

Abstract
The human dopamine transporter (hDAT) regulates the level of the neurotransmitter dopamine (DA) in the synaptic cleft and recycles DA for storage in the presynaptic vesicular pool. Many neurotransmitter transporters exist as oligomers, but the physiological role of oligomerization remains unclear; for example, it has been speculated to be a prerequisite for amphetamine-induced release and protein trafficking. Previous studies point to an oligomeric quaternary structure of hDAT; however, the exact stoichiometry and the fraction of co-existing oligomeric states are not known. Here, we used single-molecule brightness analysis to quantify the degree of oligomerization of heterologously expressed hDAT fused to monomeric GFP (mGFP–hDAT) in Chinese hamster ovary (CHO) cells. We observed that monomers and dimers of mGFP–hDAT co-exist and that higher-order molecular complexes of mGFP–hDAT are absent at the plasma membrane. The mGFP–hDAT dimers were stable over several minutes, and the fraction of dimers was independent of the mGFP–hDAT surface density. Furthermore, neither oxidation nor depletion of cholesterol had any effect on the fraction of dimers. Unlike for the human serotonin transporter (hSERT), in which direct binding of phosphatidylinositol 4,5-bisphosphate (PIP2) stabilized the oligomers, the stability of mGFP–hDAT dimers was PIP2 independent.
Keywords: membrane protein, dopamine transporter, single-molecule biophysics, dimerization, inositol phospholipid, protein–protein interaction, neurotransmitter, single-molecule brightness analysis, synaptic transmission
Introduction
The human dopamine transporter (hDAT)3 is a presynaptic protein containing 12 transmembrane helices (TMHs). It is involved in dopamine (DA) homeostasis by high-affinity reuptake of dopamine from the synaptic cleft (1, 2) thereby terminating chemical signal transduction in dopaminergic synapses. hDAT belongs to the Na+- and Cl−-dependent solute carrier 6 (SLC6) family (3, 4), which also includes the transporters for serotonin (SERT), norepinephrine (NET), γ-amino acid transporters (GAT), glycine (GlyT), and several other amino acids and osmolytes (4, 5). hDAT is a primary target for illicit drugs and psychostimulants such as cocaine and amphetamines (6, 7). Alterations in hDAT functions could lead to diseases including depression, attention deficit hyperactivity disorder, bipolar disorders, schizophrenia, or Parkinson's disease (8).
Over the last few decades, oligomerization of proteins has been an intense area of research from the perspective of protein evolution and opportunities for functional control. Oligomerization has been proposed to play a critical role in the regulation of cellular function including signal transduction and immune response (9, 10) as it may offer stability, higher order complexity, and compartmentalization of reactions (11). Oligomers could also have deleterious effects on cells as observed in the case of neurodegenerative disorders (12–14). For a large number of proteins, there are specific binding domains that facilitate or modulate oligomerization (15, 16). Lipids have also been found to be important in membrane protein folding and oligomerization (17–19). For example, we recently found a prominent role of the minor phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) for human SERT (hSERT) oligomerization and function (20, 21).
Like several members of the SLC6 family that were shown to form oligomers (22–27), including our recent studies on hSERT (20, 28), hDAT has also been reported to exhibit oligomeric quaternary organization as observed in radiation inactivation studies (29, 30), co-purification experiments with differentially tagged epitopes (31, 32), co-immunoprecipitation and fluorescence resonance energy transfer (FRET) microscopy studies in heterologous expression systems (33) and transfected neurons (34). In addition, cross-linking experiments suggest the involvement of TMHs 4 and 6 in the interfaces of symmetrical hDAT dimers (31, 35, 36). More recently, unbiased molecular dynamics simulations followed the dimerization of two randomly oriented hDAT monomers and reported the existence of a limited number of symmetric and asymmetric dimers (16). Additionally, the bundle domain comprising of TMHs 1, 2, 6, and 7 was largely excluded from the dimer interface, whereas the scaffold domain formed a significant part of the dimer interface (16).
Interestingly, the exact functional significance of such oligomerization behavior is largely unknown and has been widely speculated (22, 37). Oligomerization of SLC6 family members has been proposed to be important for the following processes. (i) As a quality control checkpoint before trafficking of correctly folded proteins from the endoplasmic reticulum (ER) and/or the Golgi to the cell surface (22, 38). It has been observed that oligomerization-deficient mutants of GAT1 are retained in the ER, but they remain capable of transporting γ-amino butyric acid (GABA) in vesicular preparations (39). Interestingly, the oligomeric state does not seem to influence the neurotransmitter uptake activity, as observed in these GAT1 mutants (39). Inversely, oligomerization of both SERT and GAT1 is not influenced by various transporter substrates and inhibitors (23). In the case of hDAT, it has been observed that formation of the oligomeric complex is essential for correct trafficking of the transporter complex to the cell membrane (31, 33, 40–43). (ii) Transporter oligomerization has been suggested to entail functional consequences (25, 44, 45) and to support the countertransport model of neurotransmitter transport (37), where amphetamine binds to one subunit of an SLC6 oligomer and the transporter-mediated efflux of neurotransmitter is subsequently performed by the neighboring subunit (46). In this model, only SLC6 complexes having at least two transporter subunits would be susceptible to amphetamine-elicited neurotransmitter efflux.
The role of membrane cholesterol in modulating hDAT function has been extensively studied (47–50). Depletion of membrane cholesterol was shown to lower the affinity of hDAT for DA and its uptake velocity in [3H]DA uptake assays (47, 48) and efflux rate (51). Although these studies support the oligomerization behavior of hDAT, they could not quantify the exact subunit stoichiometry, direct modulation by cholesterol–hDAT interaction, and the fraction of different co-existing oligomeric states in live cell membrane, their interaction kinetics, and factors influencing its oligomerization behavior.
In this study, we used single-molecule brightness analysis to probe the oligomerization behavior of hDAT fused to monomeric GFP (mGFP–hDAT) in the plasma membrane of living Chinese hamster ovary (CHO) cells. In the case of GFP it is known that n-mer complexes give rise to n-fold increased mean brightness compared with monomer constructs (52). The according brightness probability distributions of n-mers can be calculated from the GFP monomer signals (53, 54). Brightness analysis was successfully applied for characterizing, e.g. the oligomeric state of an ion channel (53), and the serotonin transporter (20, 28). Here, we observed that mGFP–hDAT in the plasma membrane predominantly showed the co-existence of monomers and dimers that were found to be independent of hDAT surface density. Furthermore, perturbation of membrane cholesterol either by oxidation or depletion had no impact on the oligomeric distribution of hDAT. Using a modified photobleaching protocol (28), we found that the mGFP–hDAT dimers were stable at least on a time scale of several minutes. In contrast to hSERT, where direct binding of PIP2-mediated stable oligomer formation (20), we obtained evidence that perturbation of PIP2 levels did not have any impact on hDAT dimerization.
Results
DAT co-exists as monomers and dimers in the live cell plasma membrane
We determined the subunit stoichiometry of mGFP–hDAT by heterologously expressing the construct as a stable mGFP–hDAT CHO cell line. Because it was shown earlier that multiple molecular determinants in the carboxyl terminus of hDAT regulate its export from the ER (42), we fused mGFP to the cytosolic N terminus of hDAT. The fluorescence signal was recorded from the plasma membrane, which is close to the glass surface, by using total internal reflection fluorescence (TIRF) microscopy. The transporter showed considerable mobility: fluorescence recovery after photobleaching (FRAP) yielded a mobile fraction of 78 ± 10% (Fig. 1A), and single-molecule tracking showed a lateral mobility of D = 0.118 ± 0.002 μm2/s (Fig. 1B). mGFP–hDAT was functional as indicated from [3H]DA uptake measurements in cells transiently transfected with mGFP–hDAT (Fig. S1). N-terminal mGFP fusion to hDAT does not have functional consequences on DA transport, as WT hDAT and mGFP–hDAT showed comparable Km values, 2.98 ± 0.38 μm for hDAT and 4.11 ± 0.85 μm for mGFP–hDAT. The difference in the Vmax arises from lower surface expression of the mGFP–DAT construct compared with the WT construct.
Figure 1.

