

Abstract
Diabetes promotes the posttranslational modification of proteins by O-linked addition of GlcNAc (O-GlcNAcylation) to Ser/Thr residues of proteins and thereby contributes to diabetic complications. In the retina of diabetic mice, the repressor of mRNA translation, eIF4E-binding protein 1 (4E-BP1), is O-GlcNAcylated, and sequestration of the cap-binding protein eukaryotic translation initiation factor (eIF4E) is enhanced. O-GlcNAcylation has also been detected on several eukaryotic translation initiation factors and ribosomal proteins. However, the functional consequence of this modification is unknown. Here, using ribosome profiling, we evaluated the effect of enhanced O-GlcNAcylation on retinal gene expression. Mice receiving thiamet G (TMG), an inhibitor of the O-GlcNAc hydrolase O-GlcNAcase, exhibited enhanced retinal protein O-GlcNAcylation. The principal effect of TMG on retinal gene expression was observed in ribosome-associated mRNAs (i.e. mRNAs undergoing translation), as less than 1% of mRNAs exhibited changes in abundance. Remarkably, ∼19% of the transcriptome exhibited TMG-induced changes in ribosome occupancy, with 1912 mRNAs having reduced and 1683 mRNAs having increased translational rates. In the retina, the effect of O-GlcNAcase inhibition on translation of specific mitochondrial proteins, including superoxide dismutase 2 (SOD2), depended on 4E-BP1/2. O-GlcNAcylation enhanced cellular respiration and promoted mitochondrial superoxide levels in WT cells, and 4E-BP1/2 deletion prevented O-GlcNAcylation–induced mitochondrial superoxide in cells in culture and in the retina. The retina of diabetic WT mice exhibited increased reactive oxygen species levels, an effect not observed in diabetic 4E-BP1/2–deficient mice. These findings provide evidence for a mechanism whereby diabetes-induced O-GlcNAcylation promotes oxidative stress in the retina by altering the selection of mRNAs for translation.
Keywords: O-linked N-acetylglucosamine (O-GlcNAc), eukaryotic translation initiation factor 4E-binding protein 1 (EIF4EBP1), eukaryotic translation initiation, diabetes, retina, oxidative stress, post-translational modification (PTM), RiboSeq, ribosome foot printing
Introduction
Despite recent advances in therapeutics, diabetic retinopathy remains the most frequent cause of new cases of blindness among adults aged 20–74 in developed countries (1). The principal underlying cause of diabetic retinopathy is hyperglycemia, as intensive glycemic control is associated with a reduction in both the onset and progression of neurovascular complications in the retina (2). Nevertheless, the molecular mechanisms whereby hyperglycemia causes neurovascular dysfunction are incompletely understood. One potential mechanism that is largely unexplored in the retina is altered gene expression in response to enhanced flux through the nutrient- and stress-sensing hexosamine biosynthetic pathway (HBP).3
Hyperglycemia and diabetes promote flux through the HBP, which converts glucose to UDP GlcNAc. Posttranslational modification of cellular proteins by enzymatic O-linked addition of the monosaccharide GlcNAc to Ser or Thr residues (O-GlcNAcylation) contributes to the pathophysiology of diabetes (3), and evidence supports a role for O-GlcNAc signaling in diabetic retinopathy (4). O-GlcNAcylation of proteins has been observed to alter function through a number of mechanisms, including changes in subcellular localization, protein-protein interactions, enzymatic activity, and degradation rates (5–9). Protein O-GlcNAcylation is catalyzed by the enzyme O-GlcNAc transferase (OGT), whereas the enzyme O-GlcNAcase (OGA) catalyzes removal of the modification. These enzymes dynamically cycle GlcNAc residues on and off of proteins in a manner that is more reminiscent of protein phosphorylation compared with more stable forms of glycosylation (10).
Enhanced O-GlcNAcylation of retinal proteins has been observed in both diabetic Akita mice (11) and streptozotocin-treated mice (12). Moreover, both of these mouse models of type 1 diabetes exhibit elevated expression of OGT. OGT-positive cells localize predominantly in the inner nuclear layer of the retina but also in the inner plexiform layer, ganglion cell layer, and photoreceptor inner segment. The O-GlcNAc cycling enzymes OGT and OGA strongly associate with cytosolic ribosomes, suggesting that they play an important role in regulating mRNA translation (13). In retinal pericytes exposed to hyperglycemic conditions, O-GlcNAcylated proteins are most heavily represented in the functional category “Protein Synthesis and Processing” (14). Indeed, O-GlcNAcylation has been detected on 34 of the ∼80 proteins that compose the mammalian ribosome (13). Among translation initiation factors, subunits of eIF2 (15), eIF3 (15, 16), eIF4 (17, 18), eIF5 (15), poly(A)-binding protein (19), and 4E-BP1 (20) are all O-GlcNAcylated; however, the functional consequence of these modifications on gene expression remains to be established.
Recruitment of ribosomes to mRNA is altered in response to hyperglycemic conditions (21) or overexpression of OGT (13). In the retina of diabetic rodents, global rates of protein synthesis are reduced; however, all mRNAs are not affected equally (22). Diabetes promotes ribosome association for a number of retinal mRNAs, including the synaptic proteins Snap25 and snaptophysin 1 (22). Importantly, hyperglycemia also promotes translation of the mRNA encoding the proangiogenic cytokine vascular endothelial growth factor (VEGF) (23). VEGF concentrations are elevated in the eyes of patients with diabetic macular edema, and the cytokine is considered to play a causal role in the proliferative stages of diabetic retinopathy. In the retina of mice lacking the translational repressor 4E-BP1, VEGF expression remains unchanged in response to diabetes (24), and diabetes-induced visual dysfunction is delayed (25).
4E-BP1 regulates the selection of mRNAs for translation through binding and sequestration of the cap-binding protein eIF4E, which recognizes the 7-methyl-GTP cap structure found at the 5′ terminus of all eukaryotic mRNAs (26). In the retina of diabetic mice, hyperglycemia promotes 4E-BP1 O-GlcNAcylation and binding to eIF4E (25). Variation in the availability of eIF4E serves as a critical regulator of gene expression patterns, as mRNAs exhibit a range of dependence on eIF4E for translation. In fact, some messages, such as the one that encodes VEGF, contain RNA elements that facilitate ribosome recruitment and translation independent of eIF4E and the 5′ cap structure (i.e. cap-independent translation) (27). Although 4E-BP1 reduces eIF4E availability and cap-dependent translation, mRNAs that are capable of recruiting ribosomal subunits independent of the 5′ cap often exhibit an increase in their relative rates of translation because of reduced competition for binding (21). Thus, altered selection of mRNAs for translation represents an intriguing target for novel clinical therapies that attempt to address the underlying molecular cause of DR.
In this study, we evaluated the hypothesis that enhanced O-GlcNAcylation alters retinal gene expression. We used next-generation sequencing to evaluate changes in retinal mRNA abundance and performed ribosome profiling to reveal which of those mRNAs were actually being translated into proteins. This technique goes beyond more commonly used methods for assessing gene expression by isolating and sequencing nuclease-resistant 28-nt ribosome-protected mRNA fragments (RPFs). Although a few genes exhibited altered mRNA abundance, the principal effect of enhanced O-GlcNAcylation on retinal gene expression was observed at the level of mRNA translation. Overall, the findings provide new insights into the impact of O-GlcNAcylation on retinal gene expression and identify a molecular network of translationally regulated mRNAs that potentially underlie dysfunctional mitochondrial respiration and superoxide production in diabetic retinopathy.
