

Abstract
We previously reported that iron down-regulates transcription of the leptin gene by increasing occupancy of phosphorylated cAMP response element-binding protein (pCREB) at two sites in the leptin gene promoter. Several nutrient-sensing pathways including O-GlcNAcylation also regulate leptin. We therefore investigated whether O-glycosylation plays a role in iron- and CREB-mediated regulation of leptin. We found that high iron decreases protein O-GlcNAcylation both in cultured 3T3-L1 adipocytes and in mice fed high-iron diets and down-regulates leptin mRNA and protein levels. Glucosamine treatment, which bypasses the rate-limiting step in the synthesis of substrate for glycosylation, increased both O-GlcNAc and leptin, whereas inhibition of O-glycosyltransferase (OGT) decreased O-GlcNAc and leptin. The increased leptin levels induced by glucosamine were susceptible to the inhibition by iron, but in the case of OGT inhibition, iron did not further decrease leptin. Mice with deletion of the O-GlcNAcase gene, either via whole-body heterozygous deletion or through adipocyte-targeted homozygous deletion, exhibited increased O-GlcNAc levels in adipose tissue and increased leptin levels that were inhibited by iron. Of note, iron increased the occupancy of pCREB and decreased the occupancy of O-GlcNAcylated CREB on the leptin promoter. These patterns observed in our experimental models suggest that iron exerts its effects on leptin by decreasing O-glycosylation and not by increasing protein deglycosylation and that neither O-GlcNAcase nor OGT mRNA and protein levels are affected by iron. We conclude that iron down-regulates leptin by decreasing CREB glycosylation, resulting in increased CREB phosphorylation and leptin promoter occupancy by pCREB.
Keywords: O-GlcNAcylation, O-linked N-acetylglucosamine (O-GlcNAc), leptin, iron, adipocyte, diabetes
Introduction
Leptin, the product of the OB gene, is one of the central regulators of energy expenditure, appetite, and fuel homeostasis (1). Appropriate to these functions, it is produced largely in adipocytes and secreted as a function of fat mass. Its transcriptional regulation occurs in part through peroxisome proliferator-activated receptor γ, CCAAT/enhancer-binding protein α, and specificity protein 1 (2–4). Activation of CREB5 during the adipocyte differentiation process suppresses leptin secretion and expression in mesenchymal stem cells (5). CREB is an attractive candidate as a regulator of leptin because of its central role in orchestrating the transition from fasting to feeding.
Another nutrient-dependent mechanism involved in leptin regulation is protein modification by O-GlcNAcylation (6–9). This modification regulates numerous metabolic pathways, is controlled through the activities of a single O-GlcNAc transferase (OGT) and a single enzyme that removes O-GlcNAc (O-GlcNAcase, OGA), and is largely nutrient-driven; synthesis of the rate-limiting substrate of OGT, UDP-GlcNAc, is limited by fluxes of the components used for its synthesis, particularly glucose and glucosamine-6-phosphate (10, 11). The regulation of leptin by O-glycosylation has been demonstrated to be at the level of transcription (12), and many transcription factors can be modified by O-GlcNAc (10, 11, 13).
Iron is a micronutrient that also plays a crucial role in metabolic regulation (14, 15). The cell's need for iron as a cofactor for fuel oxidation and energy production in the face of its potential danger as an oxidant has given rise to a complex system, coordinated across tissues, to tightly regulate its levels, distribution, and bioavailability. In yeast, for example, the switch from fermentative (glycolytic) metabolism to oxidative phosphorylation depends on the presence of iron in the environment. Conversely the potentially dangerous oxidant is not imported into the cell unless there is a need for oxidative phosphorylation. Thus, there has arisen over evolution an important link between iron homeostasis and metabolism, and we have shown that many of those connections are conserved in mammals. We have demonstrated that in humans and mouse models, iron regulates leptin, as well as another adipocyte hormone that plays an important role in fuel utilization, adiponectin (16, 17). The fact that the adipocyte expresses many specialized proteins involved in iron homeostasis led us to hypothesize that the adipocyte not only senses macronutrient status but also the essential micronutrient iron to orchestrate factors involved in lipid metabolism such as integration of metabolic processes.
In our investigation of the regulation of leptin by iron (16), we demonstrated that the down-regulation of leptin in mice on a high-iron diet was physiologically significant in that it was accompanied by increased food intake. Altered food intake was not seen in the leptin-deficient Ob/Ob mouse on a high-iron diet. We determined that iron negatively regulates leptin transcription via CREB activation and identified two potential CREB-binding sites in the mouse leptin promoter region. Mutation of both sites completely blocked the effect of iron on promoter activity. ChIP analysis revealed that binding of phosphorylated CREB is enriched at these two sites in iron-treated 3T3-L1 adipocytes compared with untreated cells. We were unable to demonstrate, however, increased activities of CREB kinases such as protein kinase A (PKA) or calcium/calmodulin-dependent kinase IV (CAMK), so the mechanism for this change in phosphorylation was not determined. Phosphorylation and O-glycosylation are often reciprocal (10, 11), in some cases because of shared sites of modification. The known regulation of leptin by O-GlcNAcylation led us, therefore, to investigate the effects of iron on O-glycosylation as a possible mechanism for its regulation of leptin.
Results
Iron treatment of 3T3-L1 adipocytes results in decreased O-GlcNAcylation of proteins
Treatment of differentiated 3T3-L1 adipocytes with 100 μg/ml ferric ammonium citrate (FAC) for 24 h resulted in decreased protein modification by O-linked GlcNAc as detected by Western blotting using an antibody that recognizes O-GlcNAc on a wide spectrum of proteins (18) (Fig. 1, A and B). As previously reported (16), the same treatment also resulted in significant down-regulation of the mRNA for leptin (Figs. 1C and 2).
Figure 1.
