sound localization, integrates excitatory and inhibitory inputs from the ipsilateral and the contralateral ear, respectively. In gerbils and rats, inhibition to the LSO reportedly shifts from GABAergic to glycinergic within the first three postnatal weeks.
Surprisingly, we found no evidence for synaptic GABA signalling during this time window in mouse LSO principal neurons. However, we found that presynaptic GABABRs modulate Ca2+ influx into medial nucleus of the trapezoid body axon terminals, resulting in reduced synaptic strength. Moreover, GABA elicited strong responses in LSO neurons that were mediated by extrasynaptic GABAARs. RNA sequencing revealed highly abundant δ subunits, which are characteristic of extrasynaptic receptors.
Whereas GABA increased the excitability of neonatal LSO neurons, it reduced the excitability around hearing onset.
Collectively, GABA appears to control the excitability of mouse LSO neurons via extrasynaptic and presynaptic signalling. Thus, GABA acts as a modulator, rather than as a classical transmitter.
Abstract
GABA and glycine mediate fast inhibitory neurotransmission and are coreleased at several synapse types. Here we assessed the contribution of GABA and glycine in synaptic transmission between the medial nucleus of the trapezoid body (MNTB) and the lateral superior olive (LSO), two nuclei involved in sound localization. Whole‐cell patch‐clamp experiments in acute mouse brainstem slices at postnatal days (P) 4 and 11 during pharmacological blockade of GABAA receptors (GABAARs) and/or glycine receptors demonstrated no GABAergic synaptic component on LSO principal neurons. A GABAergic component was absent in evoked inhibitory postsynaptic currents and miniature events. Coimmunofluorescence experiments revealed no codistribution of the presynaptic GABAergic marker GAD65/67 with gephyrin, a postsynaptic marker for GABAARs, corroborating the conclusion that GABA does not act synaptically in the mouse LSO. Imaging experiments revealed reduced Ca2+ influx into MNTB axon terminals following activation of presynaptic GABABRs. GABABR activation reduced the synaptic strength at P4 and P11. GABA appears to act on extrasynaptic GABAARs as demonstrated by application of 4,5,6,7‐tetrahydroisoxazolo[5,4‐c]pyridin‐3‐ol, a δ‐subunit‐specific GABAAR agonist. RNA sequencing showed high mRNA levels for the δ‐subunit in the LSO. Moreover, GABA transporters GAT‐1 and GAT‐3 appear to control extracellular GABA. Finally, we show an age‐dependent effect of GABA on the excitability of LSO neurons. Whereas tonic GABA increased the excitability at P4, leading to spike facilitation, it decreased the excitability at P11 via shunting inhibition through extrasynaptic GABAARs. Taken together, we demonstrate a modulatory role of GABA in the murine LSO, rather than a function as a classical synaptic transmitter.
Keywords: Synaptic transmission, extrasynaptic signaling, modulatory function of GABA, GABA transporters, GABAAR modulator pentobarbital
Key points
The lateral superior olive (LSO), a brainstem hub involved in sound localization, integrates excitatory and inhibitory inputs from the ipsilateral and the contralateral ear, respectively. In gerbils and rats, inhibition to the LSO reportedly shifts from GABAergic to glycinergic within the first three postnatal weeks.
Surprisingly, we found no evidence for synaptic GABA signalling during this time window in mouse LSO principal neurons. However, we found that presynaptic GABABRs modulate Ca2+ influx into medial nucleus of the trapezoid body axon terminals, resulting in reduced synaptic strength. Moreover, GABA elicited strong responses in LSO neurons that were mediated by extrasynaptic GABAARs. RNA sequencing revealed highly abundant δ subunits, which are characteristic of extrasynaptic receptors.
Whereas GABA increased the excitability of neonatal LSO neurons, it reduced the excitability around hearing onset.
Collectively, GABA appears to control the excitability of mouse LSO neurons via extrasynaptic and presynaptic signalling. Thus, GABA acts as a modulator, rather than as a classical transmitter.
Introduction
Fast‐acting inhibitory neurotransmission in the vertebrate central nervous system is mainly achieved by glycine and γ‐aminobutyric acid (GABA). Glycine acts via ligand‐gated Cl− channels (GlyRs) comprising only two classes of subunits (α and β), its action thus being of a relatively uniform nature (Lynch, 2004, 2009; Betz & Laube, 2006). Metabotropic glycine receptors have not been identified in mammals (Tritsch et al. 2016). By contrast, GABAergic signalling appears to be much more complex, because it is mediated through heteropentameric ionotropic receptors (GABAA/CRs) composed of eight classes of subunits (α, β, γ, δ, ε, θ, π, ρ) with a total of 19 identified isoforms (Rudolph & Möhler, 2014; Smart, 2015; Naffaa et al. 2017). Moreover, GABA can act on G protein‐coupled receptors (GABABRs), which are also very heterogeneous, displaying pronounced diversity in subcellular location, functional properties and cellular signalling (Brenowitz et al. 1998; Xu et al. 2014; Schwenk et al. 2016). Apart from targeting GABAA/CRs and GABABRs in sub‐ and presynaptic regions, GABA can escape the synaptic cleft and bind to extrasynaptic receptors, thus exerting tonic effects via a non‐synaptic path (Otis et al. 1991; Barbour & Häusser, 1997; Farrant & Nusser, 2005; Lee & Maguire, 2014). Finally, GABA may be released not only from GABAergic neurons, but also from astrocytes, thereby operating as a gliotransmitter (Yoon & Lee, 2014; Gundersen et al. 2015).
GABA and GABARs are widely distributed in the CNS. By contrast, glycine is of greater importance in the brainstem and spinal cord (Smart & Paoletti, 2012). In several neural systems, glycine and GABA coexist in presynaptic axon terminals and in synaptic vesicles from which they can be released together (Ottersen et al. 1988; Todd et al. 1996; Jonas et al. 1998; O'Brien & Berger, 1999; Keller et al. 2001; Dugué et al. 2005; Crook et al. 2006; Dufour et al. 2010; Hirtz et al. 2012; Rahman et al. 2013; Nerlich et al. 2014; Ramakrishnan et al. 2014; Vaaga et al. 2014; Moore & Trussell, 2017). Therefore, mixed GABA–glycine inhibition has been implicated (Kotak et al. 1998; Dumoulin et al. 2001; Keller et al. 2001; Muller et al. 2006; Dufour et al. 2010; Apostolides & Trussell, 2013; Ishibashi et al. 2013; Nerlich et al. 2017; but see Hnasko & Edwards, 2012).
In the present study, we analysed GABAergic effects at fast inhibitory synapses in the auditory brainstem of mice. In mature animals, these synapses are glycinergic (Caspary & Finlayson, 1991) and tuned for resilience, reliability and temporal precision, even under sustained activation at stimulation frequencies >100 Hz (Kramer et al. 2014; Krächan et al. 2017). They connect the medial nucleus of the trapezoid body (MNTB) with the lateral superior olive (LSO) and form a crucial link in sound localization based on detecting interaural level differences (for a review, see Grothe & Pecka, 2014). Our analysis involved patch‐clamp recordings, pharmacology, coimmunolabelling, calcium imaging and RNA sequencing. We obtained no evidence for functional GABAergic synapses in the LSO or a shift from GABAergic to glycinergic neurotransmission, unlike findings in gerbils and rats (Kotak et al. 1998; Nabekura et al. 2004). Instead, our results point towards postsynaptic GABA effects at extrasynaptic GABAARs throughout development, mediating tonic activity rather than phasic inhibition. GABA also acts at MNTB axon terminals. Based on our results, it appears that ambient GABA modulates the excitability of LSO neurons. During the first postnatal week, when the internal Cl− concentration [Cl−]i is high and Cl− flux leads to depolarization (Kandler & Friauf, 1995; Ehrlich et al. 1999; Löhrke et al. 2005), GABA appears to increase the excitability. The opposite effect, namely suppression of excitation, occurs later in development, at times when Cl− flux leads to hyperpolarization. We conclude that GABA acts in concert with glycine at MNTB–LSO synapses in an activity‐dependent fashion and exerts a modulatory function, thus allowing fine‐tuning of the magnitude and duration of fast and phasic synaptic inhibition (Tritsch et al. 2016).
Methods
Ethical approval
Experiments were approved by the regional council according to the German Animal Protection Law (TSchG §4, Absatz 3) and followed the NIH Guide for the Care and Use of Laboratory Animals. Moreover, the authors understand, and the work conforms to, the principles and regulations of The Journal of Physiology (Grundy, 2015).
Animals
Most experiments were performed on C57BL/6N mice of both sexes that were raised in the animal facilities of the University of Kaiserslautern or purchased from Charles River (Sulzfeld, Germany). Animals were housed on a 12 h light–dark cycle with ad libitum access to food and water. The day of birth was designated as postnatal day (P) 0. Calcium imaging experiments were performed at the State University of New York at Buffalo on CBA/CaJ mice of both sexes which were raised in the local animal facilities. Analysis was done at P4 ± 1, P11 ± 1, or P60.
Electrophysiology
After decapitation, mouse brains were quickly removed from the skull and transferred into ice‐cold preparation solution, containing (in mM): 2.5 KCl, 1 MgCl2, 1.25 NaH2PO4, 2 sodium pyruvate, 3 myo‐inositol, 26 NaHCO3, 260 d‐glucose, 2 CaCl2 (pH 7.4, when bubbled with carbogen; 325 ± 10 mosmol/L). Coronal slices of 270 μm thickness were cut on a VT1200 S vibrating blade microtome (Leica Microsystems, Wetzlar, Germany). For recovery, they were incubated at 37°C for 55 min in artificial cerebral spinal fluid (ACSF) containing (in mM): 125 NaCl, 2.5 KCl, 1 MgCl2, 1.25 NaH2PO4, 2 sodium pyruvate, 3 myo‐inositol, 0.44 l‐ascorbic acid, 25 NaHCO3, 10 d‐glucose, 2 CaCl2 (pH 7.4, when bubbled with carbogen; 295 ± 5 mosmol/L). After another 0.5–8 h at room temperature, slices were transferred into the recording chamber. Whole‐cell patch‐clamp recordings (V hold = −70 mV) were performed with an Axioscope 2 FS microscope (Zeiss; Carl Zeiss Microscopy, Jena, Germany) or an Eclipse E600N (Nikon, Tokyo, Japan) both equipped with IR‐DIC optics (Zeiss Fluar ×5/0.25 ∞/0.17; Olympus LUMPlanFL N ×60/1.00 W ∞/0/FN26.5 (Olympus, Tokyo, Japan); Nikon ×4 CFI Achromat, 0.1 ∞; ×60 CFI Fluor W, 1.00 W ∞). Slices were continuously superfused with ACSF (1–2 ml/min, 22–27°C or 36 ± 1°C) and visualized with an Orca‐05G (Hamamatsu, Hamamatsu city, Japan) or a VX 44 (PCO computer optics) CCD camera. Principal LSO neurons were identified by their spindle‐shaped somata and electrophysiological properties (Sterenborg et al. 2010). Patch pipettes were pulled from glass capillaries (Science Products, Hofheim am Taunus, Germany) with a P‐87 horizontal puller (Sutter Instrument Co., Novato, CA, USA). Pipette resistances ranged from 3 to 6 MΩ when filled with internal solution containing (in mM): 140 potassium gluconate, 10 HEPES, 5 EGTA, 1 MgCl2, 2 Na2ATP, 0.3 Na2GTP (280 ± 10 mOsm/L). The liquid junction potential of 15.4 mV was corrected online. This internal solution was used for some P4 recordings (Figs 1, 2 and 8 A) and most P11 recordings (Figs 1, 2, 5, 7, 8 A, 10 and 11). Another set of P4 recordings (Figs 5, 7 and 12) was performed with a near‐physiological internal chloride concentration for this age, which contained (in mM): 110 potassium gluconate, 30 KCl, 10 HEPES, 5 EGTA, 1 MgCl2, 2 Na2ATP, 0.3 Na2GTP (285 ± 10 mOsm/L). In a subset of experiments (Figs 3 and 8 B) the chloride driving force was increased by using an internal solution with a high chloride concentration containing (in mM): 130 KCl, 10 HEPES, 5 EGTA, 1 MgCl2, 2 Na2ATP, 0.3 Na2GTP (280 ± 10 mOsm/L; uncorrected liquid junction potential of −3.5 mV). Recordings were obtained with an EPC9 or EPC10 amplifier and PatchMaster software (HEKA Elektronik, Lambrecht (Pfalz), Germany). Recordings were discarded if the series resistance R S changed >20%. The membrane resistance was determined as R M = R in − R S, with R in representing the input resistance. R S and R in were derived from partially compensated current traces during 1 Hz triplets of −5 mV voltage steps (Fig. 11 Bb).
Figure 1. Synaptic transmission at mouse MNTB–LSO inputs is mediated by glycine, not by GABA.
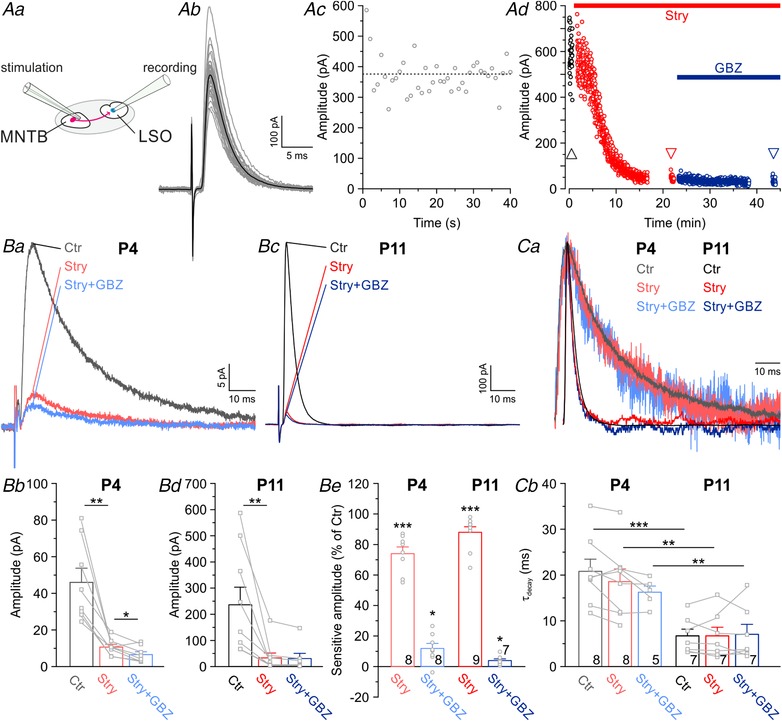
Aa, MNTB–LSO synapses were activated at 1 Hz by focal electrical stimulation of MNTB axons. Ab and c, evoked IPSCs (eIPSCs) at P11, demonstrating their robustness and stability. Grey traces represent overlays of 40 repetitive events, with the average in black. Dashed line in Ac marks mean peak amplitude. Ad, pharmacological characterization of a P11 LSO neuron. Subsequent blockade of glycine receptors (GlyRs) with strychnine (100 nM Stry) and GABAA receptors (GABAARs) with GABAzine (10 μM GBZ) abolished eIPSCs nearly completely. Ba and c, eIPSCs at P4 and P11, revealing a large glycinergic component, yet a virtually absent GABAergic component. Traces in Bc are from the same neuron shown in Ad, where arrowheads mark time points. Bb and d, statistical analysis of eIPSC peak amplitudes. Be, Stry and GBZ sensitivity of the peak amplitudes. Ca, scaled and peak‐aligned eIPSCs, revealing no drug‐induced change in decay kinetics. Same traces as in Ba and c. Cb, statistical analysis of τdecay.
Figure 2. GlyR‐mediated currents are diminished by GBZ.
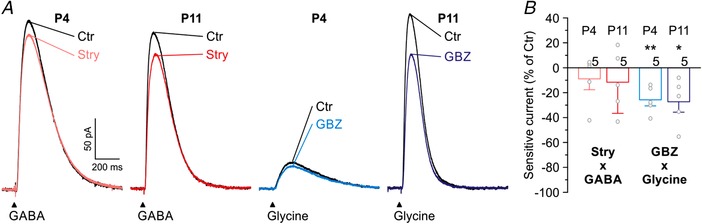
A, averaged current traces of four P4 and P11 LSO neurons (40 traces per neuron), evoked by application of GABA or glycine (100 μM; 100 ms puffs for 2 min at 0.2 Hz) in the absence or presence of receptor blockers. Black traces represent control conditions, and coloured traces represent recordings after 15 min wash‐in of 100 nM Stry or 10 μM GBZ. B, Stry‐sensitive and GBZ‐sensitive fraction of GABA‐ and glycine‐evoked currents, respectively. Strychnine at 100 nM had no effect on GABA‐evoked currents. In contrast, 10 μM GBZ reduced glycine‐evoked currents both at P4 and at P11 (20 cells, 5 at each age, 5 for each drug). Asterisks indicate differences from control.
