

Abstract
Atg101 is an autophagy-related gene identified in worms, flies, mice, and mammals, which encodes a protein that functions in autophagosome formation by associating with the ULK1-Atg13-Fip200 complex. In the last few years, the critical role of Atg101 in autophagy has been well-established through biochemical studies and the determination of its protein structure. However, Atg101's physiological role, both during development and in adulthood, remains less understood. Here, we describe the generation and characterization of an Atg101 loss-of-function mutant in Drosophila and report on the roles of Atg101 in maintaining tissue homeostasis in both adult brains and midguts. We observed that homozygous or hemizygous Atg101 mutants were semi-lethal, with only some of them surviving into adulthood. Both developmental and starvation-induced autophagy processes were defective in the Atg101 mutant animals, and Atg101 mutant adult flies had a significantly shorter lifespan and displayed a mobility defect. Moreover, we observed the accumulation of ubiquitin-positive aggregates in Atg101 mutant brains, indicating a neuronal defect. Interestingly, Atg101 mutant adult midguts were shorter and thicker and exhibited abnormal morphology with enlarged enterocytes. Detailed analysis also revealed that the differentiation from intestinal stem cells to enterocytes was impaired in these midguts. Cell type–specific rescue experiments disclosed that Atg101 had a function in enterocytes and limited their growth. In summary, the results of our study indicate that Drosophila Atg101 is essential for tissue homeostasis in both adult brains and midguts. We propose that Atg101 may have a role in age-related processes.
Keywords: autophagy, neuron, cell differentiation, homeostasis, aging, neurogenesis, Atg101, insect development, intracellular degradation, midgut
Introduction
Autophagy (macroautophagy) is a process in which cytoplasmic materials, including organelles and macromolecules, are delivered to and degraded in the lysosome (1–4). As a major intracellular degradation system, autophagy plays important roles in development, tissue homeostasis, and aging (5–7). Defects in the autophagy pathway cause various human diseases, such as cancer and neurodegenerative diseases (8–10).
Genetic studies from budding yeast have identified more than 30 Atg genes, which function at various steps during autophagy (2, 3, 11). Most of these genes are highly conserved from yeast to mammals (3, 11). Among them, the Atg1 complex acts at the initiation stage of autophagy functioning as a scaffold for the recruitment of downstream Atg2 proteins to the pre-autophagosomal structure (4, 11–13). The yeast Atg1 complex consists of Atg1, Atg13, Atg17, Atg29, and Atg31, and its mammalian counterpart is composed of ULK1 (or ULK2), Atg13, FIP200 (also known as RB1CC1) and Atg101 (4, 11–13). Mammalian ULK1 and ULK2 are homologs of Atg1 (13). FIP200 is generally considered as a homolog of yeast Atg11 and Atg17 (13). Homologs of Atg29 and Atg31 are not found in higher eukaryotes (13). In contrast, Atg101 is present in most eukaryotes, with the exception of budding yeast (13). It has been proposed that the regulatory mode of the Atg1 complex and the regulatory mechanism for controlling autophagy initiation might have evolved from yeast to animal cells (13). Because of its absence in budding yeast, Atg101 has not been studied as extensively as other components of the ULK1 complex until the recent determination of the crystal structure of the Atg13-Atg101 complex (14–20). Atg101 contains a single HORMA domain and forms a heterodimer with Atg13 via this domain (14, 15, 18–23). The interaction between Atg101 and Atg13 helps to stabilize Atg13 and ULK1 in the complex (14, 15, 18–23). Although the functional requirement for Atg101 in autophagy has been established in mammalian cells, Caenorhabditis elegans, and Drosophila, the physiological role of Atg101 remains largely unexplored (14–17).
Drosophila has increasingly become an attractive model system for studying autophagy, especially the physiological functions of autophagy in tissue homeostasis and neurogenesis (24–26). In this study, in which we generated an Atg101 loss-of-function mutant using the CRISPR/Cas9 approach in Drosophila, we report on the roles of Atg101 in regulating neuron and midgut homeostasis.
Results
Generation and characterization of Atg101 loss-of-function mutant alleles
Drosophila Atg101 is located in an intron of the S6KL gene, which encodes an S6 kinase–like protein (Fig. 1A). To explore the physiological function of Atg101, we generated an Atg101 loss-of-function mutant allele, Atg1016h, using the recently developed CRISPR/Cas9 system (Fig. 1A). Atg1016h contains a 13-nucleotide deletion in the coding region, which causes a frameshift mutation (Fig. 1A). To examine whether this Atg101 mutation affects the expression of its host gene, S6KL, we extracted total RNA from both WT and Atg1016h mutant animals and performed quantitative PCR analysis using a pair of exon-specific primers for S6KL. Our quantitative PCR results showed that the mature S6KL level was ∼1.2 times of WT level in Atg1016h mutants (Fig. S1A). Previous studies have reported that overexpression and mutation of S6KL leads to a decreased and increased bouton number at the larval neuromuscular junctions (NMJ), respectively (27, 28). To further determine whether loss of Atg101 affects S6KL function, we performed immunostaining analysis and examined larval NMJ development in Atg1016h mutants. For this analysis, an anti-CSP antibody was used to label presynaptic components at larval NMJs. Our results revealed that there were no significant changes in the number of boutons for the muscle 4 NMJ in Atg1016h mutant larvae as compared with the control (Fig. S1, B and C). This indicated that Atg1016h mutants exhibited no detectable defects during larval NMJ development and further confirmed that loss of Atg101 does not affect S6KL function.
Figure 1.

Loss of Atg101 causes defects in starvation-induced and developmental autophagy. A, schematic diagram for the Atg101 gene locus. Coding exons are in black, and noncoding exons are in white. The guide RNA target sequence is indicated in red. The PAM sequence is indicated in purple. The dotted line indicates the deleted sequence. Atg1016h has a 13-bp deletion around the gRNA target site. ChX, chromosome X. B–B″, accumulation of GFP-Atg8a and RFP-Atg8a punctate structures in fat body cells of middle stage WT third instar larvae in response to starvation. C–C″, absence of GFP-Atg8a and RFP-Atg8a punctate structures in Atg1016h mutant animals under the same growth condition as in B–B″. D, quantification of the number of RFP-Atg8a punctate spots from B′ and C′. 145 cells from 16 WT fat body samples and 114 cells from 15 Atg1016h mutant fat body samples were counted. E, quantification of the size of RFP-Atg8a punctate spots shown in B′ and C′. 1000 spots from 12 WT fat body samples and 198 spots from 16 Atg1016h mutant fat body samples were measured. F–F″, accumulation of GFP-Atg8a and RFP-Atg8a punctate structures in fat body cells of wandering-stage WT third instar larvae. G–G″, absence of GFP-Atg8a and RFP-Atg8a punctate structures in Atg1016h mutant animals. H, quantification of the number of RFP-Atg8a punctate spots shown in F′ and G′. 82 cells from 15 WT fat body samples and 162 cells from 29 Atg1016h mutant fat body samples. I, quantification of the size of RFP-Atg8a punctate spots shown in B′ and C′. 150 spots from six WT fat body samples and 410 spots from four Atg1016h mutant fat body samples were measured. Data are presented as mean ± S.E. An unpaired t test was used for statistical analysis. ***, p < 0.001. Scale bars: 50 μm.