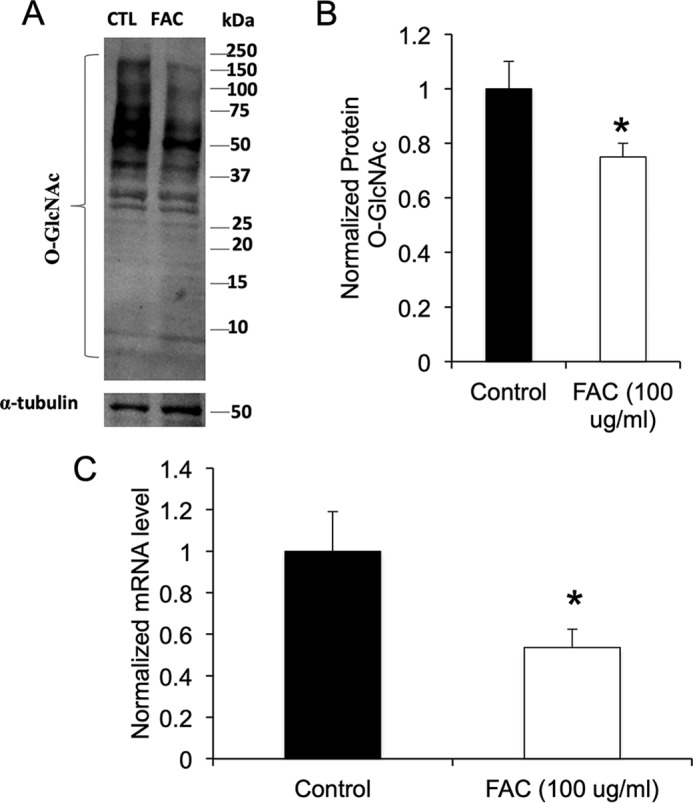
Iron treatment decreases total O-GlcNAcylation in 3T3-L1 adipocytes and down-regulates leptin. A, total protein O-GlcNAcylation as revealed by Western blotting using a pan-O-GlcNAc antibody in control (CTL) cells and cells treated with 100 μg/ml FAC. B, quantification of Western blots normalized to α-tubulin (n = 3 independent determinations). C, leptin (Ob) mRNA in cells treated with or without FAC (n = 6 independent determinations). *, p < 0.05 using by two-tailed Student's t test. The data represent means ± S.E.
Figure 2.
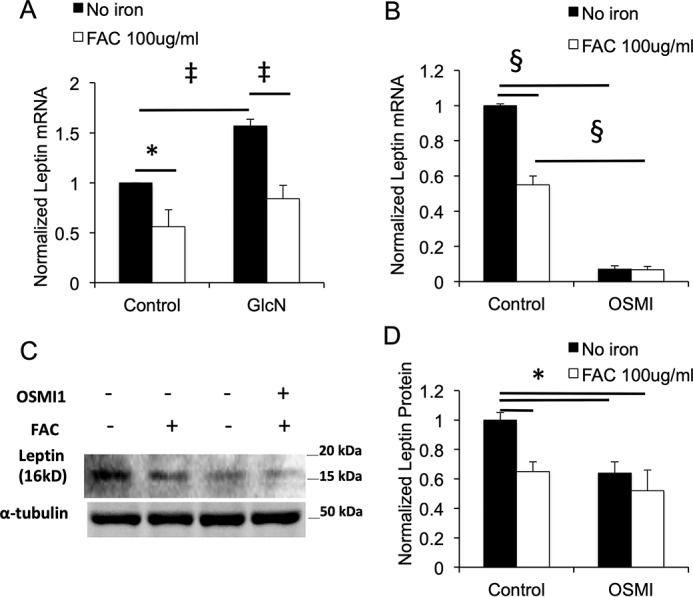
Effects of hexosamine pathway activation and inhibition on leptin in 3T3-L1 adipocytes. A, effects of GlcN (2 mm, 24 h) and/or iron (100 μg/ml FAC) on leptin mRNA levels in 3T3-L1 cells (n = 7 independent determinations, normalized to β-actin mRNA). B, effects of treatment of 3T3-L1 adipocytes with an OGT inhibitor (OSMI-1, 50 μmol/liter, 24 h) and/or iron on leptin mRNA (n = 7 independent determinations, normalized to β-actin mRNA). C and D, effects of treatment of 3T3-L1 adipocytes with OSMI-1 on leptin protein. The cells were harvested after the 24-h treatment and leptin quantified by Western blotting. C, representative Western blotting. D, quantification of blots, n = 4 independent determinations, normalized to α-tubulin. Shown are means ± S.E. *, p < 0.05; ‡, p < 0.01; §, p < 0.001 by ANOVA.
Experimental modulation of O-GlcNAcylation regulates leptin in 3T3-L1 adipocytes in an iron-dependent manner
Glucosamine (GlcN) enters cells through glucose transporters and is phosphorylated to GlcN-6-phosphate by hexokinases, thus bypassing the rate-limiting enzyme in the hexosamine biosynthesis, glutamine:fructose-6-phosphate transaminase (GFPT). Glucosamine treatment of cells has been shown by this mechanism to increase both the levels of the substrate for O-glycosylation (UDP-GlcNAc) and global O-GlcNAcylation (6, 7, 19). Glucosamine treatment of 3T3-L1 adipocytes increased leptin mRNA by 57% (p < 0.01) (Fig. 2A). Iron decreased leptin mRNA in control cells by 44% (p < 0.01) and blunted the increase caused by glucosamine to a similar degree (46%, p < 0.01) (Fig. 2A). Inhibition of OGT with a pharmacologic inhibitor (αR-α-[[(1,2-dihydro-2-oxo-6-quinolinyl)sulfonyl]amino]-N-(2-furanylmethyl)-2-methoxy-N-(2-thienylmethyl)-benzeneacetamide, OSMI-1 (20)) significantly decreased leptin mRNA (Fig. 2B), although iron had no further effect to inhibit leptin expression in OSMI-1–treated cells. The effects of iron and OSMI-1 on leptin mRNA were paralleled by their effects on leptin protein levels (Fig. 2C).
Leptin levels in a mouse model with increased adipocyte O-GlcNAc modification
To demonstrate that the regulation of leptin by iron is operating through O-GlcNAc modification in an animal model, we first studied mice heterozygous for whole-body deletion of the gene encoding the enzyme responsible for removing O-GlcNAc from proteins, OGA. Homozygous insufficiency of OGA is lethal, so OGA+/− heterozygous knockout (KO) mice were fed a normal chow with 35 mg/kg iron (the level in the standard American Institute of Nutrition chow AIN-93G) or the same chow supplemented to 2000 mg/kg iron. Levels of global O-GlcNAcylation detected by Western blotting were increased in the OGA+/− heterozygotes compared with WT mice, and both groups showed decreased O-GlcNAc when placed on the high-iron diet (Fig. 3, A and B). The high-iron diet resulted in a 57% decrease in serum leptin in the WT mice (Fig. 3C, p < 0.05). On the lower-iron diet, the OGA+/− heterozygous KO mice exhibited a 43% increase in serum leptin compared with WT, although that result was only significant when corrected for an observed 24% decrease in epididymal fat pad weight on the lower-iron diet (Fig. 3C, p < 0.05). The OGA+/− heterozygotes on high iron had a 58% decrease in serum leptin compared with low iron, and their fat pads had the same weight as the WT on high iron. The results for serum leptin were paralleled by changes of similar magnitude in leptin mRNA (data not shown).