Mobility of mGFP–hDAT at the live cell plasma membrane. A, to determine the mobile fraction of mGFP–hDAT, FRAP experiments were performed on cells expressing mGFP–hDAT. The integrated intensity of the bleached area (I) was normalized to the fluorescence intensity before bleaching (I0) and plotted over time (black circles, showing a representative recovery curve). The average over 11 cells yielded a mean mobile fraction of m = 78 ± 10%. B, using single molecule tracking, the diffusion coefficient D was determined. Mean square displacements were calculated for a range of time lags (t-lag) and D was determined from the first two points in the MSD plot according to MSD = 4Dtlag + offset, yielding D = 0.118 ± 0.002 μm2/s. Error bars show the mean ± S.E.
At typical expression levels, fluorescently labeled membrane proteins are present at high surface density, so that single-molecule signals cannot be separated. We hence made use of a method termed “Thinning out clusters while conserving stoichiometry of labeling” (TOCCSL) (54). In conjunction with single molecule brightness analysis, TOCCSL allows for the quantification of the oligomeric state of fluorescently labeled mobile membrane constituents. After recording a pre-bleach image for control, a distinct small area of the cell membrane is irreversibly photobleached. The photobleached region is confined by imaging an aperture onto the sample. Ideally, the protein complexes are either entirely photobleached (inside the analysis region) or remain entirely fluorescent (outside the analysis region), which can be assessed by a control image recorded immediately after the bleaching pulse. Due to Brownian motion, unbleached molecules diffuse into the bleached analysis region. TOCCSL makes use of the very onset of the recovery process: individual molecules or clusters diffuse into the bleached area and can be resolved as single, clearly distinguishable fluorescent spots. Fig. 2A shows the brightness distribution obtained in the TOCCSL images where the brightness distribution is plotted as a probability density function depicted in black, whereas the fit and the n-mer contributions are shown in red and blue, respectively. Fig. 2B reveals the oligomeric distribution of mGFP–hDAT as obtained by our single-molecule brightness analysis. Interestingly, we observe only the presence of monomers (∼55%) and dimers (∼35%) co-existing in the live cell plasma membrane. Marginal fractions of higher order aggregates are below the detection sensitivity of TOCCSL (55).
Figure 2.
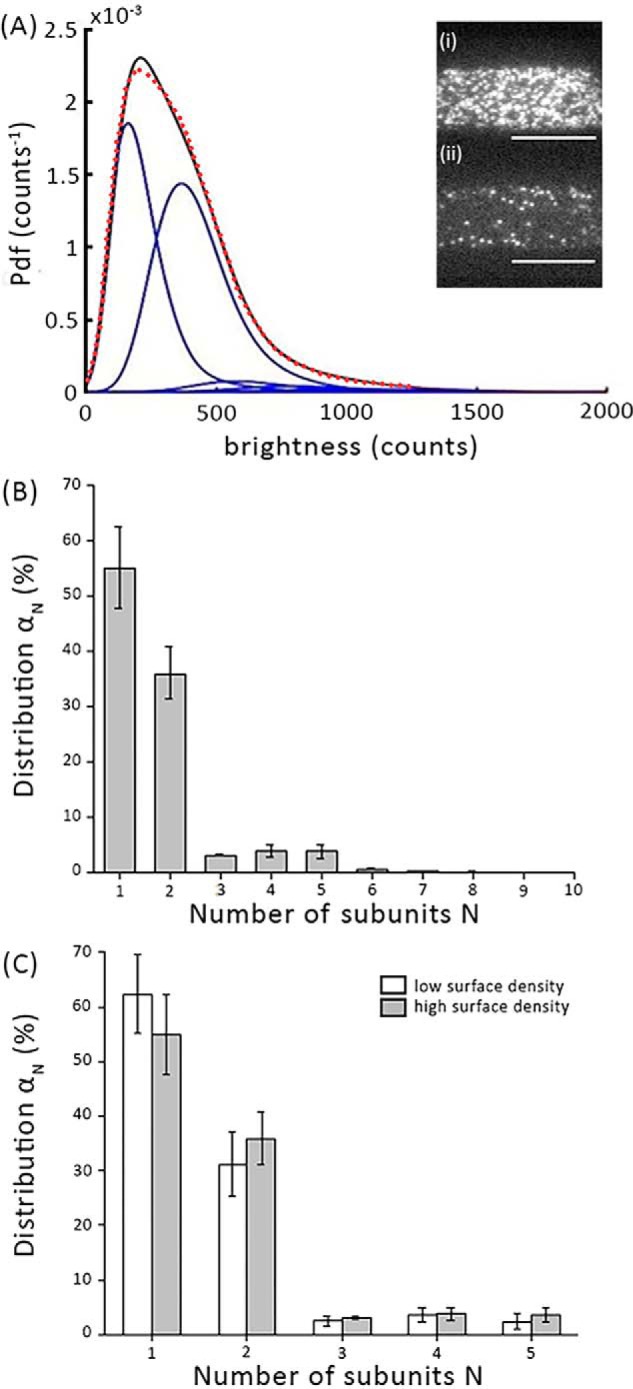
Determination of mGFP–hDAT oligomer distribution on the plasma membrane using single molecule brightness analysis. A, the obtained brightness distribution of the oligomeric fractions are plotted as probability density function (Pdf) (black line): the fit is shown as a dashed red line and the n-mer contributions as blue lines. Inset, representative pre-bleach (i) and post-bleach (ii) fluorescence images are shown from a region of the plasma membrane. Scale bar, 10 μm. B, the distribution of oligomeric states (gray bars) of mGFP–hDAT shows the presence of monomers (∼55%) and dimers (∼35%) co-existing at the plasma membrane (n = 103 cells). C, the oligomeric distribution is independent of the density of mGFP–hDAT at the plasma membrane when evaluated at a mean density of ∼5 mGFP–hDAT/μm2 for low surface density and ∼40 mGFP–hDAT/μm2 for high surface density (n = 90 cells). Error bars show the mean ± S.E.
Next, we were interested to probe whether the process of dimer formation equilibrates at the plasma membrane, as observed for the oligomerization of G protein–coupled receptors (56). In this case, the higher surface density of mGFP–hDAT would drive the equilibrium toward higher oligomeric structures and vice versa. For this, we separately analyzed data from cells showing either low (∼5 molecules of mGFP–hDAT/μm2) or high (∼40 molecules of mGFP–hDAT/μm2) expression levels. Although the two groups differed by a factor of eight in their surface densities, we did not observe a substantial difference in their oligomeric size (Fig. 2C). Unfortunately, for technical reasons it was not possible to analyze cells with higher expression levels of mGFP–hDAT because of intracellular retention of hDAT, leading to strong background signals that prevented single molecule imaging. Our data suggests that mGFP–hDAT oligomer association and dissociation is not a continuous process at the plasma membrane.
mGFP–hDAT forms stable dimers
To discriminate between rapid subunit exchange and stable association, we have earlier designed a modified protocol based on repetitive TOCCSL runs on the very same cells (28). Here, the TOCCSL run is repeated every 5 min for 50 min on the same cell. After each TOCCSL run, the oligomeric distribution is monitored on the recovery image using single-molecule brightness analysis. Pooling the data obtained from multiple cells provides brightness distributions as a function of time. Two scenarios can be discriminated: if the interaction of subunits within a given complex was stable, repetitive TOCCSL runs would reduce the total number of observed fluorescent spots on the plasma membrane, but would not alter the brightness distribution. In contrast, if the exchange rate of subunits between dimers was high, bleached subunits from the previous TOCCSL experiments would rearrange with unbleached subunits. Over time, this mixing would increase the number of complexes comprised of both dark and fluorescent subunits, thereby shifting the apparent distribution toward monomers (Fig. 3A). At the plasma membrane, we observed massive depletion of fluorescent molecules after 10 repeats (i.e. after 50 min) (Fig. S2), whereas the mGFP–hDAT dimer fraction in subsequent TOCCSL runs (Fig. 3B) remained unchanged indicating that no exchange of dimer subunits took place within the 50-min time scale. We therefore infer that the dimers are stable at least over several minutes at the plasma membrane.
Figure 3.
