

Abstract
AMPA-type glutamate receptors (AMPARs) mediate fast excitatory neurotransmission in the mammalian central nervous system. Preferential AMPAR subunit assembly favors heteromeric GluA1/GluA2 complexes. The presence of the GluA2 subunit generates Ca2+-impermeable (CI) AMPARs that have linear current-voltage (I-V) relationships. However, diverse forms of synaptic plasticity and pathophysiological conditions are associated with shifts from CI to inwardly rectifying, GluA2-lacking, Ca2+-permeable (CP) AMPARs on time scales ranging from minutes to days. These shifts have been linked to GluA1 phosphorylation at Ser-845, a protein kinase A (PKA)-targeted site within its intracellular C-terminal tail, often in conjunction with protein kinase A anchoring protein 79 (AKAP79; AKAP150 in rodents), which targets PKA to GluA1. However, AKAP79 may impact GluA1 phosphorylation at other sites by interacting with other signaling enzymes. Here, we evaluated the ability of AKAP79, its signaling components, and GluA1 phosphorylation sites to induce CP-AMPARs under conditions in which CI-AMPARs normally predominate. We found that GluA1 phosphorylation at Ser-831 is sufficient for the appearance of CP-AMPARs and that AKAP79-anchored protein kinase C (PKC) primarily drives the appearance of these receptors via this site. In contrast, other AKAP79-signaling components and C-terminal tail GluA1 phosphorylation sites exhibited a permissive role, limiting the extent to which AKAP79 promotes CP-AMPARs. This may reflect the need for these sites to undergo active phosphorylation/dephosphorylation cycles that control their residency within distinct subcellular compartments. These findings suggest that AKAP79, by orchestrating phosphorylation, represents a key to a GluA1 phosphorylation passcode, which allows the GluA1 subunit to escape GluA2 dominance and promote the appearance of CP-AMPARs.
Keywords: A-kinase anchoring protein (AKAP); alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA receptor, AMPAR); phosphorylation; synaptic plasticity; protein kinase C (PKC); calcineurin; protein kinase A (PKA); calcium; ion channel; subunit composition
Introduction
The GluA1 AMPAR2 intracellular C-terminal tail harbors multiple phosphorylation sites that are intimately linked with synaptic plasticity based on their ability to control the receptor's biophysical and trafficking properties (1). Ser-845, a PKA/PKG site (2, 3), is linked with GluA1 open probability and surface expression (4, 5), whereas Ser-831, a CaMKII/PKC site (6, 7), controls GluA1 single channel conductance (8). Ser-818 and Thr-840 are additional PKC sites that modify single-channel conductance (9). However, Ser-818 additionally contributes to the synaptic incorporation of AMPARs during synaptic plasticity (10). Native AMPARs are predominantly CI due to the presence of the GluA2 subunit (11). Previous studies indicated that incorporation of GluA2 into GluA1/GluA2 heteromers disables the impact of CaMKII-mediated Ser-831 phosphorylation on single-channel conductance (12), an effect alleviated by transmembrane AMPAR regulatory protein (TARP) family members (13). Because PKC can phosphorylate multiple sites within GluA1, as well as within GluA2 (14), we were interested in determining the impact of PKC infusion on GluA1/GluA2 heteromers. Moreover, multiple lines of investigation have implicated AKAP79/150 as a key regulatory element for various forms of synaptic plasticity by virtue of its ability to target PKA, PKC, and the Ca2+-dependent phosphatase calcineurin (CaN) to GluA1 (15–25). In particular, an expanding body of literature suggests that AKAP79/150 via Ser-845 phosphorylation may exert its effects through its ability to promote the appearance of presumably homomeric GluA1 CP-AMPARs (18, 24–27). However, GluA1 phosphorylation at Ser-831 has also been associated with the appearance of CP-AMPARs (28–38). As such, using HEK 293 cells as a model system, we set out to systematically examine by whole-cell electrophysiology whether AMPARs generated by co-expression of GluA1 and GluA2 were impacted by the presence of AKAP79, with a particular focus on AMPAR subunit composition. In addition, we sought to determine whether any AKAP79-dependent effects could be linked to its well-described interactions with PKA, PKC, or CaN and/or the GluA1 phosphorylation state. Additional biochemical studies were performed to assess the impact of AKAP79 on GluA1 phosphorylation in the context of GluA2 co-expression as well as to examine the impact of phosphorylation on GluA1 accessibility to the cell surface. Collectively, these studies revealed that the ability of AKAP79 to target PKC to GluA1 is most tightly aligned with the ability of AKAP79 to foster the appearance of CP-AMPARs. Nonetheless, the coordinated activities of AKAP79-anchored PKA and CaN influence the extent to which AKAP79 promotes the appearance CP-AMPARs. The underlying basis for these findings is an apparent hierarchy among the GluA1 C-terminal phosphorylation sites, whereby phosphomimetic substitution at Ser-831 suffices to promote the generation of CP-AMPA receptors. In contrast, other GluA1 C-terminal phosphorylation sites exhibit a permissive quality that likely relies on their ability to undergo active phosphorylation/dephosphorylation cycles.
Results
GluA1/GluA2 heteromers are resistant to augmentation by acute elevation of PKC
AKAP79 directs PKC to GluA1, enabling efficient phosphorylation of Ser-831 and concomitant enhancement of homomeric GluA1 receptor currents, via this site, with properties resembling that of CaMKII (20, 39). Interestingly, CaMKII-mediated enhancement of recombinant AMPARs, due to Ser-831 phosphorylation, is hindered within the context of heteromeric GluA1/GluA2 receptors (in the absence of TARP family members) (12, 13). Because additional PKC phosphorylation sites exist within GluA1 and GluA2 that control either the biophysical or trafficking properties of these subunits (9, 10, 14, 40, 41), the possibility existed that these sites could contribute to the functional modulation of GluA1/GluA2 heteromers, particularly when PKC is localized to these receptors via AKAP79. As such, we examined whether infusion (4 nm) of the constitutively active PKC fragment (PKM), generated by trypsinization of rat brain conventional PKC isoforms, modulated recombinant GluA1/GluA2 heteromers. GluA1 and GluA2 were co-transfected in a 1:1 ratio, which generates predominantly CI-AMPARs with linear I-V relationships that are distinct from inwardly rectifying CP-AMPARs typified by GluA1 homomers (42). A voltage ramp protocol was used to initiate each experiment and ensure that the I-V relationships of the currents largely conformed to GluA1/GluA2 heteromers. Cells that exhibited a visually noticeable degree of nonlinearity and were subsequently confirmed as having a ratio of outward current at +40 mV to inward current at −60 mV of less than 0.50 were excluded from this analysis. GluA1/GluA2 heteromers were resistant to the acute elevation of PKC regardless of the presence of AKAP79-GFP, as there was only a minimal decline in the amplitude of the AMPAR current (GluA1/GluA2: 0.91 ± 0.07, n = 5; +AKAP79-GFP: 0.90 ± 0.10, n = 6; Fig. 1A) and no obvious changes in the I-V relationship (Fig. 1B). This contrasts with a ∼30–40% increase in homomeric GluA1 receptor currents under identical conditions (20, 39). Thus, similar to its impact on CaMKII-mediated regulation, the presence of the GluA2 subunit shunts the ability of acute elevation of PKC to up-regulate heteromeric GluA1-containing AMPARs.
Figure 1.

GluA1/GluA2 heteromers are resistant to acute elevation PKC regardless of the presence of AKAP79. A, HEK 293 cells were transfected with GluA1/GluA2 with or without AKAP79-GFP. The catalytic fragment of PKC (PKM) was included in the pipette recording solution. Currents were evoked every 30 s in response to glutamate (1 mm) in the presence of cyclothiazide (100 μm). Shown is a summary time course for glutamate-evoked currents. Currents for each cell were normalized to their initial amplitude. Only cells with minimal rectification indicative of heteromers were included in the analysis. B, averaged I-V relationships for cells expressing GluA1/GluA2 with or without AKAP79-GFP obtained via ramp protocol at the beginning and end of each recording. Error bars, S.E.
AKAP79 promotes the expression of GluA1 homomers
Interestingly, recordings from cells expressing AKAP79-GFP were frequently terminated because they exhibited, at the onset of the experiment, a varying degree of inward rectification (Fig. 2A), such that fraction of current at +40 mV normalized to that at −60 mV was significantly reduced for cells expressing AKAP79 (0.52 ± 0.02, n = 14 (GluA1/GluA2) versus 0.40 ± 0.03, n = 23 (+AKAP79); p < 0.05; Fig. 2B). Application of 1-naphthylacetylspermine (10 μm), a selective blocker of GluA2-lacking AMPARs, completely eliminated the inwardly rectifying component in cells expressing AKAP79-GFP, consistent with AKAP79 promoting an increase in the fraction of current carried by GluA1 homomers (Fig. 2C). Commensurate with these findings, AKAP79-GFP co-expression nearly doubled the current density at −60 mV (−264.8 ± 59.3 pA/pF, n = 14 (GluA1/GluA2) versus −516.2 ± 99.1 pA/pF, n = 23 (+AKAP79); p < 0.05; Fig. 2D), but only modestly increased the current at +40 mV (144.4 ± 34.2 pA/pF, n = 14 (GluA1/GluA2) versus 176.1 ± 32.3 pA/pF, n = 23 (+AKAP79); Fig. 2D). It is important to note that the AKAP79-dependent increases in rectification and current density at −60 mV did not require the inclusion of PKM within the recording solution, as these effects were evident in a subset of cells in which PKM was omitted (Figs. 2D and 6). Although AKAP79 increased rectification and current density, no clear correlation between these parameters could be ascertained, suggesting that these likely represent separate phenomena. Based on linear extrapolation of the outward component to negative potentials, we estimated that ∼14.7% of the current, at −60 mV, was carried by GluA1 homomers in GluA1/GluA2-expressing cells, similar to previous estimates upon co-expression of these subunits in a 1:1 ratio (42). Given the conductance difference between GluA1 homomers and GluA1/GluA2 heteromers (12, 13), this is consistent with GluA1 homomers comprising only ∼2–4% of the surface receptors. In contrast, ∼36.6% of the current was carried by GluA1 homomers in cells expressing AKAP79-GFP. This ∼150% increase in GluA1 homomer activity is greater than that expected from the effects of phosphorylation on channel properties, particularly given that these sites often exhibit substantial basal phosphorylation (43). Thus, phosphorylation-mediated amplification of an existing pool of GluA1 homomers is unlikely to solely account for this effect of AKAP79. Rather, these findings suggest that AKAP-GFP expression fosters the appearance of a substantial population of GluA1 homomers under conditions in which GluA1/GluA2 heteromers normally prevail.