Figure 8. LSO principal neurons possess extrasynaptic δ‐GABAARs.
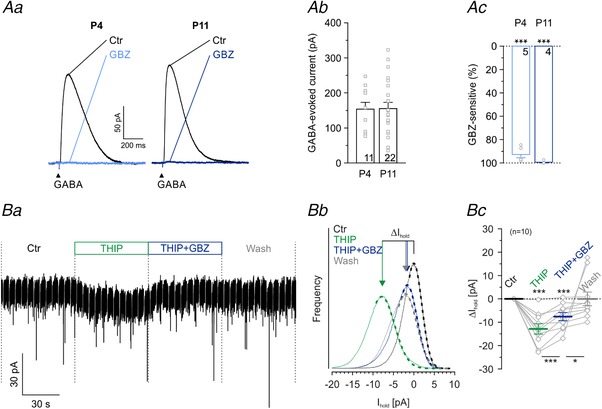
Aa, averaged current traces (n = 40) of a P4 and a P11 LSO neuron, evoked by GABA application (100 μM; 100 ms puffs for 2 min at 0.2 Hz). Black traces represent control conditions, and grey traces represent recordings after 15 min wash‐in of 10 μM GBZ. Ab, amplitude distribution of GABA‐evoked currents at P4 and P11. Ac, Sensitivity to GBZ at P4 and P11. Ba, current traces during puff application of the δ‐GABAAR agonist THIP (1 μM), demonstrating a long‐lasting inward current that was partially blocked with GBZ. Significance levels were Šidák corrected in case of two comparisons. Bb, same cell as in Ba. The holding current (I hold) was determined from Gaussian fits (dashed lines) to all‐point histograms (solid lines) of the different pharmacological conditions. Due to the high internal chloride concentration, both inhibitory and excitatory spontaneous synaptic current events are inward directed. To exclude those inward‐directed events, Gaussian fitting was done at the right side of the all‐point histograms. Bc, statistics of THIP‐evoked currents and sensitivity to GBZ (n = 10 cells). Asterisks above dashed line depict differences from control. Significance levels were Šidák corrected in case of two comparisons.
Figure 5. LSO principal neurons possess functional GABABRs.
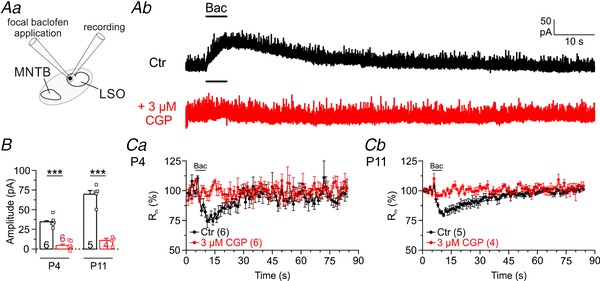
Aa, scheme of the experimental set‐up. Ab, original traces (P11) with focal baclofen application (10 μM, 5 s) at the indicated time points in control (Ctr, black,) and CGP (3 μM, red). B, statistical analysis of peak amplitude. C and D, time course of membrane resistance.
Figure 7. Activation of GABABRs decreases eIPSC amplitudes at MNTB–LSO synapses.
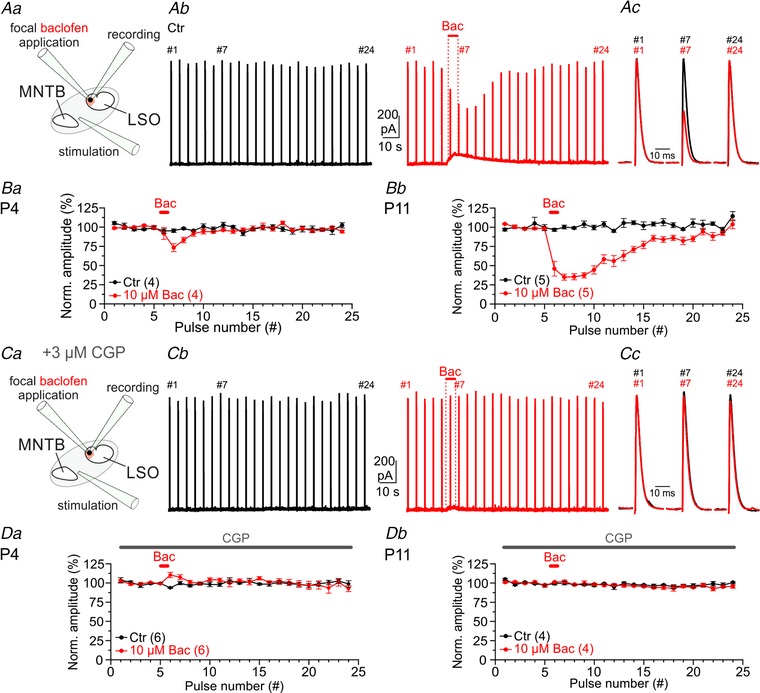
Aa, scheme of the experimental set‐up. Ab, MNTB fibre stimulation at P11 (24 pulses, 0.2 Hz) in control (Ctr, black) and focal baclofen application (10 μM Bac, 5 s, red). Traces are averages of three repeats. Ac, close ups of eIPSCs at the pulse no. 1, 7 and 24 also denoted in Ab. B, quantification of normalized eIPSC amplitudes (mean of first five pulses set to 100%) at P4 (Ba) and P11 (Bb). BC and D, same as A and B, but in the presence of bath‐applied 3 μM CGP.
Figure 10. Extracellular GABA is synergistically controlled by GABA transporters GAT‐1 and GAT‐3.
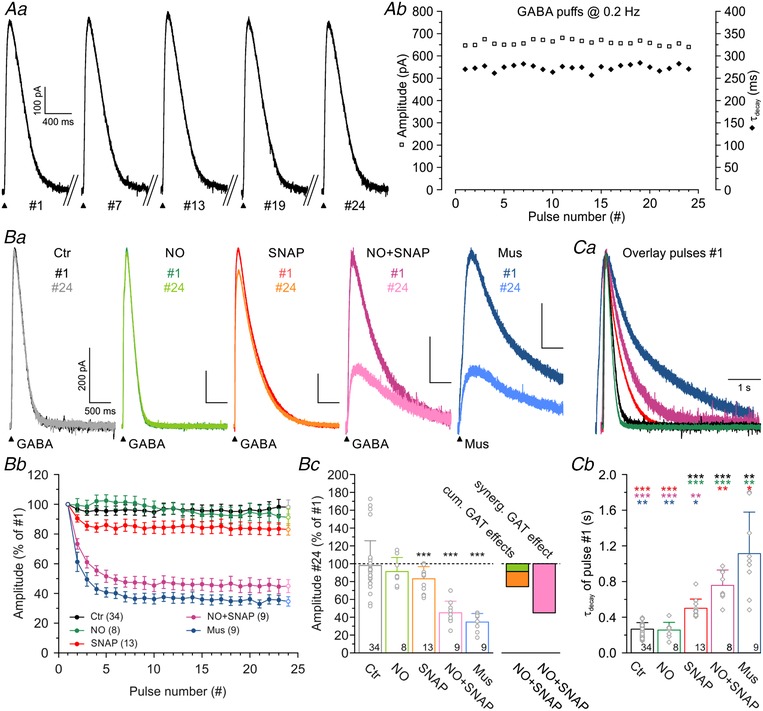
Aa, current traces of a P11 LSO neuron, evoked by GABA application (100 μM, 100 ms puffs for 2 min at 0.2 Hz). The pulse number (#) is provided under each trace. Ab, peak amplitudes and τdecay of the evoked events as a function of time, demonstrating low amplitude jitter (squares) and stable decay kinetics (diamonds). Ba, events from pulse no. 1 to pulse no. 24 under various pharmacological conditions. Drugs were bath‐applied 10 min before puff applications and were also contained in the puff pipettes. Bb, peak amplitudes did not change in the control condition. Blockade of GAT‐1 (10 μM NO‐711) caused no amplitude reduction and blockade of GAT‐3 (40 μM SNAP‐5114) caused a weak one. Coapplication of the two blockers, however, revealed a synergistic effect. Substitution of muscimol (Mus, 100 μM) for GABA mimicked the effect of coapplied NO‐711 and SNAP‑5114. Bc, statistics for the data shown in Bb. Ca, scaled and peak‐aligned IPSCs under various pharmacological conditions, illustrating the differences in decay kinetics. Same colour code as in Ba. Cb, statistical analysis of τdecay of pulse no. 1. Colours of asterisks mark the dataset to which the comparison was made. Significance levels were Šidák corrected in case of four comparisons.
Figure 11. Tonic GABA reduces the excitability of P11 LSO neurons through extrasynaptic GABAARs.
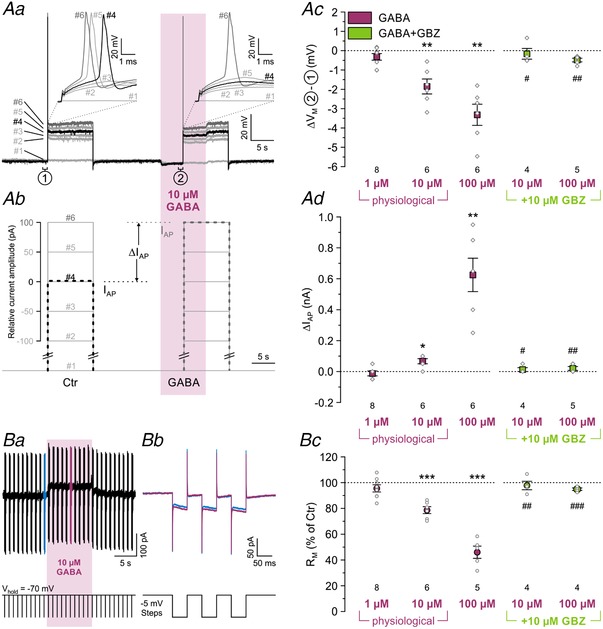
Aa and b, voltage responses of a representative P11 LSO neuron to rectangular current injections with increasing amplitude (10 s duration, 6 steps). In controls, an action potential could first be elicited at pulse no. 4 (black trace), whereas a higher current amplitude was required in the presence of GABA (10 s puff; purple zone), namely at pulse no. 6 (dark grey trace). I AP depicts the increase in current amplitude required to elicit an action potential; (1) and (2) mark the 1 s intervals during which the resting membrane potential (V M) was determined. Notably, GABA also hyperpolarized the neuron. Ac, statistical analysis of ΔV M at three GABA concentrations and during coapplication of GABA+GBZ (green). Ad, statistical analysis of I AP. Ba, exemplifying 30 s current trace (top), demonstrating the effect of tonic GABA (10 μM, 10 s puff, purple zone). To assess R M, hyperpolarizing voltage steps of 5 mV were applied at 1 Hz (bottom) in triplets (enlargements in Bb, details in Methods) in the absence and presence of GABA (blue and purple, offset‐corrected in Bb). Bc, statistical analysis of R M. Asterisks indicate significance levels between GABA and control; hashtags indicate significance levels between GBZ+GABA and GABA.
Figure 12. Tonic GABA increases the excitability of P4 LSO neurons through extrasynaptic GABAARs.
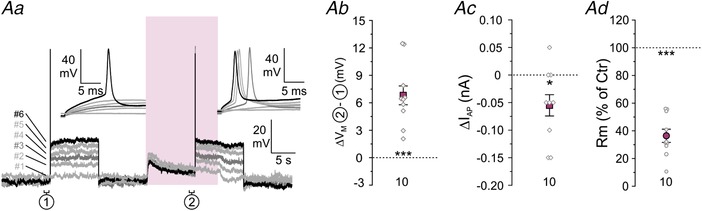
Aa, voltage responses of a representative P4 LSO neuron to rectangular current injections with increasing amplitude. In controls, an action potential could first be elicited at pulse no. 6 (black trace), whereas a lower current amplitude was required in the presence of 10 μM GABA (15 s puff; purple zone), namely at pulse no. 3 (dark grey trace). Notably, GABA also depolarized the neuron. Ab–d, statistical analysis of ΔV M (Ab), ΔI AP (Ac) and R M (Ac).
Figure 3. All inhibitory synaptic inputs to LSO neurons are mediated by glycine, not by GABA.
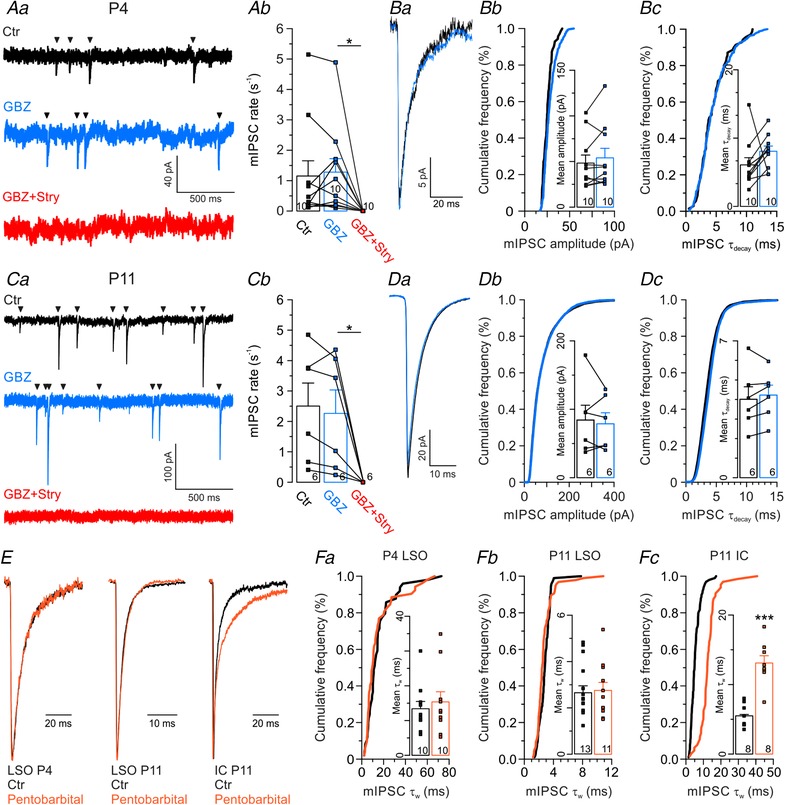
Aa, miniature IPSCs (mIPSCs, arrowheads) from a representative P4 LSO neuron under control conditions (Ctr, black), followed by consecutive wash‐in of GBZ (10 μM, blue) and Stry (1 μM, red). Ab, statistical analysis of mIPSC rate. Ba, averaged peak‐aligned mIPSCs recordings of a representative P4 LSO neuron under control conditions and in GBZ. Bb and c, statistical analysis of amplitude and τdecay. Cumulative plots show a representative cell, bar diagrams show the sample. C and D same as in A and B, but for P11. E, average peak‐scaled mIPSCs (n = 41–147) under control conditions (black) and in the presence of pentobarbital (30 μM, orange) from P4 LSO (left), P11 LSO (middle) and P11 inferior colliculus (IC, right). Fa–c, statistical analysis of τw.
For focal electrical stimulation of MNTB input fibres, TST150 theta electrodes (WPI, Sarasota, FL, USA) or patch pipette with tip diameters of ∼20 and ∼5 μm, respectively, were filled with ACSF, connected to a STG4004 (Multichannel Systems, Reutlingen, Germany) or Master8/Isoflex stimulator (A.M.P.I., Jerusalem, Israel) and placed at the lateral edge of the ipsilateral MNTB. Bipolar or monopolar rectangular current pulses (100–200 μs) of 20–16,000 μA intensity were applied at 0.2–1 Hz, evoking inhibitory postsynaptic currents (eIPSCs) of short latencies (P4: 2–5 ms; P11: 1–3 ms). In some experiments, 3 μM CGP55845 was bath‐applied to prevent GABABR activation. GlyRs and GABAARs were blocked with 100 nM strychnine and 10 μM GABAzine (GBZ alias SR95531), respectively.