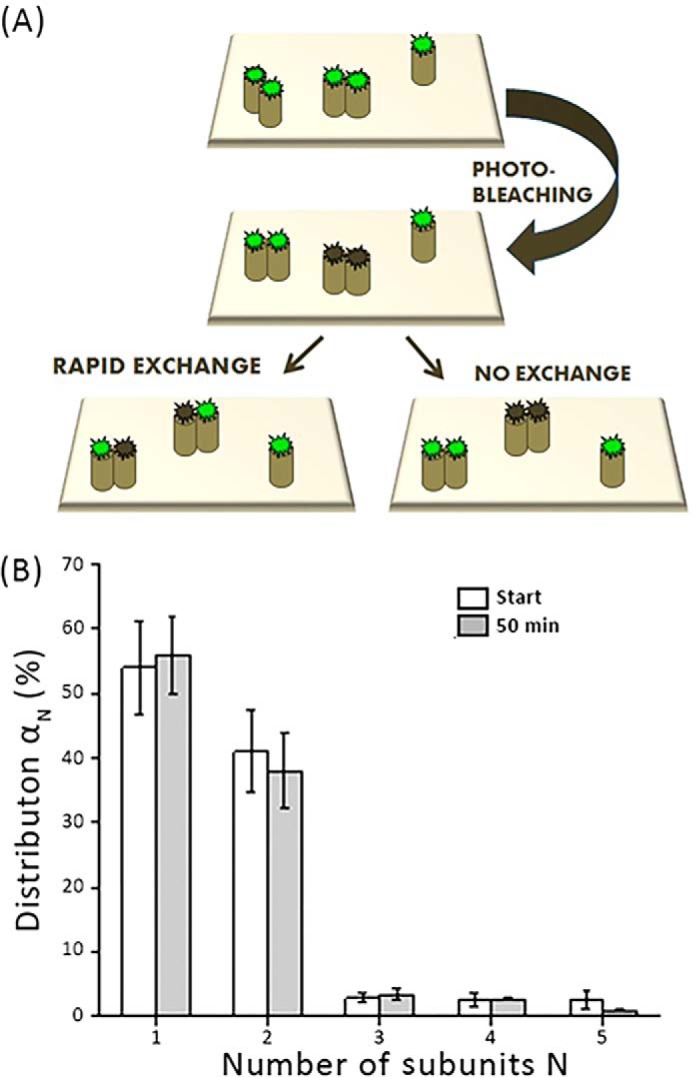
To evaluate the stability of the mGFP–hDAT dimers. A, the transporter complexes in a cell were photobleached using a sequence of 10 TOCCSL experiments, and the oligomeric distribution was monitored for 50 min. A, the sketch shows two potential cases: transient interaction of mGFP–hDAT would lead to a rearrangement after photobleaching, resulting in a mixed population of bleached (gray) and unbleached (green) molecules per dimer; in this case, the number of unbleached dyes per dimer would be reduced (left). In contrast, stable interaction would produce either completely bleached or unbleached dimers, without altering the distribution (right). B, dimeric distribution αN at the start of the experiment (white bars), and after 10 consecutive TOCCSL experiments performed every 5 min (gray bars) (n = 37 cells). No change in the dimeric distribution was observed. Error bars show the mean ± S.E.
Oxidation or depletion of plasma membrane cholesterol does not affect the dimer distribution
It has been reported earlier that depletion of membrane cholesterol reduced both DA uptake and efflux rates (51). We hence studied the effect of cholesterol on the dimer distribution of mGFP–hDAT. Cholesterol oxidase oxidizes the hydroxyl group of membrane cholesterol to a keto group (57, 58). When we perturbed membrane cholesterol with the cholesterol oxidase (2 units/ml for 30 min) we found no effect on the dimer fraction (∼35%) of mGFP–hDAT (Fig. 4A). This was found to be independent of plasma membrane expression of mGFP–hDAT (Fig. S3).
Figure 4.
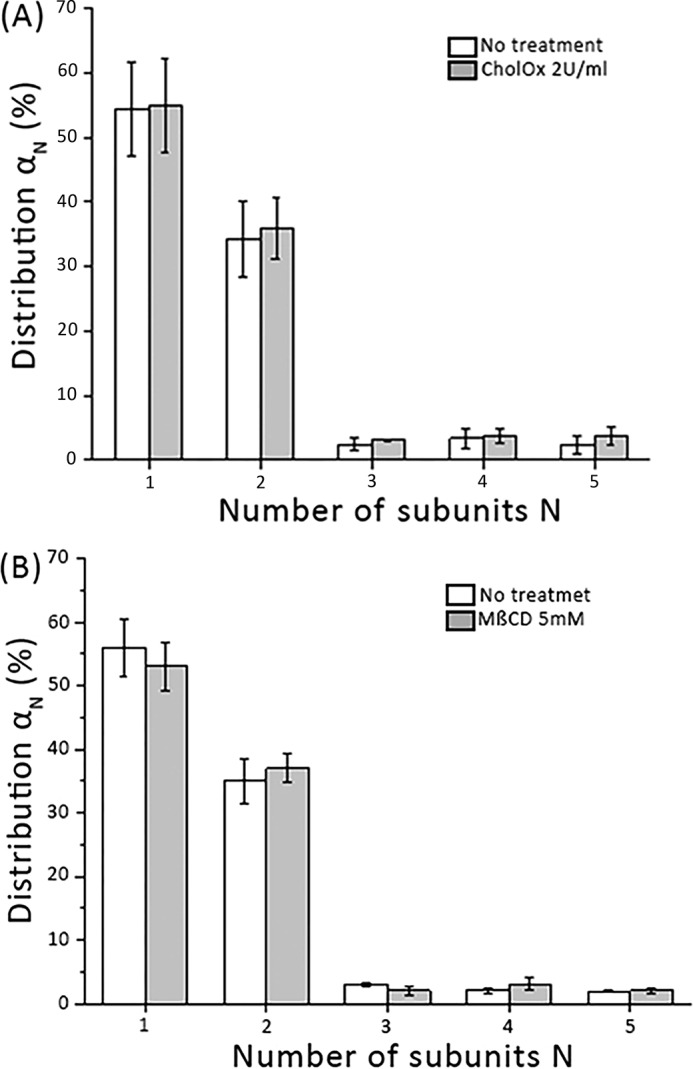
mGFP–hDAT dimeric distribution is not affected by perturbation of cholesterol. A, comparison of the dimeric mGFP–hDAT distributions in the unaltered lipid environment of the plasma membrane (white bars) and after cholesterol oxidation with cholesterol oxidase (2 units/ml for 30 min). No apparent influence of the cholesterol content is observable (n = 61 cells). B, cholesterol depletion by MβCD (5 mm for 1 h) does not affect the dimer distribution (gray bars) when compared with the unaltered lipid environment of the plasma membrane (white bars) (n = 43 cells). Error bars show the mean S.E.
In an alternative approach, we depleted cholesterol using methyl-β-cyclodextrin (MβCD) (59) (5 mm for 1 h), however, we found that the dimer fraction remained unchanged when compared with untreated cells (Fig. 4B), which was again found to be independent of the density of mGFP–hDAT on the cell surface (Fig. S4). These data show that perturbing the membrane cholesterol concentration either by oxidation or by depletion has no effect on the dimer distribution of mGFP–hDAT.
Stability of the mGFP–hDAT dimers does not depend on PIP2 availability
It has been observed that the hDAT N terminus interacts electrostatically with PIP2 and that this interaction impairs DA efflux, but not uptake (60). We hence probed whether PIP2 contributes to the stability of mGFP–hDAT dimers. PLCγ is a highly specific enzyme for PIP2 hydrolysis, leaving other phosphoinositides virtually unaffected (61). We used the direct PLCγ-activator 2,4,6-trimethyl-N-(3-trifluoromethylphenyl)benzenesulfonamide (m-3M3FBS) (25 μm) to enzymatically deplete PIP2 at the plasma membrane. Fig. 5A shows that PIP2 depletion had no influence on the dimer distribution of mGFP–hDAT. The mean surface density of the transporter was not affected upon treatment with m-3M3FBS (Fig. S5). The lack of effect was consistent among all tested surface densities of mGFP–hDAT (Fig. 5B). Taken together, mGFP–hDAT forms stable dimers at the plasma membrane in a PIP2-independent manner.
Figure 5.