Figure 2.

AKAP79 promotes the expression of homomeric GluA1 CP-AMPARs. HEK 293 cells were transfected with GluA1/GluA2 with or without AKAP79. A, individual I-V relationships for cells expressing GluA1/GluA2 with or without AKAP79-GFP. Data are normalized to the current at −60 mV. B, averaged data for sweeps in A. C, representative experiment demonstrating that the inwardly rectifying component in AKAP79-expressing cells is completely blocked by the CP-AMPAR blocker 1-naphthylacetylspermine (NASPM). D, summary graph of initial current density measured at −60 and +40 mV obtained from a ramp protocol. Individual cells for each condition are shown. Open symbols, cells in which PKM was included in the pipette solution; filled symbols, cells in which PKM was not included. *, p < 0.05, compared with GluA1/GluA2. Error bars, S.E.
Figure 6.

Domain analysis suggests that AKAP79-anchored PKC primarily controls the generation of GluA1 homomers. HEK 293 cells were transfected with GluA1/GluA2 with or without AKAP79-GFP or various AKAP79 mutants deficient for PKC, CaN, or PKA. A, summary graph of the rectification index obtained from currents evoked by a ramp protocol. Individual cells for each condition are shown. Open symbols, cells in which PKM was included in the pipette solution; filled symbols, cells in which PKM was not included. Note that only the PKC-deficient mutant of AKAP79 completely blocks the rectification. B, summary graph (data for individual cells overlaid) of initial current density measured at −60 and +40 mV for the cells shown in A. Note that the CaN- and PKA-deficient AKAP79 constructs both appear to limit the AKAP79-induced increase in current density and rectification to similar extents, suggesting that they act via a common site. *, p < 0.05, compared with GluA1/GluA2 + AKAP79. Error bars, S.E.
Recently, the extensiveness of GluA1 phosphorylation in neurons has been questioned (44). However, it is important to note that AKAP79-mediated kinase/phosphatase signaling controls recombinant GluA1 currents, via regulation of Ser-831 and Ser-845 phosphorylation states, across a substantial fraction of the modulatory range linked to these sites (4, 6, 8, 17, 20). Basal phosphorylation likely constrains the ability of AKAP79 to engage the full dynamic range. Given the strong concordance between AKAP79-mediated regulation of recombinant GluA1 phosphorylation and function and that observed within native systems (17, 20, 39), it seems likely that neurons similarly utilize this dynamic range to regulate the population of GluA1-containing CP-AMPARs.
AKAP79-induced increase in GluA1 homomers is not due to selective changes in subunit abundance
Complementary biochemical experiments were initiated to assess whether AKAP79 expression led to a selective alteration in AMPA subunit abundance as a basis for the AKAP79-dependent increase in GluA1 homomers. However, immunoblot analysis revealed that cells expressing AKAP79-GFP exhibited a largely parallel increase in GluA1 and GluA2 abundance, irrespective of whether β-tubulin or Ponceau S staining was used a means for normalizing the data (GluA1: 40.0 ± 9.2%, n = 8; p < 0.01 compared with GluA1/GluA2; GluA2: 52.7 ± 14.2%, n = 8; p < 0.01 compared with GluA1/GluA2; Fig. 3). Whereas AKAP79-induced alterations in GluA1 or GluA2 abundance are insufficient to readily explain the increase in GluA1 homomers, these data are nonetheless consistent with the ability of AKAP79/150 to interact with and influence GluA2-containing AMPARs (22, 23), in addition to GluA2-lacking AMPARs. Importantly, these data suggest that AKAP79 favors surface expression and/or activity of GluA1 homomers.
Figure 3.

AKAP79 causes a parallel increase in GluA1 and GluA2 abundance. HEK 293 cells were transfected with GluA1/GluA2 with or without AKAP79-GFP. A, representative Western blot analysis (IB) of GluA1 and GluA2 abundance in the absence and presence of AKAP79-GFP. For these experiments, β-tubulin was used to normalize for loading differences. B, similar Western blot analysis except that Ponceau S staining was used for normalization. C, amalgamated summary graph showing percentage increase in GluA1 and GluA2 subunit abundance due to AKAP79-GFP co-expression. Data from individual experiments are shown and were normalized to β-tubulin (open circles) or by Ponceau S staining (filled circles). Both methods yielded similar results. Error bars, S.E.
GluA1 basal phosphorylation is elevated by GluA2 regardless of AKAP79 co-expression
AKAP79 enhances the basal phosphorylation of recombinant homomeric GluA1 at Ser-845 and favors Ser-831 phosphorylation upon treatment of cells with PKC-elevating agents, similar to that observed in native systems where GluA1/GluA2 heteromers predominate (15, 17, 20, 39). The degree to which phosphorylation at these sites, within native systems, emanates from GluA1 homomers or GluA1/GluA2 heteromers is unclear. Thus, it was of interest to ascertain whether AKAP79 differentially alters the basal phosphorylation of GluA1 when GluA2 is present. To address this issue, we employed phosphospecific antibodies to GluA1 at Ser-831 and Ser-845. We independently validated that these antibodies specifically detect basal phosphorylation at these sites based on their ability to recognize recombinant GluA1 on immunoblots but are devoid of signal when probed against their corresponding phosphodeficient alanine mutant (Fig. S1). Attempts to biochemically assess the impact of AKAP79 on the phosphorylation state of GluA1 at Ser-818 and Thr-840 were abandoned, due to the unavailability of commercially available antibodies to these sites that satisfied these criteria. Basal phosphorylation at Ser-831 for GluA1 homomers was modestly, but not significantly, increased by AKAP79-GFP (A1 + AKAP79: 1.15 ± 0.04, n = 6; Fig. 4A), as described previously (20). The increase in Ser-845 for GluA1 homomers due to AKAP79-GFP was more readily apparent (A1 + AKAP79: 1.28 ± 0.07, n = 6; p < 0.05 compared with GluA1 alone; Fig. 4B), as documented previously (15, 17). Surprisingly, basal GluA1 phosphorylation was elevated at Ser-831 and Ser-845 upon co-expression of GluA2, compared with GluA1 alone (S831P: 1.29 ± 0.01, n = 6; p < 0.01 compared with GluA1 alone; S845P: 1.55 ± 0.13, n = 6; p < 0.01 compared with GluA1 alone; Fig. 4, A and B, respectively). This increase in basal phosphorylation may be related to the reduced number of GluA1 subunits in GluA1/GluA2 heteromers compared with GluA1 homomers. Indeed, if basal phosphorylation primarily occurs on a single GluA1 subunit within a given tetrameric assembly, then the proportion of GluA1 subunits basally phosphorylated would be expected to increase in the GluA1/Glu2 heteromers relative to GluA1 homomers. Importantly, AKAP79-GFP did not alter the extent of basal phosphorylation for cells expressing GluA1/GluA2 (S831P: 1.35 ± 0.13, n = 6; S845P: 1.67 ± 0.11, n = 6; Fig. 4, A and B, respectively), suggesting an apparent uncoupling of basal phosphorylation from AKAP79 when GluA2 is present. Collectively, these data suggest that a differential change in the basal GluA1 phosphorylation at either site appears unable to readily explain the ability of AKAP79 to drive the appearance of GluA1 homomers. Whereas these findings appear at odds with the known influence of AKAP79 on GluA1 phosphorylation, they could instead be reflective of a role for AKAP79 in the maintenance of GluA1 phosphorylation within discrete populations.
Figure 4.

GluA1 basal phosphorylation is elevated by GluA2 co-expression and apparently uncoupled from AKAP79. HEK 293 cells were transfected with either GluA1 or GluA1/GluA2 with or without AKAP79-GFP. A (top), representative Western blotting (IB) for S831P and GluA1 for each of the indicated conditions. Bottom, summary graph showing the ratio of S831P/GluA1 for multiple experiments. Data for each experiment were normalized to those obtained for GluA1 alone. Circles, data from individual experiments. B, parallel analysis of Ser-845/GluA1 ratio is shown. *, p < 0.05; **, p < 0.01, compared with GluA1 alone. Error bars, S.E.