Figure 3.

Effects of iron and genetic manipulation of OGA on leptin in mice. A, representative Western blotting for global protein O-GlcNAcylation in epididymal fat pads of WT (control) and OGA heterozygous (Het or het) KO mice fed chow containing 35 or 2000 mg/kg iron. B, quantitation of data from A, each lane normalized to α-tubulin, and all the results were normalized to WT (Control) mice on 35 mg/kg chow. C, serum leptin levels in the mice (n = 4–5 determinations/group). D, protein O-GlcNAcylation in adipose tissue of mice with adipocyte-specific deletion of OGA (OGA adipoKO) fed normal and high-iron diets and normalized to α-tubulin, and all results were normalized to WT (control) mice on 35 mg/kg chow. E, leptin mRNA in OGA adipoKO mice fed normal and high-iron diets. The results (n = 4/group) are normalized to β-actin mRNA for each determination and then normalized to values of control mice on normal iron. F, food intake (g/day) was measured for 4 weeks, beginning 8 weeks after starting diets, n = 3–4 mice/group. Shown are means ± S.E. *, p < 0.05 by ANOVA for indicated differences; ‡, p < 0.01; §, p < 0.001.
Because of potential effects on metabolic regulation of changes in O-GlcNAc in nonadipose tissues, we also examined mice with adipocyte-specific knockout of OGA (OGA adipoKO). C57BL6/J mice with a flox'ed OGA allele (21) were crossed with mice on the same background expressing cre recombinase under the adiponectin promoter (AdipoQ-cre, generously provided by Dr. Philipp Scherer, University of Texas Southwestern, Dallas, TX). Mice homozygous for OGAflox/flox and either heterozygous for AdipoQ-cre (OGA adipoKO) or mice not expressing cre (Control) were exposed to the normal or high-iron diets for 8 weeks. The mice with deletion of the OGA gene in adipocytes had increased levels of protein O-GlcNAcylation, and both groups showed decreased O-GlcNAc modification of proteins in fat tissue on the high-iron diet (Fig. 3D). On normal iron, OGA adipoKO mice had increased leptin mRNA levels compared with WT control mice on the same diet, and both groups exhibited decreased leptin mRNA on high iron compared with the same group on low iron (Fig. 3E). The effect of the high-iron diet to decrease leptin mRNA was relatively greater in the OGA adipoKO mice (56%) than in the WT mice (39%). Parallel effects were noted for serum leptin, namely a 34% increase in in the OGA adipoKO mice compared with controls on normal iron, and 59 and 67% decreases in control and OGA adipoKO mice on high iron compared with normal iron (not shown). These results are significant even when not corrected for fat pad weight, which would further exaggerate the differences: the increased leptin in the OGA adipoKO mice occurred despite decreases in epididymal fat pad weight (26% decrease in OGA adipoKO on normal iron (p < 0.05) and 39% decrease on high iron (p = 0.001) compared with WT, not shown). The effects of iron on leptin were accompanied by the expected inverse relationship with food intake (Fig. 3F).
Decreased CREB glycosylation and decreased occupancy of the leptin promoter by O-GlcNAcylated CREB after iron treatment of 3T3-L1 adipocytes
We previously demonstrated that iron-mediated phosphorylation of CREB was involved in its regulation of leptin and that iron treatment of 3T3-L1 adipocytes increased pCREB occupancy of two inhibitory CREB-binding sites in the leptin promoter (16). We therefore measured the effect of iron on glycosylation of CREB. Iron decreased levels of O-GlcNAcylated CREB in 3T3-L1 adipocytes by 15% (p < 0.05), in parallel with a 23% increase in pCREB (Fig. 4A, p < 0.05, quantification of blot not shown). ChIP analysis revealed that iron treatment resulted in decreased occupancy of the previously identified CREB sites in the leptin promoter by O-GlcNAcylated CREB (Fig. 4B). Total CREB (tCREB) occupancy of the CRE sites was also increased by iron treatment, as was pCREB occupancy, as previously reported (Fig. 4C) (16). After immunoprecipitation by the tCREB antibody, subsequent immunoprecipitation with the O-GlcNAc antibody RL2 yielded no signal (Fig. 4C).
Figure 4.

Effect of iron on O-glycosylation of CREB, occupancy of the leptin promoter by O-GlcNAcylated CREB, and on modulation of pCREB and leptin by mTOR inhibition. A, posttranslational modification of CREB in 3T3-L1 adipocytes treated or not with 100 μg/ml FAC. After treatment of cells for 24 h, extracts were immunoprecipitated with an antibody to total CREB. After SDS-PAGE, the blots were probed with an antibody specific for pCREB and for glycosylated CREB using the RL2 antibody. B, ChIP of O-GlcNAc–modified CREB on the two inhibitory CRE sites on the leptin promoter. C, ChIP of total CREB (tCREB) and pCREB from the CRE sites on the leptin promoter. In this panel, data from the two CRE sites were averaged together. D–F, effects of iron and mTOR inhibition by rapamycin (RAP or Rap) on phosphorylated P70S6 kinase (pP70S6K) (D), pCREB (E), and leptin (F) mRNA (normalized to β-actin), with all results further normalized to untreated control (CTL) cells. For all studies, cells were treated with and without FAC or 25 μmol/liter rapamycin for 24 h prior to ChIP analysis. ‡, p < 0.01; *, p < 0.05.