Transmitters (100 μM GABA or 100 μM glycine) or agonists (1 μM 4,5,6,7‐tetrahydoisoxazolo[5,4‐c]pyridin‐3‐ol (THIP; alias gaboxadol or OV101) or 10 μM baclofen) were puff‐applied 10–20 μm distant from patched somata via theta electrodes (TST150, WPI) or patch electrodes (tip diameters: ∼2–5 μm). To do so, puffs (5–10 psi) were generated with a Multi‐channel Picospritzer (Science Products) or a PDES‐02DX (NPI Electronic, Tamm, Germany). In some experiments, 10 μM NO‐711, a specific GAT‐1 blocker, or 40 μM SNAP‐5114, a specific GAT‐3 blocker, were bath applied (in the following denoted as NO and SNAP, respectively). To avoid ‘washout’ of bath‐applied blockers by puffing GABAAR agonists, the second channel of the theta electrode was filled with the agonist plus the blocker. In order to address possible changes in action potential generation due to tonic GABAergic inhibition, GABA (1–100 μM) was puff‐applied for 10 s, starting 5 s prior to depolarizing current injections (see Fig. 11 Aa). Tonic inhibition was achieved within 3–5 s, when the GABA‐induced hyperpolarization had reached a steady state. When steady‐state depolarization was not reached early enough, puff application was prolonged to 15 s.
Glutamatergic events were blocked with 5 mM kynurenic acid or 20 μM CNQX. Miniature IPSCs (mIPSCs) were isolated with 1 μM tetrodotoxin (TTX). Glycinergic or GABAergic mIPSCs were identified in the presence of bath‐applied 10 μM GBZ or 1 μM strychnine, respectively. In one series of experiments, mIPSCs were recorded from LSO or inferior colliculus neurons with the GABAAR modulator pentobarbital (30 μM) in the bath. mIPSCs were analysed with MiniAnalysis 6.0.3 (Synaptosoft, Fort Lee, NJ, USA) when their decay phase could be fitted with an R 2 > 0.85. mIPSC decay times under pentobarbital were determined as follows. Because of the possibility of mixed transmitters, we did not assume a single exponential decay for the mIPSCs in pentobarbital. Instead, the decay course was fitted to a double‐exponential function:
where t is time, A 1 and A 2 are the peak amplitudes of the fast and the slow decay components at t = 0, and τ1 and τ2 are fast and slow decay time constants, respectively. From this, we calculated the weighted decay time constant τw:
Files were analysed using IGOR Pro 6.3 (Wavemetrics, Lake Oswego, OR, USA), running Patcher's Power Tools (Max Planck‐Institute for Membrane Biophysics) with customized routines.
Calcium imaging
Brainstem slices were prepared and stored as described above. The preparation solution contained (in mM): 76 NaCl, 25 NaHCO3, 75 sucrose, 25 glucose, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2; and the ACSF contained (in mM): 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 1 MgCl2, 1.5 CaCl2, 20 glucose, 4 sodium l‐lactate, 2 sodium pyruvate, 0.4 sodium l‐ascorbate. MNTB somata/axons were loaded using a borosilicate pipette (5–8 μm tip diameter) filled with ACSF, containing 0.05% fast‐green (Sigma‐Aldrich, St. Louis, MO, USA) and 110 μM magnesium‐green (Mg‐green, M3735, Thermo Fisher Scientific, Carlsbad, CA, USA). Axonal uptake of the dye was achieved by continuous pressure application at the dorsolateral edge of the MNTB for 30–45 min. A second pipette with a tip diameter of 20–30 μm was placed at the ventrolateral edge of the MNTB to immediately suck away excess dye (Regehr & Atluri, 1995). After loading, slices were stored in ACSF at room temperature for at least 1 h to allow diffusion of the dye along the axons. Electrical stimulation was performed as described above. Calcium signals were visualized with a ×60 objective attached to an Olympus BX51WI microscope equipped with a 150 W xenon light source (Optiquip Model 770; OPTIQUIP, Rochester, UK) and a FITC U‐N41001 (Olympus) filter set. Recordings with a Sensicam QE (Cooke Corp., Auburn Hills, MI, USA) at 2 × 2 binning allowed a spatial resolution of ∼0.1 μm/pixel. For identification of loaded presynaptic terminals, a 7–9 px Gaussian filter was utilized in a test stimulation prior to the actual experiment. High temporal resolution recordings at 5 kHz were performed with a custom‐made single photodiode system (Hamamatsu S1336‐BK) connected to a BNC‐2090 interface (National Instruments, Austin, TX, USA). To improve the signal to noise ratio of the photodiode, the field of view was constrained to 40–70 μm with a diaphragm and 30 traces were averaged (20 s interstimulus intervals). Camera settings, shutter and stimulation timings, data acquisition as well as analysis were performed with IGOR Pro 6.2 (Wavemetrics), running SIDX drivers (Bruxton Corp., Seattle, WA, USA), and custom software (mafPC).
Immunohistochemistry
Deeply anaesthetized mice (7% chloral hydrate, 0.01 ml/g body weight, i.p.) were transcardially perfused with 15 mM phosphate‐buffered saline (PBS; pH 7.4, room temperature), followed by ice‐cold 4% paraformaldehyde for 20 min (Ecoline VC‐360 pump, IsmaTec, Wertheim, Germany). Brains were removed from the skull, postfixed for 2 h in 4% paraformaldehyde, and stored overnight in 30% sucrose–PBS. Coronal brainstem slices were cut at 40 μm with a sliding microtome (HM 430, Thermo Fisher Scientific) and transferred into 15% sucrose–PBS for 5 min; 3 × 10 min rinse steps in PBS followed at room temperature. Antibodies against gephyrin (1:500, host: mouse, Synaptic Systems, Göttingen, Germany), GlyT2 (1:10,000, host: guinea pig, Chemicon, Limburg an der Lahn, Germany) and GAD65/67 (1:1,000, host: rabbit, Abcam, Cambridge, UK) were applied free‐floating at 4°C overnight in blocking solution (0.3% Triton X‐100, 5% goat serum, 1% BSA in PBS), followed by rinsing 3 × 10 min in PBS at room temperature. Slices were then incubated in the dark for 2 h in blocking solution and secondary antibody (1:1000; goat‐anti‐mouse, Alexa Fluor 647, Thermo Fisher Scientific; goat‐anti‐guinea pig, Alexa Fluor 488, Thermo Fisher Scientific; goat‐anti‐rabbit, Alexa Fluor 488, Thermo Fisher Scientific) and rinsed 3 × 10 min in PBS. Cell nuclei were stained with propidium iodide (1 μg/ml in PBS, Sigma‐Aldrich) for 10 min and slices were rinsed again 3 × 10 min in PBS. Slices were then mounted on gelatin‐coated glass slides and covered with mounting medium containing 2.5% 1,4‐diazabicyclo[2.2.2]octane (DABCO; Sigma‐Aldrich).
Images were acquired with a confocal microscope equipped with an EC Plan‐Neofluar ×40/1.3 oil objective (LSM700, Zeiss). Intensity profiles across inhibitory synapses were determined in ZEN2012 (blue edition, Zeiss) by scan lines, drawn from the centre of the cell soma across membrane‐associated gephyrin signals (see Fig. 4 A). We analysed 7–18 and 1–3 intensity profiles per cell at P11 and P4, respectively. The position of maximal gephyrin signal was set to zero; negative and positive scan positions indicate intracellular and extracellular location, respectively. Prior to codistribution analysis (Bolte & Cordelieres, 2006), a rolling ball background subtraction (radius 20) was applied with Fiji ImageJ 1.48 (Schindelin et al. 2012), followed by brightness adjustment. Regions of interest (ROIs) outreached the cell somata by 1–2 μm, and Pearson's coefficient, together with the corresponding Costes P‐value (P Costes, 200 repetitions), was determined with the Fiji plugin Coloc2. Codistribution became significant with P Costes ≥ 0.95.
Figure 4. GlyT2, but not GAD, codistributes with gephyrin at somata of LSO principal neurons.
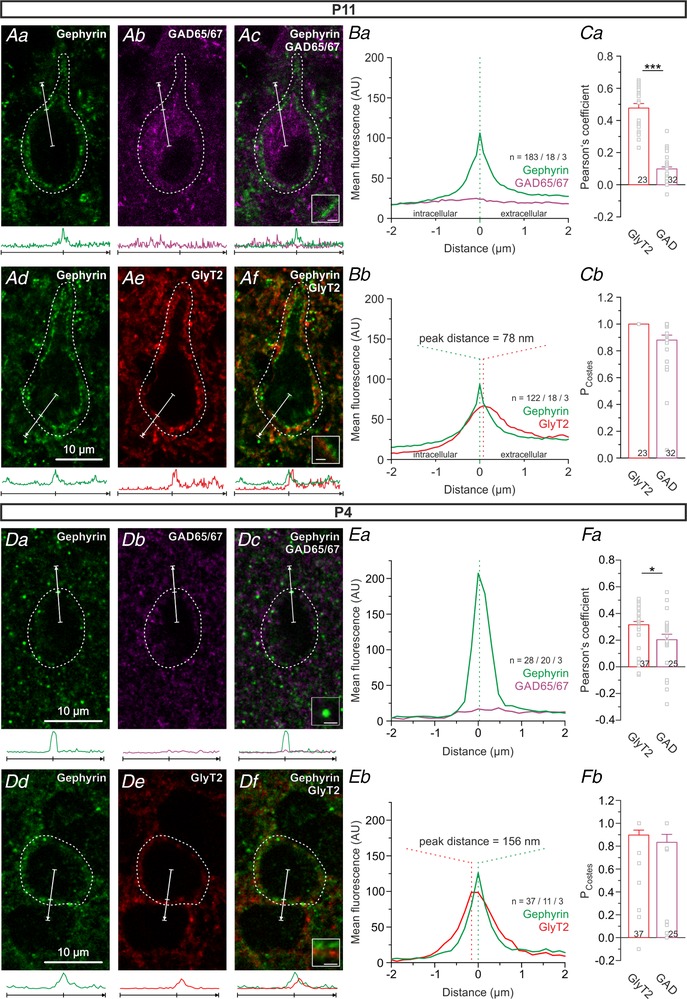
Aa–c, immunohistochemical detection of the inhibitory postsynaptic marker gephyrin and the presynaptic GABA marker GAD65/67 at P11. Ad–f, same as a–c, but with gephyrin and the presynaptic glycinergic marker GlyT2. Merged images in panels ac and Af. Intensity profiles of the immunosignals across inhibitory synapses (bottom of Aa–f) determined by line scans originating from the soma centre and crossing membrane‐associated gephyrin signals (white arrows in Aa–f, centre tick marks gephyrin signal). Ba and b, spatial distribution of gephyrin with GAD65/67 or GlyT2 as visualized by average intensity profiles. n, number of profiles/cells/animals; AU, arbitrary units. Before averaging, each profile was aligned to the position of the maximal gephyrin signal. Negative μm values demonstrate an intracellular signal, positive μm values an extracellular one; 78 nm equals the pixel width, thus indicating the closest detectable distance. Ca and b, to assess a somatic codistribution of gephyrin with GlyT2 or GAD65/67, ROIs were drawn 1–2 μm distal to the plasma membrane (dashed lines in Aa–f) and analysed for Pearson's coefficient and corresponding P Costes values. Codistribution is evidenced by P Costes > 0.95. D–F, same as A–C, but for P4. Pixel width is 156 nm instead of 78 nm.
RNA sequencing
Coronal brainstem cryosections of 30 μm thickness from P60 mice were placed on PEN membrane glass slides (Leica Microsystems) and dehydrated twice in ascending ethanol (75%, 95%, 100%, 1 min each). LSO tissue was collected using an LMD6500/DM6000B laser microdissection system (Leica Microsystems). For RNA extraction, we used an Arcturus PicoPure RNA Isolation Kit (Thermo Fisher Scientific). RNA quality control and cDNA synthesis were done as described earlier (Picelli et al. 2013). Libraries were prepared with the Nextera DNA Library Preparation Kit (Illumina, Inc., San Diego, CA, USA) and sequenced 1 × 100 bp on a HiSeq 2500 (Illumina). Reads were trimmed for adapter sequences and low quality (phred score < 20) with Trim Galore! (v0.4.2; http://www.bioinformatics.babraham.ac.uk/projects/trim_galore), aligned to the mm10 mouse reference genome using two‐step STAR (v2.5.2a) alignment (Dobin & Gingeras, 2015) and marked for PCR duplicates with Picard tools (MarkDuplicate v1.115; http://broadinstitute.github.io/picard/). Gene‐wise read counts were summarized with featureCounts (Liao et al. 2014) using Gencode annotation vM2 and normalized as counts per million (CPM) to set a threshold for expressed genes (CPM ≥ 0.5), and as reads per kilobase per million (RPKM) to compare expression levels.
Statistics
Statistical analysis was performed with the Microsoft Excel plugin Winstat 2012.1 (R. Fitch Software). Gaussian distributed (Kolmogorov–Smirnov test) samples were compared in paired or unpaired two‐tailed Studnet's t test, and a Wilcoxon signed rank test or a Mann–Whitney U test was applied otherwise. Equality of variances in unpaired t tests was determined with F test, and a homo‐ or heteroskedastic test was performed when appropriate. Bar charts are presented as the mean ± SEM with single data points depicted by dots (connected if paired). Significance levels are indicated as follows: P < 0.05*, P < 0.01 **, P < 0.001 ***; no symbols if P > 0.05. In Figs 8 Bc and 10 Cb, critical α values were post hoc Šidák corrected (Abdi, 2007).
Results
No GABA contribution to synaptic transmission at mouse MNTB–LSO inputs
In order to assess the transmitter phenotype of MNTB–LSO synapses in immature mice, we performed whole‐cell recordings from LSO neurons at P4 and P11 while electrically stimulating MNTB fibres (Fig. 1 Aa and b). Averaged eIPSC amplitudes in response to 1 Hz stimulation were robust over time (Fig. 1 Ac) and revealed an ∼5‐fold amplitude increase between P4 and P11 (Fig. 1 Ba–d; P4: 46 ± 8 pA, n = 8; P11: 236 ± 67 pA, n = 9; P = 0.022, unpaired t test). Likewise, decay kinetics accelerated 3.3‐fold with age (Fig. 1 Ca and b; P4: 22.1 ± 2.9 ms, n = 8; P11: 6.7 ± 1.5 ms, n = 7; P = 6.3 × 10−4, unpaired t test). GABAergic and glycinergic inputs were pharmacologically separated at the putatively mixed MNTB–LSO synapses (Fig. 1 Ad). Wash‐in of the GlyR antagonist strychnine (100 nM) blocked eIPSCs by 74 ± 4% at P4 and by 88 ± 4% at P11, demonstrating a very prominent glycinergic component at both ages (Fig. 1 Ba–e; P4: n = 8, P = 5.9 × 10−7; P11: n = 9, P = 1.0 × 10−8; paired t tests). At 100 nM, strychnine is insufficient to completely block GlyRs, but non‐specific inhibition of GABAARs is avoided (Jonas et al. 1998; see also Fig. 2 B). Thus, we most likely underestimated the prominent glycinergic component. Additional wash‐in of the high‐affinity GABAAR antagonist GBZ (10 μM; K d = 0.15 μM) further reduced eIPSC amplitudes only slightly by 12 ± 3% at P4 and by as little as 4 ± 1% at P11 (Fig. 1 Ba–e; P4: n = 8, P = 0.009; P11: n = 7, P = 0.016; paired t tests). When normalized to the amplitude obtained under strychnine, the reduction accounted for 40 ± 11% at P4 and 31 ± 5% at P11 (data not shown). As ∼30% of glycine‐induced currents were also blocked by 10 μM GBZ in puff application experiments (Fig. 2 B), we conclude that the reduction of synaptic currents by GBZ was due to non‐specific inhibition of GlyRs that had remained unblocked by 100 nM strychnine. Indeed, increasing the strychnine concentration to 1 μM completely blocked the residual currents (those remaining after wash‐in of 100 nM strychnine and 10 μM GBZ; n = 4, data not shown). As kinetic properties of glycine‐mediated currents are faster than GABA‐mediated currents (Jonas et al. 1998; O'Brien & Berger, 1999; Russier et al. 2002; Awatramani et al. 2005; Ishibashi et al. 2013), one would expect a slow‐down of the decay at mixed synapses after wash‐in of strychnine. However, in line with our results pointing towards virtually purely glycinergic synapses, we observed no change in τdecay upon strychnine or GBZ application, neither at P4 nor at P11 (Fig. 1 C). Taken together, the results demonstrate a negligible GABAergic contribution to synaptic transmission and are thus in clear contrast to the earlier reports of mixed MNTB–LSO synapses in gerbils and rats (Kotak et al. 1998; Nabekura et al. 2004). However, they are in line with data obtained in juvenile mice (Giugovaz‐Tropper et al. 2011). We conclude that exclusively glycine mediates phasic inhibitory synaptic transmission at mouse MNTB–LSO synapses, both at P4 and at P11.