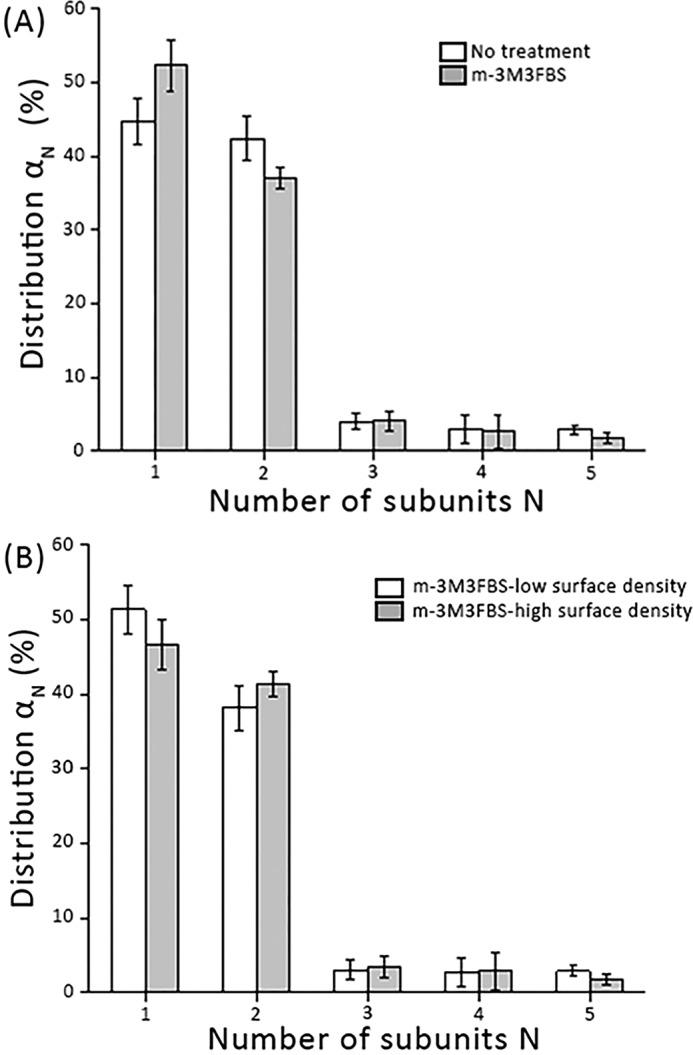
mGFP–hDAT dimerization at the plasma membrane is independent of PIP2 levels. A, we enzymatically depleted PIP2 at the plasma membrane via activation of PLCγ by incubating cells for 20 min with the direct PLCγ activator m-3M3FBS (25 μm). m-3M3FBS did not yield any effect on the oligomeric distribution (gray bars) compared with the untreated cells (white bars) (n > 80 cells per experimental condition). mGFP–hDAT surface densities were similar: 35 ± 4 μm2 (white bars) and 33 ± 5 μm2 (gray bars). B, the oligomeric distribution is plotted for various mGFP–hDAT surface density (n > 40 cells) following PIP2 depletion via m-3M3FBS; mGFP–hDAT densities are ∼7 mGFP–hDAT/μm2 for low surface density and ∼40 mGFP–hDAT/μm2 for high surface density. Error bars show the mean S.E.
Discussion
The physiological role of hDAT is to maintain DA homeostasis in the synaptic nerve terminals by rapid reuptake of DA from the synaptic cleft for subsequent storage and release (62). Oligomerization is a phenomenon observed for a significant fraction of cellular proteins (11) including members of the monoamine transporter family (22). It is generally assumed that oligomerization of transporters of the SLC6 family plays a role in such diverse processes as membrane trafficking, transporter function, regulation, and substrate turnover (63). Concerning trafficking, protein oligomerization serves as a quality control mechanism to enable the export from the ER and/or the Golgi apparatus to the plasma membrane (38). Concerning function, transporter oligomerization has been suggested to be of importance for forward transport (25, 44, 45) and to establish the basis for the countertransport model proposed for transporter-mediated efflux of neurotransmitters (37, 46). In support of this latter model, superresolution microscopy revealed the presence of hDAT nanodomains in the plasma membrane (64). This could have functional implications such as regulation of transporter internalization (50) and amphetamine-induced dopamine efflux (47, 65).
Here, we examined the homo-oligomerization behavior of mGFP–hDAT in the plasma membrane of CHO cells by determining the oligomeric distribution and the interaction kinetics of the protomers using single-molecule brightness analysis. The mGFP was fused to the N terminus instead of the C terminus as it is critical for surface expression of hDAT (22, 66–68) and is vital for hDAT recognition by SEC24 as part of the ER export machinery (68, 69). We tried to compare and contrast the oligomerization behavior of hDAT with our previous findings with the hSERT to comprehend the nature of molecular association of members of the SLC6 family. Because the TOCCSL illumination protocol is blind to the immobile fraction, all our observations pertaining to the single-molecule analysis comprise only the mobile fraction which represents ∼80% of hDAT molecules at the plasma membrane (Fig. 1A). Surprisingly, we observed that mGFP–hDAT formed only monomers (∼55%) and dimers (∼35%), whereas higher order oligomers were absent (Fig. 2B). This is different to our previous observations for hSERT, where significant fractions of higher oligomeric structures of at least up to pentamers were observed in the plasma membrane (28).
A modified bleaching protocol in conjunction with the brightness analysis (referred in the study as repeat TOCCSL) allowed us to study the exchange kinetics of subunits within the multimers. Our group had earlier observed that mGFP–hSERT oligomers exhibited a dynamic equilibrium at the ER membrane due to its ability to rapidly exchange subunits within oligomers, which became kinetically trapped after trafficking to the plasma membrane, indicating a spatial decoupling of the oligomerization process at various sites within the cell (20). At the plasma membrane, the degree of oligomerization was found to be independent of hSERT surface density (28). Similar to what was observed earlier with hSERT, the distribution of mGFP–hDAT dimers is insusceptible to expression levels of mGFP–hDAT (Fig. 2C) indicating that formation and dissociation of mGFP–hDAT dimers does not occur at the plasma membrane. Consistently, the repeated TOCCSL protocol to determine the stability of mGFP–hDAT dimers showed that they are stable just like hSERT oligomers at least for a time window of 50 min (Fig. 3B). Taken together, the results indicate that the dimers behave similar to the hSERT oligomers in terms of stability.
Earlier it was reported that partitioning of hSERT and hDAT into lipid microdomains modulates its uptake activity (48, 70). We wanted to probe whether the perturbation of cholesterol at the plasma membrane had any influence on the dimeric distribution of mGFP–hDAT. Cholesterol oxidase catalyzes the oxidation of cholesterol resulting in the formation of cholestenone (cholest-4-en-3-one) (57), whereas methyl-β-cyclodextrin depletes membrane cholesterol (59). Currently, this is regarded as the canonical test for the involvement of plasma membrane rafts for the interaction process (71). We observed that the dimer fraction of mGFP–hDAT is affected neither by the action of cholesterol oxidase (Fig. 4A, Fig. S3) nor by cholesterol depletion from the cell surface using MβCD (Fig. 4B) at any tested expression level (Fig. S4). This indicates that liquid ordered “raft”-like plasma membrane phases are not relevant for the formation of mGFP–hDAT dimers.
Previous observations from us indicated that in the case of hSERT, stability and kinetic trapping at the plasma membrane is mediated by its direct binding to PIP2, which promotes spatial decoupling of oligomer formation and provides cells with the ability to define quaternary structures independent of expression levels (20). We anticipated that similar to hSERT, PIP2 would also play a role in the stability of mGFP–hDAT dimers. We were surprised to find that mGFP–hDAT dimer distribution was not altered by changes in PIP2 levels at the plasma membrane (Fig. 5) indicating that unlike hSERT, mGFP–hDAT forms stable dimers in a PIP2-independent manner. It has been reported that the N terminus of monoamine transporters acts as a lever in amphetamine-induced efflux (72) and impacts on the overall conformational equilibrium (73). Hence, it is not surprising that the direct binding of PIP2 to the N terminus of hDAT plays an important role in amphetamine-induced, hDAT-mediated DA efflux (74). Whether the stable quaternary equilibrium of monomeric and dimeric species, unaffected by PIP2 manipulation, plays a functional role in amphetamine-induced substrate efflux awaits further experimental verification.