Homozygous or hemizygous Atg1016h mutants were found to be semi-lethal (Table S1). To determine whether the lethality we observed in Atg1016h mutants was caused by the specific loss of Atg101 function, we made a UAS-Atg101 rescue transgene. Overexpression of UAS-Atg101 with a ubiquitously expressed Daughterless-Gal4 (Da-Gal4) in Atg1016h mutants restored the viability of the mutant animals, indicating that the lethality is a likely consequence of loss of Atg101 (Table S1).
To analyze the lethality phenotype in more detail, we first examined the viability of WT and Atg1016h mutants at different developmental stages under growth control conditions. Reduced viability was observed in both embryonic and post-embryonic stages in Atg1016h mutants compared with the control, although the reduction of viability during the larval and pupal stages was subtle (Fig. S1D). Loss of Atg101 resulted in lethality before or during eclosion, similar to previously reported Atg17 mutants (Fig. S1, E and F) (29). Atg1016h mutant adult flies also displayed an abnormal wing posture, which has been described in Atg17 mutants (Fig. S1, G and F) (29). This wing posture defect was rescued by overexpressing Atg101 with Da-Gal4 (Fig. S11). Some of the newly enclosed Atg1016h mutant flies fell down easily into the food at the bottom of the vial (data not shown). In addition, we found that Atg1016h mutant animals developed relatively slower compared with the control and the time needed from the newly hatched first instar larvae to become pupae or adults was increased by 12–24 h on average (Fig. S1, J and K).
We also determined the expression level of Atg101 at different developmental stages by performing RT-PCR analysis on RNA prepared from embryos, larvae, pupae, and adults. Atg101 mRNA was expressed at all developmental stages, with the highest level in 0–12–h embryos (Fig. S11). In addition, the expression level of Atg101 was comparable with the levels of Atg1 and Atg8a but higher than the levels of Atg3, Atg4a, and Atg7 in 0–12–h embryos (Fig. S1M).
Atg101 mutants exhibit defects in starvation-induced and developmental autophagy
Atg101 is an important component of the ULK1/Atg1 kinase complex in higher eukaryotes and plays essential roles in the initiation of autophagy by interacting with Atg13 (13–15). To demonstrate the role of Atg101 during the process of autophagy in vivo, we analyzed starvation-induced and developmentally triggered autophagy in the Atg1016h mutant and WT control larval fat bodies. The dual-tagged GFP-RFP-Atg8a reporter is commonly used to follow autophagic flux and monitor autophagic activities (30). In this system, GFP fluorescence is normally quenched in the acidic environment of the autolysosome, but RFP fluorescence is pH-independent. Thus, autophagosomes can be labeled by both GFP and RFP signals and appear as yellow. Autolysosomes are positive only for RFP and appear as red. We took this approach and examined the effects of Atg101 on autophagic activities. Loss of Atg101 prevented the accumulation of both yellow and red punctate structures in fat body cells of both starved third instar larvae and late wandering third instar larvae, consistent with a previous report in which knockdown of Atg101 by RNAi blocked both starvation-induced and developmental autophagy (Fig. 1, B–I) (17). It has been shown that Atg8a-positive structures are larger in size upon Atg101 knockdown. However, we found that the size of Atg8a punctate structures was smaller in Atg101 mutant fat body cells than in the control (Fig. 1, B–I). It is possible that some remaining Atg101 activities might affect the size of Atg8a-positive structures.
Previous studies have shown that inhibition of autophagy leads to a delay in larval midgut cell death during Drosophila metamorphosis. Consistent with this finding, morphological analysis of Atg1016h mutant midguts revealed a delay in midgut cell death, as the gastric caeca still persisted at 4 h RPF (after puparium formation) in Atg1016h mutant animals (Fig. S2, A and B). Together, these data confirmed that animals lacking Atg101 function are impaired in their abilities to induce autophagy during normal development and in response to starvation.
Atg101 mutant flies show decreased lifespans and impaired mobility
Mutations in several Drosophila autophagy-related genes, including Atg7, Atg8, and Atg17, lead to reduced lifespans in adult flies (29, 31–33). We therefore examined the effect on Drosophila lifespan when Atg101 was deleted. 50–70% of the Atg1016h mutants were able to develop to adulthood, and these flies were used for lifespan measurement. Compared with the control, Atg1016h mutant flies exhibited a reduced adult lifespan, and most of mutant animals died at 3 weeks of age (Fig. 2, A and B). In addition, we noticed that Atg1016h mutant animals also showed a movement disorder, and most of them were found at the bottom of vials (data not shown). To confirm this finding, we performed a negative geotaxis assay with WT and mutant flies. When tapped to the bottom of a vial, WT flies responded by climbing to the top (Fig. 2C). However, most Atg1016h mutant flies failed to do so (Fig. 2C). At 2 days of age, 70–80% of the Atg1016h mutant flies had impaired climbing ability in a climbing assay (Fig. 2D). Their performance was much worse at day 10, with 100% of the flies impaired (Fig. 2D). These mobility defects were restored in the rescued flies (Fig. 2, C and D). Collectively, these data indicate that loss of Atg101 leads to a reduced lifespan and impaired mobility in adult flies.
Figure 2.
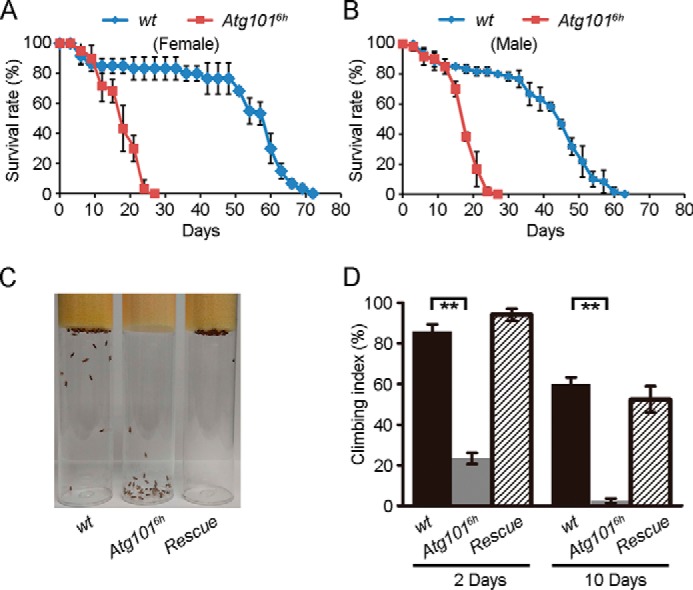
Loss of Atg101 leads to decreased adult lifespan and impaired locomotion. A, lifespan of Atg1016h mutant females is reduced compared with control females. The survival rates were calculated as the percentage of the number of surviving females versus the total number of females at the indicated days. Data are presented as mean ± S.D. B, lifespan of Atg1016h mutant males is reduced compared with control males. The survival rates were calculated as the percentage of the number of surviving males versus the total number of males at the indicated days. Data are presented as mean ± S.D. C, photograph of vials containing 7-day-old male flies with the indicated genotypes taken 15 s after taping the vials. D, groups of male flies with the indicated genotypes were tested for locomotor activity at day 2 and day 10. Data are presented as mean ± S.E. A paired t test was used for statistical analysis. **, p < 0.01.