Results
O-GlcNAcase inhibition alters mRNA translation
To evaluate the effect of enhanced protein O-GlcNAcylation on retinal gene expression, mice were administered the O-GlcNAcase inhibitor thiamet G (TMG). Twenty-four hours later, global protein O-GlcNAcylation levels were enhanced in the retina (Fig. 1A). To evaluate the impact of TMG on gene expression, we used next-generation sequencing to assess changes in retinal mRNA abundance and ribosome-bound mRNAs undergoing translation via RNA-Seq and ribosome profiling (Ribo-Seq), respectively (Fig. 1B). Overall, sequencing reads in the total mRNA and RPF that was obtained following nuclease digestion and ribosome isolation from whole retina were mapped to principal transcripts of 19,411 genes (Table S1). For both TMG and PBS administration, replicates from two independent runs were highly similar in total mRNA abundance (R2 ≥ 0.95, Fig. S1A) and RPF (R2 ≥ 0.95, Fig. S1B). In response to TMG, the majority of transcripts observed in the retina did not exhibit a change in total mRNA abundance or RPF. We observed minimal differences in mean mRNA abundances from the retina of PBS control versus TMG-exposed mice (Fig. 1C, R2 = 0.96). In total, 148 retinal mRNAs exhibited a change in abundance 24 h after administration of TMG compared with vehicle. Alternatively, the principal effect of TMG on retinal gene expression was observed at the level of mRNA translation (R2 = 0.74, Fig. 1D). Specifically, ∼19% of the transcriptome showed significant changes in ribosome density, as 1683 mRNAs exhibited increased and 1912 mRNAs exhibited reduced ribosome association with TMG administration (Fig. 1E, Table S1). To assess the effects on mRNA translational efficiency (TE), we compared the relative RPF and total mRNA abundance observations between retinas of mice receiving TMG and vehicle (Fig. 1F, Table S1) using the Riborex package (28). Although the TE of most mRNAs was not different, 3185 mRNAs exhibited altered TE in the retina of mice receiving TMG compared with vehicle (adjusted p ≤ 0.01, Table S1). Specifically, 1360 mRNAs had enhanced TE, and 1825 mRNAs had attenuated TE in the retinas of mice receiving TMG compared with vehicle. For orthogonal validation, we performed polysome fractionation by sucrose density gradient centrifugation and analyzed mRNA distribution profiles. Actively translating mRNAs associate with multiple ribosomes to form heavy polysomes, whereas poorly translated mRNAs are associated with few or no ribosomes and localize to the subpolysomal fraction. Consistent with the results of next-generation sequencing (Fig. 2A), mRNAs encoding Nr6a1 (nuclear receptor subfamily 6 group A member 1) and Fam122a (family with sequence similarity 122A) dramatically shifted from the polysome fraction so that they were almost exclusively observed in the subpolysomal fraction in the retinas of mice administered TMG (Fig. 2, B–D). In addition, mRNAs encoding Elmsan1 (ELM2 and Myb/SANT domain–containing 1), Mettl14 (methyltransferase-like 14), Nat8l (N-acetyltransferase 8–like), and IL1Rap (interleukin 1 receptor accessory protein) exhibited shifts with polysome profiling that supported the results obtained from ribosome profiling (Fig. 2D).
Figure 1.

O-GlcNAcase inhibition alters the selection of mRNAs for translation in the retina. Mice were administered the O-GlcNAcase inhibitor TMG or PBS vehicle as a control. A, total protein O-GlcNAcylation was assessed in retinal lysates by Western blot analysis 24 h after TMG administration. Protein molecular mass (in kilodaltons) is indicated on the left. B, workflow for analysis of retinal gene expression to assess changes in total mRNA abundance by RNA-Seq and nuclease-resistant RPFs by Ribo-Seq. C and D, retinal gene expression changes were assessed in two independent runs by RNA-Seq in combination with Ribo-Seq to compare mean variation in retinal mRNA abundance (C) and mRNA translation (D), respectively. E, changes in mRNA abundance and mRNA translation following TMG administration were compared. Translational efficiency as assessed by mean ribosome density was compared in mice administered either TMG or PBS vehicle. In C, D, and F, significant differences (adjusted p ≤ 0.01) are indicated in red. In E, significant differences are indicated in purple (translation), green (transcription), or blue (both translation and transcription). TPM, transcripts per million mapped reads.
Figure 2.

The effect of O-GlcNAcase inhibition on polysome association is consistent with sequencing analysis. Mice were administered TMG or PBS vehicle. A, summary of results from the sequencing analysis described in Fig. 1 for mRNAs encoding Nr6a1, Fam122a, Elmsan1, Mettl14, Nat8l, and Il1Rap. B–D, ribosomes were separated into either subpolysomal (SP) or polysomal (P) fractions via sucrose density gradient centrifugation. The distribution of mRNAs encoding Nr6a1, Fam122a, Elmsan1, Mettl14, Nat8l, and Il1Rap were assessed in subpolysomal or polysomal fractions via RT-PCR. Results are expressed as a relative percentage of the mRNA in the polysomal fraction and are representative of two independent experiments. Values are means ± S.E. for three replicates, assessing a pooled sample obtained from 10 retinas. *, p < 0.05 versus PBS vehicle.
O-GlcNAcase inhibition alters the translation of mitochondrial proteins
To assess the potential impact of the observed gene expression changes, functional relationships between genes that exhibited altered TE in the retina following O-GlcNAcase inhibition were investigated. The top canonical pathways identified by Ingenuity Pathways Analysis as being altered by TMG included acute phase response signaling, pattern recognition receptors, coagulation system, and LXR/RXR activation (Table S3). This analysis also identified toxicologic list pathways associated with mitochondrial dysfunction and oxidative stress (Fig. S2). GO enrichment analysis showed our gene set to be enriched 1.66-fold for genes associated with the annotation mitochondrion organization and 1.55-fold for genes associated with the annotation mitochondrial membrane (Table S3). Top-scoring mRNAs encoding mitochondrial proteins with TMG-induced changes in translation are listed in Fig. 3A and Table S4.
Figure 3.

O-GlcNAcase inhibition alters the translation of mRNAs encoding mitochondrial proteins in a 4E-BP1/2–dependent manner. Mice were administered TMG or PBS vehicle. A, the sequencing analysis described in Fig. 1 identified TMG-induced changes in translation of mRNAs encoding mitochondrial proteins. B, the interaction of 4E-BP1 and eIF4G with eIF4E was examined by immunoprecipitating eIF4E from retinal supernatants and measuring the amount of eIF4G and 4E-BP1 in the immunoprecipitate (IP) by Western blot analysis. Protein molecular mass (in kilodaltons) is indicated at the left. C, the interaction of 4E-BP1 with eIF4E in B was quantified. D, Western blot analysis was used to evaluate eIF4G, eIF4E, and 4E-BP1 protein expression in retinal lysates. E–I, WT and 4E-BP1/2 DKO mice were administered TMG or PBS vehicle as described previously. E, retinal protein O-GlcNAcylation as well as eIF4G, tubulin, eIF4E, and 4E-BP1 protein expression were assessed by Western blot analysis. F, the interaction of 4E-BP1 and eIF4G with eIF4E was examined as described in B. G–I, ribosomes from the retinas of WT and DKO mice administered either TMG or PBS vehicle were separated into either subpolysomal or polysomal fractions as described in Fig. 2. The distribution of mRNAs encoding the mitochondrial proteins TufM (G), Mrpl47 (H), and SOD2 (I) were assessed in subpolysomal and polysomal fractions via RT-PCR. Results are expressed as a relative percentage of the mRNA in the polysomal fraction and are representative of two independent experiments. Values are means ± S.E. for three replicates assessing a pooled sample obtained from 10 retinas. *, p < 0.05 versus PBS; #, p < 0.05 versus WT.
O-GlcNAcase inhibition regulates translation of mitochondrial proteins via 4E-BP1
4E-BP–dependent translational control has been implicated previously in regulating the expression of mitochondrion-related proteins (29). Moreover, we previously demonstrated that elevated O-GlcNAcylation promotes sequestration of eIF4E by 4E-BP1 in cells in culture (20). To determine whether enhanced O-GlcNAcylation had a similar effect in the retina, we performed immunoprecipitation of eIF4E from retinal lysates. Retinas from mice administered TMG exhibited enhanced interaction of 4E-BP1 and reduced interaction of eIF4G with eIF4E (Fig. 3, B–D). We previously demonstrated that hyperglycemia promotes 4E-BP1 O-GlcNAcylation and alters the translation of specific mRNAs in a manner that depends on 4E-BP1 (21). To evaluate the role of 4E-BP1 O-GlcNAcylation in regulating the translation of mRNAs encoding mitochondrial proteins, WT and 4E-BP1/2 (Eif4ebp1; Eif4ebp2) knockout mice were administered TMG. In the retinas of both WT and 4E-BP1/2–deficient mice, TMG enhanced global protein O-GlcNAcylation (Fig. 3E). Consistent with Fig. 3, B–D, TMG promoted 4E-BP1 co-immunoprecipitation with eIF4E in the retina of WT mice, whereas the protein could not be detected in 4E-BP1/2–deficient mice (Fig. 3, E and F). Next-generation sequencing demonstrated down-regulated translation of mRNAs encoding the mitochondrial proteins Tufm (Tu translation elongation factor, mitochondrial), Mrpl47 (mitochondrial ribosomal protein L47), and SOD2 (superoxide dismutase 2) in the retina of WT mice with enhanced protein O-GlcNAcylation (Fig. 3A, Table S1). In polysome profiles, Tufm and Mrpl47 were observed in the polysomal fraction obtained from the retina of WT mice receiving PBS (Fig. 3, G and H, respectively). TMG caused both mRNAs to shift almost exclusively into the subpolysomal fraction. In the retina of 4E-BP1/2–deficient mice, the ratio of Tufm and Mrpl47 observed in the polysomal fraction was similar with PBS and TMG administration. Similarly, the mRNA encoding SOD2 shifted out of the polysomal fraction with enhanced O-GlcNAcylation in WT mice, but a similar effect was not observed in mice deficient for 4E-BP1/2 (Fig. 3I).