We previously reported that iron had no effect on the activity of two principal CREB kinases, PKA and CAMK. The mechanistic target of rapamycin (mTOR) pathway can also affect CREB phosphorylation (22), CREB-mediated transcription (23), and leptin regulation (24). We therefore asked whether iron affects leptin transcription by changing mTOR activity. Iron did have a modest but significant effect on mTOR activity in 3T3-L1 adipocytes, resulting in increased p70S6 kinase (p70S6K) phosphorylation that was blocked by the mTOR inhibitor rapamycin (Fig. 4D). A similar degree of stimulation of p70S6K phosphorylation was observed in adipose tissue of mice fed a high-iron diet (23 ± 11% increase, not shown). However, rapamycin increased pCREB and did not affect the increase in pCREB observed with iron (Fig. 4E). Despite increasing pCREB, rapamycin increased leptin mRNA levels and did not affect the degree to which iron inhibits leptin (Fig. 4F), suggesting that the mTOR pathway is not involved in the effect of iron on leptin. Inhibition of mTOR activity with rapamycin also did not affect global or CREB O-GlcNAcyation or its decrease by iron (not shown).
Iron has limited effects on OGA and OGT gene expression but decreases GFPT gene transcription
To probe possible mechanisms for the iron-induced decrease in O-GlcNAc modification, we examined transcript and protein levels for OGA, OGT, and GFPT, the rate-limiting enzyme for synthesis of the substrate for O-glycosylation, UDP-GlcNAc. Neither OGT nor OGA transcript levels in 3T3-L1 adipocytes were significantly affected by iron, but GFPT was down-regulated by 30% (Fig. 5A). Because O-GlcNAcylation can affect protein stability (25), and OGT and OGA themselves can be glycosylated (26, 27), we also measured protein levels. Iron had no discernable effect on expression of OGA, OGT, or GFPT1 protein in 3T3-L1 adipocytes (Fig. 5B). Similar results were seen in WT mice fed the high-iron diet: mRNAs for OGT and OGA were unaffected, and GFPT was decreased by 27% (Fig. 5C).
Figure 5.

Effect of iron on expression of OGT, OGA, and GFPT1. A, mRNA levels treated with or without 100 μg/ml FAC, samples normalized to β-actin and each mRNA normalized to control (n = 6–12 samples/group). B, representative Western blots for OGA, OGT, and GFAT protein in 3T3-L1 adipocytes. C, quantification of blots for OGA, OGT, and GFAT in 3T3-L1 adipocytes, normalized to α-tubulin, and expressed as relative density (n = 3–4 samples/group). *, p < 0.05. D, RNA levels in adipose tissue of WT mice fed either normal (35 mg/kg chow) or high (2000 mg/kg) iron diets, normalized to β-actin (n = 4–6 samples/group).
Discussion
We have previously shown that iron down-regulates leptin expression in mice and 3T3-L1 adipocytes and that these changes in leptin are physiologically significant insofar as they are reflected in feeding behavior of mice (16). We had also previously shown that the nutrient-sensing hexosamine biosynthesis pathway, which supplies the substrate for O-GlcNAcylation of proteins, regulates leptin synthesis and secretion (6–9). We have now linked these two findings, demonstrating that iron decreases levels of protein O-glycosylation and that decreasing O-GlcNAcylation is sufficient to down-regulate leptin. Conversely, pharmacologic or genetic manipulations of the hexosamine biosynthetic and O-GlcNAcylation pathways that increase levels of O-GlcNAc in cultured adipocytes and intact mice are sufficient to up-regulate leptin, and these manipulations are iron-sensitive. We had also previously demonstrated increased site occupancy of pCREB at inhibitory CRE sites on the leptin promoter in cultured cells and mice exposed to high iron and show herein that this is also accompanied by decreased occupancy of the promoter by O-GlcNAcylated CREB.
Other work on the regulation of metabolism by iron has demonstrated that iron regulates multiple pathways to coordinate availability of that metal, which is necessary for oxidative phosphorylation among many other reactions in cells, with glucose and fat metabolism (14, 15). It is important to note that this regulation occurs through the broad range of normal tissue iron levels (17). The diets used for the mice in this study both support normal growth, erythropoiesis, and reproduction, and the 2000-mg/kg diet results in a 2–3-fold increase in hepatic iron compared with the 35 mg/kg diet, well within the 4–6-fold range seen in normal humans (28). These diets were previously shown by us to result in a similar range of differential iron loading of adipocytes (16). Similarly, we have determined that treatment of 3T3-L1 cells with 100 μg/ml of FAC results in physiologic-range modulation of intracellular iron responses such as mRNA levels of the transferrin receptor (17). Thus, we believe these effects of iron are important in understanding normal physiology, as well as the effects of pathologic iron overload or deficiency.
We previously demonstrated that dominant-negative CREB was sufficient to block the effects of iron on leptin. Furthermore, activation of CREB by cAMP or PKA pathway agonists blunted the effect of iron on leptin, suggesting that changes in phosphorylation of CREB were involved (16). Increased levels of pCREB were detected in cells and mice exposed to high iron, but we did not detect increased levels of the CREB-modifying kinases PKA or CAMK. We further show in this manuscript that another pathway that affects leptin and CREB phosphorylation, mTOR, is also unlikely to be cause of the changes in CREB that are induced by iron. Namely, although iron modestly increased mTOR activity, inhibition of mTOR increased pCREB levels rather than decreasing them. Furthermore, inhibiting mTOR resulted in increased leptin mRNA, the opposite of what would be predicted from its effect on pCREB, and iron was still effective in modulating leptin in the face of mTOR inhibition. Previous work showing the involvement of O-GlcNAc in nutrient regulation of leptin, and the often reciprocal relationship between protein phosphorylation and O-linked glycosylation, led us to explore CREB O-GlcNAcylation.
O-GlcNAcylation of proteins is quantitatively as common as phosphorylation and is highly dynamic (10, 11). Signaling by O-linked GlcNAc modification of proteins serves as an integrative nutrient-responsive regulatory system whose function was originally studied largely in metabolic regulation, for example the control of insulin sensitivity (9, 19). Since then, however, O-GlcNAc signaling has been implicated in coordinating nutrient signaling with pathways as diverse as the regulation of growth and cell division, differentiation, circadian rhythms, and neuronal function (10, 11, 29, 30). Thus, O-GlcNAc signaling is an appropriate link also to mechanisms that connect regulation of appetite and metabolism by leptin to the availability of iron.