Atg101 mutants show neurodegeneration defects
The age-related decline of mobility could be a reflection of neurodegeneration in the adult brain. Previous studies have shown that an accumulation of ubiquitinated proteins is associated with progressive neurodegeneration in Atg5 and Atg7 mutant mice as well as Atg7, Atg8, and Atg17 mutant flies (29, 31–35). To determine whether the mobility defects in Atg1016h mutant were attributable to neurodegeneration, we used an antibody against ubiquitin to examine Atg1016h mutants. Ubiquitinated proteins accumulated in 1-week-old Atg1016h mutant fly head extracts as compared with the WT control (Fig. 3A). To confirm the disruption of autophagy in the Atg101 mutant fly head, we also examined the protein level of Ref(2)p in both WT and mutant fly heads. Consistently, Atg1016h mutants had an obvious increase in the Ref(2) level as compared with the control (Fig. 3B). Furthermore, our immunofluorescence staining also revealed that the ubiquitin and Ref(2)p punctate structures had accumulated in the mutant brain, suggesting the formation of protein aggregates in the central nervous system of Atg1016h mutants (Fig. 3, C–D′ and G–H′, quantified in F and J). Partial co-localization between Ref(2)p and ubiquitin was also observed in Atg1016h mutants (Fig. S3, A–B″). These protein aggregate defects were rescued by the overexpression of Atg101 (Fig. 3, E, E′, I, and I′, quantified in F and J). To further determine whether Ref(2)p accumulation occurs in neurons or glial cells, we double-stained Atg1016h mutant adult brains with anti-Ref(2)p and antibodies against Elav or Repo, which mark neurons or glia, respectively. Colocalization of Ref(2)p-positive cells with Elav and Repo in Atg1016h mutant brains showed Ref(2)p accumulation in both neurons and glial cells (Fig. 3, K–L′). Taken together, these data indicate that Atg101 function is important for the elimination of protein aggregates and neuron homeostasis in the adult brain.
Figure 3.

Accumulation of ubiquitinated proteins and Ref(2) aggregates in Atg101 mutant brains. A, Western blotting reveals that the level of ubiquitinated proteins is increased in Atg1016h mutant fly heads. 7-day-old WT and Atg1016h mutant flies were used. Tubulin was used as a loading control. B, increased Ref(2) protein levels in Atg1016h mutant fly heads. 7-day-old WT and Atg1016h mutant flies were used. GAPDH was used as a loading control. C–E′, aggregates of ubiquitinated proteins accumulate in Atg1016h mutant brains. Shown are confocal images of Drosophila brains of 7-day-old WT and Atg1016h mutant and rescue flies stained with anti-ubiquitin antibody. Scale bars: 50 μm. F, quantification of the number of ubiquitin-positive spots in WT, Atg1016h mutant, and rescue fly brains. n = 12, 11, and 15, respectively. *, p < 0.05. G–I′, aggregates of Ref(2)p proteins accumulated in Atg1016h mutant brains. Shown are confocal images of Drosophila brains of 7-day-old WT, Atg1016h mutant, and rescue flies stained with anti-Ref(2)p antibody. Scale bars: 50 μm. J, quantification of the total areas of Ref(2)p-positive spots in WT, Atg1016h mutant, and rescue fly brains. n = 6. *, p < 0.05. K–K‴, colocalization of Ref(2)p-positive cells with Elav, a marker of neuronal nuclei. Phalloidin was used to label F-actin. Scale bars: 50 μm. L–L‴, colocalization of Ref(2)p-positive cells with Repo, a marker of glial nuclei. Phalloidin was used to label F-actin. Scale bars: 50 μm.
Atg101 maintains midgut homeostasis
Defects in the maintenance of intestinal stem cell homeostasis could result in a short lifespan in adult flies (36–39). During our study, we noticed that the Atg1016h mutant abdomen was enlarged as compared with the WT animals, which indicates a possible defect in the midgut tissue (Fig. S4, A and B). This defect was also rescued by overexpressing Atg101 (Fig. S4C). We therefore extended our phenotypical analysis to the adult midgut tissue in Atg1016h mutants. Interestingly, the midgut in Atg1016h mutants was significantly shorter and thicker than that in WT controls (Fig. 4A, quantified in B). This defect was suppressed in the rescued flies, confirming that the midgut phenotype was specifically caused by the loss of Atg101 (Fig. 4A, quantified in B). To begin understanding the basis for these defects, we then focused on the posterior midgut and stained the tissue with phalloidin and DAPI to examine the posterior midgut morphology. Our phalloidin staining revealed that the regular organization of the visceral muscles was disrupted in Atg1016h mutant midguts, which suggests that the peristalsis was less efficient in the mutants (Fig. 4, C–D″). Cells with different sized nuclei are present in the posterior midgut. The number of polyploid enterocytes with large nuclei was reduced, whereas the number of diploid cells with small nuclei increased (Fig. 4, C–D″, quantified in E). We also noticed that the nuclear size of enterocyte cells was larger in the mutant midguts compared with the control (Fig. 4, C′ and D′, quantified in F). Consistently, the enterocyte cell size was enlarged in Atg1016h mutant midguts (Fig. 4, G and H). In addition, the midgut epithelium was thickened and the lumen size was increased in the Atg101 mutants (Fig. 4, I and J). Overexpression of Atg101 largely rescued all of these posterior midgut defects (Fig. 4, C–M). It is likely that food digestion and nutrient absorption were less efficient in the mutant midgut because of the irregular organization of the visceral muscles and reduced number of polyploid enterocytes. Together, these data demonstrate that Atg101 is required for the maintenance of adult midgut homeostasis.
Figure 4.

Loss of Atg101 results in adult midgut defects. A, Atg1016h mutant adult midguts are shorter and thicker than the WT control. Shown are images of midguts from adult flies with the indicated genotypes. B, quantification of the length of entire midguts and the width of posterior midguts from adult flies with the indicated genotypes. Black lines indicate the location for measuring the width of posterior midguts. 10 midguts were used for each genotype. Data are presented as mean ± S.E. An unpaired t test was used for statistical analysis. **, p < 0.01. C–E″, Z-projection confocal images of WT, Atg1016h mutant, and rescue fly posterior midguts stained for DNA (using DAPI) and actin (using phalloidin) as indicated. Top right boxed areas in C′, D′, and E′ are higher magnifications of the smaller boxed areas. Stars indicate the nuclei of EC cells. F, quantification of the number of cells with large nuclei. Cells in 18 defined regions from nine WT midguts, 18 defined regions from ten Atg1016h mutant midguts, and ten defined regions from five rescue fly midguts were counted. Data are presented as mean ± S.E. An unpaired t test was used for statistical analysis. **, p < 0.01. G, quantification of the size of the large nuclei. Sizes of 60 nuclei from six WT midguts, 90 nuclei from nine Atg1016h mutant midguts, and 90 nuclei from nine rescue fly midguts were measured. Data are presented as mean ± S.E. An unpaired t test was used for statistical analysis. **, p < 0.01. H–J, enlarged enterocytes in Atg1016h mutant midguts. Single-plane confocal images of WT, Atg1016h mutant, and rescue fly posterior midguts stained for anti-Dlg, which labels the cell membrane. Scale bars: 10 μm. K–M, cross-section of midgut epithelium from WT, Atg1016h mutant, and rescue adult flies. The white double-headed arrows indicate the intestinal lumen, and the white lines indicate the intestinal wall. Scale bars: 50 μm.