AKAP79 expands the population of GluA2-lacking GluA1 subunits
To further address this issue, we carried out GluA2 immunodepletion (ID), combined with cell surface biotinylation, as a means to examine whether AKAP79 alters the total or surface fraction of GluA1 subunits unassociated with GluA2 and to examine the GluA1 phosphorylation state within these pools. HEK 293 cells transfected with GluA1 and GluA2 subunits, either without or with AKAP79-GFP, were subjected to cell surface biotinylation, and the resulting extracts were subjected to two rounds of immunoprecipitation with anti-GluA2 antibodies. Probing the resulting supernatant for GluA2 revealed that this procedure effectively removed the vast majority (∼95%) of GluA2 subunits (A1/A2: 0.05 ± 0.01 of pre-ID, n = 5; A1/A2 + AKAP79: 0.06 ± 0.02 of pre-ID, n = 5; Fig. 5, A and B). Quantitation of the immunoblots for GluA1 revealed that AKAP79 significantly expanded the population of GluA2-lacking GluA1 subunits by ∼40% (0.39 ± 0.03 of pre-ID, n = 5 (A1/A2) versus 0.53 ± 0.05 of pre-ID, n = 5 (A1/A2 + AKAP79); p < 0.05; Fig. 5, A and C). The surface population of this largely GluA2-lacking pool of GluA1 subunits was subsequently isolated by incubation with streptavidin-coupled beads and then subjected to immunoblot analysis to assess the phosphorylation state of GluA1 at Ser-831 and Ser-845 within the surface and total fractions. The phosphorylation state of GluA1 at Ser-831 (A1/A2surface: 5.57 ± 0.57, n = 5, p < 0.01 compared with A1/A2total; A1/A2 + AKAP79surface: 6.23 ± 0.65, n = 5, p < 0.01 compared with A1/A2 + AKAP79total; Fig. 5, D and E) and Ser-845 (A1/A2surface: 4.34 ± 1.01, n = 5, p < 0.05 compared with A1/A2total; A1/A2 + AKAP79surface: 4.81 ± 1.04, n = 5, p < 0.05 compared with A1/A2 + AKAP79total; Fig. 5, D and F) was dramatically elevated within the surface pool of presumptive GluA1 homomers compared with the total fraction of GluA1 subunits. This is consistent with phosphorylation primarily serving as a signal linked to GluA1 plasma membrane residency. Interestingly, AKAP79 co-expression now significantly enhanced GluA1 phosphorylation, within the total fraction, at Ser-831 (A1/A2 + AKAP79total: 1.41 ± 0.10, n = 5; p < 0.05 compared with A1/A2total; Fig. 5, D and E). In contrast, the AKAP79-induced increase in Ser-845 phosphorylation did not achieve statistical significance due to an increased variability (A1/A2 + AKAP79total: 1.47 ± 0.20, n = 5; Fig. 5, D and F). This increase in Ser-831 phosphorylation within the GluA2-lacking pool of GluA1 subunits contrasts with the relatively modest ability of AKAP79 to regulate basal phosphorylation at Ser-831 when only obligate GluA1 homomers are expressed (Fig. 4A), suggesting that AKAP79 may more readily couple to GluA1 subunits in homomers when the presence of GluA2 normally limits the abundance of GluA1 homomers. Of note, the fraction of GluA1 subunits at the cell surface within this GluA2-immunodepleted pool was not impacted by AKAP79 expression (A1/A2: 13.1 ± 3.2%, n = 5; A1/A2 + AKAP79: 14.6 ± 4.4%, n = 5; Fig. 5, D and G). Collectively, this suggests that AKAP79-directed Ser-831 phosphorylation may signal expansion of the pool of GluA1 subunits and by extension GluA1 homomers, but once entry to this pool is gained, GluA1 homomers access the cell surface with a similar efficiency regardless of the presence of AKAP79.
Figure 5.

AKAP79 expands the population of GluA1 subunits unassociated with GluA2. HEK 293 cells were transfected with either GluA1 or GluA1/GluA2 with or without AKAP79-GFP. Cells were biotinylated then subjected to immunodepletion with GluA2/3 antibodies. A, representative Western blots (IB) of samples from cells transfected with GluA1 with or without AKAP79-GFP prior to and following immunodepletion probed for GluA2 (top), GluA1 (middle), and β-tubulin (bottom). B, summary graph showing the post-ID/pre-ID ratio for GluA2 for multiple experiments, demonstrating effective immunodepletion of the GluA2-containing fraction. Each value was normalized for loading based on β-tubulin staining. Circles represent data from individual experiments. C, similar analysis except quantifying the post-ID/pre-ID ratio for GluA1. *, p < 0.05 compared with GluA1/GluA2. D, total (10% of input) and surface populations were isolated from the supernatants from the preceding ID assay. Shown are representative Western blots probed for S831P (top), S845P (middle), and GluA1 (bottom). E, summary graph showing the ratio of S831P/GluA1 for multiple experiments. Data for each experiment were normalized to that obtained for cells transfected with GluA1/GluA2. Circles represent data from individual experiments. *, p < 0.05; **, p < 0.01, compared with GluA1/GluA2 (total). #, p < 0.01, compared with GluA1/GluA2 + AKAP79 (total). F, parallel analysis of the S845P/GluA1 ratio is shown. *, p < 0.05, compared with GluA1/GluA2 (total). #, p < 0.05, compared with GluA1/GluA2 + AKAP79 (total). G, summary graph quantifying surface GluA1 in post-ID fractions as a percentage of total GluA1 subunits. Error bars, S.E.
AKAP79-anchored PKC primarily controls the generation of GluA1 homomers
To further examine the impact of AKAP79 signaling components and GluA1 phosphorylation sites on the ability of AKAP79 to promote the appearance of GluA1 homomers, we used the rectification index (RI), the ratio of current at −60 mV to that at +40 mV, as a sensitive measure for assessing the appearance of CP-AMPARs. Using this convention, cells exhibiting a RI ≥ 2.5 can be considered strongly inwardly rectifying (SR) as ≥33% of the current at −60 mV would be derived from GluA1 homomers based on a current reversal potential of 2–3 mV. Indeed, replotting the data from Fig. 2 shows that AKAP79 substantially increased the RI and the proportion of SR cells dramatically (1.91 ± 0.07, 0 of 14 SR (GluA1/GluA2) versus 3.62 ± 0.58, 10 of 23 SR (+AKAP79); p < 0.05; Fig. 6A). Deletion of the PKC-binding region of AKAP79 (AKAP79ΔPKC) completely eliminated the increase in RI and the fraction of cells exhibiting overt rectification (AKAP79ΔPKC: 1.90 ± 0.07, 0 of 11 SR, p < 0.05 compared with AKAP79; Fig. 6 and Fig. S2). In contrast, deletion of either the CaN-binding or the C-terminal PKA-binding regions of AKAP79 (AKAP79ΔCaN and AKAPΔPKA, respectively) partially reduced the RI and the proportion of cells exhibiting overt rectification, but not as completely as observed for the AKAP79ΔPKC mutant (AKAP79ΔCaN: 2.18 ± 0.25, 3 of 11 SR; AKAPΔPKA: 2.32 ± 0.23, 3 of 11 SR; Fig. 6A and Fig. S2). None of the AKAP79 deletion mutants achieved statistical significance with respect to inhibiting the AKAP79-induced increase in current density, yet each mutant yielded current densities that were no longer significantly different from cells expressing only GluA1 and GluA2. This likely stems from the inherent variability, spanning a range up to 2 orders of magnitude within each group, associated with this measure. Nonetheless, it is interesting to note that the current densities for the AKAP79ΔCaN and AKAP79ΔPKA mutants at −60 mV and +40 mV were nearly identical to that observed for GluA1/GluA2 in the absence of AKAP79 (Fig. 6B), whereas the AKAP79ΔPKC mutant appeared to have a limited impact on current density at both potentials. Collectively, these data strongly support the idea that AKAP79-anchored PKC primarily contributes to the generation of GluA1 homomers and suggest that AKAP-anchored PKA and CaN secondarily limit the degree of rectification while possibly influencing overall subunit abundance. This last idea is consistent with a necessity for balanced opposition of AKAP79-anchored CaN and PKA in controlling Ser-845 phosphorylation and CP-AMPARs during synaptic plasticity (24, 25).
Ser-831 primarily controls the generation of GluA1 homomers
Given that the appearance of GluA1 homomers differentially relied on the ability of AKAP79 to bind to its associated signaling enzymes, we surmised that this may ultimately be mediated via regulation of the phosphorylation state of the GluA1 C-terminal tail in light of the role of AKAP79 in coordinating GluA1 phosphorylation. Thus, we systematically examined the ability of phosphodeficient alanine (A) and phosphomimetic aspartate (D) mutants at each of the four sites within the GluA1 C-terminal tail to alter the RI of AMPAR currents when expressed with GluA2 either alone or in the presence of AKAP79-GFP. Phosphomimetic and phosphodeficient substitution at each of these sites has been shown to modulate the biophysical properties of recombinant GluA1 receptor currents in a manner consistent with phosphorylation and dephosphorylation of GluA1 (4, 8, 9, 12, 45), supporting the validity of the approach. Of the four sites, only Ser-831 displayed a pattern consistent with its phosphorylation state serving as the primary signal for generation of GluA1 homomers when expressed with GluA2 subunits. Indeed, phosphodeficient mutation of this site (S831A) did not alter the RI, compared with the WT, yet blocked the AKAP79-induced increase in RI (S831A/GluA2: 1.98 ± 0.08, 0 of 11 SR; S831A/GluA2 + AKAP79: 1.90 ± 0.07, 0 of 10 SR; p < 0.05 compared with GluA1/GluA2 + AKAP79; Fig. 7 and Fig. S3). In contrast, the phosphomimetic S831D mutant sufficed to increase the RI and proportion of SR cells and occluded any additional effect of AKAP79 (S831D/GluA2: 3.54 ± 0.70, 6 of 14 SR; p < 0.01 compared with GluA1/GluA2; S831D/GluA2 + AKAP79: 3.83 ± 0.80, 9 of 17 SR; Fig. 7 and Fig. S3). In conjunction with our previous results, these data support the idea that AKAP79-anchored PKC drives an increase in homomeric GluA1 via Ser-831 phosphorylation. Remarkably, manipulation of the three remaining sites all yielded similar results, whereby phosphodeficient and phosphomimetic mutants at each site largely prevented the AKAP79-induced increase in GluA1 homomers, without any substantial effect on the RI index in the absence of AKAP79 (S818A/GluA2: 2.09 ± 0.19, 2 of 9 SR; S818A/GluA2 + AKAP79: 2.15 ± 0.34, 1 of 10 SR; S818D/GluA2: 1.85 ± 0.09, 1 of 13 SR; S818D/GluA2 + AKAP79: 1.83 ± 0.11, 1 of 11 SR; p < 0.05 compared with GluA1/GluA2 + AKAP79; T840A/GluA2: 1.96 ± 0.25, 1 of 10 SR; T840A/GluA2 + AKAP79: 2.19 ± 0.32, 2 of 9 SR; T840D/GluA2: 1.84 ± 0.03, 0 of 12 SR; T840D/GluA2 + AKAP79: 1.96 ± 0.16, 1 of 12 SR; p < 0.05 compared with GluA1/GluA2 + AKAP79; S845A/GluA2: 2.28 ± 0.24, 5 of 14 SR; S845A/GluA2 + AKAP79: 2.28 ± 0.24, 4 of 15 SR; S845D/GluA2: 2.30 ± 0.38, 1 of 11 SR; S845D/GluA2 + AKAP79: 1.83 ± 0.13, 2 of 11 SR; p < 0.05 compared with GluA1/GluA2 + AKAP79; Fig. 7 and Fig. S3). It was notable that the phosphomimetic mutants of Ser-818, Thr-840, and Ser-845 all exhibited a more reliable suppression of the ability of AKAP79 to induce rectification than their phosphodeficient counterparts. These data suggest a permissive role, in the generation or stability of GluA1 homomers, for these sites provided that they are phosphorylatable. Locking each site into a phosphodeficient or phosphomimetic state hinders the ability of AKAP79 to induce the appearance of GluA1 homomers, suggesting that active cycles of phosphorylation and dephosphorylation and/or interactions among sites critically regulates the pool of GluA1 homomers. Indeed, the double S831D/S845A mutant prevented the ability of S831D to promote the appearance of GluA1 homomers and was not substantially altered by AKAP79-GFP (S831D/S845A/GluA2: 2.02 ± 0.10, 0 of 10 SR; p < 0.05 compared with S831D/GluA2; S831D/S845A + AKAP79: 2.22 ± 0.10, 2 of 11 SR; Fig. 7 and Fig. S3). Interestingly, a double S831D/S845D mutant tended to limit the extent of rectification in SR cells but not necessarily the proportion of SR cells relative to the S831D single mutant (S831D/S845D: 2.30 ± 0.17, 4 of 10 SR; Fig. 7 and Fig. S3), but upon AKAP79 co-expression, the magnitude of rectification in SR cells tended to increase such that these cells exhibited an intermediate RI (S831D/S845D + AKAP79: 2.72 ± 0.44, 4 of 11 SR; Fig. 7 and Fig. S3). Together, this suggest that AKAP79 may secondarily rely on the Ser-845 phosphorylation state to expand the pool of GluA1 homomers.