Let us compare our results with molecular dynamics simulations of hDAT oligomerization. hDAT shows a small number of dimeric interfaces and dimer conformations, when simulated in a cholesterol-free membrane containing only palmitoyl oleoyl-phosphatidylcholine lipids revealed (16). Some of these interfaces were mutually exclusive, others were of low abundance. The dimer conformations that showed a low probability may have been too weak to be detectable by our TOCCSL approach. Also, the employed Martini coarse-grained force field was recently shown to overstabilize protein dimers in membranes (75). The largest clusters of dimers observed in the simulations can be classified into two groups. (i) Dimers, which showed a large interaction interface between transmembrane helices all across the membrane. The stability of these dimers is dominated by direct protein–protein contacts, from which it can be inferred, that a modulatory role of lipids might be minor. (ii) Dimers with interfaces, to which lipids make a large contribution and involve extra- and intracellular loops. These can be expected to be strongly dependent on lipid composition and/or membrane thickness. At this stage, it may be premature to speculate about the mechanism that stabilizes the experimentally observed hDAT dimers: whereas it appears attractive to ascribe this to the first group, one should note that the membrane composition of the computational and the present study is very different, and depletion of cholesterol as probed here, is likely not sufficient to mirror the single lipid species used in the simulation experiments. Although the nature of the attractive forces that connect the hDAT dimers is not known, high molecular weight DAT complexes were observed in synaptosomal preparations from rats pre-exposed to multiple doses of methamphetamine, which got attenuated but not abolished upon exposure to β-mercaptoethanol (76). In line with that, hDAT in the plasma membrane is natively not cross-linked, but can be cross-linked (Cys-306) to form stable dimers by the flexible cross-linker bis-EA or by CuP-generated oxidation such that the apparent molecular mass with nonreducing SDS-PAGE increased from ∼85 to ∼195 kDa (36). Hence, it may well be that disulfide bridges are also involved in the dimer formation.
In conclusion, hDAT forms stable dimers at the plasma membrane in a PIP2-independent manner. What leads to the stability of mGFP–hDAT dimers is a matter of further study but it would be interesting to discuss some of the possibilities. (i) mGFP–hDAT interacts with other hitherto unknown membrane components that stabilize the dimers. (ii) The dimerization process of mGFP–hDAT does not depend on membrane lipids and is solely driven by the protein–protein interaction of the two interacting monomers. Furthermore, unbiased molecular dynamics simulation studies of hDAT revealed the presence of a limited number of symmetric and asymmetric dimers that involved the scaffold domain (16). The interaction interfaces differed in shape and involved transmembrane helices with some contacts through the entire membrane, whereas other formed contacts only in one membrane leaflet.
Experimental procedures
DAT construct
Mutagenesis of eGFP to mGFP was performed by the mutation A206K using the QuikChange Mutagenesis Kit (Agilent) in a peGFP-C1 vector (Clontech) with the forward primer, 5′-TAC CTG AGC ACC CAG TCC AAA CTG AGC AAA GAC CCC AAC-3′, and the reverse primer, 5′-GTT GGG GTC TTT GCT CAG TTT GGA CTG GGT GCT CAG GTA-3′. A XhoI/HindIII fragment of hDAT cDNA was ligated into XhoI/HindIII-digested mGFP-C1 to produce mGFP-hDAT.
Cell culture
WT CHO and hDAT expressing stable CHO cells were cultured at 37 °C and 5% CO2 in Dulbecco's modified Eagle's and F-12 Ham's (DMEM F-12, Sigma) medium supplemented with 10% FBS (Sigma), penicillin and streptomycin (Sigma). Cell lines are tested monthly for mycoplasma contamination using MycoAlert (Lonza).
Generation of stable cell line
We performed viral infection to generate CHO cells stably expressing mGFP fused to the N terminus of hDAT. Briefly, we cloned mEGFP–hDAT into the viral expression vector pIB2 (gift from J. B. Huppa, Medical University Vienna) using Gibson Assembly (New England Biolabs) with 5′-ccgcggcggccgcggtccggATGGTGAGCAAGGGCGAGG-3′ as forward primer and 5′-CAATTGTTTTACGTATCTCGCTACACCTTGAGCCAGTGGCG-3′ as reverse primer. Viral supernatants were produced in HEK293 cells co-transfecting pIB2-mEGFP-hDAT and helper plasmids coding for viral proteins (G. Nolan, Stanford University) using Turbofect. 48 h post-transfection, viral supernatants were harvested and centrifuged at 1000 × g for 5 min at 21 °C to remove cell debris. CHO cells were then transduced in 24-well plates in the presence of 10 μg/ml of Polybrene (Sigma) for 24 h. Positive cells were selected with blasticidine (Invivogen, Toulouse, France) for 1 week and expanded.
Transient transfections and uptake experiments
HEK293 cells were cultured in Dulbecco's modified Eagle's medium with high glucose (4.5 g/liter) and l-glutamine (584 mg/liter), supplemented with 10% fetal calf serum (FCS), 100 units/ml of penicillin, and 100 μg/ml of streptomycin. The cells were maintained at 37 °C in a humidified atmosphere of 5% CO2, 95% air on standard plastic cultureware. The cells were transiently transfected with 5 μg of plasmid DNA as required, using jetPRIME® as transfection reagent. Cells were seeded at 0.8 × 105 cells/0.5 ml or 0.8 × 105 cells/well densities into poly-d-lysine–coated 48-well plates 24 h after transfection with cDNAs of either WT–hDAT or mGFP–hDAT. Uptakes were performed as described previously (77). In brief, the cell medium was aspirated and the cells were washed once with 0.5 ml of Krebs-HEPES buffer at room temperature (23 °C; Krebs-HEPES buffer, composition: HEPES, 10 mm; NaCl, 120 mm; KCl, 3 mm; CaCl2, 2 mm; MgCl2, 2 mm; glucose, 20 mm; final pH 7.3–7.4). The cells were incubated with [3H]DA (200 nm) and various concentrations of unlabeled DA (range: 0.2-60 μm; final volume 0.1 ml/well) and substrate uptake was stopped after 1 min by exchange of the substrate containing buffer with 0.5 ml of ice-cold Krebs-HEPES buffer. The cells were lysed in 0.5 ml of 1% SDS, transferred to scintillation vials, and assayed for [3H] content by liquid scintillation counting. Nonspecific uptake was determined in the presence of 100 μm cocaine.
Sample preparation
For all cell imaging experiments, 8-well glass bottom plates were used (Thermo Fisher Scientific, Nunc) and coated with poly-d-lysine (Sigma). Cleaned slides were incubated with 0.1 mg/ml of poly-d-lysine for 1 h at 37 °C and unbound poly-d-lysine were washed three times before use.
Cholesterol oxidation
To oxidize the cholesterol in the plasma membrane, the stable cells were incubated with 2 units/ml of cholesterol oxidase (Invitrogen) in Hanks' balanced salt solution for 30 min at 37 °C.
Cholesterol depletion
To deplete cholesterol from the plasma membrane, stable cells were incubated with 5 mm methyl-β-cyclodextrin (Sigma) in Hanks' balanced salt solution for 1 h at 37 °C.
PIP2 depletion
For activation of phospholipase Cγ, cells were incubated for 20 min at 37 °C with 25 μm m-3M3FBS (Sigma) in Hanks' balanced salt solution, with calcium and magnesium (Sigma) supplemented with 2% FBS. m-3M3FBS remained in the imaging buffer during measurements.
Microscopy
A 488-nm laser (SAPPHIRE HP, Coherent Inc.) was mode-cleaned using a pinhole and the illumination intensity and timing were adjusted with an acousto-optical modulator (model 1205, Isomet) using custom written software (Labview, National Instruments). The laser beam was focused onto the back-focal plane of a TIRF objective (NA 1.46, ×100 Plan APOCHROMAT, Zeiss) mounted on an inverted Zeiss Axiovert 200 microscope. The emission light was filtered using appropriate filter sets for GFP and imaged with a back-illuminated liquid nitrogen-cooled CCD camera (Micro Max 1300-PB, Roper Scientific). To restrict the excitation and photobleaching area an adjustable slit aperture (Zeiss) was used as field stop. All experiments were performed at room temperature. Imaging during all experiments was performed using an objective-type TIRF excitation with an excitation power of ∼0.6–0.8 kW/cm2 (determined in epi-configuration) and stroboscopic illumination with excitation times of 5 ms.
FRAP
To determine the mobile fraction of mGFP–hDAT, a ∼7 × 7-μm area of the bottom plasma membrane was irreversibly photobleached, and the fluorescence recovery over time was monitored (n >10 cells). Photobleaching and readout were performed in TIRF configuration. Data were analyzed using in-house algorithms implemented in Matlab (Mathworks). The central part of the bleached region was evaluated by integrating all counts and normalizing to the pre-bleach image. To calculate the mobile fraction of mGFP–hDAT, the resulting curve was fitted with I/I0 = α(1-exp(t/τD)) yielding the mobile fraction α of mGFP–hDAT.