Atg101 functions to promote intestinal stem cell differentiation
To examine the posterior midgut defect in more detail, we next performed immunofluorescence staining with antibodies against various markers for different types of posterior midgut cells. Escargot (esg)-GFP is specifically expressed in intestinal stem cells (ISC) and enteroblasts (EB), referred to collectively as midgut precursor cells (40). In Atg1016h mutant midguts, the number of esg-GFP–expressing cells was increased and cells often clustered (Fig. 5, A–B′, quantified in C and D). The increasing number of esg-GFP–positive cells could be a result of overproliferation of intestinal stem cell, a blockage of stem cell differentiation, or both. To further discriminate among these possibilities, we used anti-phospho-Ser10-Histone H3 (PH3) antibodies to label cells in mitosis. In Atg1016h mutant midguts, the number of cells labeling for PH3 was comparable with the controls, indicating that the expansion of esg-GFP–positive cells was not likely to be attributable to the increase in ISC proliferation (Fig. 5, E–F′, quantified in G). We then considered the possibility that cell differentiation was blocked in Atg1016h mutants. To test this possibility, we used anti-Prospero and anti-Pdm1 antibodies to stain for enteroendocrine (EE) and enterocyte (EC) cells, respectively, and found a significant reduction in the number of cells for EC cell types in Atg101 mutant midguts but not for the EE cell types (Fig. 5, H–I′ and K–L′, quantified in J and M) (40–42). Altogether, these analyses reveals that Atg101 is required for intestinal stem cell differentiation, especially for the differentiation of EC cell lineage in adult midguts.
Figure 5.
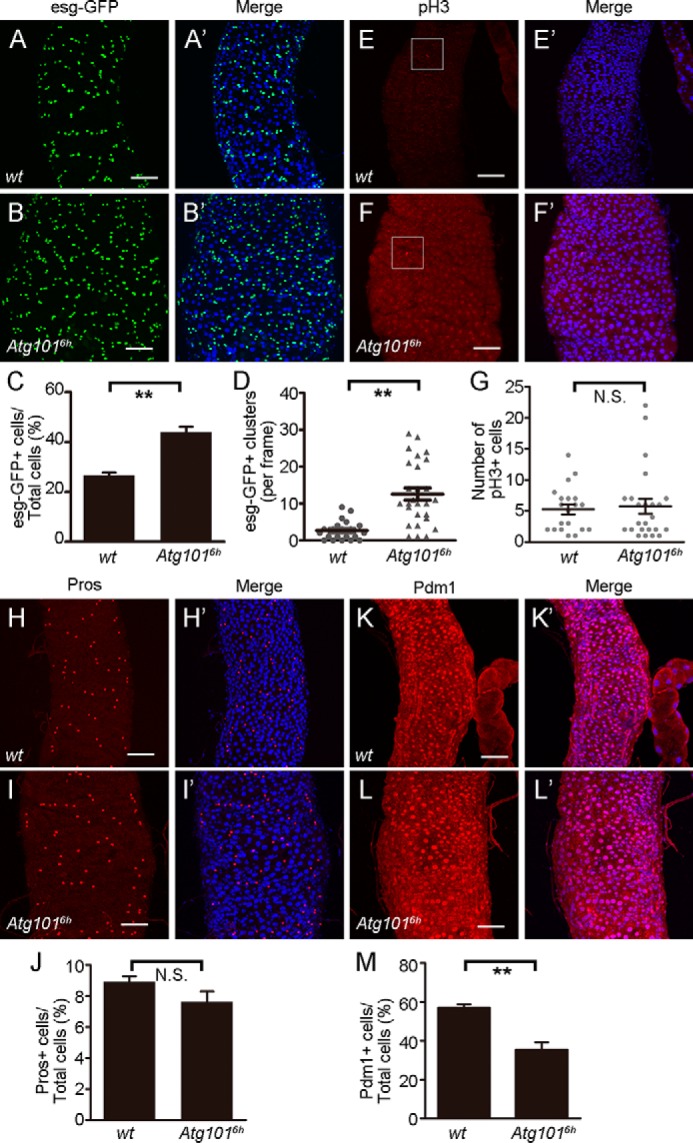
Atg101 is required for the differentiation of the intestinal stem cell. A–B′, Z-projection confocal images of the posterior midgut of WT and Atg1016h mutant flies expressing esg-GFP, which labels both ISC and EB cells. C, quantification of esg-GFP–positive cells. For quantification, Z-projection confocal images of the posterior midgut were acquired, and then the number of esg-GFP–positive cells and all other cells were counted in a defined region. For each defined region, at least 100 cells were present. Cells in eight defined regions from seven WT midguts and 15 defined regions from nine Atg1016h mutant midguts were counted. Data are presented as mean ± S.E. An unpaired t test was used for statistical analysis. **, p < 0.01. D, quantification of esg-GFP–positive cell clusters. Quantification was performed as described in C. Cell clusters (at least three cells) in 21 defined regions from WT midguts and 27 defined regions from Atg1016h mutant midguts were counted. Data are presented as mean ± S.E. An unpaired t test was used for statistical analysis. **, p < 0.01. E–F′, Z-projection confocal images of the posterior midgut of WT and Atg1016h mutant flies stained for anti-PH3, which labels mitotic ISCs. G, quantification of PH3-positive cells within the entire gut. 20 WT and 24 Atg1016h mutant midguts were used for the quantification. Data are presented as mean ± S.E. An unpaired t test was used for statistical analysis. N.S., not significant. DAPI was used to visualize DNA. Scale bars: 50 μm. H–I′, Z-projection confocal images of WT and Atg1016h mutant posterior midguts stained for anti-Prospero (Pros), which labels EB cells. J, quantification of Pros-positive cells. Quantification was performed as described in C. Cells in 27 defined regions from 25 WT midguts and 23 defined regions from 14 Atg1016h mutant midguts were counted. Data are presented as mean ± S.E. An unpaired t test was used for statistical analysis. K–L′, Z-projection confocal images of WT and Atg1016h mutant posterior midguts stained for anti-Pdm1, which labels EC cells. M, quantification of Pdm1-positive cells. Quantification was performed as described in C. Cells in ten defined regions from ten WT midguts and nine defined regions from nine Atg1016h mutant midguts were counted. Data are presented as mean ± S.E. An unpaired t test was used for statistical analysis. **, p < 0.01.
Atg101 acts in ECs to limit cell growth
Having shown that the loss of Atg101 causes defects in adult midgut homeostasis, including enlarged EC cell size and reduced intestinal stem cell differentiation, we next sought to identify the cell types in which Atg101 might function. For this purpose, we performed a rescue experiment with the UAS-Atg101 transgene using several cell type–specific Gal4 drivers in adult midguts. First, esg-Gal4 combined with a temperature-sensitive GAL80 was used to overexpress Atg101 specifically in the adult ISC and EB populations of Atg1016h mutant flies. To activate transgene expression, adult flies were shifted to the nonpermissive temperature. The results showed that there was no rescue effect on the enterocyte size when expressing UAS-Atg101 by esg-Gal4 as compared with Atg1016h mutant alone (Fig. 6, A–C). We then used the EC-specific Myo1A-Gal4 combined with Gal80ts to restrict the expression of UAS-Atg101 to ECs in the adult midgut. Interestingly, Myo1A-Gal4–driven Atg101 expression in Atg1016h mutant flies displayed a rescue of the enlarged enterocyte size normally seen in Atg1016h mutants (Fig. 6, D–F). Furthermore, no obvious rescue was observed when an EE-specific pros-Gal4 was used to drive Atg101 expression in Atg1016h mutant midguts (Fig. 6, G–I). Thus, we concluded that Atg101 has a cell autonomous effect in ECs to limit cell growth.