Pressure‐release of transmitter molecules via puff electrodes allows distinct neurotransmitter application, contrary to the scenario with coreleased GABA and glycine. We took advantage of this possibility and assessed the specificity of the blockers strychnine and GBZ. To do so, we puff‐applied GABA or glycine (100 μM each) and analysed the specificity of the blockers strychnine and GBZ (Fig. 2 A). Strychnine at 100 nM did not significantly affect GABA‐evoked currents (Fig. 2 B; P4: n = 5, P = 0.614; P11: n = 5, P = 0.359; paired t tests). By contrast, 10 μM GBZ reduced glycine‐induced currents (Fig. 2 B; P4: 26 ± 5%, n = 5, P = 0.006; P11: 27 ± 8%, n = 5, P = 0.030; paired t tests). We interpret this as a non‐specific action of GBZ at GlyRs, consistent with previous results of GBZ‐mediated GlyR blockade with an IC50 of ∼50 μM (Wang & Slaughter, 2005; Beato et al. 2007; Li & Slaughter, 2007). Consequently, results obtained with GBZ, similar to bicuculline‐based results, need to be interpreted with some caution.
Although we found no evidence for a GABAergic component at mouse MNTB–LSO synapses, it is possible that LSO principal cells receive GABAergic input from sources other than the MNTB. In order to address a potential synaptic contribution of GABA stemming from other sources, we recorded mIPSCs from LSO principal neurons at P4 and P11. At P4, we observed mIPSCs at a rate of 1.2 ± 0.5 s−1 with peak amplitudes of 48 ± 9 pA and a τdecay of 6.1 ± 1.1 ms (Fig. 3 A and B). GBZ (10 μM) affected neither the mIPSC rate (Fig. 3 Aa and b; n = 10; P = 0.55, paired t test), nor the peak amplitude (Fig. 3 Ba and b; n = 10; P = 0.20, paired t test), nor τdecay (Fig. 3 Ba and c; n = 10; P = 0.19, paired t test). In contrast, bath application of strychnine (1 μM) completely abolished mIPSCs. We obtained similar results at P11. Under control conditions, the mIPSC rate amounted to 2.5 ± 0.8 s−1, with a peak amplitude of 84 ± 22 pA and a τdecay of 4.0 ± 0.6 ms (Fig. 3 C and D). In the presence of GBZ (10 μM), we observed an unchanged mIPSC rate (Fig. 3 Ca and b; n = 6; P = 0.272, paired t test), an unchanged peak amplitude (Fig. 3 Da and b; n = 6; P = 0.625, paired t test), and an unchanged τdecay (Fig. 3 Da and c; n = 6; P = 0.325, paired t test). mIPSCs were completely abolished in 1 μM strychnine. These results suggest an absence of functional synaptic GABAARs at principal LSO neurons.
In order to assess further whether functional synaptic GABAARs are absent at LSO principal neurons, we recorded mIPSCs in the presence of the GABAAR modulator pentobarbital (30 μM bath‐applied). One can thus avoid the side effects of GBZ, namely cross reactivity at the counterpart receptor molecule GlyR. To determine the decay time, we fitted a double exponential function to individual mIPSC traces, thereby revealing τfast and τslow and the respective amplitudes. From these values, we calculated the weighted τw (see Methods). We found no difference in τw between Ctr and drug, neither at P4 nor at P11 (Fig. 3 E and Fa and b; P4: Ctr: 13.4 ± 2.1 ms, pentobarbital: 15.4 ± 2.9 ms, P = 0.61; P11: Ctr: 2.6 ± 0.3 ms, pentobarbital: 2.8 ± 0.3 ms, P = 0.81; unpaired t tests). The results further corroborate our conclusion that GlyRs, but not GABAARs, mediate synaptic inhibition of LSO principal cells. To check for the functional integrity of the drug, we performed additional experiments in the inferior colliculus, where pentobarbital effects have been described (Moore & Trussell, 2017). Pentobarbital prolonged τw of mIPSCs >2‐fold (Fig. 3 E and Fc; 5.5 ± 0.2 ms vs. 13.1 ± 1.0 ms, P = 3.3 × 10−5, unpaired t test), demonstrating that the drug was indeed pharmacologically functional.
The results shown in Figs 1, 2, 3 demonstrate an absence of GABAAR‐mediated synaptic transmission in the mouse LSO. We asked ourselves whether there are GABAergic synapses at all in the LSO, which, if they exist, would be silent (Charpier et al. 1995; Bekkers, 2005). We addressed this possibility by switching to a histological approach, in which we combined immunohistochemistry with confocal microscopy in order to check for a codistribution of gephyrin with GAD65/67. Gephyrin is a ubiquitous postsynaptic marker protein for inhibitory synapses (Kneussel & Loebrich, 2007; Specht et al. 2013; Tyagarajan & Fritschy, 2014), whereas GAD65/67 (the GABA‐synthesizing glutamate decarboxylases GAD65 and GAD67) are presynaptic proteins at GABAergic synapses. The analysis revealed no codistribution of the two markers (Fig. 4 Aa–c and Ba). We also looked for codistribution of gephyrin with GlyT2, the glycine transporter type 2, which is a presynaptic marker for glycinergic axon terminals (Friauf et al. 1999; Eulenburg et al. 2005). In contrast to the GAD65/67–gephyrin results, GlyT2 and gephyrin immunosignals were in very close proximity of as little as 78 nm (Fig. 4 Ad–f and Bb). Further quantification by Pearson's coefficient and the P Costes value (a measure of reliability) confirmed the absence of GAD65/67–gephyrin codistribution (Pearson's coefficient: 0.10 ± 0.01; n = 32; P Costes = 0.88 ± 0.04), yet the presence of GlyT2–gephyrin codistribution (Pearson's coefficient: 0.48 ± 0.03; n = 23; P Costes = 1.00 ± 0.00). Hence, there is no evidence for GABAergic synapses in the mouse LSO at P11, when hearing onset occurs. In order to check whether this is also the case during early development, we performed the same series of experiments at P4. We also detected gephyrin, GlyT2 and GAD65/67 (Fig. 4 D). Similar to P11, fluorescence of gephyrin and GlyT2 peaked in close proximity (Fig. 4 Ea). In contrast, there was no match between GAD65/67 and gephyrin fluorescence (Fig. 4 Eb). Accordingly, Pearson's coefficient and P Costes values showed a codistribution trend for gephyrin and GlyT2 (Fig. 4 Fa and b; Pearson's coefficient: 0.32 ± 0.02; n = 37; P Costes = 0.90 ± 0.04), but no evidence towards a codistribution for gephyrin and GAD65/67 (Fig. 4 Fa and b; Pearson: 0.20 ± 0.04; n = 25; P Costes = 0.83 ± 0.07).
LSO principal neurons possess functional GABABRs
As synaptic GABAARs are absent in MNTB–LSO inputs, we wondered whether other types of GABA receptors might be involved in GABA signalling and checked for pre‐ and postsynaptic GABABRs. To assess the presence of GABABRs, we analysed responses of LSO principal neurons to focally applied baclofen, a GABABR agonist (Fig. 5 Aa). We observed robust outward currents with slow kinetics (Fig. 5 Ab) and peak amplitudes of 35 ± 1 pA at P4 and 70 ± 5 pA at P11 (Fig. 5 Ba). These baclofen‐induced outward currents were virtually abolished when the GABABR blocker CGP was present (Fig. 5 Ab), with peak amplitudes as low as 5 ± 2 pA at P4 and 10 ± 2 pA at P11 (Fig. 5 Ba). The fraction of the blocked current was ∼90% at both ages. Outward currents with slow kinetics are indicative of K+ outflux mediated by G protein‐coupled inward‐rectifying K+ channels (Lüscher & Slesinger, 2010). To assess channel contribution, we monitored R m. During baclofen application, R m was reduced to 74 ± 3% at P4 and to 79 ± 1% at P11 (Fig. 5 C and D). R m was unchanged when baclofen was applied in the presence of CGP (Fig. 5 C and D; 104 ± 3% at P4; 101 ± 2% at P11). Together, these data suggest that LSO principal neurons possess functional GABABRs, at least during prehearing development and at hearing onset.
Ca2+ influx into MNTB axon terminals is reduced via presynaptic GABABRs
Next, we investigated the presence of presynaptic GABABRs and a possible presynaptic modulation of glycinergic MNTB–LSO synapses by these receptors, as seen in gerbils (Magnusson et al. 2008). Presynaptic GABABRs commonly suppress neurotransmitter release by inhibiting voltage‐activated Ca2+ channels and reducing Ca2+ influx (Wu & Saggau, 1995; Dittman & Regehr, 1996; Takahashi et al. 1998; Chanda et al. 2011; Gassmann & Bettler, 2012). To study whether GABABR activation results in reduced Ca2+ influx into MNTB axon terminals, we performed Ca2+ imaging using Mg‐green (Fig. 6 A and B; see Methods for details). Electrical stimulation of P11 MNTB axons resulted in increased fluorescence at healthy presynaptic terminals (Fig. 6 Ab and B, green ROI). No increase occurred at structures showing a fluorescent bleb or at loaded axons (Fig. 6 Ab and B, blue ROI). In a similar series of experiments, we obtained recordings from the complete field of view with a photodiode, enabling us to do Ca2+ imaging at sub‐millisecond temporal resolution from one to five loaded boutons at a time (Fig. 6 C and D). A rapid increase of the Ca2+ signal occurred upon stimulation, followed by a fast decline with a τdecay of 15.3 ± 2.4 ms (Fig. 6 D; n = 5). Bath application of baclofen (100 μM) reversibly diminished the stimulus‐evoked peak calcium response to 80 ± 3% (Fig. 6 Da and b; n = 5, P = 0.003, paired t test) but did not affect τdecay (Fig. 6 Dc; n = 5, P = 0.896, paired t test). In conclusion, functional GABABRs are present on MNTB axon terminals in the LSO and their activation leads to reduced Ca2+ influx.
Figure 6. Presynaptic Ca2+ influx at MNTB–LSO synapses is controlled by GABABRs.
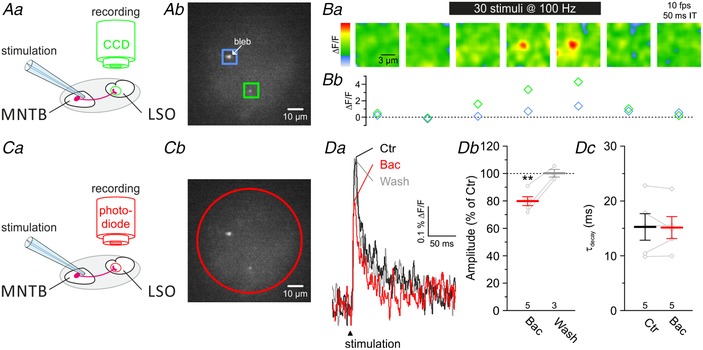
A and C, loading of MNTB fibres with the Ca2+ indicator Mg‐green resulted in labelled presynaptic axon terminals in the LSO (Ab and Cb). Ca2+ imaging during electrical stimulation of MNTB–LSO fibres (Aa) enables spatial recordings from labelled presynaptic terminals (Ab, green and blue ROIs) in the LSO with a temporal resolution of 10 frames s−1 (fps, Ba). Integration time (IT) for a single frame was 50 ms. The green ROI in Ab frames a Mg‐green positive, healthy presynaptic terminal, which is also shown during stimulation in Ba. The relative change in fluorescence (ΔF/F) of this terminal upon 100 Hz stimulation is depicted in Bb (green), together with a large bleb (blue, blue frame in Ab). Noise in Ba was removed with a Gaussian filter (radius 6 pixels). Photodiode recordings (Ca and red circle in Cb) allowed Ca2+ imaging in the sub‐millisecond range. D, average Ca2+ responses (n = 40 in each case) upon MNTB–LSO fibre stimulation at 0.2 Hz (Da) under control conditions (Ctr, black) became reversibly diminished in amplitude (Db) in the presence of 100 μM baclofen (+Bac, red). Wash‐out (wash) of 15 min of Bac is depicted in grey. Kinetics of Ca2+ influx was unaffected in the presence of Bac (Dc).
GABABR activation in the LSO results in reduced glycinergic eIPSCs
Reduced presynaptic Ca2+ influx upon GABABR activation reduces the release probability for synaptic vesicles, which finally results in smaller ePSC amplitudes and, likewise, weaker synaptic strength (Zucker & Regehr, 2002; Friauf et al. 2015). GABABR activation at MNTB axon terminals may thus reduce the amount of released glycine and therefore result in smaller glycinergic eIPSC amplitudes in LSO neurons. We addressed this point and recorded from P4 and P11 LSO principal neurons while electrically stimulating MNTB fibres (trains of 24 pulses at 0.2 Hz). During the trains, we focally applied the GABABR agonist baclofen (10 μM for 5 s; Fig. 7 Aa–c). Under Ctr conditions, MNTB fibre stimulation elicited robust and stable eIPSCs, most likely mediated by glycine (Fig. 7 Ab). In contrast, baclofen application resulted in substantially reduced synaptic strength (Fig. 7 Ab and c). Amplitudes were reduced to 73 ± 5% at P4 and to 35 ± 4% at P11 (Fig. 7 Ba and b; n = 4, 5). In the presence of 3 μM CGP, eIPSC amplitudes remained unchanged upon baclofen application (Fig. 7 C and D). Collectively, we provide pharmacological evidence that GABABR activation reduces the synaptic strength (Fig. 7 Da and b). Hence, we conclude that GABA modulates glycinergic synaptic transmission at mouse MNTB–LSO synapses through activating presynaptic GABABRs (see Giugovaz‐Tropper et al. 2011).
GABAAR‐mediated inhibition is extrasynaptic
Although we obtained no evidence for a participation of GABA in synaptic transmission at mouse MNTB–LSO synapses, GABA may still be able to act at extrasynaptic GABAARs (Farrant & Nusser, 2005; Brickley & Mody, 2012). To check for the presence of functional extrasynaptic GABAARs in principal LSO neurons, we puff‐applied GABA (100 μM) while recording whole‐cell currents. Such GABA puffs consistently evoked robust responses of very similar peak amplitudes at P4 and P11 (Fig. 8 Aa and b; P4: 157 ± 20 pA, n = 11; P11: 155 ± 18 pA, n = 22; P = 0.962, unpaired t test). When GABA was coapplied with GBZ, responses were almost completely suppressed (Fig. 8 Ac; P4: 93 ± 3%, n = 5, P = 0.062; P11: 99 ± 1%, n = 4, P = 0.391; paired t tests compared to 100%).
GABA is a potent full agonist at GABAARs, yet it is merely a partial agonist at extrasynaptic receptor isoforms, particularly those containing δ subunits (δ‐GABAARs). In contrast, THIP is a much more efficient agonist at δ‐GABAARs (Krogsgaard‐Larsen et al. 1994; Krogsgaard‐Larsen et al. 2002; Orser, 2006; Olsen & Sieghart, 2008; Mortensen et al. 2010), displaying ‘super agonist’ behaviour with an E max of ∼160% (Brown et al. 2002; Wohlfarth et al. 2002; Krogsgaard‐Larsen et al. 2004; Drasbek & Jensen, 2006). THIP has a lower EC50 value and evokes a larger maximal current at α4β3δ receptors than at α4β3γ2 receptors (Brown et al. 2002). In order to check for functional δ‐GABAARs, we puff‐applied THIP onto somata of P11 LSO principal neurons. In contrast to others (Hoestgaard‐Jensen et al. 2014, 100 μM), we used a much lower concentration of 1 μM to minimize side effects such as THIP's antagonistic activity at ρ1–3 subunits (Johnston et al. 2003; Alexander et al. 2017). Despite the low concentration, much lower than the EC50 of 44 μM (Drasbek & Jensen, 2006), we observed tonic inward currents of −13 ± 2 pA (Fig. 8 B; n = 10, P = 1.2 × 10−4, paired t test). Their peak amplitudes were in the expected range for δ‐GABAARs under high internal Cl− (Meera et al. 2011). The addition of GBZ (100 μM) reduced the tonic inward currents to −8 ± 2 pA (n = 10, P = 4.7 × 10−4, paired t test), and washout conditions were again indistinguishable from controls (n = 10, P = 0.499, paired t test). Based on these pharmacological results, we conclude that LSO principal neurons possess functional extrasynaptic GABAARs.