TOCCSL
TOCCSL experiments were performed as described previously (28). A pre-bleach image was recorded, which was used for determination of the mGFP–hDAT surface density. After a tpre = 50 ms, a confined region of the cell membrane was photobleached for tbleach = 800 ms with a high laser power of ∼8–10 kW/cm2. To check for complete bleaching, a post-bleach image was recorded tpost = 40 ms after the bleach pulse. Finally, the TOCCSL image was recorded after an adjustable recovery time of trecovery = 15,000–20,000 ms. Images were acquired at low excitation power of ∼0.6–0.8 kW/cm2 (all excitation intensities were determined in epiconfiguration).
Repetitive TOCCSL experiment
For the hDAT dimer stability studies, a repetitive TOCCSL protocol was applied. One TOCCSL run every 5 min repeated over 50 min starting from the first bleach pulse was performed on the same region of each cell; following a modification of the timing protocol used earlier (28). Both bleaching and image acquisition were obtained in the TIRF mode. The TOCCSL image of each run was used for brightness analysis.
Brightness analysis
For single-molecule brightness analysis, images were analyzed in Matlab using an in-house algorithm. The pixel counts obtained were subsequently converted to photon by subtracting the offset and multiplying with the inverse gain (as per specification of the Roper camera). Individual diffraction-limited fluorescent signals from the regions of interest were selected and fitted with a Gaussian intensity function. The fitting routine yielded the single spot brightness B, which was then used to determine the oligomeric state of mGFP–hDAT. Next, the obtained brightness values of each fluorescent spot in the TOCCSL image were plotted as a probability density function, ρ(B). The cells were extensively photobleached to obtain the brightness distribution of single mGFP molecules ρ1(B), such that each potential mGFP–hDAT oligomer contained only one active fluorophore. Using autoconvolution, the monomer brightness distribution was used to calculate the brightness distributions for dimers ρ2(B), trimers ρ3(B), and so on. The overall single spot brightness distribution ρ(B) was then fitted by a linear combination of ρ1(B), ρ2(B), ρ3(B) and so on.
| (Eq. 1) |
Fitting ρ(B) in the above equation yielded the fractions αN of the different numbers of co-diffusing active mGFP molecules. To obtain error bars during our analysis, we followed a bootstrapping method. Briefly, randomly chosen subsamples containing 50% of the data were analyzed using Equation 1; the error bars represent the obtained standard deviation from 100 repetitions for each oligomeric size divided by √2.
Matlab code
The Matlab Source code for TOCCSL analysis is available at https://github.com/schuetzgroup/TOCCSL_analysis.4
Author contributions
A. K. D., H. H. S., and G. J. S. conceptualization; A. K. D. formal analysis; A. K. D., F. B., and K. J. investigation; A. K. D., O. K., F. B., and G. J. S. methodology; A. K. D. writing-original draft; T. S., H. H. S., and G. J. S. supervision; T. S., H. H. S., and G. J. S. writing-review and editing; H. H. S. and G. J. S. funding acquisition; G. J. S. resources; G. J. S. software; G. J. S. project administration.
Supplementary Material
Acknowledgment
We thank Elena Parkinson for assistance in cell culture.
This work was supported by Austrian Science Fund/FWF F35 (to T. S., H. H. S., and G. J. S) and Vienna Science and Technology Fund Grant CS15-033 (to H. H. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- hDAT
- human dopamine transporter
- DA
- dopamine
- GAT
- γ-amino acid transporter
- SERT
- serotonin transporter
- NET
- norepinephrine transporter
- ER
- endoplasmic reticulum
- mGFP
- monomeric GFP
- TIRF
- total internal reflection fluorescence
- TOCCSL
- thinning out clusters while conserving stoichiometry of labeling
- FRAP
- fluorescence recovery after photobleaching
- FBS
- fetal bovine serum
- m-3M3FBS
- 2,4,6-trimethyl-N-(3-trifluoromethylphenyl)benzenesulfonamide
- TMH
- transmembrane helices
- SLC6
- solute carrier 6
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- CHO
- Chinese hamster ovary
- MβCD
- methyl-β-cyclodextrin
- eGFP
- enhanced green fluorescent protein
- hSERT
- human serotonin transporter.
References
- 1. Giros B., and Caron M. G. (1993) Molecular characterization of the dopamine transporter. Trends Pharmacol. Sci. 14, 43–49 10.1016/0165-6147(93)90029-J [DOI] [PubMed] [Google Scholar]
- 2. Rothman R. B., and Baumann M. H. (2003) Monoamine transporters and psychostimulant drugs. Eur. J. Pharmacol. 479, 23–40 10.1016/j.ejphar.2003.08.054 [DOI] [PubMed] [Google Scholar]
- 3. Saier M. H. Jr, Tran C. V., and Barabote R. D. (2006) TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 34, D181–186 10.1093/nar/gkj001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kristensen A. S., Andersen J., Jørgensen T. N., Sørensen L., Eriksen J., Loland C. J., Strømgaard K., and Gether U. (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585–640 10.1124/pr.108.000869 [DOI] [PubMed] [Google Scholar]
- 5. Masson J., Sagné C., Hamon M., and El Mestikawy S. (1999) Neurotransmitter transporters in the central nervous system. Pharmacol. Rev. 51, 439–464 [PubMed] [Google Scholar]
- 6. Kuhar M. J., Ritz M. C., and Boja J. W. (1991) The dopamine hypothesis of the reinforcing properties of cocaine. Trends Neurosci. 14, 299–302 10.1016/0166-2236(91)90141-G [DOI] [PubMed] [Google Scholar]
- 7. Ritz M. C., Lamb R. J., Goldberg S. R., and Kuhar M. J. (1987) Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science 237, 1219–1223 10.1126/science.2820058 [DOI] [PubMed] [Google Scholar]
- 8. Torres G. E., Gainetdinov R. R., and Caron M. G. (2003) Plasma membrane monoamine transporters: structure, regulation and function. Nat. Rev. Neurosci. 4, 13–25 10.1038/nrn1008 [DOI] [PubMed] [Google Scholar]
- 9. Ullrich A., and Schlessinger J. (1990) Signal transduction by receptors with tyrosine kinase activity. Cell 61, 203–212 10.1016/0092-8674(90)90801-K [DOI] [PubMed] [Google Scholar]
- 10. Metzger H. (1992) Transmembrane signaling: the joy of aggregation. J. Immunol. 149, 1477–1487 [PubMed] [Google Scholar]
- 11. Ali M. H., and Imperiali B. (2005) Protein oligomerization: how and why. Bioorgan Med. Chem. 13, 5013–5020 10.1016/j.bmc.2005.05.037 [DOI] [PubMed] [Google Scholar]
- 12. Sarkar B., Das A. K., and Maiti S. (2013) Thermodynamically stable amyloid-β monomers have much lower membrane affinity than the small oligomers. Front. Physiol. 4, 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Das A. K., Pandit R., and Maiti S. (2015) Effect of amyloids on the vesicular machinery: implications for somatic neurotransmission. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, pii: 20140187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 10.1074/jbc.R800016200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Akiva E., Itzhaki Z., and Margalit H. (2008) Built-in loops allow versatility in domain-domain interactions: lessons from self-interacting domains. Proc. Natl. Acad. Sci. U.S.A. 105, 13292–13297 10.1073/pnas.0801207105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jayaraman K., Morley A. N., Szöllöosi D., Wassenaar T. A., Sitte H. H., and Stockner T. (2018) Dopamine transporter oligomerization involves the scaffold domain, but spares the bundle domain. Plos Comput. Biol. 14, e1006229 10.1371/journal.pcbi.1006229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palsdottir H., and Hunte C. (2004) Lipids in membrane protein structures. Biochim. Biophys. Acta 1666, 2–18 10.1016/j.bbamem.2004.06.012 [DOI] [PubMed] [Google Scholar]
- 18. Barrera N. P., Zhou M., and Robinson C. V. (2013) The role of lipids in defining membrane protein interactions: insights from mass spectrometry. Trends Cell Biol. 23, 1–8 10.1016/j.tcb.2012.08.007 [DOI] [PubMed] [Google Scholar]