Figure 6.
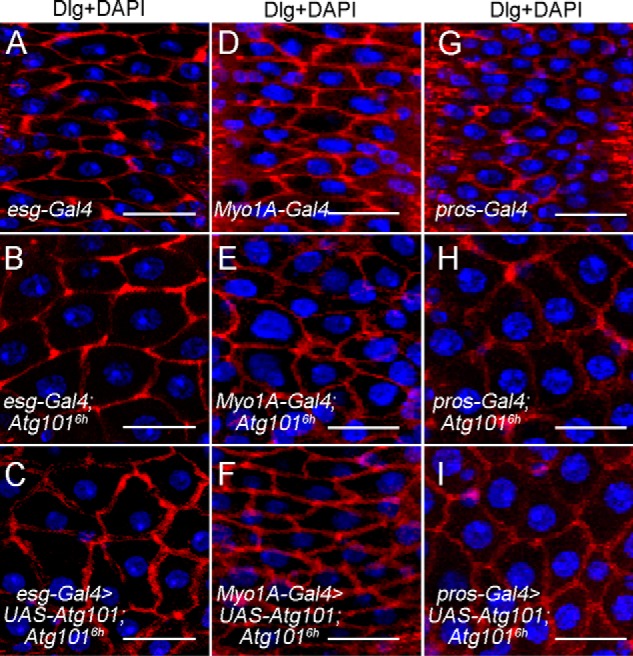
Atg101 functions in ECs to regulate cell growth. A–C, esg-Gal4–driven Atg101 expression in both the ISC and EB populations failed to rescue the enlarged enterocyte size defects in Atg101 mutants. Shown are confocal images of the esg-Gal4 control, Atg1016h mutant, and rescued fly posterior midguts stained for DNA and Dlg as indicated. For conditional induction of Atg101, esg-Gal4 UAS-GFP; Gal80ts was used in the rescue experiment. D–F, Myo1A-Gal4–driven Atg101 expression in EC cells was able to rescue the enlarged enterocyte size defects in Atg101 mutants. Shown are confocal images of the Myo1A-Gal4 control, Atg1016h mutant and rescued fly posterior midguts stained for DNA and Dlg as indicated. For conditional induction of Atg101, Myo1A-Gal4 UAS-GFP; Gal80ts was used in the rescue experiment. G–I, pros-Gal4–driven Atg101 expression in EE cells failed to rescue the enlarged enterocyte size defects in Atg101 mutants. Shown are confocal images of the pros-Gal4 control, Atg1016h mutant, and rescued fly posterior midguts stained for DNA and Dlg as indicated. pros-Gal4 was used in the rescue experiment. Scale bars: 20 μm.
Discussion
Here we generated and characterized an Atg101 loss-of-function mutant in Drosophila. The Atg101 mutant showed reduced viability in the embryonic, larval, and pupal stages. Most mutant animals can survive to adult stages but have a short lifespan. Our study also provides genetic evidence that Atg101 has a key role in maintaining neuron and midgut homeostasis.
Atg101 is a core subunit of the Atg1 complex, which is essential for autophagosome formation (13, 23). Studies in mammalian cells have identified the role of Atg101 in autophagy initiation (14, 15). Knockdown of Atg101 by RNAi in Drosophila also leads to autophagy defects (17). Consistent with this, the lack of Atg101 function causes defects in both starvation-induced and developmental autophagy in Drosophila third instar larval fat body tissues. In addition, we observed a variety of phenotypes in adult flies, such as reduced lifespan, impaired locomotion, accumulation of ubiquitinated proteins, and blockage of intestinal stem cell differentiation.
Autophagy has been implicated in the process of aging (43). Suppression of autophagy disrupts age-dependent tissue homeostasis in various organs (43). In flies, the loss of Atg7, Atg8a, or Atg17 in the entire organism leads to a reduced lifespan as well as the accumulation of ubiquitinated protein aggregates in the brains (29, 31–33). It has been proposed that the basal level of autophagy in the nervous system is required for the clearance of toxic proteins or damaged organelles (43, 44). Similar to other Atg mutants, Atg101 mutant flies also have a shorter lifespan. Protein aggregates were evident in Atg101 mutant fly brains. The decline of locomotion ability in Atg101 mutants during aging further demonstrates a neurondegeneration defect in the absence of autophagy function.
Recently, several reports indicate that autophagy-related genes also regulate intestine homeostasis. A core autophagy gene, Atg16L1, has been shown to be associated with Crohn disease (45). Later studies performed in mice show that Paneth cells are abnormal in Atg16L1 or Atg5 knockout mutant animals (46). In flies, Atg9 has been shown to be required for Jun N-terminal kinase (JNK)-mediated intestinal stem cell proliferation (47). Interestingly, the induction of autophagy can block stress-induced ISC proliferation (47). In addition, Atg9 is also required for midgut homeostasis under normal physiological condition, and it specifically acts in enterocytes to control cell growth by limiting TOR signaling (48). Analysis of various Atg genes in the Drosophila midgut has demonstrated that the Atg1 complex components, including Atg1, Atg13, and Atg17, play crucial roles in controlling enterocytes cell growth (48). Furthermore, it appears that the Atg1 complex and Atg9 regulate TOR signaling via different mechanisms during enterocyte cell growth (48). Interestingly, other core autophagy-related genes, such as Atg7, Atg12, Atg16, Atg18, and Vps32, are not required for enterocyte cell growth in Drosophila adult midguts, indicating that the role of the Atg1 complex and Atg9 in maintaining midgut homeostasis might be specific (48). Consistent with the reported phenotype, upon knocking down Atg1, Atg13, and Atg17, we found that loss of Atg101 caused defects in adult midgut homeostasis and resulted in abnormal midgut morphology with enlarged enterocytes. Interestingly, the number of intestinal progenitor cells was increased in Atg101 mutant midguts. However, the number of dividing intestinal stem cells when stained with anti-PH3 remained the same as in the control. In contrast, we observed reduced numbers of differentiating enterocyte cells. These data strongly indicate that Atg101 plays an important role in intestinal stem cell differentiation but not proliferation. In addition, our cell type–specific rescue experiments revealed that Atg101 functions in ECs to limit cell growth autonomously. It has been reported that apoptotic enterocytes promote intestinal stem cell division nonautonomously (49). It remains unclear whether the overgrowth of ECs has an effect on intestinal stem cell function. In summary, these findings indicate that Atg101 plays essential roles in maintaining neuron and midgut homeostasis, both of which may affect the aging process. A recent study also reveals that the activation of autophagy in the adult brain by expression of either AMPK or Atg1 induces autophagy in the intestine and leads to an increased lifespan (50). Further studies on the connection between the brain and midgut homeostasis in Drosophila Atg101 mutant animals will likely provide novel insights into the cross-talk between the brain and the midgut.
Experimental procedures
Drosophila stocks
We used the following fly stocks: w1118, Daughterless-Gal4, esg-GFPP01986/Cyo, Cg-Gal4 UAS-GFP-RFP-Atg8a/Cyo, Esg-Gal4 UAS-GFP/Cyo; Gal80ts/TM6B, Myo1A-Gal4; Gal80ts, pros-Gal4. For experiments using Gal80ts, crosses were set up and cultured at 18 °C to limit Gal4 activities. To inactivate Gal80ts, 2–4-day-old F1 adult flies were shifted to 29 °C for 7 days before dissection.