We next assessed the relative abundance of all GABAAR subunit transcripts in the LSO. To do so, we coupled laser capture microscopy with global transcriptome profiling. We detected 13 of the 19 different transcripts (Fig. 9). Gabra5, which codes for the α5 subunit, was most abundant, followed by Gabrd, which codes for the δ subunit. Both subunits are components of extrasynaptic GABAARs (Brickley & Mody, 2012; Knoflach et al. 2016) and mediate tonic inhibition (Thomas et al. 2005; Glykys et al. 2008). δ‐GABAARs are exclusively located at perisynaptic and extrasynaptic sites (Wei et al. 2003; Semyanov et al. 2004; Farrant & Nusser, 2005; Mody, 2005), where they form α4/α6‐containing receptors (Karim et al. 2012). α5 subunits are typically associated with γ2 and β1 subunits (Walker & Semyanov, 2008), and Gabrg2 and Gabrb1 transcripts coding for the latter subunits were among the most abundant counts. Together, our profiling results provide further evidence for the existence of extrasynaptic GABAARs in the LSO.
Figure 9. Transcripts Gabra5 and Gabrd, coding for the extrasynaptic GABAAR subunits α5 and δ, are most abundant in the mouse LSO.
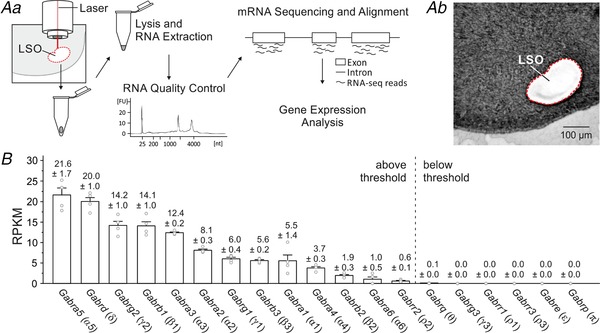
Aa, LSO tissue was collected via laser capture microdissection. After lysis and total RNA extraction, RNA quality control, mRNA sequencing, alignment and gene expression analysis were performed. FU, fluorescence units; nt, nucleotides. Bb, coronal brainstem slice after microdissection of the LSO. B, RPKM values (reads per kilobase per million) of 19 transcripts coding for GABAAR subunits in the LSO (n = 4 for each transcript). Numbers above columns depict mean RPKM values ± SEM; corresponding protein subunits are depicted in brackets. Genes positioned left of the dashed line were expressed above threshold.
Extrasynaptic GABAergic inhibition depends on GAT‐1/3 activity
GATs are responsible for GABA reuptake, but these transporters can also work in the reverse direction, thereby elevating extracellular GABA levels (Richerson & Wu, 2003; Heja et al. 2009; Unichenko et al. 2013). As functional GAT‐1 and GAT‐3 are present in the LSO (Stephan & Friauf, 2014), we hypothesized that GAT‐1/3 activity may affect extrasynaptic GABAergic currents at LSO neurons. If GATs take up GABA, GAT blockade should foster extracellular GABA buildup, causing GABAAR desensitization and prolonged activation of GABAAR (Jones & Westbrook, 1995; Brickley et al. 1999; Mortensen et al. 2010). When we puff‐applied GABA (100 μM; 24 pulses at 0.2 Hz), we observed robust inhibitory currents of 820 ± 71 pA that did not change between pulse no. 1 and no. 24 (Fig. 10 Aa and b and Bc; n = 34, P = 0.104, paired t test). The τdecay was 267 ± 12 ms (Fig. 10 Cb) and remained constant during the 2 min stimulus period (Fig. 10 Ab; n = 34, P = 0.088, paired t test). Inhibition of GAT‐1 with 10 μM NO‐711 affected neither the amplitude (Fig. 10 Bb and c; n = 8, P = 0.691, paired t test) nor τdecay of pulse no. 1 (Fig. 10 Ca and b; n = 8, P = 0.711, unpaired t test). By contrast, inhibition of GAT‐3 with 40 μM SNAP‐5114 reduced amplitudes by 17 ± 4% from pulse no. 1 to no. 24 (Fig. 10 Ba–c; n = 13, P = 7.9 × 10−4, paired t test) and caused a 1.9‐fold slower decay (Fig. 10 Ca and b; τdecay = 500 ± 29 ms, n = 13, P = 2.9 × 10−11, unpaired t test). A possible mechanism for this reduction is increased GABAAR desensitization by prolonged elevated GABA levels, which are strongly regulated by GAT‐3 (Dalby, 2000). Coapplication of NO‐711 with SNAP‐5114 caused yet stronger reduction of GABA currents by 55 ± 4% (Fig. 10 Ba–c; n = 9, P = 1.4 × 10−6, paired t test) from pulse no. 1 to no. 24 and a concomitant increase of τdecay of pulse no. 1 to 757 ± 61 ms (Fig. 10 Ca and b; n = 8, P = 6.4 × 10−5, unpaired t test). The more than additive effect points to a synergistic action of GAT‐1 and GAT‐3 in regulating GABA. To test whether the reduction was a result of GABAAR desensitization, rather than a different GABA target, we tested the effects of 100 μM muscimol, a GABAAR agonist not transported by GAT‐1 and GAT‐3 (Keros & Hablitz, 2005; Madsen et al. 2010). Puff application of muscimol mimicked the situation of NO‐711 and SNAP‐5114 coapplication. Amplitudes were depressed by 75 ± 5% from pulse no. 1 to no. 24 (Fig. 10 Ba–c; n = 9, P = 3.3 × 10−8, paired t test) and τdecay of pulse no. 1 increased to 1.11 ± 0.15 s (Fig. 10 Ca and b; n = 9, P = 5.8 × 10−4, unpaired t test). These results suggest that receptor desensitization, resulting from prolonged agonist exposure, specifically causes the reduction. Collectively, they demonstrate a functional role of astrocytic GATs in regulating extracellular GABA levels in the mouse LSO.
Extrasynaptic GABA decreases the excitability of LSO neurons with low [Cl−]i
In contrast to subsynaptic GABAARs, extrasynaptic GABAARs respond to low concentrations of ambient GABA, thus generating a tonic conductance (Farrant & Nusser, 2005; Brickley & Mody, 2012). To complement our view of the action of inhibitory transmitters in the LSO, we finally addressed the impact of tonic GABAergic inhibition on the excitability of LSO neurons. To do so, we injected 10 s current pulses, stepwise increasing the current amplitude until an action potential was elicited. We did this in control conditions and during GABA puffs at various concentrations (Fig. 11 Aa and b). GABA at 1 μM was insufficient to cause changes in the resting membrane potential (V M) or in I AP, the current amplitude required to elicit an action potential (Fig. 11 Ac and d; P = 0.451 and P = 0.102, n = 8, paired t test). In the presence of 10 μM GABA, however, the firing threshold was reached at a higher current amplitude than in controls (Fig. 11 Aa and d). Notably, 1 μM and 10 μM GABA are in the physiological range (Attwell et al. 1993; Morishima et al. 2010). We observed a tonic hyperpolarization (ΔV M) of −1.9 ± 0.4 mV (n = 6, P = 0.005, paired t test) and a concurrent shift of I AP (ΔI AP) amounting to 67 ± 17 pA (n = 6, P = 0.010, paired t test). The effects were stronger at 100 μM GABA (Fig. 11 Ac and d; ΔI AP: 625 ± 108 pA; ΔV M: −3.3 ± 0.5 mV; P = 0.002 and P = 0.002, n = 6, paired t test). All effects were abolished when GABA was coapplied with GBZ (Fig. 11 Ac and d). Compared to 10 μM GABA, 100 μM GABA led to a 1.7‐fold increased ΔV M, yet to a 9.3‐fold increased ΔI AP. As the disproportionally high increase of ΔI AP may be due to a reduced membrane resistance (R M), we calculated R M during control condition and during GABA application (Fig. 11 Ba and b). Under control conditions, R M averaged 55 ± 8 MΩ (n = 19), but it dropped by 21 ± 3% during application of 10 μM GABA (Fig. 11 Bb and c; n = 6, P = 5.1 × 10−4, paired t test). When GABA was raised to 100 μM, R M dropped by 54 ± 5% (n = 5, P = 3.4 × 10−4, paired t test). The effect was abolished upon coapplication of GABA with GBZ. Collectively, these results show that the GABA‐induced decrease of R M is in line with a decreased excitability of LSO neurons mediated through GABAARs.
Extrasynaptic GABA increases the excitability of LSO neurons with high [Cl−]i
GABA and glycine can be depolarizing and even excitatory in immature neurons (Kasyanov et al. 2004). The effect is also observed in the LSO and can be attributed to a high [Cl−]i and a reversal potential (E Cl) more positive than V M (Ehrlich et al. 1999; Balakrishnan et al. 2003; Löhrke et al. 2005). We therefore wondered whether tonic, extrasynaptic GABA effects might increase the excitability at early developmental stages. We addressed the question by performing experiments at P4 similar to the P11 experiments illustrated in Fig. 11. To mimic the native intracellular Cl− concentration (Ehrlich et al. 1999), patch pipettes contained 32 mM Cl− (consequently, E Cl = −35 mV). As expected for such conditions, GABA puffs (10 μM) depolarized V M by 6.8 ± 1.0 mV (Fig. 12 A and B; n = 10, P = 0.0001, paired t test). Moreover, the excitability of LSO neurons increased during GABA application, as evidenced by lower current amplitudes required to elicit an action potential (Fig. 12 A and C; ΔI AP = −55.0 ± 19.3 pA, n = 10, P = 0.020, paired t test). The increased excitability was observed although R M dropped by 63.5 ± 4.5% (Fig. 12 D; n = 10, P = 3.1 × 10−7, paired t test), arguing against shunting inhibition as a major effect of ambient GABA at immature LSO neurons.
Discussion
Five major results have emerged from the present study (Fig. 13). First, fast inhibitory synaptic transmission in the mouse LSO appears to be mediated exclusively by glycine. Second, synaptic GABAARs appear to be missing in the MNTB–LSO circuit. Third, GABA modulates presynaptic Ca2+ influx through presynaptic GABABRs, which results in weaker synaptic strength. Fourth, postsynaptic GABABRs mediate slow outward currents and GAT‐1 and GAT‐3 synergistically control extracellular GABA levels. Fifth, ambient GABA reduces the excitability of P11 LSO neurons, presumably via extrasynaptic α5/δ‐GABAARs, resulting in spike suppression. By contrast, ambient GABA increases the excitability at P4, resulting in spike facilitation. Notably, the increased excitability occurs when the immature network undergoes synaptic refinement and Cl−‐mediated neurotransmission is still depolarizing.
Figure 13. Schematic summary illustrating transmission at MNTB–LSO synapses of mice.
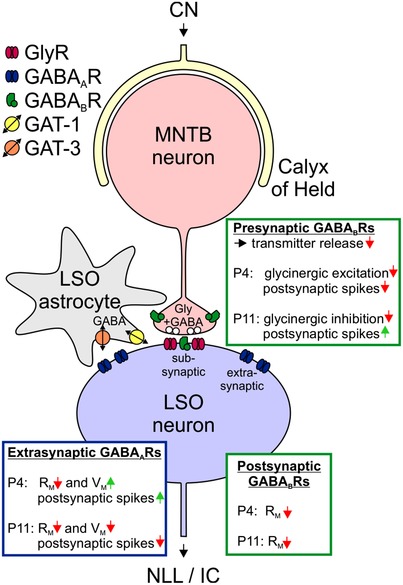
MNTB neurons (red) receive excitatory input from the contralateral CN via a calyx of Held (light yellow) and convey inhibitory information to LSO principal neurons (blue) via bouton‐type synapses. LSO neurons in turn innervate mesencephalic auditory nuclei like the nucleus of the lateral lemniscus (NLL) and the inferior colliculus (IC). GABA and glycine may be coreleased from MNTB–LSO synapses, but synaptic signalling appears to be purely glycinergic, i.e. subsynaptic receptors are GlyRs, but not GABAARs. MNTB axon terminals possess presynaptic GABABRs, LSO neurons possess extrasynaptic GABAARs and GABABRs. Extrasynaptic GABAARs may be activated by GABA spillover from the synaptic cleft or by reverse transport activity of astrocytic GAT‐1/3. Age‐specific effects mediated through presynaptic and postsynaptic GABABRs as well as extrasynaptic GABAARs are summarized in the green and blue boxes. R M, membrane resistance; V M, membrane potential.
Fast synaptic inhibition in the mouse LSO is purely glycinergic
We found glycine‐mediated synaptic inhibition of LSO neurons at P4 as well as P11, in agreement with others (Kandler & Friauf, 1995; Kotak et al. 1998; Nabekura et al. 2004; Kim & Kandler, 2010). However, our results that glycine alone, without the contribution of GABA, mediates fast inhibitory synaptic transmission at mouse MNTB–LSO synapses are in contrast to findings in rats and gerbils. There, predominant GABA release has been reported in neonates, and a developmental shift (Kotak et al. 1998) or switch (Nabekura et al. 2004) from GABA to glycine has been postulated. We obtained no evidence for such a shift at mouse MNTB–LSO synapses, and our immunohistochemical data, which demonstrate no GAD65/67 codistribution with gephyrin, regardless of age, corroborate the physiological results, as they point towards a lack of GABAergic synapses in the LSO.
Pharmacological considerations
So‐called GABAAR and GlyR antagonists (GBZ, bicuculline; strychnine) also act non‐specifically at the counterpart receptor molecules. Therefore, separation of GABAergic and glycinergic contributions at mixed synapses is a challenging task. Due to non‐specific inhibition of GlyRs by 10 μM bicuculline (Jonas et al. 1998; Nabekura et al. 2004), we reason that Nabekura and colleagues may have overestimated the GABA‐mediated component at rat MNTB–LSO synapses. Evaluating their findings (cf. their Fig. 1c) leads us to conclude that these synapses appear to lose most, if not all, of their GABAergic characteristics before P7. In gerbils, only the medial, high‐frequency limb of the LSO showed a GABA‐mediated component (Kotak et al. 1998). Again, the authors possibly overestimated this component due to the non‐specific action of bicuculline. Results from the pharmacological experiments by Kotak and collaborators, in which 2 μM strychnine was applied initially, are more conclusive. At this concentration, strychnine most likely blocked all GlyRs and some of the GABAARs. The remaining current was subsequently abolished completely by 10 μM bicuculline, providing compelling evidence for the existence of a GABAergic component in the medial limb of the gerbil LSO until P8–11. In the present study, we applied strychnine not only at 100 nM, but also 10‐fold higher. As GABAARs are only partially blocked by 1 μM strychnine (Jonas et al. 1998), a residual GABA‐mediated component would have remained if GABAARs were present. This, however, was not the case. Instead, we observed a complete blockade of eIPSCs upon 1 μM strychnine (not shown). Interestingly, in contrast to Kotak and colleagues, others (Walcher et al. 2011) found almost no effect on MNTB–LSO transmission in P10 gerbils with 10 μM GBZ, indicating only a minor contribution of GABAARs at this age, if at all. The virtual absence of GABA‐mediated currents in the lateral, low‐frequency limb of the gerbil LSO (Kotak et al. 1998) is consistent with our results. Nevertheless, we observed no GABA‐mediated synaptic currents along the tonotopic axis of the LSO (data not shown). Taken together, there are some discrepancies in the literature, which we cannot completely resolve at present.