O-GlcNAcylation of CREB was originally identified in the brain (31), where its glycosylation is involved in memory formation (32). The functional consequences of CREB glycosylation have since been demonstrated in nonneuronal tissues as well (33), although it remains little studied. Cross-talk between phosphorylation and O-GlcNAcylation of CREB, however, has been shown to modulate its activity (34). In the brain, O-GlcNAcylation of CREB does not affect binding to DNA but decreases its interaction with CREB-regulated transcriptional coactivator (CRTC/TORC) (32). This mechanism is consistent with the ChIP data (Fig. 4), which clearly show binding of O-GlcNAc-CREB to the CRE sites and do not provide evidence consistent with competitive binding.
In general, O-GlcNAcylation of transcription factors can affect transcription in multiple ways, including 1) augmenting translocation to the nucleus; 2) facilitating interactions with coactivators, including of CREB with CREB-binding protein; and 3) promoting protein stability (13). The possibility that O-GlcNAcylated CREB is excluded from the nucleus is not likely given that most transcription factors are, if anything, preferentially translocated to the nucleus when glycosylated (13). The lack of a change in total CREB protein seen with iron treatment (Fig. 4) also suggests that the effect of iron is also not to differentially modulate stability of glycosylated CREB. Thus, it is most likely that the balance of phosphorylation and glycosylation are affecting differential association of CREB with transcriptional coactivators/corepressors.
Protein glycosylation and phosphorylation typically have a reciprocal “yin/yang” relationship; namely decreased glycosylation is associated with higher levels of phosphorylation (10, 11). In many cases the sharing or at least proximity of sites for the two modifications dictates that loss of one modification is necessary for the other modification to occur. In the case of CREB, the mechanistic basis for the reciprocal relationship is not known. Certain kinases themselves are regulated by O-GlcNAcylation, including the CREB kinase PKA, but in that case the effect of decreased O-GlcNAcylation is to decrease its kinase activity (35) and hence is unlikely to explain increased CREB phosphorylation by iron. Furthermore, in our previous study we were not able to demonstrate increased CREB kinase activity with iron treatment.
Our results argue instead that the direct target of iron is the O-GlcNAc pathway. Iron is a known oxidant, but nonspecific oxidant stress is probably not the mediator of the results presented herein insofar as oxidant stress, in general, increases levels of O-GlcNAcylation (36). Iron did not affect mRNA or protein levels of OGA, and the fact that iron exerts its full effect in mice with deletion of the OGA gene in adipocytes demonstrates that the decreased glycosylation is not due to increased OGA activity. Thus, rather than increasing deglycosylation, iron is more likely to exert its effects by decreasing glycosylation. This is consistent with the lack of additive effects of iron and pharmacologic OGT inhibition on leptin mRNA, although the nearly total inhibition of leptin transcription by that inhibitor may limit the ability to see further inhibition by iron. OGT protein and mRNA were not affected by iron, although the mRNA for the rate-limiting enzyme in producing the substrate for O-GlcNAcylation, GFPT, was modestly down-regulated. That is unlikely to be the entire explanation for decreased glycosylation, however, insofar as iron was able to counter the effect of glucosamine treatment, and glucosamine bypasses GFPT to increase O-GlcNAc levels.
The fact that global O-GlcNAcylation decreases with iron also suggests that iron is not affecting targeting of OGT to specific proteins, which has been demonstrated in several cases to be based on protein–protein interactions of OGT with a variety of binding partners (37). OGT activity is regulated by phosphorylation, including by the nutrient-sensing AMP-dependent kinase (38) and CAMK (39), which also happens to be a CREB kinase. We have shown that some of the effects of iron on metabolic regulation are mediated by AMP-dependent kinase (40), so this candidate mechanism merits further exploration.
In sum, the current results demonstrate that adipocytes serve to integrate not only the nutrient status, but also the iron status of the organism to modulate metabolism, appetite, and energy homeostasis. Presumably, this would help maintain food-seeking behavior in an iron-rich environment that would support high rates of oxidative metabolism, energy production, growth, and reproduction. The ability of the adipocyte to respond to iron availability and adjust expression of adipokines related to metabolism make it well-suited to perform its broader nutrient-sensing function.
Experimental procedures
Experimental animals
Dietary iron manipulations were accomplished with diets containing 60% carbohydrate, 17.7% protein, and 7.2% fat by weight, and either 35 mg/kg (TD. 10211, Harlan Teklad) or 2 g/kg carbonyl iron (TD. 10214) for a period of 2 months before tissue analysis. The mice were weaned at 21 days and maintained on the 35 mg/kg iron diet until the experimental period. WT mice were of the C57BL6/J strain. Mice with total body deletion of the O-GlcNAcase gene (21) were the generous gift of Dr. John Hanover (National Institutes of Health, Bethesda, MD) and used in the heterozygous state because homozygosity for the whole-body deletion is embryonically lethal. C57BL6/J mice with a flox'ed OGA allele, also provided by Dr. Hanover (21), were crossed with mice on the same background expressing cre recombinase under the adiponectin promoter (AdipoQ-cre, generously provided by Dr. Philipp Scherer, University of Texas Southwestern, Dallas, TX). Studies were approved by the Institutional Animal Care and Use Committees of the University of Utah and Wake Forest School of Medicine.
Reagents and assays
Reagents were purchased from Sigma–Aldrich unless otherwise noted. Serum leptin levels were measured by ELISA kit (Millipore).
3T3-L1 adipocyte culture and differentiation
3T3-L1 adipocytes (ATCC, Manassas, VA) were maintained in high-glucose DMEM (HG-DMEM, Invitrogen) supplemented with 10% bovine calf serum (Hyclone) and penicillin/streptomycin (Invitrogen). For differentiation (41), the cells were incubated in HG-DMEM with 10% bovine calf serum for 48 h after confluence. The cells were then cultured in differentiation medium I (HG-DMEM, 10% fetal bovine serum (FBS), 1 μg/ml insulin, 0.25 μg/ml dexamethasone, 0.5 mm 3-isobutyl-1-methylxanthine, 4 μm ciglitazone) for 4 days, followed by differentiation medium II (HG-DMEM, 10% FBS, 10 μg/ml insulin) for 48 h. Prior to experiments, the cells were cultured overnight in DMEM (Invitrogen) with 0.5% BSA. All experiments were performed in minimum essential medium-α, which is iron-free, supplemented with 10% FBS. Iron treatments were performed with 100 μg/ml ferric ammonium citrate for 24 h prior to assay.