Figure 7.
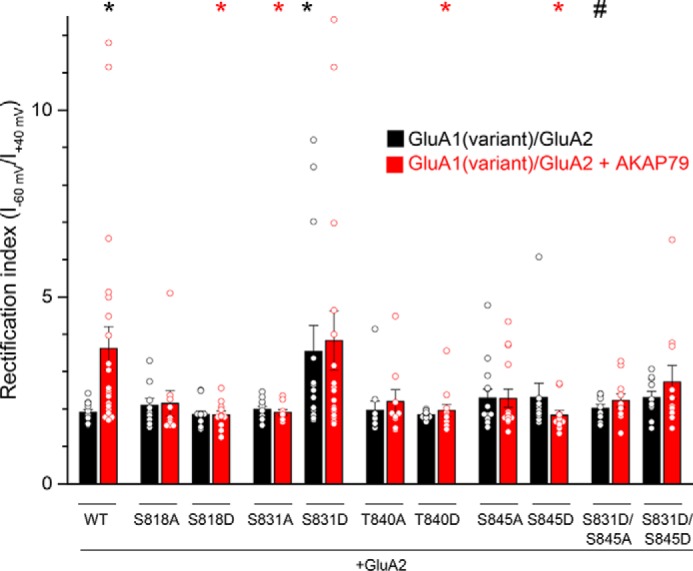
Mutational analysis of GluA1 phosphorylation sites reveals that Ser-831 uniquely controls the generation of GluA1 homomers. HEK 293 cells were transfected with WT GluA1 or phosphodeficient alanine or phosphomimetic aspartate mutants/GluA2 with or without AKAP79-GFP. Shown is a summary graph of the rectification index obtained from currents evoked by a ramp protocol. Data corresponding to individual cells for each condition are indicated by the open circles. Note that manipulation of Ser-831 was the only site in which the phosphodeficient S831A blocked the effect of AKAP79, whereas the corresponding phosphomimetic S831D mimicked the effect of AKAP79. Black asterisk, p < 0.05, compared with GluA1/GluA2. Red asterisk, p < 0.05 compared with GluA1/GluA2 + AKAP79. #, p < 0.05, compared with S831D/GluA2. Error bars, S.E.
Surface expression of GluA1 homomers relies on phosphorylation cycling
Ser-845 phosphorylation (38, 46–56) in conjunction with AKAP79/150 (18, 24–27) has been extensively linked to the presence of presumably homomeric GluA1 CP-AMPARs. However, phosphomimetic mutation of this site was largely inhibitory to the appearance of GluA1 homomers. Thus, we were interested in ascertaining whether constraining phosphorylation at this site alters the degree to which obligate GluA1 homomers are expressed at the cell surface and whether AKAP79 has an independent modulatory role. Using cell surface biotinylation, the phosphodeficient S845A mutant exhibited a slightly, but nonsignificantly, reduced ratio of surface to total GluA1 compared with WT GluA1 (0.88 ± 0.11, n = 11; Fig. 8A). Both WT and S845A exhibited a similar degree of surface expression in the presence of AKAP79 (GluA1 + AKAP79: 0.89 ± 0.09, n = 11; S845A + AKAP79: 0.84 ± 0.11, n = 11; Fig. 8A). These findings are generally consistent with a lack of impact of ablation of the Ser-845 site on GluA1 surface expression in transgenic animals (57, 58). In contrast, the phosphomimetic S845D mutant showed an over 50% reduction in surface expression compared with WT GluA1, regardless of the presence of AKAP79, suggesting that persistent phosphorylation of GluA1 at Ser-845 limits cell surface expression (S845D: 0.47 ± 0.07, n = 7; p < 0.01 compared with GluA1; S845D + AKAP79: 0.43 ± 0.09, n = 7; p < 0.05 compared with GluA1 + AKAP79; Fig. 8B). Down-regulation of GluA1 homomers by AKAP79-anchored CaN appears exclusively reliant on Ser-845 (17). Accordingly, AKAP79ΔCaN co-expression with WT GluA1, to strongly favor of PKA activity at the receptor (25, 27), led to a similar reduction in GluA1 surface expression, further supporting this idea (GluA1 + AKAP79ΔCaN: 0.54 ± 0.09, n = 7; p < 0.01 compared with GluA1; Fig. 8C). Moreover, AKAP79ΔCaN co-expression did not further impair the surface expression of the S845D mutant (S845D: 0.39 ± 0.04, n = 7; p < 0.01 compared with GluA1; S845D + AKAP79ΔCaN: 0.33 ± 0.01, n = 7; Fig. 8C). These data further suggest that the restriction of surface expression due to phosphomimetic mutation at Ser-845 is not simply an artifact arising from the presence of a nonphosphorylatable residue, but rather this process is engaged when this site is phosphorylated by endogenous mechanisms.
Figure 8.

Phosphorylation cycling regulates GluA1 surface expression. A, HEK 293 cells were transfected with GluA1 or S845A with or without AKAP79-mCherry and subjected to cell surface biotinylation, followed by immunoblot analysis for GluA1. Top, representative Western blotting. Bottom, summary graph quantifying the results from multiple experiments. Data are normalized to the surface/total ratio for GluA1 alone. Individual data points for each experiment are shown. B, similar analysis comparing the phosphomimetic S845D with WT GluA1, demonstrating that mimicking phosphorylation constrains surface expression of obligate homomers. **, p < 0.01, compared with GluA1; *, p < 0.05, compared with GluA1 + AKAP79. C, complementary experiment in which the CaN-deficient mutant was co-expressed with either GluA1 or the S845D mutant, suggesting that “overphosphorylation” of WT GluA1 similarly impairs surface expression. **, p < 0.01, compared with GluA1; *, p < 0.05, compared with GluA1 + AKAP79ΔCaN. D and E, parallel analysis examining the impact of phosphodeficient S831A (D) and the phosphomimetic S831D (E) mutations on the surface expression of obligate GluA1 homomers. *, p < 0.05; **, p < 0.01, compared with GluA1.
Given that Ser-831 phosphorylation uniquely promoted the appearance of GluA1 homomers, complementary surface biotinylation experiments were performed to assess whether constraining phosphorylation at this site commensurately altered the surface expression of obligate GluA1 homomers. Similar to the S845A mutant, the surface expression of the phosphodeficient S831A mutant was not significantly different from WT GluA1 (S845A: 0.91 ± 0.16, n = 3; Fig. 8D). However, there was a relatively small but significant decrease of surface expression in the presence of AKAP79 (S831A + AKAP79: 0.78 ± 0.03, n = 3; p < 0.05 compared with GluA1; Fig. 8D). This presumably aligns with the ability of this mutant, when expressed with GluA2, to impair the AKAP79-dependent shift in subunit composition toward homomers. Surprisingly, the phosphomimetic S831D mutant dramatically reduced surface expression compared with WT GluA1, either in the absence or presence of AKAP79 (S831D: 0.31 ± 0.11, n = 3, p < 0.05 compared with GluA1; S831D + AKAP79: 0.24 ± 0.28, n = 3, p < 0.05 compared with GluA1; Fig. 8E). This behavior was similar to the S845D mutant, further supporting the idea that precluding active phosphorylation within the C-terminal region limits the ability of GluA1 to access the cell surface. Remarkably, this ability of the S831D mutant to hinder obligate homomeric surface expression (Fig. 8E) yet favor homomeric activity when expressed with GluA2 (Fig. 7) suggests that the surface residency and/or itinerary of GluA1 homomers may be contingent on the context in which they were generated. This parallels our finding that AKAP79-directed Ser-831 phosphorylation is more readily apparent when GluA1 is segregated from GluA2-containing receptors (Fig. 5E) than when expressed as obligate homomers (Fig. 4A). Nonetheless, these data lend further credence to the notion that whereas phosphorylation of Ser-831 serves as a primary signal for GluA1 to escape GluA2 dominance, the ability to undergo active phosphorylation at C-terminal phosphorylation sites likely provides a permissive signal for maintaining homomers at the cell surface.
Discussion
Theoretically, phosphorylation-dependent enhancement of the conductance of a small pool of GluA1 homomers, even in the absence AKAP79, could lead to an increase in the proportion of current carried by GluA1 homomers and may contribute to our findings. However, if this was the primary mode for the increase in the proportion of GluA1 activity in cells expressing AKAP79, we would have expected S818D and T840D to exhibit an increase in rectification similar to the S831D mutant, because phosphorylation of these sites has an equivalent impact on homomeric GluA1 conductance (9). On the other hand, an inverse relationship between the GluA1 phosphorylation state at Thr-840 and Ser-845 may exist (59, 60), which could mitigate the effects of single phosphomimetic substitution at either site. Regardless, the disparity in effects tied to these PKC sites points to a distinct role for phosphorylation at Ser-831 in expanding the population of membrane-accessible GluA1 homomers.