Generation of Atg101 mutant and transgenic fly lines
CRISPR-mediated mutagenesis was performed according to a previous report (51). Briefly, the Atg101 target sequence and PAM site were determined using http://tools.flycrispr.molbio.wisc.edu/targetFinder/ (52).3 After identifying the target region, two primers (TAATACGACTCACTATAGGAGGTGTGGACGGTGCACCGTTTTAGAGCTAGAAATAGC and AAAAAAAGCACCGACTCGGTGCCAC) were used to amplify the DNA fragment from the pMD19-T gRNA scaffold vector. The PCR products were used for gRNA in vitro transcription with the RiboMAX Large Scale RNA Production Systems T7 kit. To synthesize Cas9 mRNA, the pSP6–2sNLS-spCas9 plasmid was first cut by XbaI and then purified. Transcription was performed using the Sp6 mMESSAGE mMACHINE kit (Ambion). The poly(A) tails were added to the 3′-end of Cas9 mRNAs using Escherichia coli poly(A) polymerase kit (New England Biolabs). Cas9 mRNAs and Atg101 gRNA were then mixed and injected into w1118 fly embryos. Genomic DNA from dead embryos were used for PCR amplification and sequencing to determine the efficiency and usefulness of gRNA. The primers used for amplifying the target region were 5′-TTTCACACCGTCCTCTTCCAC-3′ and 5′-ATGATGGGAGGATTTGCGTTC-3′. The detection of a string of “double peaks” in the sequencing chromatogram indicates the mismatched region and the usefulness of gRNA. Single flies were selected and balanced over FM6B. Exact deletions were determined by PCR screening and sequencing for individual flies. Atg1016h mutants have a 13-bp deletion (from ChX 18792261 to ChX 18792273).
For the generation of UAS-Atg101-HA transgenic flies, an Atg101 cDNA fragment was amplified using the primers 5′-CGGCGGCCGCATGAACGCGCGTTCGCAGGT-3′ and 5′-CGCTCGAGCATTGCGAGCGTTTCCTTGA-3′ and cloned into a modified pUAST vector with a 3HA tag at the C-terminal. The pUAST-Atg101-HA construct was then injected into the ZF-25C landing site on chromosome II using standard methods.
Quantitative PCR and RT-PCR
Total RNA was extracted using the TRIzol reagent (Ambion, 15596-026). cDNA was synthesized with PrimeScript RTase (TaKaRa, PrimeScriptTM II 1st Strand cDNA synthesis kit, code 621A). Quantitative PCR was performed on the ABI 7900HT Fast Real-Time PCR system using the following primers: S6KL, 5′-GTCAGATGCTGACGCAG-3′ and 5′-GCCATCACACTGCGGATAC-3′; and rp49, 5′-GCTAAGCTGTCGCACAAA-3′ and 5′-TCCGGTGGGCAGCATGTG-3′.
RT-PCR was performed using the following primers: Rp49, 5′-GCTAAGCTGTCGCACAAA-3′ and 5′-TCCGGTGGGCAGCATGTG-3′; Atg101, 5′-GAGGTGTGGACGGTGCACC-3′ and 5′-GATGTGTCGAAGATCAG-3′; Atg1, 5′-GGATTTTGGGTTTGCGCGAT-3′ and 5′-CAGAGATCCGCCTTGGAGTC-3′; Atg3, 5′-CGCCCGTTTTGAAGGAATCG-3′ and 5′-TGTCTTGGTCTCATCGCCAG-3′; Atg4a, 5′-TAGTGCGCTTCGATGACTGG-3′ and 5′-TATGAGCAGCAGTGGCTTCC-3′; Atg7, 5′-GATGTTACGGCCCCTGGAAA-3′ and 5′-GCCAGCTCCTTACGAGGATG-3′; and Atg8a, 5′-TCATTCGCAAGCGCATCCA-3′ and 5′-AGTCCTCCTCGTGATGTTCC-3′.
Western blotting
Adult fly heads were collected and lysed in modified radioimmune precipitation assay lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% (v/v) IGEPAL CA-630, and 0.5% (w/v) sodium deoxycholate) with protease inhibitor mixture (Roche, catalog No. 04693132001) and PhosStop phosphatase inhibitor mixture (Roche, catalog No. 4906845001). Samples were then subjected to SDS-PAGE and transferred to polyvinylidene fluoride membrane. Membranes were immunoblotted with mouse anti-ubiquitin (1:1000, P4D1, Cell Signaling Technology catalog No. 3936S), rabbit anti-Ref(2)p (1:500, Abcam, ab178440), mouse anti-GAPDH (1:1000, Goodhere Biotechnology Co., AB-M-M001), and mouse anti-tubulin (1:1000, Beyotime Biotech, AT819-1). Detection of proteins was performed using the ChemiLucentTM ECL detection reagents (Millipore, WBKLS0500). Images were taken using the chemiluminescence imaging system (Clinx Science Instruments, Shanghai).
Immunostaining and microscopy
Drosophila adult midguts were dissected in SD medium and fixed with 4% paraformaldehyde in PBS for 40 min with rocking at room temperature. Midguts were washed three times in PBST (0.1% Triton X-100 in PBS) before blocking for 1 h in PBST plus 3% BSA buffer at room temperature. Next, midguts were incubated with the primary antibodies overnight at 4 °C. After four washes in PBST, the midguts were incubated with secondary antibodies for 2 h at room temperature with rocking. DAPI was added for the last 20 min. After four further washes with PBST, the midguts were mounted in Vectashield mounting medium. 7-day-old adult males were used in all analyses. The posterior region of the midgut was chosen for imaging. Drosophila adult brains and larval NMJs were dissected in SD medium. Samples were then fixed with 4% paraformaldehyde in PBS for 20 min and stained as described above. The following primary antibodies were used: chicken anti-GFP (1:2000; ab13970, Abcam), mouse anti-CSP (1:50; 6D6, DSHB), mouse anti-ubiquitin (1:400; P4D1, Cell Signaling Technology, catalog No. 3936S), rabbit anti-Ref(2)p (1:500; ab178440, Abcam), rat anti-Elav (1:50; 7E8A10, DSHB), mouse anti-Repo (1:50; 8D12, DSHB), rabbit anti-PH3 (1:500, Millipore), mouse anti-Prospero (1:100), rabbit anti-Pdm1 (1:500), and mouse anti-Dlg (1:500; DHSB, 4F3). Fluorescent secondary antibodies (Alexa Fluor 488– or Alexa Fluor 555–conjugated anti-rabbit, anti-mouse, and anti-chicken) were obtained from Molecular Probes (1:500). Phalloidin (phalloidin 568, Invitrogen A12380, 1033926) was used in a 1:1000 dilution. DNA was labeled with DAPI (1 μg/ml, Sigma). To induce starvation, middle L3 stage larvae were collected and transferred to a 20% sucrose solution for 4 h. For live imaging, larval fat body tissues were dissected in SD medium. Imaging was performed on an Olympus FV1000 confocal microscope, and images were processed using ImageJ and Adobe Photoshop.