Quantification of transcripts
Quantitative RT-PCR was performed as described previously (42). Briefly, mRNA was extracted from primary or 3T3-L1 adipocytes or epididymal fat pads using TRIzol (Invitrogen), purified using an RNeasy column (QIAgen), and synthesized into cDNA using a first-strand cDNA synthesis kit (Invitrogen). The fat cake in TRIzol was cleared by transferring the infranatant to a clean tube using a glass syringe before extraction. Real-time PCR was performed with a QuantStudioTM real-time PCR system (Applied Biosystems). cDNA products were quantified using the relative standard curve method. mRNA levels of specific genes were normalized to RPL13A or β-actin.
Western blotting
Cell lysate protein concentrations were determined using a Pierce BCA protein assay kit. The lysate proteins were separated by 4–15% SDS-PAGE, and the resolved proteins were transferred to polyvinylidene difluoride membranes. The blots were blocked with TBST containing 5% (w/v) nonfat dried milk for 1 h at room temperature. 5% (w/v) BSA was used in lieu of dried milk for detection with the anti-O-GlcNAc antibodies. The blots were incubated with primary antibodies overnight at 4 °C. After washing, the blots were incubated with the appropriate horseradish peroxidase–conjugated secondary antibody for 1 h at room temperature. The blots were imaged by treatment with Super Signal West Femto Maximum Sensitivity Substrate kit and exposure to PXI imager using Genesys software to scan the bands. Scans were analyzed using National Institutes of Health ImageJ. For studies of global O-GlcNAcylation, the entire lane was scanned vertically. In all of the experiments, α-tubulin was used to normalize for gel loading and then normalized to control values.
Antibodies
Antibodies used included phosphor-CREB (pCREB, Ser-133, 87G3) and CREB (86B10) from Cell Signaling Inc. (Danvers, MA), Ob (A20, sc842) from Santa Cruz Inc. (Dallas, TX), anti-O-linked GlcNAc antibody RL-2 (43) (ab2739) from Abcam Inc. (Cambridge, UK), anti-O-linked GlcNAc antibody CTD 110.6 from Covance (Durham, NC), and α-tubulin (T9026) from Sigma–Aldrich.
Immunoprecipitation
Well-differentiated adipocytes were washed with PBS once and collected in lysis buffer with protease inhibitor (Roche Complete Tablet 11836153001) and phosphatase inhibitor (Sigma P2850). After centrifugation at 15,000 rpm for 10 min, the supernatant was transferred to a new tube, and protein concentration was measured. The lysates (600 μg) were immunoprecipitated with 4 μg of CREB antibody (Cell Signaling) at 4 °C for overnight and added with 100 μl of (50% slurry) protein A–agarose beads (Millipore) overnight after being precleared with 20 μl of free protein A–agarose beads (Millipore) for 2 h. The supernatant was discard, and the immune complex on beads was washed with lysis buffer with protease inhibitor and phosphatase inhibitor three times and boiled in sample buffer. The immunoprecipitated products were subjected to 7.5% SDS-PAGE, transferred onto the membrane, and immunoblotted with the antibodies RL-2 and pCREB.
ChIP assay
ChIP studies were performed as described with minor modification (16). Chromatin was extracted from 3T3-L1 adipocytes on days 8–10 after differentiation using the SimpleChIP kit (catalog no. 9003, Cell Signaling Technology) according to the manufacturer's instructions. Briefly, the cells were treated with 100 μg/ml of FAC for 24 h prior to cross-linking for 10 min with 1% formaldehyde. The cells were then lysed by micrococcal nuclease and sonicated three times for 20 s using a sonic dismembrator (Fisher Scientific). RL2 was then immunoprecipitated from precleared lysates with the Chip-Grade protein G magnetic beads. DNA was released from protein–DNA complexes by proteinase K digestion and then subjected to quantitative PCR using power SYBR green kit (Applied Biosystems). The following primers were used to amplify the CRE-1 site: forward, 5′-GCA CGA TGT AAC CAC GAA TG-3′, and reverse, 5′-ACG TCC ATT CAG CAA AAA CC-3′. The following primers were used to amplify the CRE-2 site: forward, 5′-GGC GAA AGG CAA ACA TAA GA-3′, and reverse, 5′-TTC CCG CTC TGA CAT TCT TT-3′. ChIP– quantitative PCR data were normalized for amount of chromatin by input samples and normal IgG samples.
Statistics
Descriptive statistics in the text and figures are represented as averages ± S.E. An unpaired two-tailed Student's t test was used to determine significance between controls and individual experimental groups. One-way ANOVA was used to compare series of data. p < 0.05 was considered significant for all tests. All statistical analyses were performed with STATA.
Author contributions
Y. G., F. R. L., and D. A. M. conceptualization; Y. G., J. L., S. S., and D. A. M. data curation; Y. G., J. L., Z. B., S. S., C. Z., and D. A. M. investigation; Y. G., J. L., and D. A. M. writing-original draft; Y. G., J. L., Z. B., S. S., C. Z., F. R. L., and D. A. M. writing-review and editing; J. L., Z. B., C. Z., F. R. L., and D. A. M. formal analysis; S. S., F. R. L., and D. A. M. project administration; D. A. M. resources; D. A. M. supervision; D. A. M. funding acquisition; D. A. M. validation.
This work was supported by National Institutes of Health Grant 1R01 DK081842 and Research Service of the Department of Veterans Affairs Grant 2I01 BX001140. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CREB
- cyclic AMP response element binding protein
- OGT
- O-glycosyltransferase
- OGA
- O-GlcNAcase
- GFPT1
- glutamine:fructose-6-phosphate transaminase
- FAC
- ferric ammonium citrate
- PKA
- protein kinase A
- AMPK
- AMP-dependent kinase
- pCREB
- phosphorylated CREB
- CAMK
- calcium/calmodulin-dependent kinase IV
- FAC
- ferric ammonium citrate
- GlcN
- glucosamine
- KO
- knockout
- mTOR
- mechanistic target of rapamycin
- HG
- high-glucose
- FBS
- fetal bovine serum
- ANOVA
- analysis of variance.