Multiple lines of evidence point to AKAP79-anchored PKC as critical for the appearance of GluA1 homomers via Ser-831 phosphorylation. However, we failed to see a corresponding current develop upon acute elevation of PKC activity by inclusion of PKM within the patch pipette, either in the absence or presence of AKAP79. Several factors likely contribute to this finding. First is the fact that we selected cells for these experiments that were largely devoid of surface homomers based on their linear I-V relationships. Such heteromeric GluA1/GluA2 currents are resistant to acute modulation through phosphorylation of this site in the absence of a TARP (12, 13). Second, even if some GluA1 homomers were present on the cell surface, they were more likely to exhibit substantial basal phosphorylation (Fig. 5, D and E). Finally, our data suggest that AKAP79-anchored PKC-driven phosphorylation of Ser-831 may be primarily ushering the expansion of an intracellular pool of GluA1 homomers. The time frame over which this process occurs may exceed that of our recordings, which were performed with extensive Ca2+ buffering that likely precludes our ability to resolve delivery of subunits to the plasma membrane. The critical signals, sites, and ordering of steps required for eventual delivery of GluA1 homomers remain incompletely understood but likely involve the coordination of multiple signaling pathways extending beyond GluA1 resident sites (25, 61–65).
Due to the preferential assembly of GluA2-containing AMPARs (42, 66), the vast majority of hippocampal and cortical AMPARs are CI (11). However, an expanding body of literature indicates that GluA2-lacking, inwardly rectifying CP-AMPARs appear at synapses following various forms of synaptic plasticity and across varying time scales (67–69). GluA1 homomers are generally thought to provide the basis for these CP-AMPARs. How GluA1 homomers are formed, maintained, and granted synaptic access is not entirely clear. Whereas differential alteration in subunit synthesis can shift AMPAR subunit composition in favor of GluA1 homomers (70, 71), we found that AKAP79 reliably increased both subunits by similar amounts. AKAP79 is targeted to GluA1 by synapse-associated protein 97 (SAP97) (15), which can interact with GluA1-containing receptors early in the biosynthetic pathway (72). However, it seems unlikely that the ability of AKAP79 to increase the abundance of both subunits, yet alter composition, reflects a change in the initial assembly and/or delivery of subunits. Rather, it appears more plausible that AKAP79 acts in a regulatory fashion, controlling post-endocytic sorting of GluA1 homomers to recycling pools and/or precluding GluA1 homomers from entering degradative pathways. In agreement, our collective data (Figs. 2 and 5–7) suggests a scenario in which Ser-831 phosphorylation provides a signal for preferential intracellular accumulation of GluA1 homomers, particularly in the presence of GluA1/GluA2 heteromers, with AKAP79 serving as facilitator of this action by virtue of its ability to recruit PKC to these receptors. Indeed, AKAP79/150 and its signaling constituents are exquisitely positioned to operate in this capacity, as they are found in recycling endosomes and the plasma membrane within heterologous expression systems and native tissue (63, 73). Moreover, phosphorylation within the GluA1 C-terminal tail can impair its lysosomal degradation (5, 74). Whereas these pathways could support the AKAP79-driven accumulation of GluA1 homomers, the mechanistic details underlying these processes remain obscure. Our analysis reveals a previously unrecognized role for AKAP79-anchored PKC in expanding the population of GluA1 homomers via the phosphorylation state at Ser-831. Although multiple studies have implicated phosphorylation of this site in the appearance of CP-AMPARs (28–38), until now, no clear demonstration existed indicating that shifts in AMPAR composition can be readily reconstituted either via manipulation of the phosphorylation state of this site or by targeting a relevant kinase to this site.
Substantial evidence implicates AKAP150 and its associated PKA and CaN as principal elements governing the appearance of GluA1-containing CP-AMPARs via the phosphorylation of GluA1 at Ser-845 (18, 24–27). Here, using a reductionist approach, we have recapitulated and validated several previous findings. In particular, we have demonstrated that co-expression of AKAP79 suffices to promote the appearance of GluA1 homomers under conditions in which heteromers normally prevail and that deletion of the PKA-binding domain of AKAP79 or the corresponding phosphodeficient S845A mutant hinders the expression of GluA1 homomers. However, contrary to expectations, we found that deletion of the CaN-binding domain of AKAP79 and the corresponding phosphomimetic S845D mutant reduced the AKAP-driven appearance of GluA1 homomers. However, these latter findings may be mitigated by our evidence that constraining the phosphorylation state of GluA1 limits the ability of homomers to optimally populate the plasma membrane. Despite previous positive correlations between Ser-845 phosphorylation and surface expression (38, 46–56), the S845D mutant was expressed at the cell surface to a lesser extent than the S845A mutant. As a means to reconcile these findings, we suggest that active phosphorylation may favor successful insertion/reinsertion from this pool, but active dephosphorylation may ensure its continued access into the recycling pathway. Commensurate with this idea, the CaN-deficient mutant of AKAP150 favors basal expression of presumably homomeric GluA1 CP-AMPARs, yet this population is resistant to further augmentation (25). As such, extensive or persistent phosphorylation at Ser-845 may preclude successive entry into the recycling pathway and thereby ensure the transient appearance of GluA1 homomers under certain plasticity-inducing paradigms (24, 75). This suggests that the recycling capacity for GluA1 homomers may be limited. A capacity limit may explain the cell-to-cell variability in the presence of GluA1 homomers in cells expressing either AKAP79 or S831D along with GluA2.
Whereas our evidence clearly supports the idea that AKAP79-induced shifts in the GluA1 phosphorylation state at multiple sites drive the appearance of GluA1 homomers, we only obtained complementary biochemical evidence upon immunodepletion of the heteromeric population. This may stem from a more extensive population of GluA1/GluA2 heteromers, which predominate the surface in our heterologous expression system and within native systems (76, 77) and consequently obscure the population of GluA1 subunits unassociated with GluA2. Thus, the influence of specific GluA1 phosphorylation sites on the appearance CP-AMPARs in response to various plasticity paradigms may be underestimated if determined solely on the basis of biochemical measures from a total population of subunits. Nonetheless, selective alterations in the GluA1 phosphorylation state at each site have been documented under a variety of conditions linked with changes in subunit composition. Whether species, brain region, tissue, or stimulation paradigm differences contribute to the selective stabilization of one phosphorylation site over another remains unclear. The most parsimonious explanation for these differences may lie with the concept that a dynamic phosphorylation passcode exists for GluA1 homomers, which enables these receptors to sort into distinct regulated endosomal compartments, with each harboring unique spatial and/or temporal trajectories for repopulating the surface. As such, biochemical measures of phosphorylation state may be reflective of the fraction of receptors participating in these recycling trajectories. In support of this idea, mimicking phosphorylation within the GluA1 C-terminal tail enables subunits to accumulate within intracellular dendritic compartments, which can nonetheless be delivered to the cell surface with appropriate additional signals including other phosphorylation sites (10, 74).
Our studies suggest that dynamic, rather than static, phosphorylation within the GluA1 C-terminal tail appears necessary for functional GluA1 homomers to populate the cell surface. Nonetheless, the sufficiency of the phosphomimetic S831D mutant for promoting a population of GluA1 homomers in the presence of GluA2 strongly suggests that phosphorylation at this site is a chief signal enabling accumulation of GluA1 homomers. Indeed, Ser-831 is transiently phosphorylated in response to learning paradigms with a time course that overlaps with the appearance of CP-AMPARs (78, 79), suggesting that active cycling in its phosphorylation state contributes to the appearance and disappearance of these receptors. In hippocampal neurons, phosphorylation of this site along with Ser-845 appears necessary for Ser-818 phosphorylation to drive synaptic incorporation of GluA1 homomers (10), suggesting that multisite, and possibly sequential, GluA1 C-terminal tail phosphorylation may be required to allow GluA1 homomers access to the cell surface. At present, it is unclear whether the ability of multiple phosphomutants to impair the AKAP79-induced expression of GluA1 homomers actually reflects the necessity of each site to undergo concerted cycles of phosphorylation/dephosphorylation. Rather, each of these sites may impose unique contingencies that limit one or more of the multitude of steps needed for GluA1 homomers to become membrane-resident. Indeed, multiple positive and negative interactions among AMPAR posttranslational modifications have been noted (80). Nonetheless, qualitatively similar outcomes are largely obtained by constraining the phosphorylation state at these sites, with regard to the ability of AKAP79 to promote the appearance of GluA1 homomers. This suggests that each site must be at least available for modification and that some coordinated cycling of phosphorylation and dephosphorylation among these sites gates the ability of GluA1 homomers to eventually populate the cell surface.
In summary, our analysis reveals a previously unrecognized role for AKAP79-anchored PKC in expanding the population of GluA1 homomers that is exquisitely controlled via the phosphorylation state at Ser-831. As such, our results suggest that AKAP79, by controlling local kinase and phosphatase signaling, plays a critical role in guiding GluA1 through a series of phosphorylation site checkpoints, which allows it to escape from GluA2 dominance and thereby promote the appearance of GluA1 homomers. This pathway likely contributes to the appearance of CP-AMPARs under physiological and maladaptive forms of plasticity associated with AKAP79/150.
Experimental procedures
Molecular constructs and site-directed mutagenesis
GluA1flip in pRK5 was described previously (17). The edited form of GluA2(R)flip was generously provided by Peter Seeburg (Max Planck Institute, Heidelberg, Germany). AKAP79-GFP, AKAP79(75–427)-GFP denoted as AKAP79ΔPKC, and AKAP79(1–360)-GFP denoted as AKAP79ΔPKA have been described previously (17, 81). AKAP79-(del337–343)-mCherry denoted as AKAP79ΔCaN was generously provided by Mark Dell'Acqua (University of Colorado School of Medicine, Aurora, CO) and described previously (82). Site-directed mutagenesis for all phosphomutants was performed using the GluA1flip as template using the QuikChange Lightning mutagenesis kit (Agilent) according to the manufacturer's instructions. Incorporation of the appropriate mutations was verified by automated sequencing performed by the University of Tennessee Health Science Center's Molecular Resource Center. Upon sequence verification, the construct was digested with AfeI and HindIII, and the resulting fragment, corresponding to the C-terminal region, was subsequently ligated back into the corresponding position of the parent template to ensure that no unwanted PCR errors were propagated outside the mutagenized region. Constructs were resequenced to verify ligation success.