Typically, GABAergic currents decay more slowly than those mediated by glycine (Russier et al. 2002; but see Moore & Trussell, 2017). Remarkably, Nabekura and colleagues (2004) reported extremely long‐lasting eIPSCs in rat LSO neurons (≥500 ms at P7, ≥200 ms at P14; no information about temperature). Drastically shorter durations were demonstrated at MNTB–LSO synapses of gerbils (>160 ms at P4, ∼80 ms at P14; Kotak et al. 1998; ∼20 ms at P8–15; Sanes, 1993; ∼20 ms at P10; Walcher et al. 2011). Shorter durations were also described in mice (∼10 ms at P10; Walcher et al. 2011; ∼10 ms at P11; Kramer et al. 2014; ∼20 ms at P11–15; Giugovaz‐Tropper et al. 2011; ∼100 ms at P4, ∼20 ms at P11, present study, Fig. 1). The values vary widely, even within the same species, but appear to decrease with age. Thus, there is some further discrepancy concerning the nature of these IPSCs that we cannot explain. We like to emphasize that GABA‐mediated and glycine‐mediated IPSCs are not mutually exclusively associated with long and short decay times, respectively. Rather, long decay times of ∼80 ms, and thus long‐lasting IPSCs, are also compatible with GlyRs containing α2 subunits (Takahashi et al. 1992; Ghavanini et al. 2006). Such α2‐GlyRs represent the ‘neonatal’ isoform and are characterized by low binding affinity for strychnine (Becker et al. 1988). During postnatal development of the brainstem, α2‐GlyRs are replaced by α1‐GlyRs (Friauf et al. 1997; Kungel & Friauf, 1997; Piechotta et al. 2001), and the replacement is accompanied by rapidly decaying synaptic responses and, consequently, speeding of glycinergic transmission (Singer et al. 1998; Pilati et al. 2016). Thus, an age‐dependent acceleration of IPSCs does not necessarily imply a decrease in the GABAergic component. It is also compatible with our observation of purely glycinergic inhibition in the developing mouse MNTB–LSO synapses. In summary, developing MNTB–LSO synapses may well display species‐specific differences in the transmitter phenotype, yet we think that the GABA contribution is overestimated and less general than previously anticipated. One way to address the issue would be to perform pentobarbital experiments in gerbils and rats as we have done in the present study (Fig. 3).
GABA is a modulator
Instead of acting synaptically, GABA acts as a modulator at the glycinergic MNTB–LSO circuit. The modulation occurs via three pathways: (1) via extrasynaptic GABAARs, the excitability of LSO neurons is changed in an age‐dependent manner; (2) via presynaptic GABABRs, the transmitter release from MNTB axons is reduced; and (3) via postsynaptic GABABRs, the membrane resistance of LSO neurons is changed. It appears that GABAergic modulation in the MNTB–LSO circuit is not a unique property of mice. In vivo activation of GABABRs at gerbil MNTB axon terminals shifts the ILD function by suppressing eIPSCs via a mechanism similar to the one demonstrated in the present study (Magnusson et al. 2008). eIPSC suppression mediated by presynaptic GABABRs is present in the projection from the MNTB to the medial superior olive of gerbils (Stange et al. 2013). These modulatory effects in gerbils, together with the overestimation of the synaptic GABA component (Nabekura and Kotak studies), imply that modulatory effects of GABA seem to be general in the auditory brainstem.
Sources of GABA in mouse LSO
What might be the main source of ambient GABA responsible for tonic activity in the mouse LSO? It is unlikely that MNTB axon terminals corelease GABA and glycine. First, we did not observe GABAAR‐mediated eIPSCs or mIPSCs in mouse LSO neurons (Figs 1 and 3). Such corelease can occur in other systems (see Introduction), and synaptic release can lead to activation of extrasynaptic GABARs elsewhere (Vaaga et al. 2014; Tritsch et al. 2016). However, we saw no GABAR activation upon stimulating MNTB–LSO synapses. Second, our immunohistochemical results show codistribution of gephyrin with GlyT2, but not with GAD65/67 (Fig. 4). The latter finding is supportive of an absence of GABA from presynaptic terminals, including terminals of MNTB axons (see, however, Weisz et al. 2016). Double immunolabelling for GAD65/67 and GlyT2 may directly address codistribution of both proteins in a given presynaptic terminal. Preliminary data from our group show a mixed phenotype, namely GlyT2 plus GAD65/67 immunosignals, in only 4% of boutons contacting somata of LSO principal cells in P12 mice (Hirtz et al. 2012). Whether the ratio is higher at younger ages needs to be determined.
In the gerbil LSO, immunohistochemical labelling for GABA is medium to intense between P4 and P21 in boutons contacting principal cells (Korada & Schwartz, 1999). Staining for GABAAR subunits β2 and β3, however, decreases from intense at P4 to light at P14. These data indicate that axon boutons may release GABA onto gerbil LSO neurons throughout development, whereas synaptic GABAAR signalling declines with age. In conclusion, extrasynaptic GABAergic transmission may prevail at older ages in this species as well.
Aside from MNTB axon terminals, another GABA source may be LSO neurons themselves. We found GAD65/67 immunosignals within their somata (Fig. 4), confirming previous reports of GABAergic LSO neurons in rodents (Moore & Moore, 1987; Helfert et al. 1989; Korada & Schwartz, 1999; Jenkins & Simmons, 2006). This immunoreactivity is also consistent with vesicular release of GABA from axon terminals in the various target nuclei of LSO neurons. Retrograde GABA release from LSO neurons during spiking activity and subsequent modulation of synaptic input strength has been demonstrated in juvenile gerbils (Magnusson et al. 2008). It is unknown whether GABAergic LSO neurons have axon collaterals that terminate in the LSO, either on neighbouring neurons or through autaptic feedback, as shown in the superior paraolivary nucleus adjacent to the LSO (Pollak et al. 2011). However, preliminary results in rats indicate that intra‐nuclear synaptic connections between LSO neurons are absent (Kim & Kandler, 2003).
Effects of extrasynaptic GABA
Low ambient GABA levels at extrasynaptic GABAAR sites and a concomitant activation of uninterrupted, tonic conductances occur in many CNS areas (Yeung et al. 2003; Semyanov et al. 2004). Likewise, tonic GABA conductances are increasingly recognized as important regulators of cell and neuronal network excitability (Glykys & Mody, 2007a ; Patel et al. 2016), and a multitude of neurological disorders are associated with imbalanced tonic inhibition (Belelli et al. 2009; Cope et al. 2009; Clarkson et al. 2010; Brickley & Mody, 2012; Wu et al. 2014). GABA levels can briefly exceed 1 mM at synaptic sites (Cherubini & Conti, 2001; Patel et al. 2016). Extrasynaptic GABA concentrations, however, are likely to be in the 10 nM to a few micromolar range (Lerma et al. 1986; Attwell et al. 1993; Juhasz et al. 1997; Maex & De Schutter, 1998; Huang et al. 2008; Dvorzhak et al. 2010; Morishima et al. 2010; Karim et al. 2012; Numata et al. 2012). Thus, the GABA concentrations used in the present study that were sufficient to successfully elicit tonic responses were probably more than 100‐fold lower than they are at synaptic sites (1–10 μM; Figs 11 and 12). Furthermore, we argue that ambient GABA concentrations are very low in the LSO in a synaptically silent slice preparation (nanomolar range), too low to enable the recording of tonic inhibition. Concentrations rise above detection level only during spike activity (see Glykys & Mody, 2007b ; Tang et al. 2011). Consequently, constitutively active GABAergic inputs to the LSO appear to be absent in vitro. The assumption is consistent with previous reports demonstrating a dependency of ambient GABA levels on neuronal activity (Lerma et al. 1986; Rowley et al. 1995; Kennedy et al. 2002; de Groote & Linthorst, 2007; Vanini et al. 2011). Because of the activity dependency, measuring ambient GABA is challenging, and the diversity of experimental conditions and analytical methods makes comparisons of tonic inhibition between studies quite difficult (Bright & Smart, 2013; Christensen et al. 2014).
A common thought is that α1–3 subunits of GABAARs are localized at synapses and mediate phasic inhibition, whereas α4–6 subunits are prevalent outside the synapse and mediate tonic inhibition (Karim et al. 2012). Our mRNA profiling analysis in the LSO identified 13 transcripts encoding for GABAAR subunits (Fig. 9). Although some of them, e.g. α3 and β3 (position 5 and 8), are typically associated with synaptic‐type α3β3γ2‐GABAARs (Mortensen et al. 2012), α3 and β3 can as well combine into binary extrasynaptic α3β3‐GABAARs (Mortensen et al. 2012). Likewise, α1 and β2 subunits (position 9 and 11), which are often compounds of ternary synaptic receptors (α1β1γ2, α1β2γ2, α1β3γ2), can as well coassemble in binary extrasynaptic α1β2‐ or ternary extrasynaptic α1β2δ‐GABAARs. In general, GABA is less potent on ternary GABAARs (α1–6β1–3γ2) than on those with binary combinations (α1–6β1–3; (Ducic et al. 1995). We also identified transcripts for the γ1 and γ2 subunits (position 7 and 3). γ subunits preferentially coassemble with α4 and α6 subunits and predominate at peri‐ and extrasynaptic locations (Wei et al. 2003). The marginal desensitization associated with such receptors makes them ideal for being activated by ambient GABA levels (Mody, 2001), and indeed, α6γ2‐containing receptors were shown to be most sensitive to GABA (Karim et al. 2013).
To our knowledge, the α2 subunit is not associated with extrasynaptic GABAARs, but rather restricted to synaptic ones (α2β3γ2). Remarkably, α2 transcripts were relatively abundant in the LSO (position 6). Our explanation for this surprising result is that α2 subunits may contribute to synaptic GABAARs on non‐principal LSO cells, for example lateral olivo‐cochlear neurons. Mouse lateral olivo‐cochlear neurons receive inhibitory input that is blocked by bicuculline and strychnine (Archakov et al. 2003; Sterenborg et al. 2010). The slow kinetics of this input are consistent with a GABAergic character.
Some of the most abundant extrasynaptic GABAARs are composed of α4βδ and α6βδ subunits (Farrant & Nusser, 2005; Jia et al. 2005; Belelli et al. 2009). Moreover, there is evidence for extrasynaptic α4β‐ and α6β‐GABAARs (Bencsits et al. 1999; Sinkkonen et al. 2004). Our RNA sequencing results are in harmony with the idea of an extrasynaptic location (α4 position 10, α6 position 12, δ position 2). Notably, GABA has the highest potency at α6‐containing receptors and the lowest at α2‐ and α3‐containing ones, in accordance with low GABA concentrations at extrasynaptic receptor sites (Mortensen et al. 2012). As the range of potency is of paramount importance for the activation of GABAARs, it affects the role these receptors play in controlling excitability.
Notably, our RNA sequencing analysis identified transcripts for the ρ2 subunit, albeit at a low level (position 13). ρ subunits assemble into ionotropic homomeric receptors originally named GABACRs, but after reclassification they are seen as a specialized set of GABAARs often referred to as GABAA‐ρ receptors (Naffaa et al. 2017). GABAA‐ρ receptors are insensitive to GABAAR modulators like benzodiazepines, barbiturates and steroids (Bormann & Feigenspan, 1995; Bormann, 2000). Moreover, GABA's potency at homomeric GABAA‐ρ receptors is 10‐ to 100‐fold higher than at heteromeric GABAARs, with slow activation and deactivation and less desensitization (Bormann & Feigenspan, 1995; Bormann, 2000). Again, these features are hallmarks of extrasynaptic receptors. Interestingly, and of some relevance for pharmacological experiments with THIP, besides being an agonist at δ‐GABAARs, THIP is also a GABAA‐ρ receptor antagonist (Johnston et al. 2003). Collectively, with the exception of the α1 subunit, all subunits identified via transcript profiling can be a component of extrasynaptic GABAARs. Many open issues remain concerning the abundance, composition and function of extrasynaptic GABAARs in the LSO, and these issues require future research, including very high‐resolution studies of gene expression patterns such as single‐cell RNA sequencing (Patch‐seq; Cadwell et al. 2016).
Tonic activation of extrasynaptic GABAARs regulates neuronal excitability (Otis et al. 1991; Salin & Prince, 1996; Bautista et al. 2010), yet the impact varies across brain region and cell type (Lee & Maguire, 2014). Moreover, tonic activation does not necessarily imply tonic inhibition and spike suppression. With relatively positive Cl− reversal potentials (E Cl > V M) and a strong enough conductance, the action of high‐affinity extrasynaptic GABAARs may be excitatory (Song et al. 2011; Bright & Smart, 2013). Indeed, increased excitability has been described upon tonic activation of GABAARs (Kilb et al. 2013; Yu et al. 2013). Our findings of persistent hyperpolarizing or depolarizing effects when patch pipettes were filled with 2 mM or 32 mM [Cl−], respectively, are in full agreement with this conclusion (Figs 11 and 12). By controlling developmental activity patterns in the prenatal and neonatal brain, the modulatory role of GABA appears to be closely associated with the formation of neuronal circuits (Kilb et al. 2013).
Conclusions
Our results imply that mouse MNTB–LSO inputs lack synaptic GABA signalling. Nevertheless, GABA causes presynaptic effects on axon terminals of MNTB neurons via GABABRs where it reduces activity‐evoked Ca2+ influx. The lower Ca2+ levels will probably reduce transmitter release from axon terminals and thus dampen glycinergic transmission in the MNTB–LSO projection. Moreover, GABA causes postsynaptic effects on LSO principal neurons via extrasynaptic GABAARs likely to contain α5 and δ subunits. Extrasynaptic GABA signalling increases the excitability and facilitates spike generation in immature LSO neurons, which have a high [Cl−]i. In contrast, it decreases the excitability in mature neurons, which have a low [Cl−]i, and thereby suppresses spike generation. Collectively, we conclude that GABA is a modulatory transmitter in the mouse LSO rather than playing a direct role in fast inhibitory neurotransmission. Because of the multitude of GABA receptor subunits, GABA appears to be in a better position to function as a modulator than glycine, which acts on receptors of relatively uniform nature. Whether the scenario is also present in other species or unique to the mouse MNTB–LSO circuit needs to be determined.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
A.U.F.: experimental design and project conception, electrophysiology, calcium imaging and immunofluorescence experiments, analysis and interpretation of data, writing of manuscript; N.I.C.M.: experimental design, electrophysiology, analysis and interpretation of data, writing of manuscript; T.D. and D.D.T.: laser microdissection, RNA sequencing; J.O.F.: electrophysiology, analysis of data; D.G.: experimental design and project conception, interpretation of data; K.K., A.M. and J.W.: experimental design, RNA sequencing, analysis and interpretation of data; V.R.: immunofluorescence, analysis and interpretation of data; M.A.X.‐F.: experimental design, calcium imaging, interpretation of data, manuscript writing; E.F.: experimental design and project conception, interpretation of data, manuscript writing. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Priority Program 1608 ‘Ultrafast and temporally precise information processing: normal and dysfunctional hearing’ of the Deutsche Forschungsgemeinschaft (grant Fr 1784/17‐1 to E.F.) and the Research Initiative Membrane Biology RIMB (to D.G. and E.F.). Further support was provided by the Erasmus Mundus Program Auditory Cognitive Neuroscience CAN (to A.U.F.) and by the Center for Cognitive Science, a research initiative of the Federal State of Rhineland–Palatine.
Acknowledgements
We thank Jennifer Winkelhoff, Ralph Reiss and Tina Kehrwald for excellent technical assistance, Erik Frotscher for input on miniature synaptic events, and Lucille A. Moore for valuable tips concerning pentobarbital. We are grateful to Dr Jochen W. Deitmer, who provided access to the confocal microscope. Finally, we thank Elisa G. Krächan and Dr Jan J. Hirtz for helpful comments on the manuscript.
Biographies
Alexander Fischer received his PhD in Biology at the University of Kaiserslautern, where he worked on fast synaptic transmission in the auditory system, with special interest in short‐term plasticity of inhibitory synapses. He recently headed to industry and follows his interest in deep analysis of data in the area of natural language understanding, utilizing principals of artificial intelligence.

Nicolas Müller received an MSc in Cell and Neurobiology and is currently finishing his PhD thesis in the lab of Eckhard Friauf (University of Kaiserslautern). In his thesis, he studied synaptic elimination and strengthening of the MNTB–LSO projection upon peripheral and central manipulations of the auditory system. By doing so, he gained mechanistic insight in the elimination and strengthening process.