References
- 1. Friedman J. M., and Halaas J. L. (1998) Leptin and the regulation of body weight in mammals. Nature 395, 763–770 10.1038/27376 [DOI] [PubMed] [Google Scholar]
- 2. Kallen C. B., and Lazar M. A. (1996) Antidiabetic thiazolidinediones inhibit leptin (ob) gene expression in 3T3-L1 adipocytes. Proc. Natl. Acad. Sci. U.S.A. 93, 5793–5796 10.1073/pnas.93.12.5793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hollenberg A. N., Susulic V. S., Madura J. P., Zhang B., Moller D. E., Tontonoz P., Sarraf P., Spiegelman B. M., and Lowell B. B. (1997) Functional antagonism between CCAAT/Enhancer binding protein-α and peroxisome proliferator-activated receptor-γ on the leptin promoter. J. Biol. Chem. 272, 5283–5290 10.1074/jbc.272.8.5283 [DOI] [PubMed] [Google Scholar]
- 4. Mason M. M., He Y., Chen H., Quon M. J., and Reitman M. (1998) Regulation of leptin promoter function by Sp1, C/EBP, and a novel factor. Endocrinology 139, 1013–1022 10.1210/endo.139.3.5792 [DOI] [PubMed] [Google Scholar]
- 5. Yang D. C., Tsay H. J., Lin S. Y., Chiou S. H., Li M. J., Chang T. J., and Hung S. C. (2008) cAMP/PKA regulates osteogenesis, adipogenesis and ratio of RANKL/OPG mRNA expression in mesenchymal stem cells by suppressing leptin. PLoS One 3, e1540 10.1371/journal.pone.0001540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang J., Liu R., Hawkins M., Barzilai N., and Rossetti L. (1998) A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature 393, 684–688 10.1038/31474 [DOI] [PubMed] [Google Scholar]
- 7. Considine R. V., Cooksey R. C., Williams L. B., Fawcett R. L., Zhang P., Ambrosius W. T., Whitfield R. M., Jones R., Inman M., Huse J., and McClain D. A. (2000) Hexosamines regulate leptin production in human subcutaneous adipocytes. J. Clin. Endocrinol. Metab. 85, 3551–3556 10.1210/jcem.85.10.6916,10.1210/jc.85.10.3551 [DOI] [PubMed] [Google Scholar]
- 8. McClain D. A., Alexander T., Cooksey R. C., and Considine R. V. (2000) Hexosamines stimulate leptin production in transgenic mice. Endocrinology 141, 1999–2002 10.1210/endo.141.6.7532 [DOI] [PubMed] [Google Scholar]
- 9. McClain D. A., Lubas W. A., Cooksey R. C., Hazel M., Parker G. J., Love D. C., and Hanover J. A. (2002) Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc. Natl. Acad. Sci. U.S.A. 99, 10695–10699 10.1073/pnas.152346899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bond M. R., and Hanover J. A. (2015) A little sugar goes a long way: the cell biology of O-GlcNAc. J. Cell Biol. 208, 869–880 10.1083/jcb.201501101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zachara N., Akimoto Y., and Hart G. W. (2015) The O-GlcNAc modification. In Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) pp. 239–251, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 12. Zhang P., Klenk E. S., Lazzaro M. A., Williams L. B., and Considine R. V. (2002) Hexosamines regulate leptin production in 3T3-L1 adipocytes through transcriptional mechanisms. Endocrinology 143, 99–106 10.1210/endo.143.1.8568 [DOI] [PubMed] [Google Scholar]
- 13. Ozcan S., Andrali S. S., and Cantrell J. E. (2010) Modulation of transcription factor function by O-GlcNAc modification. Biochim. Biophys. Acta 1799, 353–364 10.1016/j.bbagrm.2010.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simcox J. A., and McClain D. A. (2013) Iron and diabetes risk. Cell Metab. 17, 329–341 10.1016/j.cmet.2013.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fernández-Real J. M., McClain D., and Manco M. (2015) Mechanisms linking glucose homeostasis and iron metabolism toward the onset and progression of type 2 diabetes. Diabetes Care 38, 2169–2176 10.2337/dc14-3082 [DOI] [PubMed] [Google Scholar]
- 16. Gao Y., Li Z., Gabrielsen J. S., Simcox J. A., Lee S. H., Jones D., Cooksey B., Stoddard G., Cefalu W. T., and McClain D. A. (2015) Adipocyte iron regulates leptin and food intake. J. Clin. Invest. 125, 3681–3691 10.1172/JCI81860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gabrielsen J. S., Gao Y., Simcox J. A., Huang J., Thorup D., Jones D., Cooksey R. C., Gabrielsen D., Adams T. D., Hunt S. C., Hopkins P. N., Cefalu W. T., and McClain D. A. (2012) Adipocyte iron regulates adiponectin and insulin sensitivity. J. Clin. Invest. 122, 3529–3540 10.1172/JCI44421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Comer F. I., Vosseller K., Wells L., Accavitti M. A., and Hart G. W. (2001) Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine. Anal. Biochem. 293, 169–177 10.1006/abio.2001.5132 [DOI] [PubMed] [Google Scholar]
- 19. Marshall S., Bacote V., and Traxinger R. R. (1991) Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system: role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 266, 4706–4712 [PubMed] [Google Scholar]
- 20. Ortiz-Meoz R. F., Jiang J., Lazarus M. B., Orman M., Janetzko J., Fan C., Duveau D. Y., Tan Z. W., Thomas C. J., and Walker S. (2015) A small molecule that inhibits OGT activity in cells. ACS Chem. Biol. 10, 1392–1397 10.1021/acschembio.5b00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keembiyehetty C., Love D. C., Harwood K. R., Gavrilova O., Comly M. E., and Hanover J. A. (2015) Conditional knock-out reveals a requirement for O-linked N-Acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J. Biol. Chem. 290, 7097–7113 10.1074/jbc.M114.617779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Du K., and Montminy M. (1998) CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 273, 32377–32379 10.1074/jbc.273.49.32377 [DOI] [PubMed] [Google Scholar]