O-GlcNAcase inhibition promotes mitochondrial ROS via 4E-BP1
To determine whether increased O-GlcNAcylation could affect mitochondrial respiration in a manner that depends on 4E-BP1, cultures of WT and 4E-BP1/2–deficient mouse embryonic fibroblasts (MEFs) were exposed to TMG. In both WT and 4E-BP1/2–deficient MEFs, exposure to TMG enhanced protein O-GlcNAcylation (Fig. 4A) and altered cellular respiration (Fig. 4B). TMG increased the basal oxygen consumption rate (OCR) (Fig. 4C), maximal OCR (Fig. 4D), and ATP production (Fig. 4E) in both WT and 4E-BP1/2-deficient MEFs. Although TMG enhanced cellular respiration in both WT and 4E-BP1/2–deficient cells, the maximal OCR was attenuated in 4E-BP1/2–deficient MEFs in both the presence and absence of TMG compared with the WT (Fig. 4D). In addition to changes in oxidative phosphorylation, mitochondrial dysfunction can also involve the generation of reactive oxygen species (ROS). To determine whether TMG altered cellular ROS production, we assessed mitochondrial superoxide. Superoxide levels were similar in vehicle-treated WT and 4E-BP1/2-deficient MEF cultures (Fig. 4, F and G). TMG exposure increased superoxide in WT MEFs. However, the effect of TMG was absent in 4EBP1/2-deficient MEFs, as superoxide levels were similar to those observed in both WT and 4E-BP1/2–deficient MEFs exposed to vehicle alone. This supports that acute elevations in O-GlcNAcylation promote mitochondrial superoxide levels via a 4E-BP1/2–dependent mechanism. To further investigate the role of 4E-BP1 in O-GlcNAcylation–induced mitochondrial superoxide levels, we restored 4E-BP1 expression in 4E-BP1/2–deficient MEFs in culture (Fig. 5A). TMG enhanced mitochondrial superoxide in cells expressing WT 4E-BP1 (Fig. 5, A and B). However, in cells expressing a 4E-BP1 variant (Y54A) that is deficient in binding to eIF4E (30), TMG did not enhance mitochondrial superoxide levels (Fig. 5, A and B). In contrast, expression of a 4E-BP1 variant with an F113A substitution that prevents mTORC1-dependent phosphorylation and promotes eIF4E binding (31) was sufficient to enhance mitochondrial superoxide levels in WT MEFs (Fig. 5, C and D).
Figure 4.

O-GlcNAcase inhibition promotes mitochondrial ROS via 4E-BP1/2. WT and 4E-BP1/2 DKO mouse embryonic fibroblasts were cultured in the presence of TMG or vehicle (Veh) for 24 h. A, protein O-GlcNAcylation and 4E-BP1 expression were assessed by Western blot analysis. Protein staining (Protein S.) is shown as a loading control. Protein molecular mass (in kilodaltons) is indicated at the left. B, mitochondrial respiration was assessed by Seahorse XF cell mitochondrial stress test. C–E, basal respiration (C), maximal respiration (D), and ATP production (E) were compared. F and G, mitochondrial superoxide was assessed in live cells using the MitoSOX Red superoxide indicator. MitoSOX fluorescence was visualized by confocal laser microscopy (F) and quantified (G). Results are representative of three experiments. Within each experiment, two to three independent samples were analyzed. Values are means ± S.E. *, p < 0.05 versus vehicle; #, p < 0.05 versus WT. RFU, relative fluorescent units.
Figure 5.
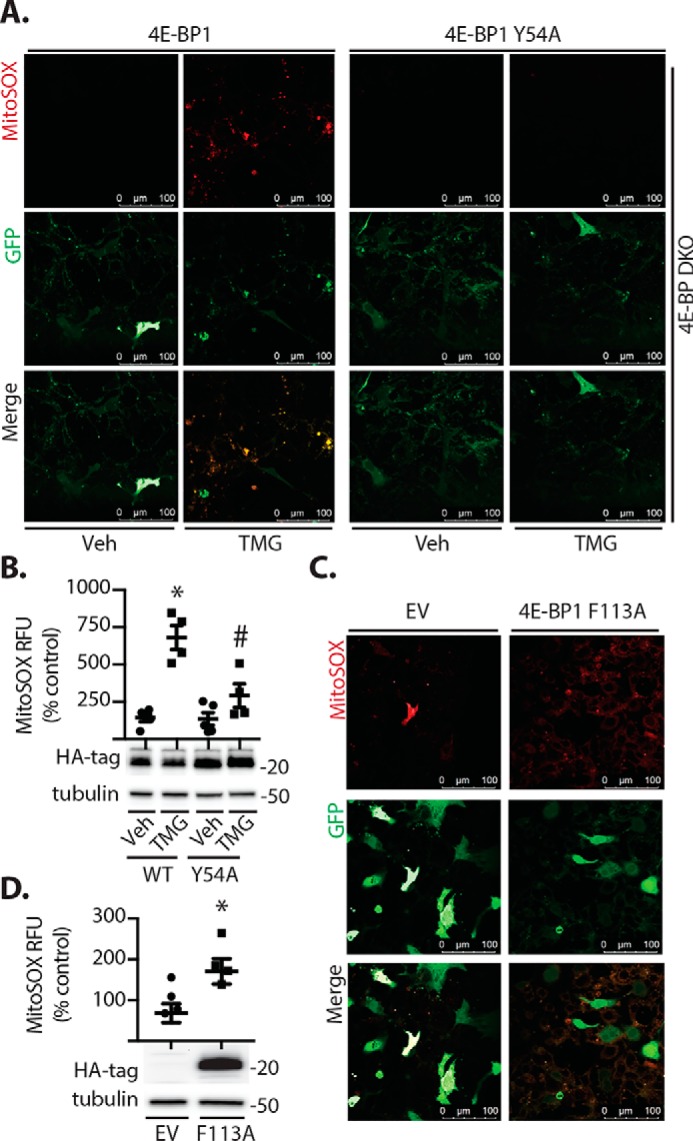
4E-BP1 regulates mitochondrial superoxide levels. A and B, WT or 4E-BP1/2 double knockout (4E-BP DKO) mouse embryonic fibroblasts were cultured in the presence of TMG or vehicle (Veh) for 24 h. Cells were co-transfected with plasmids that express GFP and either a WT 4E-BP1 or a 4E-BP1 variant (Y54A) that is deficient in binding to eIF4E. C and D, WT cells were co-transfected with plasmids that express GFP and either an empty vector (EV) control or a plasmid that expresses a 4E-BP1 F113A variant that is unable to be phosphorylated by mTORC1, resulting in constitutive eIF4E binding. MitoSOX fluorescence and GFP expression were visualized by confocal laser microscopy (A and C), and relative MitoSOX fluorescence was quantified (B and D). Values are means ± S.E. Results are representative of two experiments. Within each experiment, two to three independent samples were analyzed. *, p < 0.05 versus vehicle or empty vector; #, p < 0.05 versus WT. RFU, relative fluorescent units.