Edited by: Ian Forsythe & Maike Glitsch
A. Fischer and N. Müller contributed equally to this work.
References
- Abdi H (2007). The Bonferonni and Šidák corrections for multiple comparisons In Encyclopedia of Measurement and Statistics, ed. Salkind E, pp. 103–107. Sage, Thousand Oaks. [Google Scholar]
- Alexander SP, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA & Collaborators C (2017). The concise guide to pharmacology 2017/18: Transporters. Br J Pharmacol 174(Suppl 1), S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolides PF & Trussell LO (2013). Rapid, activity‐independent turnover of vesicular transmitter content at a mixed glycine/GABA synapse. J Neurosci 33, 4768–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archakov AI, Govorun VM, Dubanov AV, Ivanov YD, Veselovsky AV, Lewi P & Janssen P (2003). Protein‐protein interactions as a target for drugs in proteomics. Proteomics 3, 380–391. [DOI] [PubMed] [Google Scholar]
- Attwell D, Barbour B & Szatkowski M (1993). Nonvesicular release of neurotransmitter. Neuron 11, 401–407. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Turecek R & Trussell LO (2005). Staggered development of GABAergic and glycinergic transmission in the MNTB. J Neurophysiol 93, 819–828. [DOI] [PubMed] [Google Scholar]
- Balakrishnan V, Becker M, Löhrke S, Nothwang HG, Güresir E & Friauf E (2003). Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J Neurosci 23, 4134–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B & Häusser M (1997). Intersynaptic diffusion of neurotransmitter. Trends Neurosci 20, 377–384. [DOI] [PubMed] [Google Scholar]
- Bautista W, Aguilar J, Loeza‐Alcocer JE & Delgado‐Lezama R (2010). Pre‐ and postsynaptic modulation of monosynaptic reflex by GABAA receptors on turtle spinal cord. J Physiol 588, 2621–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beato M, Burzomato V & Sivilotti LG (2007). The kinetics of inhibition of rat recombinant heteromeric α1β glycine receptors by the low‐affinity antagonist SR‐95531. J Physiol 580, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker CM, Hoch W & Betz H (1988). Glycine receptor heterogeneity in rat spinal cord during postnatal development. EMBO J 7, 3717–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM (2005). Presynaptically silent GABA synapses in hippocampus. J Neurosci 25, 4031–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC & Cope DW (2009). Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci 29, 12757–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencsits E, Ebert V, Tretter V & Sieghart W (1999). A significant part of native γ‐aminobutyric acidA receptors containing α4 subunits do not contain γ or δ subunits. J Biol Chem 274, 19613–19616. [DOI] [PubMed] [Google Scholar]
- Betz H & Laube B (2006). Glycine receptors: recent insights into their structural organization and functional diversity. J Neurochem 97, 1600–1610. [DOI] [PubMed] [Google Scholar]
- Bolte S & Cordelieres FP (2006). A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224, 213–232. [DOI] [PubMed] [Google Scholar]
- Bormann J (2000). The ‘ABC’ of GABA receptors. Trends Pharmacol Sci 21, 16–19. [DOI] [PubMed] [Google Scholar]
- Bormann J & Feigenspan A (1995). GABAC receptors. Trends Neurosci 18, 515–519. [DOI] [PubMed] [Google Scholar]
- Brenowitz S, David J & Trussell L (1998). Enhancement of synaptic efficacy by presynaptic GABAB receptors. Neuron 20, 135–141. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Cull‐Candy SG & Farrant M (1999). Single‐channel properties of synaptic and extrasynaptic GABAA receptors suggest differential targeting of receptor subtypes. J Neurosci 19, 2960–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG & Mody I (2012). Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP & Smart TG (2013). Methods for recording and measuring tonic GABAA receptor‐mediated inhibition. Front Neural Circuits 7, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N, Kerby J, Bonnert TP, Whiting PJ & Wafford KA (2002). Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br J Pharmacol 136, 965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell CR, Palasantza A, Jiang X, Berens P, Deng Q, Yilmaz M, Reimer J, Shen S, Bethge M, Tolias KF, Sandberg R & Tolias AS (2016). Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch‐seq. Nat Biotechnol 34, 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary DM & Finlayson PG (1991). Superior olivary complex: Functional neuropharmacology of the principal cell types In Neurobiology of Hearing: The Central Auditory System, ed. Altschuler RA, Bobbin RP, Clopton BM. & Hoffman DW, pp. 141–161. Raven Press, New York. [Google Scholar]
- Chanda S, Oh S & Xu‐Friedman MA (2011). Calcium imaging of auditory nerve fiber terminals in the cochlear nucleus. J Neurosci Methods 195, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpier S, Behrends JC, Triller A, Faber DS & Korn H (1995). “Latent” inhibitory connections become functional during activity‐dependent plasticity. Proc Natl Acad Sci U S A 92, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E & Conti F (2001). Generating diversity at GABAergic synapses. Trends Neurosci 24, 155–162. [DOI] [PubMed] [Google Scholar]
- Christensen RK, Petersen AV, Schmitt N & Perrier JF (2014). Fast detection of extrasynaptic GABA with a whole‐cell sniffer. Front Cell Neurosci 8, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson AN, Huang BS, Macisaac SE, Mody I & Carmichael ST (2010). Reducing excessive GABA‐mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orban G, Errington AC, Lorincz ML, Gould TM, Carter DA & Crunelli V (2009). Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med 15, 1392–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook J, Hendrickson A & Robinson FR (2006). Co‐localization of glycine and GABA immunoreactivity in interneurons in Macaca monkey cerebellar cortex. Neuroscience 141, 1951–1959. [DOI] [PubMed] [Google Scholar]
- Dalby NO (2000). GABA‐level increasing and convulsive effects of three different GABA uptake inhibitors. Neuropharmacology 39, e2399–e2407. [DOI] [PubMed] [Google Scholar]
- de Groote L & Linthorst AC (2007). Exposure to novelty and forced swimming evoke stressor‐dependent changes in extracellular GABA in the rat hippocampus. Neuroscience 148, 773–782. [DOI] [PubMed] [Google Scholar]
- Dittman JS & Regehr WG (1996). Contributions of calcium‐dependent and calcium‐independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci 16, 1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A & Gingeras TR (2015). Mapping RNA‐seq reads with STAR. Curr Protoc Bioinformatics 51, 11.14.11‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drasbek KR & Jensen K (2006). THIP, a hypnotic and antinociceptive drug, enhances an extrasynaptic GABAA receptor‐mediated conductance in mouse neocortex. Cereb Cortex 16, 1134–1141. [DOI] [PubMed] [Google Scholar]
- Ducic I, Caruncho HJ, Zhu WJ, Vicini S & Costa E (1995). γ‐Aminobutyric acid gating of Cl− channels in recombinant GABAA receptors. J Pharmacol Exp Ther 272, 438–445. [PubMed] [Google Scholar]
- Dufour A, Tell F & Baude A (2010). Perinatal development of inhibitory synapses in the nucleus tractus solitarii of the rat. Eur J Neurosci 32, 538–549. [DOI] [PubMed] [Google Scholar]
- Dugué GP, Dumoulin A, Triller A & Dieudonné S (2005). Target‐dependent use of co‐released inhibitory transmitters at central synapses. J Neurosci 25, 6490–6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumoulin A, Triller A & Dieudonne S (2001). IPSC kinetics at identified GABAergic and mixed GABAergic and glycinergic synapses onto cerebellar Golgi cells. J Neurosci 21, 6045–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorzhak A, Myakhar O, Unichenko P, Kirmse K & Kirischuk S (2010). Estimation of ambient GABA levels in layer I of the mouse neonatal cortex in brain slices. J Physiol 588, 2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Löhrke S & Friauf E (1999). Shift from depolarizing to hyperpolarizing glycine action in rat auditory neurons is due to age‐dependent Cl− regulation. J Physiol 520, 121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulenburg V, Armsen W, Betz H & Gomeza J (2005). Glycine transporters: essential regulators of neurotransmission. Trends Biochem Sci 30, 325–333. [DOI] [PubMed] [Google Scholar]
- Farrant M & Nusser Z (2005). Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci 6, 215–229. [DOI] [PubMed] [Google Scholar]
- Friauf E, Aragon C, Löhrke S, Westenfelder B & Zafra F (1999). Developmental expression of the glycine transporter GLYT2 in the auditory system of rats suggests involvement in synapse maturation. J Comp Neurol 412, 17–37. [PubMed] [Google Scholar]
- Friauf E, Fischer AU & Fuhr MF (2015). Synaptic plasticity in the auditory system: a review. Cell Tissue Res 361, 177–213. [DOI] [PubMed] [Google Scholar]
- Friauf E, Hammerschmidt B & Kirsch J (1997). Development of adult‐type inhibitory glycine receptors in the central auditory system of rats. J Comp Neurol 385, 117–134. [DOI] [PubMed] [Google Scholar]
- Gassmann M & Bettler B (2012). Regulation of neuronal GABAB receptor functions by subunit composition. Nat Rev Neurosci 13, 380–394. [DOI] [PubMed] [Google Scholar]
- Ghavanini AA, Mathers DA, Kim HS & Puil E (2006). Distinctive glycinergic currents with fast and slow kinetics in thalamus. J Neurophysiol 95, 3438–3448. [DOI] [PubMed] [Google Scholar]
- Giugovaz‐Tropper B, Gonzalez‐Inchauspe C, Di Guilmi MN, Urbano FJ, Forsythe ID & Uchitel OD (2011). P/Q‐type calcium channel ablation in a mice glycinergic synapse mediated by multiple types of Ca2+ channels alters transmitter release and short term plasticity. Neuroscience 192, 219–230. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mann EO & Mody I (2008). Which GABAA receptor subunits are necessary for tonic inhibition in the hippocampus? J Neurosci 28, 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J & Mody I (2007a). Activation of GABAA receptors: views from outside the synaptic cleft. Neuron 56, 763–770. [DOI] [PubMed] [Google Scholar]
- Glykys J & Mody I (2007b). The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J Physiol 582, 1163–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe B & Pecka M (2014). The natural history of sound localization in mammals – a story of neuronal inhibition. Front Neural Circuits 8, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen V, Storm‐Mathisen J & Bergersen LH (2015). Neuroglial transmission. Physiol Rev 95, 695–726. [DOI] [PubMed] [Google Scholar]
- Heja L, Barabas P, Nyitrai G, Kekesi KA, Lasztoczi B, Toke O, Tarkanyi G, Madsen K, Schousboe A, Dobolyi A, Palkovits M & Kardos J (2009). Glutamate uptake triggers transporter‐mediated GABA release from astrocytes. PLoS One 4, e7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfert RH, Bonneau JM, Wenthold RJ & Altschuler RA (1989). GABA and glycine immunoreactivity in the guinea pig superior olivary complex. Brain Res 501, 269–286. [DOI] [PubMed] [Google Scholar]
- Hirtz JJ, Braun N, Griesemer D, Hannes C, Janz K, Löhrke S, Müller B & Friauf E (2012). Synaptic refinement of an inhibitory topographic map in the auditory brainstem requires functional Cav1.3 calcium channels. J Neurosci 32, 14602–14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS & Edwards RH (2012). Neurotransmitter corelease: mechanism and physiological role. Annu Rev Physiol 74, 225–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoestgaard‐Jensen K, Dalby NO, Krall J, Hammer H, Krogsgaard‐Larsen P, Frølund B & Jensen AA (2014). Probing α4βδ GABAA receptor heterogeneity: differential regional effects of a functionally selective α4β1δ/α4β3δ receptor agonist on tonic and phasic inhibition in rat brain. J Neurosci 34, 16256–16272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Li Z, Dai J, Shahid M, Wong EH & Meltzer HY (2008). Asenapine increases dopamine, norepinephrine, and acetylcholine efflux in the rat medial prefrontal cortex and hippocampus. Neuropsychopharmacology 33, 2934–2945. [DOI] [PubMed] [Google Scholar]
- Ishibashi H, Yamaguchi J, Nakahata Y & Nabekura J (2013). Dynamic regulation of glycine–GABA co‐transmission at spinal inhibitory synapses by neuronal glutamate transporter. J Physiol 591, 3821–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SA & Simmons DD (2006). GABAergic neurons in the lateral superior olive of the hamster are distinguished by differential expression of gad isoforms during development. Brain Res 1111, 12–25. [DOI] [PubMed] [Google Scholar]
- Jia F, Pignataro L, Schofield CM, Yue M, Harrison NL & Goldstein PA (2005). An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons. J Neurophysiol 94, 4491–4501. [DOI] [PubMed] [Google Scholar]
- Johnston GA, Chebib M, Hanrahan JR & Mewett KN (2003). GABAC receptors as drug targets. Curr Drug Targets CNS Neurol Disord 2, 260–268. [DOI] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J & Sandkühler J (1998). Corelease of two fast neurotransmitters at a central synapse. Science 281, 419–424. [DOI] [PubMed] [Google Scholar]
- Jones MV & Westbrook GL (1995). Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 15, 181–191. [DOI] [PubMed] [Google Scholar]
- Juhasz G, Kekesi KA, Nyitrai G, Dobolyi A, Krogsgaard‐Larsen P & Schousboe A (1997). Differential effects of nipecotic acid and 4,5,6,7‐tetrahydroisoxazolo[4,5‐c]pyridin‐3‐ol on extracellular gamma‐aminobutyrate levels in rat thalamus. Eur J Pharmacol 331, 139–144. [DOI] [PubMed] [Google Scholar]
- Kandler K & Friauf E (1995). Development of glycinergic and glutamatergic synaptic transmission in the auditory brainstem of perinatal rats. J Neurosci 15, 6890–6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim N, Wellendorph P, Absalom N, Bang LH, Jensen ML, Hansen MM, Lee HJ, Johnston GA, Hanrahan JR & Chebib M (2012). Low nanomolar GABA effects at extrasynaptic α4β1/β3δ GABAA receptor subtypes indicate a different binding mode for GABA at these receptors. Biochem Pharmacol 84, 549–557. [DOI] [PubMed] [Google Scholar]
- Karim N, Wellendorph P, Absalom N, Johnston GA, Hanrahan JR & Chebib M (2013). Potency of GABA at human recombinant GABAA receptors expressed in Xenopus oocytes: a mini review. Amino Acids 44, 1139–1149. [DOI] [PubMed] [Google Scholar]
- Kasyanov AM, Safiulina VF, Voronin LL & Cherubini E (2004). GABA‐mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc Natl Acad Sci U S A 101, 3967–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller AF, Coull JAM, Chéry N, Poisbeau P & De Koninck Y (2001). Region‐specific developmental specialization of GABA‐glycine cosynapses in laminas I‐II of the rat spinal dorsal horn. J Neurosci 21, 7871–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy RT, Thompson JE & Vickroy TW (2002). In vivo monitoring of amino acids by direct sampling of brain extracellular fluid at ultralow flow rates and capillary electrophioresis. J Neurosci Methods 114, 39–49. [DOI] [PubMed] [Google Scholar]
- Keros S & Hablitz JJ (2005). Subtype‐specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. J Neurophysiol 94, 2073–2085. [DOI] [PubMed] [Google Scholar]
- Kilb W, Kirischuk S & Luhmann HJ (2013). Role of tonic GABAergic currents during pre‐ and early postnatal rodent development. Front Neural Circuits 7, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G & Kandler K (2003). Elimination and strengthening of glycinergic/GABAergic connections during tonotopic map formation. Nat Neurosci 6, 282–290. [DOI] [PubMed] [Google Scholar]
- Kim G & Kandler K (2010). Synaptic changes underlying the strengthening of GABA/glycinergic connections in the developing lateral superior olive. Neuroscience 171, 924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M & Loebrich S (2007). Trafficking and synaptic anchoring of ionotropic inhibitory neurotransmitter receptors. Biol Cell 99, 297–309. [DOI] [PubMed] [Google Scholar]
- Knoflach F, Hernandez MC & Bertrand D (2016). GABAA receptor‐mediated neurotransmission: not so simple after all. Biochem Pharmacol 115, 10–17. [DOI] [PubMed] [Google Scholar]
- Korada S & Schwartz IR (1999). Development of GABA, glycine, and their receptors in the auditory brainstem of gerbil: A light and electron microscopic study. J Comp Neurol 409, 664–681. [DOI] [PubMed] [Google Scholar]
- Kotak VC, Korada S, Schwartz IR & Sanes DH (1998). A developmental shift from GABAergic to glycinergic transmission in the central auditory system. J Neurosci 18, 4646–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krächan EG, Fischer AU, Franke J & Friauf E (2017). Synaptic reliability and temporal precision are achieved via high quantal content and effective replenishment: auditory brainstem versus hippocampus. J Physiol 595, 839–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer F, Griesemer D, Bakker D, Brill S, Franke J, Frotscher E & Friauf E (2014). Inhibitory glycinergic neurotransmission in the mammalian auditory brainstem upon prolonged stimulation: short‐term plasticity and synaptic reliability. Front Neural Circuits 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogsgaard‐Larsen P, Frølund B, Jørgensen FS & Schousboe A (1994). GABAA receptor agonists, partial agonists, and antagonists: design and therapeutic prospects. J Med Chem 37, 2489–2505. [DOI] [PubMed] [Google Scholar]