- 23. Zhang X., Luo Y., Wang C., Ding X., Yang X., Wu D., Silva F., Yang Z., Zhou Q., Wang L., Wang X., Zhou J., Boyd N., Spafford M., Burge M., et al. (2018) Adipose mTORC1 suppresses prostaglandin signaling and beige adipogenesis via the CRTC2–COX-2 pathway. Cell Rep. 24, 3180–3193 10.1016/j.celrep.2018.08.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roh C., Han J., Tzatsos A., and Kandror K. V. (2003) Nutrient-sensing mTOR-mediated pathway regulates leptin production in isolated rat adipocytes. Am. J. Physiol. Endocrinol. Metab. 284, E322–E330 10.1152/ajpendo.00230.2002 [DOI] [PubMed] [Google Scholar]
- 25. Zhang F., Su K., Yang X., Bowe D. B., Paterson A. J., and Kudlow J. E. (2003) O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115, 715–725 10.1016/S0092-8674(03)00974-7 [DOI] [PubMed] [Google Scholar]
- 26. Whelan S. A., Lane M. D., and Hart G. W. (2008) Regulation of the O-linked β-N-acetylglucosamine transferase by insulin signaling. J. Biol. Chem. 283, 21411–21417 10.1074/jbc.M800677200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whisenhunt T. R., Yang X., Bowe D. B., Paterson A. J., Van Tine B. A., and Kudlow J. E. (2006) Disrupting the enzyme complex regulating O-GlcNAcylation blocks signaling and development. Glycobiology 16, 551–563 10.1093/glycob/cwj096 [DOI] [PubMed] [Google Scholar]
- 28. Brissot P., Bourel M., Herry D., Verger J. P., Messner M., Beaumont C., Regnouard F., Ferrand B., and Simon M. (1981) Assessment of liver iron content in 271 patients: a reevaluation of direct and indirect methods. Gastroenterology 80, 557–565 [PubMed] [Google Scholar]
- 29. Kaasik K., Kivimäe S., Allen J. J., Chalkley R. J., Huang Y., Baer K., Kissel H., Burlingame A. L., Shokat K. M., Ptácek L. J., and Fu Y. H. (2013) Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 17, 291–302 10.1016/j.cmet.2012.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang X., and Qian K. (2017) Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 18, 452–465 10.1038/nrm.2017.22,10.1038/nrn.2017.89,10.1038/nrn.2017.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lamarre-Vincent N., and Hsieh-Wilson L. C. (2003) Dynamic glycosylation of the transcription factor CREB: a potential role in gene regulation. J. Am. Chem. Soc. 125, 6612–6613 10.1021/ja028200t [DOI] [PubMed] [Google Scholar]
- 32. Rexach J. E., Clark P. M., Mason D. E., Neve R. L., Peters E. C., and Hsieh-Wilson L. C. (2012) Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat. Chem. Biol. 8, 253–261 10.1038/nchembio.770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Groussaud D., Khair M., Tollenaere A. I., Waast L., Kuo M. S., Mangeney M., Martella C., Fardini Y., Coste S., Souidi M., Benit L., Pique C., and Issad T. (2017) Hijacking of the O-GlcNAcZYME complex by the HTLV-1 Tax oncoprotein facilitates viral transcription. PLoS Pathog. 13, e1006518 10.1371/journal.ppat.1006518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Azuma Y., Miura K., Higai K., and Matsumoto K. (2007) Protein O-N-acetylglucosaminylation modulates promoter activities of cyclic AMP response element and activator protein 1 and enhances E-selectin expression on HuH-7 human hepatoma cells. Biol. Pharm. Bull. 30, 2284–2289 10.1248/bpb.30.2284 [DOI] [PubMed] [Google Scholar]
- 35. Jin N., Ma D., Gu J., Shi J., Xu X., Iqbal K., Gong C. X., Liu F., and Chu D. (2018) O-GlcNAcylation modulates PKA-CREB signaling in a manner specific to PKA catalytic subunit isoforms. Biochem. Biophys. Res. Commun. 497, 194–199 10.1016/j.bbrc.2018.02.053 [DOI] [PubMed] [Google Scholar]
- 36. Kátai E., Pál J., Poór V. S., Purewal R., Miseta A., and Nagy T. (2016) Oxidative stress induces transient O-GlcNAc elevation and tau dephosphorylation in SH-SY5Y cells. J. Cell Mol. Med. 20, 2269–2277 10.1111/jcmm.12910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levine Z. G., and Walker S. (2016) The biochemistry of O-GlcNAc transferase: which functions make it essential in mammalian cells? Annu. Rev. Biochem. 85, 631–657 10.1146/annurev-biochem-060713-035344 [DOI] [PubMed] [Google Scholar]
- 38. Bullen J. W., Balsbaugh J. L., Chanda D., Shabanowitz J., Hunt D. F., Neumann D., and Hart G. W. (2014) Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK). J. Biol. Chem. 289, 10592–10606 10.1074/jbc.M113.523068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song M., Kim H. S., Park J. M., Kim S. H., Kim I. H., Ryu S. H., and Suh P. G. (2008) O-GlcNAc transferase is activated by CaMKIV-dependent phosphorylation under potassium chloride-induced depolarization in NG-108–15 cells. Cell Signal. 20, 94–104 10.1016/j.cellsig.2007.09.002 [DOI] [PubMed] [Google Scholar]
- 40. Huang J., Simcox J., Mitchell T. C., Jones D., Cox J., Luo B., Cooksey R. C., Boros L. G., and McClain D. A. (2013) Iron regulates glucose homeostasis in liver and muscle via AMP-activated protein kinase in mice. FASEB J. 27, 2845–2854 10.1096/fj.12-216929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Z., Torrens J. I., Anand A., Spiegelman B. M., and Friedman J. M. (2005) Krox20 stimulates adipogenesis via C/EBPβ-dependent and -independent mechanisms. Cell Metab. 1, 93–106 10.1016/j.cmet.2004.12.009 [DOI] [PubMed] [Google Scholar]
- 42. Huang J., Gabrielsen J. S., Cooksey R. C., Luo B., Boros L. G., Jones D. L., Jouihan H. A., Soesanto Y., Knecht L., Hazel M. W., Kushner J. P., and McClain D. A. (2007) Increased glucose disposal and AMP-dependent kinase signaling in a mouse model of hemochromatosis. J. Biol. Chem. 282, 37501–37507 10.1074/jbc.M703625200 [DOI] [PubMed] [Google Scholar]
- 43. Taylor R. P., Geisler T. S., Chambers J. H., and McClain D. A. (2009) Up-regulation of O-GlcNAc transferase with glucose deprivation in HepG2 cells is mediated by decreased hexosamine pathway flux. J. Biol. Chem. 284, 3425–3432 10.1074/jbc.M803198200 [DOI] [PMC free article] [PubMed] [Google Scholar]