4E-BP1/2 deletion prevents O-GlcNAc- and diabetes-induced ROS in the retina
To evaluate the role of 4E-BP1 in the effect of O-GlcNAcyaltion on the retina, WT and 4E-BP1/2–deficient mice were administered TMG. Retinal cryosections were exposed to the fluorescent ROS indicator 2,7-dichlorofluoroscein (DCF) to evaluate oxidative stress. In the retina of WT mice, TMG administration enhanced DCF fluorescence compared with vehicle (Fig. 6, A and B). Enhanced ROS levels were most obvious in the photoreceptor layers but could also be observed throughout the entire retina. DCF fluorescence was similar in the retina of WT and 4E-BP1/2–deficient mice receiving vehicle. Moreover, ROS levels were similar in the retina of 4E-BP1/2–deficient mice administered either PBS or TMG. Importantly, ROS levels were reduced in 4E-BP1/2–deficient mice administered TMG compared with WT controls administered TMG. We previously demonstrated that 4E-BP1 O-GlcNAcylation was enhanced in the retina of streptozotocin (STZ)-induced diabetic mice (25). Consistent with previous reports (e.g. 32), STZ-induced diabetes enhanced DCF fluorescence in the retina of WT mice compared with nondiabetic controls (Fig. 6, C and D). In nondiabetic 4E-BP1/2–deficient mice, DCF fluorescence was similar to that observed in diabetic 4E-BP1/2–deficient mice. Moreover, ROS levels were reduced in diabetic 4E-BP1/2–deficient mice compared with diabetic WT mice. Overall, the results support a model wherein diabetes-induced 4E-BP1 O-GlcNAcylation alters mRNA translation in a manner that causes mitochondrial oxidative stress in the retina.
Figure 6.

4E-BP1/2 deletion prevents O-GlcNAcyaltion- and diabetes-induced ROS in the retina. A and B, WT and 4E-BP1/2 double knockout (4E-BP DKO) mice were administered TMG or vehicle (Veh) as described previously. C and D, diabetes was induced in WT and 4E-BP DKO mice by administration of STZ. Nondiabetic controls were administered vehicle. Whole eyes were isolated, cryosectioned into sagittally oriented longitudinal cross-sections, stained with Hoechst, and exposed to DCF. Retinas were evaluated 24 h after TMG administration (A and B) or after 4 weeks of diabetes (C and D). DCF fluorescence and nuclear staining were evaluated by confocal laser microscopy (A and C), and relative DCF fluorescence was quantified (B and D). Results are representative of two experiments (n = 3–6). *, p < 0.05 versus vehicle; #, p < 0.05 versus WT.
Discussion
Hyperglycemia and diabetes promote flux through the HBP, which in turn results in posttranslational modification of proteins by O-GlcNAcylation. Dysregulation of O-GlcNAc cycling contributes to diabetic complications, and evidence supports a role in diabetic retinopathy (4). One potential mechanism whereby dysregulated O-GlcNAcylation contributes to diabetic retinopathy is by altering retinal gene expression, as numerous regulatory factors involved in gene transcription and mRNA translation are modified by O-GlcNAc. Thus, this study investigated the impact of O-GlcNAcylation on retinal gene expression. Assessment of retinal gene expression by deep sequencing demonstrated that TMG altered translation of a molecular network of mRNAs associated with mitochondrial function and oxidative stress. Overall, the findings support a model whereby O-GlcNAcylation enhances cellular respiration and mitochondrial superoxide production.
Gene expression is the sum of events that regulate cellular protein concentrations, including transcription and translation, as well as mRNA and protein degradation. Historically, gene expression studies have utilized techniques such as microarrays and, more recently, RNA-Seq to assess changes in mRNA abundance. However, mRNA abundance is a relatively poor correlate for protein expression (33, 34), and some studies suggest that they only account for ∼40% of the global variation in protein expression (35, 36). Translational control facilitates the selective recruitment of ribosomes to specific mRNAs to provide a rapid and reversible response in the expression of specific proteins to changes in nutrient supply, hormones, and stress (37, 38). Thus, an assessment of ribosome-protected mRNA fragments accounts for both transcriptional and translational changes that lead to variation in protein expression. A previous study from our laboratory demonstrated that hyperglycemic conditions alter the selection of mRNAs for translation (21). In this study, we performed genome-wide quantitative analysis of in vivo translation in the retina in combination with RNA-Seq to assess changes in retinal gene expression. This technique utilized nuclease footprinting to produce mRNA fragments that indicate which mRNAs were associated with ribosomes. The effect of O-GlcNAcase inhibition on retinal gene expression was largely observed at the level of mRNA translation. Remarkably, only a few genes exhibited changes in mRNA abundance (<1% of the observed transcripts) following TMG administration. However, ∼19% of the transcriptome exhibited altered mRNA translation. The extremely limited transcriptional effect of O-GlcNAcase inhibition on retinal gene expression was surprising, as the activity of a number of transcription factors is regulated by O-GlcNAcylation (40). An important caveat is that retinal gene expression was assessed 24 h after administration of the O-GlcNAcase inhibitor, and thus transcriptional effects on gene expression may be more obvious after prolonged disruption of O-GlcNAc cycling. Another important consideration of our analysis is that the contribution of protein degradation to gene expression has not been considered. Protein concentrations in a cell are determined by the relative rates of synthesis and degradation. Enhanced rates of retinal protein degradation have been observed in diabetic rodents (22); however, compared with rates of synthesis, the impact of protein stability on gene expression is relatively minor (35).
In this study, changes in ribosome density were interpreted as reflecting changes in mRNA translation. Although this is a common assumption in the field, it is only accurate when there are no systemic pauses or stalls of ribosomes on transcripts. To ensure that the effects of O-GlcNAcase inhibition on ribosome density were not due to pausing/stalls, polarity scores (41) were generated to determine whether TMG skewed ribosome density toward the 5′ or 3′ ends of mRNA. Importantly, there were no significant changes in polarity scores between the two treatments (Wilcoxon signed-rank test with continuity correction, p = 0.09, Fig. S3), indicating that the large-scale effect of TMG on mRNA translation was due to changes in initiation rates.
One of the best-characterized mechanisms for regulating translation initiation involves 4E-BP1 binding to eIF4E to prevent eIF4F complex assembly at the 5′ end of an mRNA. We initially discovered that 4E-BP1 is directly modified by O-GlcNAcylation in the liver of diabetic rats (20). A variety of conditions that increase O-GlcNAc levels (e.g. hyperglycemia, O-GlcNAcase inhibition, or glucosamine addition to the culture medium) promote binding of 4E-BP1 with eIF4E (20, 21, 25). Furthermore, 4E-BP1 O-GlcNAcylation inhibits its degradation via an E3 ubiquitin ligase complex containing CUL3. Specifically, we have provided evidence for a model wherein O-GlcNAcylation and GSK3-dependent phosphorylation competitively modify 4E-BP1 at Thr-82/Ser-86 (25). Of importance to this study is that we reported previously that 4E-BP1 O-GlcNAcylation (25) and binding to eIF4E (24) are enhanced in the retina of diabetic rodents. Here we found that, in the retina of mice receiving TMG, O-GlcNAcylation levels were enhanced concomitant with an increase in co-immunoprecipitation of 4E-BP1 with eIF4E.
TMG-induced O-GlcNAcylation altered the translation of mRNAs encoding numerous mitochondrial proteins, including those involved in oxidative phosphorylation (OXPhos), mitochondrial ribosomal subunits, mitochondrion-specific tRNA synthetases, and subunits of the translocase of inner membrane (TIM)/translocase of outer membrane (TOM) complex. Importantly, we demonstrated that 4E-BP1/2 were critical in mediating the effects of O-GlcNAcylation on the translation of specific mRNAs encoding mitochondrial proteins and mitochondrial ROS production. This observation supports previous reports implicating 4E-BP–dependent translational control in the regulation of mitochondrial function (29, 42). The mitochondrial electron transport chain compromises ∼150 distinct proteins, including 13 hydrophobic inner membrane proteins that are encoded by the mitochondrial DNA (mtDNA) and synthesized by mitochondrial ribosomes (mitoribosomes). Alternatively, the remaining majority of OXPhos proteins are synthesized by cytosolic ribosomes and imported by the TIM/TOM complex. Proper mitochondrial function requires rapid adaptation to metabolic conditions and involves coordinated expression from both the mtDNA and nuclear genome. Yeast ribosome profiling demonstrates that nuclear and mitochondrial encoded OXPhos mRNA abundances are not altered in concordance following a nutrient shift, but rather, synchronized protein expression is achieved via translational control (43). Thus it is likely that defects in mRNA translation would contribute to human pathology, as there are a variety of mitochondrial diseases that result from improper mitochondrial protein expression (reviewed in Ref. 44).