Cell culture and transfections
HEK 293 cells (catalog no. CRL-1573; ATCC, Manassas, VA) were obtained at passage 35 and used for a maximum of eight passages. Cell cultures were maintained in DMEM with 10% fetal bovine serum (Hyclone (Logan, UT) or Life Technologies, Inc.) and penicillin/streptomycin. For electrophysiology, cells were plated at low density (∼50,000 cells/ml) on 15-mm round glass coverslips in 12-well plates. Cells were transfected with 1 μg of each construct per coverslip, and 0.3 μg of pEGFP was included for control cells as above. Epifluorescence was used to confirm the expression of the corresponding AKAP. For biochemistry, HEK 293 cells were plated at ∼50% confluence on 6-well plates. HEK 293 cells were transfected by the calcium phosphate method. For these experiments, 1 μg of each construct was used. pEGFP or mCherry (0.3 μg) was included in control conditions based on the corresponding AKAP constructs. All experiments were performed 24 h after transfection.
Electrophysiology
Whole-cell recordings were made with an Axopatch 200B or Axoclamp 700A amplifier (Molecular Devices, Sunnyvale, CA). Patch pipettes (2–4 megaohms) contained 140 mm cesium methanesulfonate, 10 mm HEPES, 5 mm adenosine triphosphate (sodium salt), 5 mm MgCl2, 0.2 mm CaCl2, and 10 mm BAPTA (pH 7.4). PKM (catalog no. SE-133; Biomol, Plymouth Meeting, PA) was added to this solution from frozen stocks. The extracellular solution was 150 mm NaCl, 5 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2 10 mm HEPES, 10 mm glucose, and 100 μm cyclothiazide (LC Laboratories, Woburn, MA) (pH 7.4). Glutamate (1 mm) was added from frozen concentrated stock solutions. Cyclothiazide (Axxora (San Diego, CA) or Ascent Scientific (Cambridge, UK); 100 μm) was used to block AMPA receptor desensitization. Solution exchanges were accomplished through a series of flow pipes that were controlled by solenoid valves (Warner, Hamden, CT) and moved into position by a piezoelectric bimorph. HEK 293 cells were lifted off of the coverslip to speed the solution exchange time. Currents were digitized at 5 kHz and filtered at 1 kHz and acquired with a Digidata 1322A board and Clampex 9 or 10 software (Molecular Devices, Sunnyvale, CA). Series resistance (90–95%) and whole-cell capacitance compensation were employed. Series resistance was monitored throughout the experiments by 5-mV hyperpolarizing jumps prior to each application of glutamate. Only cells that had a series resistance < 6 megaohms and were stable throughout the recording were included for analysis. All experiments were initiated within 1 min of establishing the whole-cell configuration and were performed at a holding potential of −60 mV at 20 °C. For time course experiments, currents were normalized to the amplitude of current from the initial agonist application for each experiment. Current-voltage (I-V) relationships were generated from the difference between 250-ms voltage ramps (from −60 to 40 mV) obtained in the absence and presence of agonist and normalized to the current at −60 mV and used to derive the RI. All data are expressed as means ± S.E. and were subjected to statistical analysis using either a t test (Figs. 1 and 2) or ANOVA followed by a Dunnett's post hoc test (Figs. 6 and 7) using the SPSS software package (IBM, Armonk, NY).
Cell surface biotinylation, immunodepletion, and Western blotting
Cells were rinsed three times in PBS (Life Technologies) at 4 °C. For cell surface biotinylation, 2.5 mm CaCl2 and 1 mm MgCl2 were added to the PBS and then subsequently incubated in this solution supplemented with 1.5 mg/ml sulfo-NHS-SS-biotin (Thermo Fisher Scientific) for 20 min at 4 °C. The biotinylation reaction was quenched with 50 mm glycine in the modified PBS solution (two washes, 5 min each) and then a final rinse in PBS. All extracts were prepared by lysing cells in 250 μl of lysis buffer (10 mm phosphate, 150 mm NaCl, 5 mm EDTA, 5 mm EGTA, 1% Triton X-100) containing protease inhibitors (Sigma; 1:100 dilution) and phosphatase inhibitors (10 mm sodium pyrophosphate, 50 mm sodium fluoride, 1 mm sodium orthovanadate, 1 μm okadaic acid, 1 μm microcystin, and 1 μm cyclosporin A). Lysates were incubated on ice for 10 min and then clarified by centrifugation at 14,000 × g for 10 min at 4 °C. For immunodepletion, cells were instead lysed in 300 μl of buffer, and 20 μl of the extract was reserved to assess pre-ID input, whereas the remaining was incubated with 4 μg of rabbit polyclonal anti-GluA2/3 (EMD Millipore) coupled to protein G–coated Dynabeads (40 μl; Life Technologies) for 2 h at 4 °C with nutation. The resulting supernatant was subjected to a second round of GluA2 immunodepletion carried out overnight (∼16 h). 40 μl of the resulting supernatant was reserved for post-ID analysis and as total input for subsequent biotin-based assessment of surface receptors as indicated below. For biotinylation assays, 20 μl of the extract (corresponding to 10%) was reserved for assessment of total GluA1, and 200 μl was subsequently incubated with streptavidin-coated MyOne-T1 Dynabeads (40 μl; Life Technologies) for 1 h at 4 °C with nutation. Following three washes (5 min) with lysis buffer, biotinylated complexes were eluted in 2× Laemmli buffer. For all other lysate, 2× Laemmli buffer was added as well. All samples were boiled for 5 min and subsequently resolved on 4–20% SDS-polyacrylamide gels (Lonza, Rockland, ME) and transferred to nitrocellulose and stained with Ponceau S solution (Sigma). Membranes were blocked overnight in 3% nonfat dry milk in TBS + 0.05% Tween 20 (TBST). For analysis of GluA1 and GluA2 levels, Western blots were probed with mouse monoclonal anti-GluA1 (N-terminal) clone RH95 (1:1000; EMD Millipore), stripped, and reprobed with mouse monoclonal anti-GluA2 (N-terminal) clone 6C4 (1:1000; EMD Millipore), stripped and finally reprobed with mouse monoclonal anti-β-tubulin clone AA2 (1:1000; EMD Millipore). Signals for GluA1 and GluA2 were adjusted by the β-tubulin loading levels and then normalized to controls in the absence of AKAP79. For analysis of GluA1 phosphorylation state, blots were probed with either rabbit monoclonal to phospho-Ser-831 clone N453 (1:1000; EMD Millipore) or rabbit polyclonal to phospho-Ser-845 (1:500; Cell Signaling Technology, Danvers, MA) and then stripped and reprobed with rabbit polyclonal anti-GluA1 (C-terminal) (1:1000; catalog no. AB1504; EMD Millipore). The ratio of phospho-GluA1 to GluA1 was determined and normalized to the control condition for each experiment. For analysis of surface to total ratios, blots were probed with anti-GluA1 (N-terminal) as above. Goat anti-rabbit or anti-mouse IgG horseradish peroxidase–conjugated antibodies (EMD Millipore; 1:10,000) were used as secondary antibody. All immunoblots were visualized by enhanced chemiluminescence (Thermo Fisher Scientific). Data were digitally acquired and quantified using a Bio-Rad XRS chemiluminescence documentation system and Quantity One software. Data are expressed as means ± S.E. and were subjected to statistical analysis by t test (Figs. 3 and 5 (B, C, and G)) or ANOVA followed by either Games–Howell post hoc test (Figs. 4, 5 (E and F), and 8 (A–C)) or Dunnett's post hoc test (Fig. 8, D and E) using SPSS software.
Author contributions
K. C. S., A. S. B., and S. J. T. resources; K. C. S., A. S. B., and S. J. T. investigation; K. C. S., A. S. B., and S. J. T. writing-original draft; S. J. T. conceptualization; S. J. T. formal analysis; S. J. T. supervision; S. J. T. funding acquisition; S. J. T. methodology; S. J. T. writing-review and editing.
Supplementary Material
This work was supported by National Institutes of Health Grant NS076637 (to S. J. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- AMPAR
- AMPA-type glutamate receptor
- PKA
- PKC, PKG, and PKM, protein kinase A, C, G, and M, respectively
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- TARP
- transmembrane AMPAR regulatory protein
- CaN
- calcineurin
- CP
- Ca2+-permeable
- CI
- Ca2+-impermeable
- pF
- picofarads
- ID
- immunodepletion
- RI
- rectification index
- SR
- strongly inwardly rectifying.