Quantification and statistical analysis
In Fig. 1, D, E, H, and I, the number of RFP-Atg8a spots was counted manually, and the size of the RFP-Atg8a spots was measured with NIS-Elements. In Fig. 3, F and J, the number of ubiquitin-positive spots and the area of the Ref(2)p-positive spots were measured with ImageJ. In Fig. 4B, midgut length and width were measured with NIS-Elements from images of whole midguts acquired with a Nikon Eclipse 80i microscope. In Fig. 4, F and G, the number of nuclei was counted manually, and the size of the nuclei was measured with NIS-Elements. Posterior midgut regions R4a and R4b were chosen. In Fig. 5, C, J, and M, the number of esg-positive, pros-positive, and pdm1-positive cells were counted manually. Posterior midgut regions R4a and R4b were chosen. In Fig. 5D, the number of esg-positive cell clusters was counted manually for the entire frame. In Fig. 5G, the number of PH3-positive cells was counted manually for each midgut. In Fig. S2B, the gastric caeca size was measured with NIS-Elements. The sample size for the quantification analysis is indicated in the legends for Figs. 1, 3, 4, 5, S1 and S2. Statistical analysis was performed using GraphPad Prism 5.
Hatching rate, pupation rate, and eclosion rate
3–4-day-old flies were collected and put in cages. Embryos were collected every 2 h. To measure the hatching rate, 200 embryos were transferred to a fresh plate, and the number of hatched first instar larvae were counted. To monitor the rate and timing of pupation and eclosion, 50 first instar larvae were collected and cultured in a vial. The number of pupae and adults were counted every 12 h. The pupation rate was calculated as the percentage of the number of pupae versus the number of first instar larvae contained in each vial. The eclosion rate was calculated as the percentage of the number of eclosed adults versus the total number of pupae contained in each vial.
Climbing assay
For negative geotaxis assay, aged flies were separated by gender and grouped in cohorts of 20 animals. Before testing, the flies were transferred to a tube made by two vertically joined empty vials and allowed to rest for 1 h before the assay. After tapping the flies down to the bottom of the vial, we measured the number of flies that could climb above the 15-cm mark within 15 s. A climbing index was calculated as the percentage of the number of flies above the mark versus the total number of flies in the tube. Six replicate sets of experiments were performed for each genotype.
Lifespan assay
For the lifespan analysis, groups of 20 newly eclosed males or females were collected and transferred to vials with fresh food every 2–3 days. The number of dead flies was counted. Survival rates were calculated as the percentage of the number of surviving flies versus the total number of flies. Three replicate sets of experiments were performed for each genotype.
Author contributions
T. G. and W. G. conceptualization; T. G., C. M., X. J., W. Y., Z. W., Y. T., H. B., and P. G. investigation; T. G. and W. G. writing-original draft; T. G., X. Y., and W. G. writing-review and editing; Z. N., H. B., J. L., H. Z., Q. D., P. G., and Y. X. methodology; Y. X. and X. Y. resources; X. Y. and W. G. supervision; W. G. funding acquisition.
Supplementary Material
Acknowledgments
We thank Guanjun Gao for help with the generation of Atg101 mutants using CRISPR-Cas9 method. We are grateful to Xinhua Lin, Yongqing Zhang, Chao Tong, Jie Shen, Jian-Quan Ni, Zhouhua Li, the Developmental Studies Hybridoma Bank, the Bloomington Drosophila stock center and the Tsinghua Fly Center for fly stocks and antibodies.
This work was supported by Grant 2018YFC1003200 from the National Key Research and Development Program of China and Grant 31371319 from the National Natural Science Foundation of China. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4 and Table S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- Atg
- autophagy-related gene
- NMJ
- neuromuscular junction
- ULK1/2
- UNC-51-like autophagy–activating kinase 1/2
- Fip200
- focal adhesion kinase family–interacting protein, 200 kDa
- CRISPR
- clustered regularly interspaced short palindromic repeats
- PAM
- protospacer adjacent motif
- RFP
- red fluorescent protein
- Ref(2)p
- refractory to sigma P
- DAPI
- 4′,6-diamidino-2-phenylindole
- esg
- escargot
- ISC
- intestinal stem cell
- EB
- enteroblast
- EC
- enterocyte
- EE
- enteroendocrine cell
- PH3
- phospho-Ser10-Histone H3
- TOR
- target of rapamycin
- DSHB
- Developmental Studies Hybridoma Bank.
References
- 1. Chen Y., and Klionsky D. J. (2011) The regulation of autophagy: Unanswered questions. J. Cell Sci. 124, 161–170 10.1242/jcs.064576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Feng Y., He D., Yao Z., and Klionsky D. J. (2014) The machinery of macroautophagy. Cell Res. 24, 24–41 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mizushima N. (2007) Autophagy: process and function. Genes Dev. 21, 2861–2873 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- 4. Noda N. N., and Inagaki F. (2015) Mechanisms of autophagy. Annu. Rev. Biophys. 44, 101–122 10.1146/annurev-biophys-060414-034248 [DOI] [PubMed] [Google Scholar]
- 5. Levine B., and Kroemer G. (2009) Autophagy in aging, disease and death: The true identity of a cell death impostor. Cell Death Differ. 16, 1–2 10.1038/cdd.2008.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hale A. N., Ledbetter D. J., Gawriluk T. R., and Rucker E. B. 3rd. (2013). Autophagy: Regulation and role in development. Autophagy 9, 951–972 10.4161/auto.24273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Galluzzi L., Pietrocola F., Levine B., and Kroemer G. (2014) Metabolic control of autophagy. Cell 159, 1263–1276 10.1016/j.cell.2014.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mizushima N., and Komatsu M. (2011) Autophagy: Renovation of cells and tissues. Cell 147, 728–741 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 9. Jiang P., and Mizushima N. (2014) Autophagy and human diseases. Cell Res. 24, 69–79 10.1038/cr.2013.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levine B., and Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mizushima N., Yoshimori T., and Ohsumi Y. (2011) The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- 12. Stanley R. E., Ragusa M. J., and Hurley J. H. (2014) The beginning of the end: How scaffolds nucleate autophagosome biogenesis. Trends Cell Biol. 24, 73–81 10.1016/j.tcb.2013.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mizushima N. (2010) The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 22, 132–139 10.1016/j.ceb.2009.12.004 [DOI] [PubMed] [Google Scholar]
- 14. Hosokawa N., Sasaki T., Iemura S., Natsume T., Hara T., and Mizushima N. (2009) Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5, 973–979 10.4161/auto.5.7.9296 [DOI] [PubMed] [Google Scholar]
- 15. Mercer C. A., Kaliappan A., and Dennis P. B. (2009) A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 5, 649–662 [DOI] [PubMed] [Google Scholar]
- 16. Liang Q., Yang P., Tian E., Han J., and Zhang H. (2012) The C. elegans ATG101 homolog EPG-9 directly interacts with EPG-1/Atg13 and is essential for autophagy. Autophagy 8, 1426–1433 10.4161/auto.21163 [DOI] [PubMed] [Google Scholar]