In recent years, O-GlcNAcylation has emerged as a critical regulator of mitochondrial function (45, 46). Overexpression of the enzymes responsible for O-GlcNAc modification (i.e. OGT and OGA) alters mitochondrial protein levels, including specific proteins involved in OXPhos (45). Moreover, cells with elevated O-GlcNAc levels exhibit abnormal mitochondrial morphology and hyperpolarization (46). Numerous mitochondrial proteins are posttranslationally modified by O-GlcNAcylation directly in response to diabetes and hyperglycemic conditions (47, 48). Tan et al. (46) have suggested previously that mitochondria undergo a two-stage adaptive process in response to O-GlcNAcase inhibition. Initially, mitochondrial proteins are O-GlcNAcylated, resulting in increased cellular respiration and enhanced ROS production (49). However, with sustained elevation in O-GlcNAcylation, mitochondria undergo transcriptional reprogramming as ATP production and ROS levels decline (46). Consistent with the previous studies, we observed increased cellular respiration and elevated mitochondrial superoxide levels in cells after 24 h of exposure to culture medium containing TMG. This study extends these previous reports by demonstrating that the synthesis of mitochondrial proteins is dramatically altered by enhanced O-GlcNAcylation in response to TMG-induced O-GlcNAcase inhibition. Although the effect of TMG on cellular respiration was observed in 4E-BP1/2–deficient cells, TMG-induced mitochondrial superoxide was absent in cells lacking 4E-BP1/2. This observation supports a role for 4E-BP1/2–dependent translational control in increased ROS production during the first stage of the adaptive process that is initiated upon disruption of O-GlcNAc cycling.
The retina is among the most metabolically active tissues in the body and therefore is heavily dependent on a constant supply of energy from oxidative phosphorylation in the mitochondria. However, in diabetes, retinal mitochondria contain damaged mtDNA (50) in association with increased capillary cell apoptosis, an abnormality that occurs prior to the onset of histopathological changes in the retina (51). The dynamic nature of mitochondria necessitates tight regulation of the organelle's shape, size, formation of cristae, and transport of molecules across the mitochondrial membranes (52). In retinas of diabetic rats, this tight control is impaired as the mitochondrial ultrastructure is disrupted (53), and the enzymes required for repairing damaged retinal mtDNA fail to access the mitochondria despite an increase in their mRNA levels (50). Furthermore, the expression of small-molecule transport proteins involved in the exchange of citric acid cycle substrates as well as transport of antioxidant molecules into the mitochondria are less abundant in the mitochondria of diabetic rats and diabetic donor eyes (53). Similarly, we report in our dataset a TMG-induced decrease in translation of the outer membrane translocase complex proteins Tomm6, Tomm40, and Tomm70a, the mitochondrial amino acid transporter Slc25a29, and the mitochondrial protein transporter Metaxin3. These findings are consistent with previous reports that suggest diabetes induces impairment in mitochondrial transport machinery (e.g. Ref. 53).
Hyperglycemic conditions provide an abundance of substrate for glucose oxidation, leading to increased electron transport chain flux and a higher voltage gradient across the mitochondrial membrane (54, 55). When a critical threshold is reached, electron transfer inside complex III of the electron transport chain is obstructed, giving way to accumulation of electrons at coenzyme Q and the formation of superoxide (56, 57). Studies have shown this to be problematic in diabetic retinopathy, as retinal mitochondrial superoxide levels are elevated in diabetic rodents (58), whereas the activities of the superoxide-scavenging SOD2 (also known as Mn-SOD) and complex III of the electron transport chain are attenuated (58, 59). Overproduction of superoxides by mitochondria links the principal pathways, such as the HBP, responsible for hyperglycemia-induced tissue damage (55). Notably, long-term administration of antioxidants inhibits the development of retinopathy in diabetic rats (60), as does overexpression of SOD2 (61). In the retina of mice administered TMG, SOD2 mRNA translation was attenuated compared with controls. However, in the retina of 4E-BP1/2–deficient mice administered TMG, relative SOD2 mRNA translation was similar to that observed in 4E-BP1/2–deficient mice and WT controls administered vehicle alone. Thus, 4E-BP1 is necessary for the repressive effect of O-GlcNAcylation on SOD2 mRNA translation. ROS levels were elevated in the retina of diabetic WT mice or nondiabetic mice administered TMG. Moreover, 4E-BP1/2 deletion was sufficient to normalize ROS levels in the retina of diabetic mice as well as of those administered TMG to enhance O-GlcNAcylation.
Enhanced O-GlcNAcylation of retinal proteins has been observed in rodent models of both type 1 (11, 12) and pre/type 2 (62) diabetes. This study provides new evidence that O-GlcNAcylation regulates mitochondrial function in the retina. We report that enhanced O-GlcNAcylation alters retinal gene expression by principally acting at the level of mRNA translation. We provide evidence that the effect of O-GlcNAcylation on translation of specific mitochondrial mRNAs depends on 4E-BP1/2 and provide functional evidence that O-GlcNAcylation enhances ROS levels in the retina in a 4E-BP1/2–dependent manner. Overall, the findings are consistent with a model wherein diabetes-induced O-GlcNAcylation acts to promote oxidative stress in the retina by altering the selection of mRNAs for translation. Because of the critical role oxidative stress plays in the development of diabetic retinopathy, pharmacological targeting of O-GlcNAcylation or 4E-BP1 binding to eIF4E may represent novel targets for therapeutic intervention.
Experimental procedures
Animals
At 4 weeks of age, male and female WT and 4E-BP1/2 double knockout C57BL/6J mice received 50 mg/kg TMG or phosphate-buffered saline as a control via intraperitoneal injection. Retinas were extracted 24 h after TMG injections. Alternatively, male mice were administered 50 mg/kg streptozotocin for 5 consecutive days to induce diabetes. Control mice were injected with equivalent volumes of sodium citrate buffer. Diabetic phenotype was confirmed by blood glucose concentration (>250 mg/dL) in fasted animals. Retinas were extracted after 4 weeks of diabetes. All procedures were approved by the Pennsylvania State College of Medicine Institutional Animal Care and Use Committee.
Protein analysis
Retinas were extracted, flash-frozen in liquid nitrogen, and later homogenized in 250 μl of extraction buffer as described previously (24). The homogenate was centrifuged at 1000 × g for 5 min at 4 °C, and the supernatant was collected for analysis. A fraction of the supernatant was added to 1× SDS sample buffer, boiled for 5 min, and analyzed by Western blot analysis as described previously (24). Antibodies used included O-GlcNAc (Cell Signaling Technology, catalog no. 9875), α-actin (Cell Signaling Technology, catalog no. 4970), and GAPDH (Santa Cruz Biotechnology, catalog no. sc-32233). Preparation of the eIF4E, 4E-BP1, and eIF4G antibodies has been described previously (63, 64). Immunoprecipitations were performed by incubating supernatants of retina homogenates with monoclonal anti-eIF4E antibody as described previously (21).
Sequencing library preparation
Total RNA and RPF libraries for deep sequencing were prepared from mouse retina using the TruSeq Ribo Profile Kit (Illumina). Briefly, 10 retinas/group were Dounce-homogenized in 500 μl of polysome buffer containing 10% Triton X-100, 100 mm DTT, 100 mm EGTA, 0.5 units of DNaseI, and 50 μg of cycloheximide. For the RPF, 300 μl of retinal homogenate was treated with 0.6 units of nuclease/optical density unit measured at A260 and purified using an Illustra MicroSpin S-400 HR column (GE Healthcare Life Sciences). rRNA depletion was performed with a Ribo-Zero Gold rRNA Removal Kit (Illumina). Total RNA was prepared from the remaining retinal homogenate and heat-fragmented according to the manufacturer's instructions. RPF RNA and fragmented total RNA were end-repaired with TruSeq Ribo Profile PNK for 3′ adapter ligation, and cDNA was reverse-transcribed with EpiScript RT. cDNA libraries were PAGE-purified, circularized with CircLigase, and amplified by PCR. Libraries were purified with Agencourt AMPure XP beads (Beckman Coulter) and sequenced on an Illumina HiSeq 2500 to a depth of 15 million total reads/sample.