References
- 1. Lussier M. P., Sanz-Clemente A., and Roche K. W. (2015) Dynamic regulation of N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by posttranslational modifications. J. Biol. Chem. 290, 28596–28603 10.1074/jbc.R115.652750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roche K. W., O'Brien R. J., Mammen A. L., Bernhardt J., and Huganir R. L. (1996) Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16, 1179–1188 10.1016/S0896-6273(00)80144-0 [DOI] [PubMed] [Google Scholar]
- 3. Serulle Y., Zhang S., Ninan I., Puzzo D., McCarthy M., Khatri L., Arancio O., and Ziff E. B. (2007) A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron 56, 670–688 10.1016/j.neuron.2007.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Banke T. G., Bowie D., Lee H., Huganir R. L., Schousboe A., and Traynelis S. F. (2000) Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 20, 89–102 10.1523/JNEUROSCI.20-01-00089.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ehlers M. D. (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525 10.1016/S0896-6273(00)00129-X [DOI] [PubMed] [Google Scholar]
- 6. Barria A., Derkach V., and Soderling T. (1997) Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J. Biol. Chem. 272, 32727–32730 10.1074/jbc.272.52.32727 [DOI] [PubMed] [Google Scholar]
- 7. Mammen A. L., Kameyama K., Roche K. W., and Huganir R. L. (1997) Phosphorylation of the α-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J. Biol. Chem. 272, 32528–32533 10.1074/jbc.272.51.32528 [DOI] [PubMed] [Google Scholar]
- 8. Derkach V., Barria A., and Soderling T. R. (1999) Ca2+/calmodulin-kinase II enhances channel conductance of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 96, 3269–3274 10.1073/pnas.96.6.3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jenkins M. A., Wells G., Bachman J., Snyder J. P., Jenkins A., Huganir R. L., Oswald R. E., and Traynelis S. F. (2014) Regulation of GluA1 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor function by protein kinase C at serine-818 and threonine-840. Mol. Pharmacol. 85, 618–629 10.1124/mol.113.091488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boehm J., Kang M. G., Johnson R. C., Esteban J., Huganir R. L., and Malinow R. (2006) Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron 51, 213–225 10.1016/j.neuron.2006.06.013 [DOI] [PubMed] [Google Scholar]
- 11. Isaac J. T., Ashby M. C., and McBain C. J. (2007) The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871 10.1016/j.neuron.2007.06.001 [DOI] [PubMed] [Google Scholar]
- 12. Oh M. C., and Derkach V. A. (2005) Dominant role of the GluR2 subunit in regulation of AMPA receptors by CaMKII. Nat. Neurosci. 8, 853–854 10.1038/nn1476 [DOI] [PubMed] [Google Scholar]
- 13. Kristensen A. S., Jenkins M. A., Banke T. G., Schousboe A., Makino Y., Johnson R. C., Huganir R., and Traynelis S. F. (2011) Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat. Neurosci. 14, 727–735 10.1038/nn.2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McDonald B. J., Chung H. J., and Huganir R. L. (2001) Identification of protein kinase C phosphorylation sites within the AMPA receptor GluR2 subunit. Neuropharmacology 41, 672–679 10.1016/S0028-3908(01)00129-0 [DOI] [PubMed] [Google Scholar]
- 15. Colledge M., Dean R. A., Scott G. K., Langeberg L. K., Huganir R. L., and Scott J. D. (2000) Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron 27, 107–119 10.1016/S0896-6273(00)00013-1 [DOI] [PubMed] [Google Scholar]
- 16. Dell'Acqua M. L., Dodge K. L., Tavalin S. J., and Scott J. D. (2002) Mapping the protein phosphatase-2B anchoring site on AKAP79: binding and inhibition of phosphatase activity are mediated by residues 315–360. J. Biol. Chem. 277, 48796–48802 10.1074/jbc.M207833200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tavalin S. J., Colledge M., Hell J. W., Langeberg L. K., Huganir R. L., and Scott J. D. (2002) Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J. Neurosci. 22, 3044–3051 10.1523/JNEUROSCI.22-08-03044.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu Y., Allen M., Halt A. R., Weisenhaus M., Dallapiazza R. F., Hall D. D., Usachev Y. M., McKnight G. S., and Hell J. W. (2007) Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 26, 4879–4890 10.1038/sj.emboj.7601884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu Y., Zhang M., Lim I. A., Hall D. D., Allen M., Medvedeva Y., McKnight G. S., Usachev Y. M., and Hell J. W. (2008) AKAP150-anchored PKA activity is important for LTD during its induction phase. J. Physiol. 586, 4155–4164 10.1113/jphysiol.2008.151662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tavalin S. J. (2008) AKAP79 selectively enhances protein kinase C regulation of GluR1 at a Ca2+-calmodulin-dependent protein kinase II/protein kinase C site. J. Biol. Chem. 283, 11445–11452 10.1074/jbc.M709253200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jurado S., Biou V., and Malenka R. C. (2010) A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat. Neurosci. 13, 1053–1055 10.1038/nn.2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weisenhaus M., Allen M. L., Yang L., Lu Y., Nichols C. B., Su T., Hell J. W., and McKnight G. S. (2010) Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS One 5, e10325 10.1371/journal.pone.0010325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diering G. H., Gustina A. S., and Huganir R. L. (2014) PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 84, 790–805 10.1016/j.neuron.2014.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sanderson J. L., Gorski J. A., and Dell'Acqua M. L. (2016) NMDA receptor-dependent LTD requires transient synaptic incorporation of Ca2+-permeable AMPARs mediated by AKAP150-anchored PKA and calcineurin. Neuron 89, 1000–1015 10.1016/j.neuron.2016.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanderson J. L., Scott J. D., and Dell'Acqua M. L. (2018) Control of homeostatic synaptic plasticity by AKAP-anchored kinase and phosphatase regulation of Ca2+-permeable AMPA receptors. J. Neurosci. 38, 2863–2876 10.1523/JNEUROSCI.2362-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qiu S., Zhang M., Liu Y., Guo Y., Zhao H., Song Q., Zhao M., Huganir R. L., Luo J., Xu H., and Zhuo M. (2014) GluA1 phosphorylation contributes to postsynaptic amplification of neuropathic pain in the insular cortex. J. Neurosci. 34, 13505–13515 10.1523/JNEUROSCI.1431-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sanderson J. L., Gorski J. A., Gibson E. S., Lam P., Freund R. K., Chick W. S., and Dell'Acqua M. L. (2012) AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J. Neurosci. 32, 15036–15052 10.1523/JNEUROSCI.3326-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Banerjee B., Medda B. K., Pochiraju S., Kannampalli P., Lang I. M., Sengupta J. N., and Shaker R. (2013) AMPA receptor subunits expression and phosphorylation in cingulate cortex in rats following esophageal acid exposure. Neurogastroenterol. Motil. 25, 973–e776 10.1111/nmo.12233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caldeira M. V., Melo C. V., Pereira D. B., Carvalho R., Correia S. S., Backos D. S., Carvalho A. L., Esteban J. A., and Duarte C. B. (2007) Brain-derived neurotrophic factor regulates the expression and synaptic delivery of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J. Biol. Chem. 282, 12619–12628 10.1074/jbc.M700607200 [DOI] [PubMed] [Google Scholar]
- 30. Huie J. R., Stuck E. D., Lee K. H., Irvine K. A., Beattie M. S., Bresnahan J. C., Grau J. W., and Ferguson A. R. (2015) AMPA receptor phosphorylation and synaptic colocalization on motor neurons drive maladaptive plasticity below complete spinal cord injury. eNeuro 2, ENEURO.0091–15.2015 10.1523/ENEURO.0091-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jones T. L., and Sorkin L. S. (2005) Activated PKA and PKC, but not CaMKIIα, are required for AMPA/kainate-mediated pain behavior in the thermal stimulus model. Pain 117, 259–270 10.1016/j.pain.2005.06.003 [DOI] [PubMed] [Google Scholar]
- 32. Lee S., Song B., Kim J., Park K., Hong I., An B., Song S., Lee J., Park S., Kim J., Park D., Lee C. J., Kim K., Shin K. S., Tsien R. W., and Choi S. (2013) GluA1 phosphorylation at serine 831 in the lateral amygdala is required for fear renewal. Nat. Neurosci. 16, 1436–1444 10.1038/nn.3491 [DOI] [PubMed] [Google Scholar]
- 33. Martínez-Rivera A., Hao J., Tropea T. F., Giordano T. P., Kosovsky M., Rice R. C., Lee A., Huganir R. L., Striessnig J., Addy N. A., Han S., and Rajadhyaksha A. M. (2017) Enhancing VTA Cav1.3 L-type Ca2+ channel activity promotes cocaine and mood-related behaviors via overlapping AMPA receptor mechanisms in the nucleus accumbens. Mol. Psychiatry 22, 1735–1745 10.1038/mp.2017.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park K., Song B., Kim J., Hong I., Song S., Lee J., Park S., Kim J., An B., Lee H. W., Lee S., Kim H., Lee J. C., Lee S., and Choi S. (2014) ABA renewal involves enhancements in both GluA2-lacking AMPA receptor activity and GluA1 phosphorylation in the lateral amygdala. PLoS One 9, e100108 10.1371/journal.pone.0100108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen G., Mohamed M. S., Das P., and Tietz E. I. (2009) Positive allosteric activation of GABAA receptors bi-directionally modulates hippocampal glutamate plasticity and behaviour. Biochem. Soc. Trans. 37, 1394–1398 10.1042/BST0371394 [DOI] [PubMed] [Google Scholar]
- 36. Spaethling J., Le L., and Meaney D. F. (2012) NMDA receptor mediated phosphorylation of GluR1 subunits contributes to the appearance of calcium-permeable AMPA receptors after mechanical stretch injury. Neurobiol. Dis. 46, 646–654 10.1016/j.nbd.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tang Y., Liu S., Shu H., Xing Y., and Tao F. (2018) AMPA receptor GluA1 Ser831 phosphorylation is critical for nitroglycerin-induced migraine-like pain. Neuropharmacology 133, 462–469 10.1016/j.neuropharm.2018.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wigerblad G., Huie J. R., Yin H. Z., Leinders M., Pritchard R. A., Koehrn F. J., Xiao W. H., Bennett G. J., Huganir R. L., Ferguson A. R., Weiss J. H., Svensson C. I., and Sorkin L. S. (2017) Inflammation-induced GluA1 trafficking and membrane insertion of Ca2+ permeable AMPA receptors in dorsal horn neurons is dependent on spinal tumor necrosis factor, PI3 kinase and protein kinase A. Exp. Neurol. 293, 144–158 10.1016/j.expneurol.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brooks I. M., and Tavalin S. J. (2011) Ca2+/calmodulin-dependent protein kinase II inhibitors disrupt AKAP79-dependent PKC signaling to GluA1 AMPA receptors. J. Biol. Chem. 286, 6697–6706 10.1074/jbc.M110.183558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chung H. J., Xia J., Scannevin R. H., Zhang X., and Huganir R. L. (2000) Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J. Neurosci. 20, 7258–7267 10.1523/JNEUROSCI.20-19-07258.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee H. K., Takamiya K., Kameyama K., He K., Yu S., Rossetti L., Wilen D., and Huganir R. L. (2007) Identification and characterization of a novel phosphorylation site on the GluR1 subunit of AMPA receptors. Mol. Cell Neurosci. 36, 86–94 10.1016/j.mcn.2007.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mansour M., Nagarajan N., Nehring R. B., Clements J. D., and Rosenmund C. (2001) Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron 32, 841–853 10.1016/S0896-6273(01)00520-7 [DOI] [PubMed] [Google Scholar]