- Krogsgaard‐Larsen P, Frølund B & Liljefors T (2002). Specific GABAA agonists and partial agonists. Chem Rec 2, 419–430. [DOI] [PubMed] [Google Scholar]
- Krogsgaard‐Larsen P, Frølund B, Liljefors T & Ebert B (2004). GABAA agonists and partial agonists: THIP (Gaboxadol) as a non‐opioid analgesic and a novel type of hypnotic. Biochem Pharmacol 68, 1573–1580. [DOI] [PubMed] [Google Scholar]
- Kungel M & Friauf E (1997). Physiology and pharmacology of native glycine receptors in developing rat auditory brainstem neurons. Brain Res Dev Brain Res 102, 157–165. [DOI] [PubMed] [Google Scholar]
- Lee V & Maguire J (2014). The impact of tonic GABAA receptor‐mediated inhibition on neuronal excitability varies across brain region and cell type. Front Neural Circuits 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J, Herranz AS, Herreras O, Abraira V & Martin del Rio R (1986). In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res 384, 145–155. [DOI] [PubMed] [Google Scholar]
- Li P & Slaughter M (2007). Glycine receptor subunit composition alters the action of GABA antagonists. Vis Neurosci 24, 513–521. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK & Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Löhrke S, Srinivasan G, Oberhofer M, Doncheva E & Friauf E (2005). Shift from depolarizing to hyperpolarizing glycine action occurs at different perinatal ages in superior olivary complex nuclei. Eur J Neurosci 22, 2708–2722. [DOI] [PubMed] [Google Scholar]
- Lüscher C & Slesinger PA (2010). Emerging roles for G protein‐gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JW (2004). Molecular structure and function of the glycine receptor chloride channel. Physiol Rev 84, 1051–1095. [DOI] [PubMed] [Google Scholar]
- Lynch JW (2009). Native glycine receptor subtypes and their physiological roles. Neuropharmacology 56, 303–309. [DOI] [PubMed] [Google Scholar]
- Madsen KK, White HS & Schousboe A (2010). Neuronal and non‐neuronal GABA transporters as targets for antiepileptic drugs. Pharmacol Ther 125, 394–401. [DOI] [PubMed] [Google Scholar]
- Maex R & De Schutter E (1998). Synchronization of Golgi and granule cell firing in a detailed network model of the cerebellar granule cell layer. J Neurophysiol 80, 2521–2537. [DOI] [PubMed] [Google Scholar]
- Magnusson AK, Park TJ, Pecka M, Grothe B & Koch U (2008). Retrograde GABA signaling adjusts sound localization by balancing excitation and inhibition in the brainstem. Neuron 59, 125–137. [DOI] [PubMed] [Google Scholar]
- Meera P, Wallner M & Otis TS (2011). Molecular basis for the high THIP/gaboxadol sensitivity of extrasynaptic GABAA receptors. J Neurophysiol 106, 2057–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I (2001). Distinguishing between GABAA receptors responsible for tonic and phasic conductances Neurochem Res 26, 907–913. [DOI] [PubMed] [Google Scholar]
- Mody I (2005). Aspects of the homeostatic plasticity of GABAA receptor‐mediated inhibition. J Physiol 562, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK & Moore RY (1987). Glutamic acid decarboxylase‐like immunoreactivity in brainstem auditory nuclei of the rat. J Comp Neurol 260, 157–174. [DOI] [PubMed] [Google Scholar]
- Moore LA & Trussell LO (2017). Corelease of inhibitory neurotransmitters in the mouse auditory midbrain. J Neurosci 37, 9453–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima T, Uematsu M, Furukawa T, Yanagawa Y, Fukuda A & Yoshida S (2010). GABA imaging in brain slices using immobilized enzyme‐linked photoanalysis. Neurosci Res 67, 347–353. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Ebert B, Wafford K & Smart TG (2010). Distinct activities of GABA agonists at synaptic‐ and extrasynaptic‐type GABAA receptors. J Physiol 588, 1251–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Patel B & Smart TG (2012). GABA potency at GABAA receptors found in synaptic and extrasynaptic zones. Front Cell Neurosci 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller E, Le Corronc H, Triller A & Legendre P (2006). Developmental dissociation of presynaptic inhibitory neurotransmitter and postsynaptic receptor clustering in the hypoglossal nucleus. Mol Cell Neurosci 32, 254–273. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Katsurabayashi S, Kakazu Y, Shibata S, Matsubara A, Jinno S, Mizoguchi Y, Sasaki A & Ishibashi H (2004). Developmental switch from GABA to glycine release in single central synaptic terminals. Nat Neurosci 7, 17–23. [DOI] [PubMed] [Google Scholar]
- Naffaa MM, Hung S, Chebib M, Johnston GAR & Hanrahan JR (2017). GABA‐ρ receptors: distinctive functions and molecular pharmacology. Br J Pharmacol 174, 1881–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlich J, Kuenzel T, Keine C, Korenic A, Rübsamen R & Milenkovic I (2014). Dynamic fidelity control to the central auditory system: synergistic glycine/GABAergic inhibition in the cochlear nucleus. J Neurosci 34, 11604–11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlich J, Rübsamen R & Milenkovic I (2017). Developmental shift of inhibitory transmitter content at a central auditory synapse. Front Cell Neurosci 11, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata JM, van Brederode JF & Berger AJ (2012). Lack of an endogenous GABAA receptor‐mediated tonic current in hypoglossal motoneurons. J Physiol 590, 2965–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JA & Berger AJ (1999). Cotransmission of GABA and glycine to brain stem motoneurons. J Neurophysiol 82, 1638. [DOI] [PubMed] [Google Scholar]
- Olsen RW & Sieghart W (2008). International Union of Pharmacology. LXX. Subtypes of gamma‐aminobutyric acidA receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev 60, 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orser BA (2006). Extrasynaptic GABAA receptors are critical targets for sedative‐hypnotic drugs. J Clin Sleep Med 2, S12–18. [PubMed] [Google Scholar]
- Otis TS, Staley KJ & Mody I (1991). Perpetual inhibitory activity in mammalian brain slices generated by spontaneous GABA release. Brain Res 545, 142–150. [DOI] [PubMed] [Google Scholar]
- Ottersen OP, Storm‐Mathisen J & Somogyi P (1988). Colocalization of glycine‐like and GABA‐like immunoreactivities in Golgi cell terminals in the rat cerebellum: a postembedding light and electron microscopic study. Brain Res 450, 342–353. [DOI] [PubMed] [Google Scholar]
- Patel B, Bright DP, Mortensen M, Frølund B & Smart TG (2016). Context‐dependent modulation of GABAAR‐mediated tonic currents. J Neurosci 36, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G & Sandberg R (2013). Smart‐seq2 for sensitive full‐length transcriptome profiling in single cells. Nat Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- Piechotta K, Weth F, Harvey RJ & Friauf E (2001). Localization of rat glycine receptor α1 and α2 subunit transcripts in the developing auditory brainstem. J Comp Neurol 438, 336–352. [PubMed] [Google Scholar]
- Pilati N, Linley DM, Selvaskandan H, Uchitel O, Hennig MH, Kopp‐Scheinpflug C & Forsythe ID (2016). Acoustic trauma slows AMPA receptor‐mediated EPSCs in the auditory brainstem, reducing GluA4 subunit expression as a mechanism to rescue binaural function. J Physiol 594, 3683–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak GD, Gittelman JX, Li N & Xie RL (2011). Inhibitory projections from the ventral nucleus of the lateral lemniscus and superior paraolivary nucleus create directional selectivity of frequency modulations in the inferior colliculus: A comparison of bats with other mammals. Hear Res 273, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman J, Latal AT, Besser S, Hirrlinger J & Hülsmann S (2013). Mixed miniature postsynaptic currents resulting from co‐release of glycine and GABA recorded from glycinergic neurons in the neonatal respiratory network. Eur J Neurosci 37, 1229–1241. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan NA, Drescher MJ, Morley BJ, Kelley PM & Drescher DG (2014). Calcium regulates molecular interactions of otoferlin with soluble NSF attachment protein receptor (SNARE) proteins required for hair cell exocytosis. J Biol Chem 289, 8750–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG & Atluri PP (1995). Calcium transients in cerebellar granule cell presynaptic terminals. Biophys J 68, 2156–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richerson GB & Wu Y (2003). Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol 90, 1363–1374. [DOI] [PubMed] [Google Scholar]
- Rowley HL, Martin KF & Marsden CA (1995). Determination of in vivo amino acid neurotransmitters by high‐performance liquid chromatography with o‐phthalaldehyde‐sulphite derivatisation. J Neurosci Methods 57, 93–99. [DOI] [PubMed] [Google Scholar]
- Rudolph U & Möhler H (2014). GABAA receptor subtypes: therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu Rev Pharmacol Toxicol 54, 483–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russier M, Kopysova IL, Ankri N, Ferrand N & Debanne D (2002). GABA and glycine co‐release optimizes functional inhibition in rat brainstem motoneurons in vitro . J Physiol 541, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA & Prince DA (1996). Spontaneous GABAA receptor‐mediated inhibitory currents in adult rat somatosensory cortex. J Neurophysiol 75, 1573–1588. [DOI] [PubMed] [Google Scholar]
- Sanes DH (1993). The development of synaptic function and integration in the central auditory system. J Neurosci 13, 2627–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P & Cardona A (2012). Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Perez‐Garci E, Schneider A, Kollewe A, Gauthier‐Kemper A, Fritzius T, Raveh A, Dinamarca MC, Hanuschkin A, Bildl W, Klingauf J, Gassmann M, Schulte U, Bettler B & Fakler B (2016). Modular composition and dynamics of native GABAB receptors identified by high‐resolution proteomics. Nat Neurosci 19, 233–242. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM & Silver RA (2004). Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends Neurosci 27, 262–269. [DOI] [PubMed] [Google Scholar]
- Singer JH, Talley EM, Bayliss DA & Berger AJ (1998). Development of glycinergic synaptic transmission to rat brain stem motoneurons. J Neurophysiol 80, 2608–2620. [DOI] [PubMed] [Google Scholar]
- Sinkkonen ST, Vekovischeva OY, Moykkynen T, Ogris W, Sieghart W, Wisden W & Korpi ER (2004). Behavioural correlates of an altered balance between synaptic and extrasynaptic GABAAergic inhibition in a mouse model. Eur J Neurosci 20, 2168–2178. [DOI] [PubMed] [Google Scholar]
- Smart TG (2015). GABAA receptors. In Handbook of Ion Channels, ed. Zheng J. & Trudeau MC, pp. 345–359. CRC, Boca Raton, FL. [Google Scholar]
- Smart TG & Paoletti P (2012). Synaptic neurotransmitter‐gated receptors. Cold Spring Harb Perspect Biol 4, a009662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song I, Savtchenko L & Semyanov A (2011). Tonic excitation or inhibition is set by GABAA conductance in hippocampal interneurons. Nat Commun 2, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht CG, Izeddin I, Rodriguez PC, El Beheiry M, Rostaing P, Darzacq X, Dahan M & Triller A (2013). Quantitative nanoscopy of inhibitory synapses: counting gephyrin molecules and receptor binding sites. Neuron 79, 308–321. [DOI] [PubMed] [Google Scholar]
- Stange A, Myoga MH, Lingner A, Ford MC, Alexandrova O, Felmy F, Pecka M, Siveke I & Grothe B (2013). Adaptation in sound localization: from GABAB receptor‐mediated synaptic modulation to perception. Nat Neurosci 16, 1840–1847. [DOI] [PubMed] [Google Scholar]
- Stephan J & Friauf E (2014). Functional analysis of the inhibitory neurotransmitter transporters GlyT1, GAT‐1, and GAT‐3 in astrocytes of the lateral superior olive. Glia 62, 1992–2003. [DOI] [PubMed] [Google Scholar]
- Sterenborg JC, Pilati N, Sheridan CJ, Uchitel OD, Forsythe ID & Barnes‐Davies M (2010). Lateral olivocochlear (LOC) neurons of the mouse LSO receive excitatory and inhibitory synaptic inputs with slower kinetics than LSO principal neurons. Hear Res 270, 119–126. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y & Tsujimoto T (1998). G‐protein‐coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. J Neurosci 18, 3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Momiyama A, Hirai K, Hishinuma F & Akagi H (1992). Functional correlation of fetal and adult forms of glycine receptors with developmental changes in inhibitory synaptic receptor channels. Neuron 9, 1155–1161. [DOI] [PubMed] [Google Scholar]
- Tang ZQ, Dinh EH, Shi W & Lu Y (2011). Ambient GABA‐activated tonic inhibition sharpens auditory coincidence detection via a depolarizing shunting mechanism. J Neurosci 31, 6121–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, Mortensen M, Hosie AM & Smart TG (2005). Dynamic mobility of functional GABAA receptors at inhibitory synapses. Nat Neurosci 8, 889–897. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Watt C, Spike RC & Sieghart W (1996). Colocalization of GABA, glycine, and their receptors at synapses in the rat spinal cord. J Neurosci 16, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Granger AJ & Sabatini BL (2016). Mechanisms and functions of GABA co‐release. Nat Rev Neurosci 17, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK & Fritschy JM (2014). Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci 15, 141–156. [DOI] [PubMed] [Google Scholar]
- Unichenko P, Dvorzhak A & Kirischuk S (2013). Transporter‐mediated replacement of extracellular glutamate for GABA in the developing murine neocortex. Eur J Neurosci 38, 11. [DOI] [PubMed] [Google Scholar]
- Vaaga CE, Borisovska M & Westbrook GL (2014). Dual‐transmitter neurons: functional implications of co‐release and co‐transmission. Curr Opin Neurobiol 29, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanini G, Wathen BL, Lydic R & Baghdoyan HA (2011). Endogenous GABA levels in the pontine reticular formation are greater during wakefulness than during rapid eye movement sleep. J Neurosci 31, 2649–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walcher J, Hassfurth B, Grothe B & Koch U (2011). Comparative posthearing development of inhibitory inputs to the lateral superior olive in gerbils and mice. J Neurophysiol 106, 1443–1453. [DOI] [PubMed] [Google Scholar]
- Walker MC & Semyanov A (2008). Regulation of excitability by extrasynaptic GABAA receptors In Results and Problems in Cell Differentiation, ed. Darlison MG, pp. 29–48. Springer, Berlin, Heidelberg. [DOI] [PubMed] [Google Scholar]
- Wang P & Slaughter MM (2005). Effects of GABA receptor antagonists on retinal glycine receptors and on homomeric glycine receptor alpha subunits. J Neurophysiol 93, 3120–3126. [DOI] [PubMed] [Google Scholar]
- Wei W, Zhang N, Peng Z, Houser CR & Mody I (2003). Perisynaptic localization of δ subunit‐containing GABAA receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci 23, 10650–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz CJ, Rubio ME, Givens RS & Kandler K (2016). Excitation by axon terminal GABA spillover in a sound localization circuit. J Neurosci 36, 911–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfarth KM, Bianchi MT & Macdonald RL (2002). Enhanced neurosteroid potentiation of ternary GABAA receptors containing the δ subunit. J Neurosci 22, 1541–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG & Saggau P (1995). GABAB receptor‐mediated presynaptic inhibition in guinea‐pig hippocampus is caused by reduction of presynaptic Ca2+ influx. J Physiol 485, 649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Guo Z, Gearing M & Chen G (2014). Tonic inhibition in dentate gyrus impairs long‐term potentiation and memory in an Alzheimer's disease model. Nat Commun 5, 4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Zhang W, Rondard P, Pin JP & Liu J (2014). Complex GABAB receptor complexes: how to generate multiple functionally distinct units from a single receptor. Front Pharmacol 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung JY, Canning KJ, Zhu G, Pennefather P, MacDonald JF & Orser BA (2003). Tonically activated GABAA receptors in hippocampal neurons are high‐affinity, low‐conductance sensors for extracellular GABA. Mol Pharmacol 63, 2–8. [DOI] [PubMed] [Google Scholar]
- Yoon BE & Lee CJ (2014). GABA as a rising gliotransmitter. Front Neural Circuits 8, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Proddutur A, Elgammal FS, Ito T & Santhakumar V (2013). Status epilepticus enhances tonic GABA currents and depolarizes GABA reversal potential in dentate fast‐spiking basket cells. J Neurophysiol 109, 1746–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS & Regehr WG (2002). Short‐term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]