Sequence analysis
Sequencing reads were mapped to 19,411 principal transcripts for each gene in the APPRIS database (65) referenced by the GENCODE release M14 (GRCm38.p5) mouse genome. Principal transcripts for each gene were selected using the script_for_transcript_annotation from RiboViz toolkit package (65). Transcript and ribosome footprint abundances for each transcript/gene were estimated using Kallisto v0.45.0 (66). Given the small size of RPF and RNA-Seq inserts in our libraries (∼30 bp), Kallisto indices were generated using a lower k-mer value (−k 19). Gene-specific -fold changes in RNA and footprint abundances were estimated using DESeq2 packages in R (39) using default log -fold change shrinkage options. Changes in ribosome densities (translation efficiencies) were estimated using the Riborex package in R (28). Unless otherwise specified, significant changes in RNA, RPF, and ribosome densities were estimated at an adjusted p < 0.01 (Benjamini and Hochberg–adjusted p value). Functional analysis of GO categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were conducted using clusterProfiler packages in R (28). The data obtained from sequencing analysis were also analyzed using the Ingenuity Pathways Analysis software and Gene Ontology (GO) Enrichment Analysis (http://geneontology.org/;4 67, 68).
Polysome fractionation by sucrose density gradient centrifugation and RNA isolation
Sucrose density gradient centrifugation was employed to separate the subpolysomal from the polysomal ribosome fractions. Retinas were harvested, and 10 retinas from the respective treatment groups were pooled. Retinas were homogenized in 600 μl of buffer (50 mm HEPES, 75 mm KCl, 5 mm MgCl2, 250 mm sucrose, 2 mm DTT, 100 μg/ml cycloheximide, and 20 units/μl SUPERase-InTM RNase inhibitor (Invitrogen)). Homogenates were incubated on ice for 5 min, and 75 μl of Tween–deoxycholate solution (1.34 ml of Tween 20, 0.66 g of deoxycholate, and 18 ml of sterile water) was added for 15 min. Lysates were centrifuged at 1000 × g for 3 min at 4 °C. The resulting supernatant (550 μl) was layered on a 20–47% linear sucrose gradient (10 mm HEPES, 75 mm KCl, 5 mm MgCl2, and 0.5 mm EDTA) and centrifuged in a SW41 rotor at 34,000 rpm for 190 min at 4 °C. Following centrifugation, the gradient was displaced upward (3 ml/min) using Fluorinert (Sigma) through a spectrophotometer, and A254 nm was continuously recorded (chart speed, 150 cm/h). Two sucrose fractions representing the subpolysomal and polysomal portions of the gradient were collected directly into an equal volume of TRIzol reagent (Invitrogen). To improve the recovery of RNA from dense sucrose portions of the gradient, the polysomal fraction was diluted twice with RNase-free water (HyClone), and the appropriate amount of TRIzol was added. RNA was extracted using the standard manufacturer's protocol and resuspended in 14 μl of RNA storage solution. An equal amount of RNA from each fraction collected was converted into cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems) and subjected to quantitative real-time PCR using QuantiTect SYBR Green Master Mix (Qiagen). Primer sequences can be found in Table S5.
Cell culture
Cultures of WT and Eif4ebp1;Eif4ebp2 double knockout (4E-BP1/2 DKO) MEFs were obtained from Dr. N. Sonenberg (McGill University) and maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were cultured at 37 °C and 5% CO2. Transfections were performed with Lipofectamine 2000 (Thermo Fisher Scientific). Plasmids for expression of HA-tagged 4E-BP1, HA-tagged 4E-BP1-Y54A, and HA-tagged 4E-BP1-F113A were obtained from Dr. J. Blenis (Weill Cornell Medical College).
Bioenergetics analysis of OCR
The Seahorse XFe96 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA) was used to assess mitochondrial respiration. Prior to use of the extracellular flux analyzer, cells were seeded (30,000 cells/well, following optimization of cell seeding number) into the XF96 cell culture microplate, allowed to adhere, and then exposed to medium containing 50 nm TMG for 24 h. Cell culture medium was then replaced with XF medium (Seahorse Bioscience), and cells were placed in a non-CO2 37 °C incubator for 1 h prior to the start of the extracellular flux analysis. The Cell Mito Stress Kit was applied according to the manufacturer's instructions (Seahorse Bioscience). Prior to commencement of these experiments, dose–response curves were carried out to establish the concentration of carbonyl cyanide p-trifluoromethoxyphenylhydrazone needed to achieve maximal OCR. Additionally, dose–response curves were carried out to establish the optimal concentration of oligomycin required to inhibit oxygen consumption, as cells did not respond to the manufacturer's recommended 1 μm. Both cell lines achieved a plateau of maximal OCR at 0.5 μm carbonyl cyanide p-trifluoromethoxyphenylhydrazone and inhibition of OCR at 2 μm oligomycin.
ROS detection
The MitoSOX Red mitochondrial superoxide indicator (Invitrogen) was used in WT and 4E-BP1/2 DKO MEFs to detect mitochondrial superoxide according to the manufacturer's instructions. Briefly, cells were seeded at 300,000 cells/dish in 35-mm, no. 1.5 coverglass, poly-d-lysine–coated dishes (MatTek Corp.). Cells were then exposed to 50 nm TMG for 24 h. 1 ml of 5 μm MitoSOX diluted in Hanks Balanced Salt Solution/Ca/Mg (Life Technologies) was applied to cells. The dye-loaded cells were incubated for 10 min at 37 °C in the dark and then washed with HBSS buffer. To evaluate retinal ROS levels, whole eyes were harvested from mice, embedded in optimal cutting temperature compound (OCT, Sakura Finetek), and flash-frozen. Cryosections (10 μm) were fixed with 2% paraformaldehyde in PBS (pH 7.4). Cryosections were stained with 1.6 μm Hoechst and 10 μm DCF. Live cells and cryosections were imaged using an inverted Leica TCS SP8 confocal microscope.
Author contributions
S. K. D., S. R. K., L. S. J., and M. D. D. conceptualization; S. K. D., W. P. M., J. S. F., A. C. S., L. S. J., and M. D. D. data curation; S. K. D., W. P. M., J. S. F., P. S., A. C. S., L. S. J., and M. D. D. formal analysis; S. K. D., J. S. F., P. S., Y. I. K., A. C. S., S. R. K., L. S. J., and M. D. D. investigation; S. K. D., J. S. F., P. S., A. C. S., L. S. J., and M. D. D. writing-original draft; S. K. D., W. P. M., J. S. F., P. S., Y. I. K., S. R. K., L. S. J., and M. D. D. writing-review and editing; P. S., Y. I. K., S. R. K., L. S. J., and M. D. D. supervision; P. S., L. S. J., and M. D. D. visualization; Y. I. K., S. R. K., L. S. J., and M. D. D. methodology; S. R. K. resources; S. R. K., L. S. J., and M. D. D. funding acquisition; S. R. K., L. S. J., and M. D. D. validation; S. R. K., L. S. J., and M. D. D. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Gerald Hart (University of Georgia) and the NHLBI P01HL107153 Core C4 at Johns Hopkins University for providing thiamet G. We thank Dr. Nahum Sonenberg (McGill University) for providing 4E-BP1/2 DKO MEFs and Dr. John Blenis (Weill Cornell Medical College) for providing 4E-BP1 expression plasmids. We thank Holly Lacko, Chen Yang, and Allyson Torro for technical assistance.
This work was supported by American Diabetes Association Pathway to Stop Diabetes Grant 1-14-INI-04; NEI, National Institutes of Health Grant EY023612 and the Penn State Eye Center Frontiers in Eye and Vision Research Award (to M. D. D.); startup funds from the Human Genetics Institute of New Jersey and NIGMS, National Institutes of Health Grant GM124976 (to P. S.); and NIDDK, National Institute of Health Grants DK13499 and DK15658 (to S. R. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3 and Tables S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- HBP
- hexosamine biosynthetic pathway
- OGT
- O-GlcNAc transferase
- OGA
- O-GlcNAcase
- VEGF
- vascular endothelial growth factor
- RPF
- ribosome-protected mRNA fragment
- TMG
- thiamet G
- TE
- translational efficiency
- GO
- Gene Ontology
- MEF
- mouse embryonic fibroblast
- OCR
- oxygen consumption rate
- ROS
- reactive oxygen species
- DCF
- 2,7-dichlorofluoroscein
- STZ
- streptozotocin
- OXPhos
- oxidative phosphorylation
- mtDNA
- mitochondrial DNA
- SOD
- superoxide dismutase
- cDNA
- complementary DNA
- DKO
- double knockout.