- 43. Diering G. H., Heo S., Hussain N. K., Liu B., and Huganir R. L. (2016) Extensive phosphorylation of AMPA receptors in neurons. Proc. Natl. Acad. Sci. U.S.A. 113, E4920–E4927 10.1073/pnas.1610631113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hosokawa T., Mitsushima D., Kaneko R., and Hayashi Y. (2015) Stoichiometry and phosphoisotypes of hippocampal AMPA-type glutamate receptor phosphorylation. Neuron 85, 60–67 10.1016/j.neuron.2014.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jenkins M. A., and Traynelis S. F. (2012) PKC phosphorylates GluA1-Ser831 to enhance AMPA receptor conductance. Channels 6, 60–64 10.4161/chan.18648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen T., Wang W., Dong Y. L., Zhang M. M., Wang J., Koga K., Liao Y. H., Li J. L., Budisantoso T., Shigemoto R., Itakura M., Huganir R. L., Li Y. Q., and Zhuo M. (2014) Postsynaptic insertion of AMPA receptor onto cortical pyramidal neurons in the anterior cingulate cortex after peripheral nerve injury. Mol. Brain 7, 76 10.1186/s13041-014-0076-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Clem R. L., and Huganir R. L. (2010) Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 330, 1108–1112 10.1126/science.1195298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ferrario C. R., Loweth J. A., Milovanovic M., Ford K. A., Galiñanes G. L., Heng L. J., Tseng K. Y., and Wolf M. E. (2011) Alterations in AMPA receptor subunits and TARPs in the rat nucleus accumbens related to the formation of Ca2+-permeable AMPA receptors during the incubation of cocaine craving. Neuropharmacology 61, 1141–1151 10.1016/j.neuropharm.2011.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Incontro S., Ciruela F., Ziff E., Hofmann F., Sánchez-Prieto J., and Torres M. (2013) The type II cGMP dependent protein kinase regulates GluA1 levels at the plasma membrane of developing cerebellar granule cells. Biochim. Biophys. Acta 1833, 1820–1831 10.1016/j.bbamcr.2013.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim S., Titcombe R. F., Zhang H., Khatri L., Girma H. K., Hofmann F., Arancio O., and Ziff E. B. (2015) Network compensation of cyclic GMP-dependent protein kinase II knockout in the hippocampus by Ca2+-permeable AMPA receptors. Proc. Natl. Acad. Sci. U.S.A. 112, 3122–3127 10.1073/pnas.1417498112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim S., and Ziff E. B. (2014) Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS Biol. 12, e1001900 10.1371/journal.pbio.1001900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tukey D. S., and Ziff E. B. (2013) Ca2+-permeable AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors and dopamine D1 receptors regulate GluA1 trafficking in striatal neurons. J. Biol. Chem. 288, 35297–35306 10.1074/jbc.M113.516690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Whitcomb D. J., Hogg E. L., Regan P., Piers T., Narayan P., Whitehead G., Winters B. L., Kim D. H., Kim E., St George-Hyslop P., Klenerman D., Collingridge G. L., Jo J., and Cho K. (2015) Intracellular oligomeric amyloid-β rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci. Rep. 5, 10934 10.1038/srep10934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Whitehead G., Jo J., Hogg E. L., Piers T., Kim D. H., Seaton G., Seok H., Bru-Mercier G., Son G. H., Regan P., Hildebrandt L., Waite E., Kim B. C., Kerrigan T. L., Kim K., et al. (2013) Acute stress causes rapid synaptic insertion of Ca2+-permeable AMPA receptors to facilitate long-term potentiation in the hippocampus. Brain 136, 3753–3765 10.1093/brain/awt293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yi E. S., Oh S., Lee J. K., and Leem Y. H. (2017) Chronic stress-induced dendritic reorganization and abundance of synaptosomal PKA-dependent CP-AMPA receptor in the basolateral amygdala in a mouse model of depression. Biochem. Biophys. Res. Commun. 486, 671–678 10.1016/j.bbrc.2017.03.093 [DOI] [PubMed] [Google Scholar]
- 56. Yoo S. W., Bae M., Tovar-Y-Romo L. B., and Haughey N. J. (2017) Hippocampal encoding of interoceptive context during fear conditioning. Transl. Psychiatry 7, e991 10.1038/tp.2016.254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goel A., Xu L. W., Snyder K. P., Song L., Goenaga-Vazquez Y., Megill A., Takamiya K., Huganir R. L., and Lee H. K. (2011) Phosphorylation of AMPA receptors is required for sensory deprivation-induced homeostatic synaptic plasticity. PLoS One 6, e18264 10.1371/journal.pone.0018264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee H. K., Takamiya K., Han J. S., Man H., Kim C. H., Rumbaugh G., Yu S., Ding L., He C., Petralia R. S., Wenthold R. J., Gallagher M., and Huganir R. L. (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112, 631–643 10.1016/S0092-8674(03)00122-3 [DOI] [PubMed] [Google Scholar]
- 59. Gray E. E., Guglietta R., Khakh B. S., and O'Dell T. J. (2014) Inhibitory interactions between phosphorylation sites in the C terminus of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor GluA1 subunits. J. Biol. Chem. 289, 14600–14611 10.1074/jbc.M114.553537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Toda A. M., and Huganir R. L. (2015) Regulation of AMPA receptor phosphorylation by the neuropeptide PACAP38. Proc. Natl. Acad. Sci. U.S.A. 112, 6712–6717 10.1073/pnas.1507229112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guire E. S., Oh M. C., Soderling T. R., and Derkach V. A. (2008) Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J. Neurosci. 28, 6000–6009 10.1523/JNEUROSCI.0384-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Oh M. C., Derkach V. A., Guire E. S., and Soderling T. R. (2006) Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 281, 752–758 10.1074/jbc.M509677200 [DOI] [PubMed] [Google Scholar]
- 63. Purkey A. M., Woolfrey K. M., Crosby K. C., Stich D. G., Chick W. S., Aoto J., and Dell'Acqua M. L. (2018) AKAP150 palmitoylation regulates synaptic incorporation of Ca2+-permeable AMPA receptors to control LTP. Cell Rep. 25, 974–987.e4 10.1016/j.celrep.2018.09.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Clem R. L., Anggono V., and Huganir R. L. (2010) PICK1 regulates incorporation of calcium-permeable AMPA receptors during cortical synaptic strengthening. J. Neurosci. 30, 6360–6366 10.1523/JNEUROSCI.6276-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Terashima A., Cotton L., Dev K. K., Meyer G., Zaman S., Duprat F., Henley J. M., Collingridge G. L., and Isaac J. T. (2004) Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J. Neurosci. 24, 5381–5390 10.1523/JNEUROSCI.4378-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Greger I. H., Khatri L., Kong X., and Ziff E. B. (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40, 763–774 10.1016/S0896-6273(03)00668-8 [DOI] [PubMed] [Google Scholar]
- 67. Hanley J. G. (2014) Subunit-specific trafficking mechanisms regulating the synaptic expression of Ca2+-permeable AMPA receptors. Semin. Cell Dev. Biol. 27, 14–22 10.1016/j.semcdb.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 68. Lee H. K. (2012) Ca-permeable AMPA receptors in homeostatic synaptic plasticity. Front. Mol. Neurosci. 5, 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Man H. Y. (2011) GluA2-lacking, calcium-permeable AMPA receptors—inducers of plasticity? Curr. Opin. Neurobiol. 21, 291–298 10.1016/j.conb.2011.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ju W., Morishita W., Tsui J., Gaietta G., Deerinck T. J., Adams S. R., Garner C. C., Tsien R. Y., Ellisman M. H., and Malenka R. C. (2004) Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat. Neurosci. 7, 244–253 10.1038/nn1189 [DOI] [PubMed] [Google Scholar]
- 71. Sutton M. A., Ito H. T., Cressy P., Kempf C., Woo J. C., and Schuman E. M. (2006) Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125, 785–799 10.1016/j.cell.2006.03.040 [DOI] [PubMed] [Google Scholar]
- 72. Sans N., Racca C., Petralia R. S., Wang Y. X., McCallum J., and Wenthold R. J. (2001) Synapse-associated protein 97 selectively associates with a subset of AMPA receptors early in their biosynthetic pathway. J. Neurosci. 21, 7506–7516 10.1523/JNEUROSCI.21-19-07506.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Keith D. J., Sanderson J. L., Gibson E. S., Woolfrey K. M., Robertson H. R., Olszewski K., Kang R., El-Husseini A., and Dell'acqua M. L. (2012) Palmitoylation of A-kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J. Neurosci. 32, 7119–7136 10.1523/JNEUROSCI.0784-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kessels H. W., Kopec C. D., Klein M. E., and Malinow R. (2009) Roles of stargazin and phosphorylation in the control of AMPA receptor subcellular distribution. Nat. Neurosci. 12, 888–896 10.1038/nn.2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Plant K., Pelkey K. A., Bortolotto Z. A., Morita D., Terashima A., McBain C. J., Collingridge G. L., and Isaac J. T. (2006) Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604 10.1038/nn1678 [DOI] [PubMed] [Google Scholar]
- 76. Lu W., Shi Y., Jackson A. C., Bjorgan K., During M. J., Sprengel R., Seeburg P. H., and Nicoll R. A. (2009) Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62, 254–268 10.1016/j.neuron.2009.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wenthold R. J., Petralia R. S., Blahos J. II, and Niedzielski A. S. (1996) Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J. Neurosci. 16, 1982–1989 10.1523/JNEUROSCI.16-06-01982.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shukla K., Kim J., Blundell J., and Powell C. M. (2007) Learning-induced glutamate receptor phosphorylation resembles that induced by long term potentiation. J. Biol. Chem. 282, 18100–18107 10.1074/jbc.M702906200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Whitlock J. R., Heynen A. J., Shuler M. G., and Bear M. F. (2006) Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097 10.1126/science.1128134 [DOI] [PubMed] [Google Scholar]
- 80. Diering G. H., and Huganir R. L. (2018) The AMPA receptor code of synaptic plasticity. Neuron 100, 314–329 10.1016/j.neuron.2018.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dell'Acqua M. L., Faux M. C., Thorburn J., Thorburn A., and Scott J. D. (1998) Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4,5-bisphosphate. EMBO J. 17, 2246–2260 10.1093/emboj/17.8.2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dittmer P. J., Dell'Acqua M. L., and Sather W. A. (2014) Ca2+/calcineurin-dependent inactivation of neuronal L-type Ca2+ channels requires priming by AKAP-anchored protein kinase A. Cell Rep. 7, 1410–1416 10.1016/j.celrep.2014.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.