- 17. Hegeds K., Nagy P., Gáspári Z., and Juhász G. (2014) The putative HORMA domain protein Atg101 dimerizes and is required for starvation-induced and selective autophagy in Drosophila. Biomed. Res. Int. 2014, 470482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Michel M., Schwarten M., Decker C., Nagel-Steger L., Willbold D., and Weiergräber O. H. (2015) The mammalian autophagy initiator complex contains 2 HORMA domain proteins. Autophagy 11, 2300–2308 10.1080/15548627.2015.1076605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qi S., Kim D. J., Stjepanovic G., and Hurley J. H. (2015) Structure of the human Atg13-Atg101 HORMA heterodimer: An interaction hub within the ULK1 complex. Structure 23, 1848–1857 10.1016/j.str.2015.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki H., Kaizuka T., Mizushima N., and Noda N. N. (2015) Structure of the Atg101-Atg13 complex reveals essential roles of Atg101 in autophagy initiation. Nat. Struct. Mol. Biol. 22, 572–580 10.1038/nsmb.3036 [DOI] [PubMed] [Google Scholar]
- 21. Popelka H., and Klionsky D. J. (2015) One step closer to understanding mammalian macroautophagy initiation: Interplay of 2 HORMA architectures in the ULK1 complex. Autophagy 11, 1953–1955 10.1080/15548627.2015.1087635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzuki H., Kaizuka T., Mizushima N., and Noda N. N. (2015) Open and closed HORMAs regulate autophagy initiation. Autophagy 11, 2123–2124 10.1080/15548627.2015.1091144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noda N. N., and Mizushima N. (2016) Atg101: Not just an accessory subunit in the autophagy-initiation complex. Cell Struct. Funct. 41, 13–20 [DOI] [PubMed] [Google Scholar]
- 24. McPhee C. K., and Baehrecke E. H. (2009) Autophagy in Drosophila melanogaster. Biochim. Biophys. Acta 1793, 1452–1460 10.1016/j.bbamcr.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang Y. Y., and Neufeld T. P. (2010) Autophagy takes flight in Drosophila. FEBS Lett. 584, 1342–1349 10.1016/j.febslet.2010.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mulakkal N. C., Nagy P., Takats S., Tusco R., Juhász G., and Nezis I. P. (2014) Autophagy in Drosophila: From historical studies to current knowledge. Biomed. Res. Int. 2014, 273473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kraut R., Menon K., and Zinn K. (2001) A gain-of-function screen for genes controlling motor axon guidance and synaptogenesis in Drosophila. Curr. Biol. 11, 417–430 10.1016/S0960-9822(01)00124-5 [DOI] [PubMed] [Google Scholar]
- 28. Zhao G., Wu Y., Du L., Li W., Xiong Y., Yao A., Wang Q., and Zhang Y. Q. (2015) Drosophila S6 kinase like inhibits neuromuscular junction growth by downregulating the BMP receptor thickveins. PLoS Genet. 11, e1004984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nagy P., Kárpáti M., Varga A., Pircs K., Venkei Z., Takáts S., Varga K., Erdi B., Hegeds K., and Juhász G. (2014) Atg17/FIP200 localizes to perilysosomal Ref(2)P aggregates and promotes autophagy by activation of Atg1 in Drosophila. Autophagy 10, 453–467 10.4161/auto.27442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mauvezin C., Ayala C., Braden C. R., Kim J., and Neufeld T. P. (2014) Assays to monitor autophagy in Drosophila. Methods 68, 134–139 10.1016/j.ymeth.2014.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Juhász G., Erdi B., Sass M., and Neufeld T. P. (2007) Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 21, 3061–3066 10.1101/gad.1600707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simonsen A., Cumming R. C., Brech A., Isakson P., Schubert D. R., and Finley K. D. (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184 10.4161/auto.5269 [DOI] [PubMed] [Google Scholar]
- 33. Kim M., Park H. L., Park H. W., Ro S. H., Nam S. G., Reed J. M., Guan J. L., and Lee J. H. (2013) Drosophila Fip200 is an essential regulator of autophagy that attenuates both growth and aging. Autophagy 9, 1201–1213 10.4161/auto.24811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., and Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 10.1083/jcb.200412022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., and Mizushima N. (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- 36. Biteau B., Karpac J., Supoyo S., Degennaro M., Lehmann R., and Jasper H. (2010) Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 6, e1001159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen H., Zheng X., and Zheng Y. (2014) Age-associated loss of lamin-B leads to systemic inflammation and gut hyperplasia. Cell 159, 829–843 10.1016/j.cell.2014.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Foronda D., Weng R., Verma P., Chen Y. W., and Cohen S. M. (2014) Coordination of insulin and Notch pathway activities by microRNA miR-305 mediates adaptive homeostasis in the intestinal stem cells of the Drosophila gut. Genes Dev. 28, 2421–2431 10.1101/gad.241588.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang L., Ryoo H. D., Qi Y., and Jasper H. (2015) PERK limits Drosophila lifespan by promoting intestinal stem cell proliferation in response to ER stress. PLoS Genet. 11, e1005220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Micchelli C. A., and Perrimon N. (2006) Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature 439, 475–479 10.1038/nature04371 [DOI] [PubMed] [Google Scholar]
- 41. Ohlstein B., and Spradling A. (2006) The adult Drosophila posterior midgut is maintained by pluripotent stem cells. Nature 439, 470–474 10.1038/nature04333 [DOI] [PubMed] [Google Scholar]
- 42. Lee W. C., Beebe K., Sudmeier L., and Micchelli C. A. (2009) Adenomatous polyposis coli regulates Drosophila intestinal stem cell proliferation. Development 136, 2255–2264 10.1242/dev.035196 [DOI] [PubMed] [Google Scholar]
- 43. Rubinsztein D. C., Mariño G., and Kroemer G. (2011) Autophagy and aging. Cell 146, 682–695 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- 44. Menzies F. M., Fleming A., and Rubinsztein D. C. (2015) Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357 10.1038/nrn3961 [DOI] [PubMed] [Google Scholar]
- 45. Barrett J. C., Hansoul S., Nicolae D. L., Cho J. H., Duerr R. H., Rioux J. D., Brant S. R., Silverberg M. S., Taylor K. D., Barmada M. M., Bitton A., Dassopoulos T., Datta L. W., Green T., Griffiths A. M., et al. (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 40, 955–962 10.1038/ng.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cadwell K., Liu J. Y., Brown S. L., Miyoshi H., Loh J., Lennerz J. K., Kishi C., Kc W., Carrero J. A., Hunt S., Stone C. D., Brunt E. M., Xavier R. J., Sleckman B. P., Li E., et al. (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 10.1038/nature07416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tang H. W., Liao H. M., Peng W. H., Lin H. R., Chen C. H., and Chen G. C. (2013) Atg9 interacts with dTRAF2/TRAF6 to regulate oxidative stress-induced JNK activation and autophagy induction. Dev. Cell 27, 489–503 10.1016/j.devcel.2013.10.017 [DOI] [PubMed] [Google Scholar]
- 48. Wen J. K., Wang Y. T., Chan C. C., Hsieh C. W., Liao H. M., Hung C. C., and Chen G. C. (2017) Atg9 antagonizes TOR signaling to regulate intestinal cell growth and epithelial homeostasis in Drosophila. Elife 6, pii: e29338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liang J., Balachandra S., Ngo S., and O'Brien L. E. (2017) Feedback regulation of steady-state epithelial turnover and organ size. Nature 548, 588–591 10.1038/nature23678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ulgherait M., Rana A., Rera M., Graniel J., and Walker D. W. (2014) AMPK modulates tissue and organismal aging in a non-cell-autonomous manner. Cell Rep. 8, 1767–1780 10.1016/j.celrep.2014.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yu Z., Ren M., Wang Z., Zhang B., Rong Y. S., Jiao R., and Gao G. (2013) Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics 195, 289–291 10.1534/genetics.113.153825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gratz S. J., Ukken F. P., Rubinstein C. D., Thiede G., Donohue L. K., Cummings A. M., and O'Connor-Giles K. M. (2014) Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics 196, 961–971 10.1534/genetics.113.160713 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.