References
- 1. Lee R., Wong T. Y., and Sabanayagam C. (2015) Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. (Lond) 2, 17 10.1186/s40662-015-0026-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Diabetes Control and Complications Trial Research Group, Nathan D. M., Genuth S., Lachin J., Cleary P., Crofford O., Davis M., Rand L., and Siebert C. (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 329, 977–986 10.1056/NEJM199309303291401 [DOI] [PubMed] [Google Scholar]
- 3. Issad T., Masson E., and Pagesy P. (2010) O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 36, 423–435 10.1016/j.diabet.2010.09.001 [DOI] [PubMed] [Google Scholar]
- 4. Semba R. D., Huang H., Lutty G. A., Van Eyk J. E., and Hart G. W. (2014) The role of O-GlcNAc signaling in the pathogenesis of diabetic retinopathy. Proteomics Clin. Appl. 8, 218–231 10.1002/prca.201300076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Duverger E., Roche A. C., and Monsigny M. (1996) N-acetylglucosamine-dependent nuclear import of neoglycoproteins. Glycobiology 6, 381–386 10.1093/glycob/6.4.381 [DOI] [PubMed] [Google Scholar]
- 6. Roos M. D., Su K., Baker J. R., and Kudlow J. E. (1997) O-glycosylation of an Sp1-derived peptide blocks known Sp1 protein interactions. Mol. Cell. Biol. 17, 6472–6480 10.1128/MCB.17.11.6472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao Y., Miyazaki J., and Hart G. W. (2003) The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 β-cells. Arch. Biochem. Biophys. 415, 155–163 10.1016/S0003-9861(03)00234-0 [DOI] [PubMed] [Google Scholar]
- 8. Soesanto Y. A., Luo B., Jones D., Taylor R., Gabrielsen J. S., Parker G., and McClain D. A. (2008) Regulation of Akt signaling by O-GlcNAc in euglycemia. Am. J. Physiol. Endocrinol. Metab. 295, E974–E980 10.1152/ajpendo.90366.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han I., and Kudlow J. E. (1997) Reduced O-glycosylation of Sp1 is associated with increased proteasome susceptibility. Mol. Cell. Biol. 17, 2550–2558 10.1128/MCB.17.5.2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zeidan Q., and Hart G. W. (2010) The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J. Cell Sci. 123, 13–22 10.1242/jcs.053678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gurel Z., Sieg K. M., Shallow K. D., Sorenson C. M., and Sheibani N. (2013) Retinal O-linked N-acetylglucosamine protein modifications: implications for postnatal retinal vascularization and the pathogenesis of diabetic retinopathy. Mol. Vis. 19, 1047–1059 [PMC free article] [PubMed] [Google Scholar]
- 12. Kim S. J., Yoo W. S., Choi M., Chung I., Yoo J. M., and Choi W. S. (2016) Increased O-GlcNAcylation of NF-κB enhances retinal ganglion cell death in streptozotocin-induced diabetic retinopathy. Curr. Eye. Res. 41, 249–257 10.3109/02713683.2015.1006372 [DOI] [PubMed] [Google Scholar]
- 13. Zeidan Q., Wang Z., De Maio A., and Hart G. W. (2010) O-GlcNAc cycling enzymes associate with the translational machinery and modify core ribosomal proteins. Mol. Biol. Cell 21, 1922–1936 10.1091/mbc.e09-11-0941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gurel Z., Zaro B. W., Pratt M. R., and Sheibani N. (2014) Identification of O-GlcNAc modification targets in mouse retinal pericytes: implication of p53 in pathogenesis of diabetic retinopathy. PLoS ONE 9, e95561 10.1371/journal.pone.0095561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nandi A., Sprung R., Barma D. K., Zhao Y., Kim S. C., Falck J. R., and Zhao Y. (2006) Global identification of O-GlcNAc-modified proteins. Anal. Chem. 78, 452–458 10.1021/ac051207j [DOI] [PubMed] [Google Scholar]
- 16. Teo C. F., Ingale S., Wolfert M. A., Elsayed G. A., Nöt L. G., Chatham J. C., Wells L., and Boons G. J. (2010) Glycopeptide-specific monoclonal antibodies suggest new roles for O-GlcNAc. Nat. Chem. Biol. 6, 338–343 10.1038/nchembio.338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wells L., Vosseller K., Cole R. N., Cronshaw J. M., Matunis M. J., and Hart G. W. (2002) Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol. Cell. Proteomics 1, 791–804 10.1074/mcp.M200048-MCP200 [DOI] [PubMed] [Google Scholar]
- 18. Khidekel N., Ficarro S. B., Clark P. M., Bryan M. C., Swaney D. L., Rexach J. E., Sun Y. E., Coon J. J., Peters E. C., and Hsieh-Wilson L. C. (2007) Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat. Chem. Biol. 3, 339–348 10.1038/nchembio881 [DOI] [PubMed] [Google Scholar]
- 19. Wang Z., Pandey A., and Hart G. W. (2007) Dynamic interplay between O-linked N-acetylglucosaminylation and glycogen synthase kinase-3-dependent phosphorylation. Mol. Cell. Proteomics 6, 1365–1379 10.1074/mcp.M600453-MCP200 [DOI] [PubMed] [Google Scholar]
- 20. Dennis M. D., Schrufer T. L., Bronson S. K., Kimball S. R., and Jefferson L. S. (2011) Hyperglycemia-induced O-GlcNAcylation and truncation of 4E-BP1 protein in liver of a mouse model of type 1 diabetes. J. Biol. Chem. 286, 34286–34297 10.1074/jbc.M111.259457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dennis M. D., Shenberger J. S., Stanley B. A., Kimball S. R., and Jefferson L. S. (2013) Hyperglycemia mediates a shift from cap-dependent to cap-independent translation via a 4E-BP1-dependent mechanism. Diabetes 62, 2204–2214 10.2337/db12-1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fort P. E., Losiewicz M. K., Pennathur S., Jefferson L. S., Kimball S. R., Abcouwer S. F., and Gardner T. W. (2014) mTORC1-independent reduction of retinal protein synthesis in type 1 diabetes. Diabetes 63, 3077–3090 10.2337/db14-0235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dennis M. D., Kimball S. R., Fort P. E., and Jefferson L. S. (2015) Regulated in development and DNA damage 1 is necessary for hyperglycemia-induced vascular endothelial growth factor expression in the retina of diabetic rodents. J. Biol. Chem. 290, 3865–3874 10.1074/jbc.M114.623058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schrufer T. L., Antonetti D. A., Sonenberg N., Kimball S. R., Gardner T. W., and Jefferson L. S. (2010) Ablation of 4E-BP1/2 prevents hyperglycemia-mediated induction of VEGF expression in the rodent retina and in Muller cells in culture. Diabetes 59, 2107–2116 10.2337/db10-0148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller W. P., Mihailescu M. L., Yang C., Barber A. J., Kimball S. R., Jefferson L. S., and Dennis M. D. (2016) The translational repressor 4E-BP1 contributes to diabetes-induced visual dysfunction. Invest. Ophthalmol. Vis. Sci. 57, 1327–1337 10.1167/iovs.15-18719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ptushkina M., von der Haar T., Karim M. M., Hughes J. M., and McCarthy J. E. (1999) Repressor binding to a dorsal regulatory site traps human eIF4E in a high cap-affinity state. EMBO J. 18, 4068–4075 10.1093/emboj/18.14.4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. von der Haar T., Gross J. D., Wagner G., and McCarthy J. E. (2004) The mRNA cap-binding protein eIF4E in post-transcriptional gene expression. Nat. Struct. Mol. Biol. 11, 503–511 10.1038/nsmb779 [DOI] [PubMed] [Google Scholar]
- 28. Li W., Wang W., Uren P. J., Penalva L. O. F., and Smith A. D. (2017) Riborex: fast and flexible identification of differential translation from Ribo-seq data. Bioinformatics 33, 1735–1737 10.1093/bioinformatics/btx047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morita M., Gravel S. P., Chénard V., Sikström K., Zheng L., Alain T., Gandin V., Avizonis D., Arguello M., Zakaria C., McLaughlan S., Nouet Y., Pause A., Pollak M., Gottlieb E., et al. (2013) mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 18, 698–711 10.1016/j.cmet.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 30. Mader S., Lee H., Pause A., and Sonenberg N. (1995) The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4γ and the translational repressors 4E-binding proteins. Mol. Cell. Biol. 15, 4990–4997 10.1128/MCB.15.9.4990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schalm S. S., Fingar D. C., Sabatini D. M., and Blenis J. (2003) TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 13, 797–806 10.1016/S0960-9822(03)00329-4 [DOI] [PubMed] [Google Scholar]