- 19. de Meyer F. J., Venturoli M., and Smit B. (2008) Molecular simulations of lipid-mediated protein-protein interactions. Biophys. J. 95, 1851–1865 10.1529/biophysj.107.124164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anderluh A., Hofmaier T., Klotzsch E., Kudlacek O., Stockner T., Sitte H. H., and Schütz G. J. (2017) Direct PIP2 binding mediates stable oligomer formation of the serotonin transporter. Nat. Commun. 8, 14089 10.1038/ncomms14089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buchmayer F., Schicker K., Steinkellner T., Geier P., Stübiger G., Hamilton P. J., Jurik A., Stockner T., Yang J. W., Montgomery T., Holy M., Hofmaier T., Kudlacek O., Matthies H. J., Ecker G. F., et al. (2013) Amphetamine actions at the serotonin transporter rely on the availability of phosphatidylinositol-4,5-bisphosphate. Proc. Natl. Acad. Sci. U.S.A. 110, 11642–11647 10.1073/pnas.1220552110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sitte H. H., Farhan H., and Javitch J. A. (2004) Sodium-dependent neurotransmitter transporters: oligomerization as a determinant of transporter function and trafficking. Mol. Interv. 4, 38–47 10.1124/mi.4.1.38 [DOI] [PubMed] [Google Scholar]
- 23. Schmid J. A., Scholze P., Kudlacek O., Freissmuth M., Singer E. A., and Sitte H. H. (2001) Oligomerization of the human serotonin transporter and of the rat GABA transporter 1 visualized by fluorescence resonance energy transfer microscopy in living cells. J. Biol. Chem. 276, 3805–3810 10.1074/jbc.M007357200 [DOI] [PubMed] [Google Scholar]
- 24. Just H., Sitte H. H., Schmid J. A., Freissmuth M., and Kudlacek O. (2004) Identification of an additional interaction domain in transmembrane domains 11 and 12 that supports oligomer formation in the human serotonin transporter. J. Biol. Chem. 279, 6650–6657 10.1074/jbc.M306092200 [DOI] [PubMed] [Google Scholar]
- 25. Kilic F., and Rudnick G. (2000) Oligomerization of serotonin transporter and its functional consequences. Proc. Natl. Acad. Sci. U.S.A. 97, 3106–3111 10.1073/pnas.97.7.3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jess U., Betz H., and Schloss P. (1996) The membrane-bound rat serotonin transporter, SERT1, is an oligomeric protein. FEBS Lett. 394, 44–46 10.1016/0014-5793(96)00916-7 [DOI] [PubMed] [Google Scholar]
- 27. Fjorback A. W., Pla P., Müller H. K., Wiborg O., Saudou F., and Nyengaard J. R. (2009) Serotonin transporter oligomerization documented in RN46A cells and neurons by sensitized acceptor emission FRET and fluorescence lifetime imaging microscopy. Biochem. Biophys. Res. Commun. 380, 724–728 10.1016/j.bbrc.2009.01.128 [DOI] [PubMed] [Google Scholar]
- 28. Anderluh A., Klotzsch E., Reismann A. W., Brameshuber M., Kudlacek O., Newman A. H., Sitte H. H., and Schütz G. J. (2014) Single molecule analysis reveals coexistence of stable serotonin transporter monomers and oligomers in the live cell plasma membrane. J. Biol. Chem. 289, 4387–4394 10.1074/jbc.M113.531632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berger S. P., Farrell K., Conant D., Kempner E. S., and Paul S. M. (1994) Radiation inactivation studies of the dopamine reuptake transporter protein. Mol. Pharmacol. 46, 726–731 [PubMed] [Google Scholar]
- 30. Milner H. E., Béliveau R., and Jarvis S. M. (1994) The in situ size of the dopamine transporter is a tetramer as estimated by radiation inactivation. Biochim. Biophys. Acta 1190, 185–187 10.1016/0005-2736(94)90051-5 [DOI] [PubMed] [Google Scholar]
- 31. Torres G. E., Carneiro A., Seamans K., Fiorentini C., Sweeney A., Yao W. D., and Caron M. G. (2003) Oligomerization and trafficking of the human dopamine transporter: mutational analysis identifies critical domains important for the functional expression of the transporter. J. Biol. Chem. 278, 2731–2739 10.1074/jbc.M201926200 [DOI] [PubMed] [Google Scholar]
- 32. Chen N., and Reith M. E. (2008) Substrates dissociate dopamine transporter oligomers. J. Neurochem. 105, 910–920 10.1111/j.1471-4159.2007.05195.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sorkina T., Doolen S., Galperin E., Zahniser N. R., and Sorkin A. (2003) Oligomerization of dopamine transporters visualized in living cells by fluorescence resonance energy transfer microscopy. J. Biol. Chem. 278, 28274–28283 10.1074/jbc.M210652200 [DOI] [PubMed] [Google Scholar]
- 34. Egaña L. A., Cuevas R. A., Baust T. B., Parra L. A., Leak R. K., Hochendoner S., Peña K., Quiroz M., Hong W. C., Dorostkar M. M., Janz R., Sitte H. H., and Torres G. E. (2009) Physical and functional interaction between the dopamine transporter and the synaptic vesicle protein synaptogyrin-3. J. Neurosci. 29, 4592–4604 10.1523/JNEUROSCI.4559-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hastrup H., Sen N., and Javitch J. A. (2003) The human dopamine transporter forms a tetramer in the plasma membrane: cross-linking of a cysteine in the fourth transmembrane segment is sensitive to cocaine analogs. J. Biol. Chem. 278, 45045–45048 10.1074/jbc.C300349200 [DOI] [PubMed] [Google Scholar]
- 36. Hastrup H., Karlin A., and Javitch J. A. (2001) Symmetrical dimer of the human dopamine transporter revealed by cross-linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc. Natl. Acad. Sci. U.S.A. 98, 10055–10060 10.1073/pnas.181344298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seidel S., Singer E. A., Just H., Farhan H., Scholze P., Kudlacek O., Holy M., Koppatz K., Krivanek P., Freissmuth M., and Sitte H. H. (2005) Amphetamines take two to tango: an oligomer-based counter-transport model of neurotransmitter transport explores the amphetamine action. Mol. Pharmacol. 67, 140–151 [DOI] [PubMed] [Google Scholar]
- 38. Farhan H., Freissmuth M., and Sitte H. H. (2006) Oligomerization of neurotransmitter transporters: a ticket from the endoplasmic reticulum to the plasma membrane. Handb. Exp. Pharmacol. 2006, 233–249 [DOI] [PubMed] [Google Scholar]
- 39. Scholze P., Freissmuth M., and Sitte H. H. (2002) Mutations within an intramembrane leucine heptad repeat disrupt oligomer formation of the rat GABA transporter 1. J. Biol. Chem. 277, 43682–43690 10.1074/jbc.M205602200 [DOI] [PubMed] [Google Scholar]
- 40. Block E. R., Nuttle J., Balcita-Pedicino J. J., Caltagarone J., Watkins S. C., Sesack S. R., and Sorkin A. (2015) Brain region-specific trafficking of the dopamine transporter. J. Neurosci. 35, 12845–12858 10.1523/JNEUROSCI.1391-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma S., Cheng M. H., Guthrie D. A., Newman A. H., Bahar I., and Sorkin A. (2017) Targeting of dopamine transporter to filopodia requires an outward-facing conformation of the transporter. Sci. Rep. 7, 5399 10.1038/s41598-017-05637-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miranda M., Sorkina T., Grammatopoulos T. N., Zawada W. M., and Sorkin A. (2004) Multiple molecular determinants in the carboxyl terminus regulate dopamine transporter export from endoplasmic reticulum. J. Biol. Chem. 279, 30760–30770 10.1074/jbc.M312774200 [DOI] [PubMed] [Google Scholar]
- 43. Zahniser N. R., and Sorkin A. (2009) Trafficking of dopamine transporters in psychostimulant actions. Semin. Cell Dev. Biol. 20, 411–417 10.1016/j.semcdb.2009.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kocabas A. M., Rudnick G., and Kilic F. (2003) Functional consequences of homo- but not hetero-oligomerization between transporters for the biogenic amine neurotransmitters. J. Neurochem. 85, 1513–1520 10.1046/j.1471-4159.2003.01793.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhen J., and Reith M. E. A. (2018) Functional properties of dopamine transporter oligomers after copper linking. J. Neurochem. 144, 162–171 10.1111/jnc.14259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sitte H. H., and Freissmuth M. (2010) The reverse operation of Na+/Cl−-coupled neurotransmitter transporters: why amphetamines take two to tango. J. Neurochem. 112, 340–355 10.1111/j.1471-4159.2009.06474.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cremona M. L., Matthies H. J., Pau K., Bowton E., Speed N., Lute B. J., Anderson M., Sen N., Robertson S. D., Vaughan R. A., Rothman J. E., Galli A., Javitch J. A., and Yamamoto A. (2011) Flotillin-1 is essential for PKC-triggered endocytosis and membrane microdomain localization of DAT. Nat. Neurosci. 14, 469–477 10.1038/nn.2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adkins E. M., Samuvel D. J., Fog J. U., Eriksen J., Jayanthi L. D., Vaegter C. B., Ramamoorthy S., and Gether U. (2007) Membrane mobility and microdomain association of the dopamine transporter studied with fluorescence correlation spectroscopy and fluorescence recovery after photobleaching. Biochemistry 46, 10484–10497 10.1021/bi700429z [DOI] [PubMed] [Google Scholar]