- 32. Du Y., Veenstra A., Palczewski K., and Kern T. S. (2013) Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. U.S.A. 110, 16586–16591 10.1073/pnas.1314575110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Komili S., and Silver P. A. (2008) Coupling and coordination in gene expression processes: a systems biology view. Nat. Rev. Genet. 9, 38–48 10.1038/nrg2223 [DOI] [PubMed] [Google Scholar]
- 34. Maier T., Güell M., and Serrano L. (2009) Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583, 3966–3973 10.1016/j.febslet.2009.10.036 [DOI] [PubMed] [Google Scholar]
- 35. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- 36. Jovanovic M., Rooney M. S., Mertins P., Przybylski D., Chevrier N., Satija R., Rodriguez E. H., Fields A. P., Schwartz S., Raychowdhury R., Mumbach M. R., Eisenhaure T., Rabani M., Gennert D., Lu D., et al. (2015) Immunogenetic: dynamic profiling of the protein life cycle in response to pathogens. Science 347, 1259038 10.1126/science.1259038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holcik M., and Sonenberg N. (2005) Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 6, 318–327 10.1038/nrm1618 [DOI] [PubMed] [Google Scholar]
- 38. Spriggs K. A., Bushell M., and Willis A. E. (2010) Translational regulation of gene expression during conditions of cell stress. Mol. Cell 40, 228–237 10.1016/j.molcel.2010.09.028 [DOI] [PubMed] [Google Scholar]
- 39. Love M. I., Huber W., and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ozcan S., Andrali S. S., and Cantrell J. E. (2010) Modulation of transcription factor function by O-GlcNAc modification. Biochim. Biophys. Acta 1799, 353–364 10.1016/j.bbagrm.2010.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schuller A. P., Wu C. C., Dever T. E., Buskirk A. R., and Green R. (2017) eIF5A functions globally in translation elongation and termination. Mol. Cell 66, 194–205.e5 10.1016/j.molcel.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zid B. M., Rogers A. N., Katewa S. D., Vargas M. A., Kolipinski M. C., Lu T. A., Benzer S., and Kapahi P. (2009) 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 139, 149–160 10.1016/j.cell.2009.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Couvillion M. T., Soto I. C., Shipkovenska G., and Churchman L. S. (2016) Synchronized mitochondrial and cytosolic translation programs. Nature 533, 499–503 10.1038/nature18015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van Haute L., Pearce S. F., Powell C. A., D'Souza A. R., Nicholls T. J., and Minczuk M. (2015) Mitochondrial transcript maturation and its disorders. J. Inherit. Metab. Dis. 38, 655–680 10.1007/s10545-015-9859-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tan E. P., Villar M. T., E. L., Lu J., Selfridge J. E., Artigues A., Swerdlow R. H., and Slawson C. (2014) Altering O-linked β-N-acetylglucosamine cycling disrupts mitochondrial function. J. Biol. Chem. 289, 14719–14730 10.1074/jbc.M113.525790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tan E. P., McGreal S. R., Graw S., Tessman R., Koppel S. J., Dhakal P., Zhang Z., Machacek M., Zachara N. E., Koestler D. C., Peterson K. R., Thyfault J. P., Swerdlow R. H., Krishnamurthy P., DiTacchio L., et al. (2017) Sustained O-GlcNAcylation reprograms mitochondrial function to regulate energy metabolism. J. Biol. Chem. 292, 14940–14962 10.1074/jbc.M117.797944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ma J., Liu T., Wei A. C., Banerjee P., O'Rourke B., and Hart G. W. (2015) O-GlcNAcomic profiling identifies widespread O-linked β-N-acetylglucosamine modification (O-GlcNAcylation) in oxidative phosphorylation system regulating cardiac mitochondrial function. J. Biol. Chem. 290, 29141–29153 10.1074/jbc.M115.691741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma J., Banerjee P., Whelan S. A., Liu T., Wei A. C., Ramirez-Correa G., McComb M. E., Costello C. E., O'Rourke B., Murphy A., and Hart G. W. (2016) Comparative proteomics reveals dysregulated mitochondrial O-GlcNAcylation in diabetic hearts. J. Proteome Res. 15, 2254–2264 10.1021/acs.jproteome.6b00250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Banerjee P. S., Ma J., and Hart G. W. (2015) Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc. Natl. Acad. Sci. U.S.A. 112, 6050–6055 10.1073/pnas.1424017112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Madsen-Bouterse S. A., Mohammad G., Kanwar M., and Kowluru R. A. (2010) Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid. Redox Signal. 13, 797–805 10.1089/ars.2009.2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kern T. S., Tang J., Mizutani M., Kowluru R. A., Nagaraj R. H., Romeo G., Podesta F., and Lorenzi M. (2000) Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest. Ophthalmol. Vis. Sci. 41, 3972–3978 [PubMed] [Google Scholar]
- 52. Benard G., and Rossignol R. (2008) Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid. Redox Signal. 10, 1313–1342 10.1089/ars.2007.2000 [DOI] [PubMed] [Google Scholar]
- 53. Zhong Q., and Kowluru R. A. (2011) Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Invest. Ophthalmol. Vis. Sci. 52, 8739–8746 10.1167/iovs.11-8045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Du X. L., Edelstein D., Dimmeler S., Ju Q., Sui C., and Brownlee M. (2001) Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Invest. 108, 1341–1348 10.1172/JCI11235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brownlee M. (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820 10.1038/414813a [DOI] [PubMed] [Google Scholar]
- 56. Korshunov S. S., Skulachev V. P., and Starkov A. A. (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 416, 15–18 10.1016/S0014-5793(97)01159-9 [DOI] [PubMed] [Google Scholar]
- 57. Young T. A., Cunningham C. C., and Bailey S. M. (2002) Reactive oxygen species production by the mitochondrial respiratory chain in isolated rat hepatocytes and liver mitochondria: studies using myxothiazol. Arch. Biochem. Biophys. 405, 65–72 10.1016/S0003-9861(02)00338-7 [DOI] [PubMed] [Google Scholar]
- 58. Kanwar M., Chan P. S., Kern T. S., and Kowluru R. A. (2007) Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest. Ophthalmol. Vis. Sci. 48, 3805–3811 10.1167/iovs.06-1280 [DOI] [PubMed] [Google Scholar]
- 59. Kowluru R. A., Atasi L., and Ho Y. S. (2006) Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 47, 1594–1599 10.1167/iovs.05-1276 [DOI] [PubMed] [Google Scholar]
- 60. Kowluru R. A., Tang J., and Kern T. S. (2001) Abnormalities of retinal metabolism in diabetes and experimental galactosemia: VII: effect of long-term administration of antioxidants on the development of retinopathy. Diabetes 50, 1938–1942 10.2337/diabetes.50.8.1938 [DOI] [PubMed] [Google Scholar]
- 61. Kowluru R. A., Kowluru V., Xiong Y., and Ho Y. S. (2006) Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic. Biol. Med. 41, 1191–1196 10.1016/j.freeradbiomed.2006.01.012 [DOI] [PubMed] [Google Scholar]
- 62. Dai W., Dierschke S. K., Toro A. L., and Dennis M. D. (2018) Consumption of a high fat diet promotes protein O-GlcNAcylation in mouse retina via NR4A1-dependent GFAT2 expression. Biochim. Biophys. Acta Mol. Basis. Dis. 1864, 3568–3576 10.1016/j.bbadis.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kimball S. R., Jurasinski C. V., Lawrence J. C. Jr, and Jefferson L. S. (1997) Insulin stimulates protein synthesis in skeletal muscle by enhancing the association of eIF-4E and eIF-4G. Am. J. Physiol. 272, C754–C759 10.1152/ajpcell.1997.272.2.C754 [DOI] [PubMed] [Google Scholar]
- 64. Kimball S. R., Horetsky R. L., and Jefferson L. S. (1998) Implication of eIF2B rather than eIF4E in the regulation of global protein synthesis by amino acids in L6 myoblasts. J. Biol. Chem. 273, 30945–30953 10.1074/jbc.273.47.30945 [DOI] [PubMed] [Google Scholar]
- 65. Rodriguez J. M., Maietta P., Ezkurdia I., Pietrelli A., Wesselink J. J., Lopez G., Valencia A., and Tress M. L. (2013) APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res. 41, D110–D117 10.1093/nar/gks1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bray N. L., Pimentel H., Melsted P., and Pachter L. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- 67. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., et al. (2000) Gene ontology: Tool for the unification of biology: The Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. The Gene Ontology Consortium (2017) Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–DD338 10.1093/nar/gkw1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.