- 49. Hong W. C., and Amara S. G. (2010) Membrane cholesterol modulates the outward facing conformation of the dopamine transporter and alters cocaine binding. J. Biol. Chem. 285, 32616–32626 10.1074/jbc.M110.150565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Foster J. D., Adkins S. D., Lever J. R., and Vaughan R. A. (2008) Phorbol ester induced trafficking-independent regulation and enhanced phosphorylation of the dopamine transporter associated with membrane rafts and cholesterol. J. Neurochem. 105, 1683–1699 10.1111/j.1471-4159.2008.05262.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jones K. T., Zhen J., and Reith M. E. (2012) Importance of cholesterol in dopamine transporter function. J. Neurochem. 123, 700–715 10.1111/jnc.12007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ulbrich M. H., and Isacoff E. Y. (2007) Subunit counting in membrane-bound proteins. Nat. Methods 4, 319–321 10.1038/nmeth1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Madl J., Weghuber J., Fritsch R., Derler I., Fahrner M., Frischauf I., Lackner B., Romanin C., and Schütz G. J. (2010) Resting state Orai1 diffuses as homotetramer in the plasma membrane of live mammalian cells. J. Biol. Chem. 285, 41135–41142 10.1074/jbc.M110.177881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moertelmaier M., Brameshuber M., Linimeier M., Schutz G. J., and Stockinger H. (2005) Thinning out clusters while conserving stoichiometry of labeling. Appl. Phys. Lett. 87, 263903 10.1063/1.2158031 [DOI] [Google Scholar]
- 55. Brameshuber M., and Schütz G. J. (2012) Detection and quantification of biomolecular association in living cells using single-molecule microscopy. Methods Enzymol. 505, 159–186 10.1016/B978-0-12-388448-0.00017-6 [DOI] [PubMed] [Google Scholar]
- 56. Calebiro D., Rieken F., Wagner J., Sungkaworn T., Zabel U., Borzi A., Cocucci E., Zürn A., and Lohse M. J. (2013) Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl. Acad. Sci. U.S.A. 110, 743–748 10.1073/pnas.1205798110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sampson N. S., and Vrielink A. (2003) Cholesterol oxidases: a study of nature's approach to protein design. Acc. Chem. Res. 36, 713–722 10.1021/ar9800587 [DOI] [PubMed] [Google Scholar]
- 58. Molnár I., Hayashi N., Choi K. P., Yamamoto H., Yamashita M., and Murooka Y. (1993) Bacterial cholesterol oxidases are able to act as flavoprotein-linked ketosteroid monooxygenases that catalyse the hydroxylation of cholesterol to 4-cholesten-6-ol-3-one. Mol. Microbiol. 7, 419–428 10.1111/j.1365-2958.1993.tb01133.x [DOI] [PubMed] [Google Scholar]
- 59. Christian A. E., Haynes M. P., Phillips M. C., and Rothblat G. H. (1997) Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38, 2264–2272 [PubMed] [Google Scholar]
- 60. Hamilton P. J., Belovich A. N., Khelashvili G., Saunders C., Erreger K., Javitch J. A., Sitte H. H., Weinstein H., Matthies H. J. G., and Galli A. (2014) PIP2 regulates psychostimulant behaviors through its interaction with a membrane protein. Nat. Chem. Biol. 10, 582–589 10.1038/nchembio.1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kadamur G., and Ross E. M. (2013) Mammalian phospholipase C. Annu. Rev. Physiol. 75, 127–154 10.1146/annurev-physiol-030212-183750 [DOI] [PubMed] [Google Scholar]
- 62. Giros B., Jaber M., Jones S. R., Wightman R. M., and Caron M. G. (1996) Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379, 606–612 10.1038/379606a0 [DOI] [PubMed] [Google Scholar]
- 63. Alguel Y., Cameron A. D., Diallinas G., and Byrne B. (2016) Transporter oligomerization: form and function. Biochem. Soc. Trans. 44, 1737–1744 10.1042/BST20160217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rahbek-Clemmensen T., Lycas M. D., Erlendsson S., Eriksen J., Apuschkin M., Vilhardt F., Jørgensen T. N., Hansen F. H., and Gether U. (2017) Super-resolution microscopy reveals functional organization of dopamine transporters into cholesterol and neuronal activity-dependent nanodomains. Nat. Commun. 8, 740 10.1038/s41467-017-00790-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pizzo A. B., Karam C. S., Zhang Y., Yano H., Freyberg R. J., Karam D. S., Freyberg Z., Yamamoto A., McCabe B. D., and Javitch J. A. (2013) The membrane raft protein Flotillin-1 is essential in dopamine neurons for amphetamine-induced behavior in Drosophila. Mol. Psychiatry 18, 824–833 10.1038/mp.2012.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sitte H. H., and Freissmuth M. (2015) Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol. Sci. 36, 41–50 10.1016/j.tips.2014.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vaughan R. A., and Foster J. D. (2013) Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol. Sci. 34, 489–496 10.1016/j.tips.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sucic S., El-Kasaby A., Kudlacek O., Sarker S., Sitte H. H., Marin P., and Freissmuth M. (2011) The serotonin transporter is an exclusive client of the coat protein complex II (COPII) component SEC24C. J. Biol. Chem. 286, 16482–16490 10.1074/jbc.M111.230037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sucic S., Koban F., El-Kasaby A., Kudlacek O., Stockner T., Sitte H. H., and Freissmuth M. (2013) Switching the clientele: a lysine residing in the C terminus of the serotonin transporter specifies its preference for the coat protein complex II component SEC24C. J. Biol. Chem. 288, 5330–5341 10.1074/jbc.M112.408237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Magnani F., Tate C. G., Wynne S., Williams C., and Haase J. (2004) Partitioning of the serotonin transporter into lipid microdomains modulates transport of serotonin. J. Biol. Chem. 279, 38770–38778 10.1074/jbc.M400831200 [DOI] [PubMed] [Google Scholar]
- 71. Sevcsik E., and Schütz G. J. (2016) With or without rafts? alternative views on cell membranes. Bioessays 38, 129–139 10.1002/bies.201500150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sucic S., Dallinger S., Zdrazil B., Weissensteiner R., Jørgensen T. N., Holy M., Kudlacek O., Seidel S., Cha J. H., Gether U., Newman A. H., Ecker G. F., Freissmuth M., and Sitte H. H. (2010) The N terminus of monoamine transporters is a lever required for the action of amphetamines. J. Biol. Chem. 285, 10924–10938 10.1074/jbc.M109.083154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sweeney C. G., Tremblay B. P., Stockner T., Sitte H. H., and Melikian H. E. (2017) Dopamine transporter amino and carboxyl termini synergistically contribute to substrate and inhibitor affinities. J. Biol. Chem. 292, 1302–1309 10.1074/jbc.M116.762872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Khelashvili G., Stanley N., Sahai M. A., Medina J., LeVine M. V., Shi L., De Fabritiis G., and Weinstein H. (2015) Spontaneous inward opening of the dopamine transporter is triggered by PIP2-regulated dynamics of the N-terminus. ACS Chem. Neurosci. 6, 1825–1837 10.1021/acschemneuro.5b00179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Javanainen M., Martinez-Seara H., and Vattulainen I. (2017) Excessive aggregation of membrane proteins in the Martini model. PLOS ONE 12, e0187936 10.1371/journal.pone.0187936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Baucum A. J. 2nd, Rau K. S., Riddle E. L., Hanson G. R., and Fleckenstein A. E. (2004) Methamphetamine increases dopamine transporter higher molecular weight complex formation via a dopamine- and hyperthermia-associated mechanism. J. Neurosci. 24, 3436–3443 10.1523/JNEUROSCI.0387-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Scholze P., Nørregaard L., Singer E. A., Freissmuth M., Gether U., and Sitte H. H. (2002) The role of zinc ions in reverse transport mediated by monoamine transporters. J. Biol. Chem. 277, 21505–21513 10.1074/jbc.M112265200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.