

Abstract
Most clinically available antipsychotic drugs (APDs) bind dopamine D2 receptors (D2R) at therapeutic concentrations, and it is thought that they suppress psychotic symptoms by serving as competitive antagonists of dopamine at D2R. Here, we present data that demonstrate that APDs act independently of dopamine at an intracellular pool of D2R to enhance transport of D2R to the cell surface and suggest that APDs can act as pharmacological chaperones at D2R. Among the first- and second-generation APDs that we tested, clozapine exhibited the lowest efficacy for translocating D2R to the cell surface. Thus, our observations could provide a cellular explanation for some of the distinct therapeutic characteristics of clozapine in schizophrenia. They also suggest that differential intracellular actions of APDs at their common G protein–coupled receptor (GPCR) target, D2R, could contribute to differences in their clinical profiles.
Keywords: D2 dopamine receptor, protein misfolding, G protein-coupled receptor (GPCR), receptor trafficking, schizophrenia, antipsychotic drug, clozapine, pharmacological chaperone, pharmacoperone, receptor up-regulation, receptor supersensitivity
Introduction
All antipsychotic drugs (APDs)2 presently approved for treating schizophrenia are competitive antagonists of dopamine at dopamine D2 receptors (D2R) at therapeutic concentrations (1–4). First-generation APDs, although effective at suppressing positive symptoms of schizophrenia, have modest efficacy against negative and cognitive symptoms (5). In addition, APD binding to D2R expressed in the basal ganglia produces extrapyramidal motor side effects (EPS). These include both acute motor symptoms such as Parkinsonism and the potentially irreversible hyperkinetic movement disorder, tardive dyskinesia (TD) (5).
The introduction of clozapine in the 1970s was a major milestone in the pharmacotherapy of schizophrenia because clozapine appeared to demonstrate superior efficacy against both positive and negative symptoms, with significantly minimized risk of EPS (5–7). However, clozapine-induced agranulocytosis and metabolic side effects that increase risk for cardiovascular morbidity (5, 7) catalyzed efforts to develop safer drugs that attempted to replicate clozapine's unique efficacy and reduced risk of EPS. Several second-generation APDs were developed by searching for molecules that shared pharmacological properties of clozapine that were distinct relative to the preceding first-generation drugs. A well-described property that was utilized in that effort is that clozapine displays reduced binding affinity for D2R and enhanced binding affinity for 5-HT2A serotonin receptors (5). Other mechanisms that have been proposed to differentiate first- and second-generation drugs include binding to subtypes of muscarinic acetylcholine receptors, adrenergic receptors, or glutamate receptors as well as fast dissociation kinetics, low affinity, and transient occupancy at D2R (6, 8).
However, evidence from both clinical trials and practice, reviewed in numerous papers (7, 9, 10), demonstrates that clozapine outperforms both first- and second-generation APDs with respect to several aspects of schizophrenia pharmacotherapy. For example, clozapine has the strongest evidence for utility in treatment-resistant schizophrenia and produces the greatest suppression of mortality in schizophrenia (5, 7, 11). A recent meta-analysis, and one of the largest conducted with psychotropic drugs, showed that clozapine was the most effective APD at suppressing schizophrenia symptoms (12). The same analysis also concluded that clozapine has the lowest liability for producing EPS. The favorable motor side-effect profile of clozapine is further highlighted by the fact that it is used more than any other APD for suppressing psychoses in Parkinson's disease (13). It has been said that, “patients treated with clozapine may occasionally experience an 'awakening' characterized by a return to a near normal level of cognitive, interpersonal, and vocational functioning” (14). Accordingly, clozapine has been referred to as the “gold standard” for the treatment of schizophrenia (7, 10, 14).
Thus, the molecular explanations proposed for distinguishing first- and second-generation APDs cannot account for the distinct therapeutic profile of clozapine because they are properties shared by multiple first- and second-generation drugs. For instance, one proposed hypothesis for clozapine's unique mechanism of clinical action is that it has relatively rapid dissociation kinetics from D2R (8). However, data from multiple studies indicate that the dissociation rate of clozapine from D2R is not significantly slower than that reported for some other APDs, including quetiapine, remoxipride, amisulpride, and sulpiride (8, 15–17).
It is now well-established that a G protein–coupled receptor (GPCR) can exist in more than just an “on” or “off” conformation. Moreover, different ligands could differentially stabilize each of these multiple GPCR conformations even though they bind to the same orthosteric site in a GPCR as described by the phenomenon of biased agonism (18). Such distinct conformations may differently engage downstream effectors, but they could as well differentially modulate other biochemical properties, such as expression and trafficking of GPCRs. D2R supersensitivity or increases in brain D2R after chronic treatment with APDs have been suggested as underlying mechanisms for the development of resistance to pharmacotherapy in schizophrenia (19, 20). In addition, APD-induced up-regulation of D2R has been implicated in TD (21, 22). Therefore, here we have investigated the actions of APDs on cell-surface up-regulation of D2R. We report that every APD that we tested, in the absence of dopamine, enhanced either cell-surface levels or cell-surface insertion of D2R relative to both clozapine and vehicle. Thus, our data suggest that further investigating the distinctive actions of clozapine on the trafficking and cellular processing of D2R, the common target of all APDs, could be a useful avenue for research into understanding the molecular basis for the distinct therapeutic profile of clozapine.
Results
APD treatment enhances cell-surface levels of D2R endogenously expressed in the pituitary MMQ cell line
D2R is known to be endogenously expressed in the prolactin-secreting, pituitary-derived MMQ cell line (23, 24). We measured cell-surface levels of D2R, endogenously expressed in the MMQ cell after treatment with the first-generation APD, haloperidol, or with clozapine (10 μm, 24 h). The surface levels of endogenous D2R were evaluated by isolating cell-surface proteins through specific biotinylation of these proteins. D2R present in the pool of cell-surface proteins was then quantified using an anti-D2R antibody that was previously validated with tissue from D2R knockout and WT mice (25). We observed that, even in the absence of dopamine, haloperidol, but not clozapine, significantly enhanced surface levels of D2R (Fig. 1).
Figure 1.
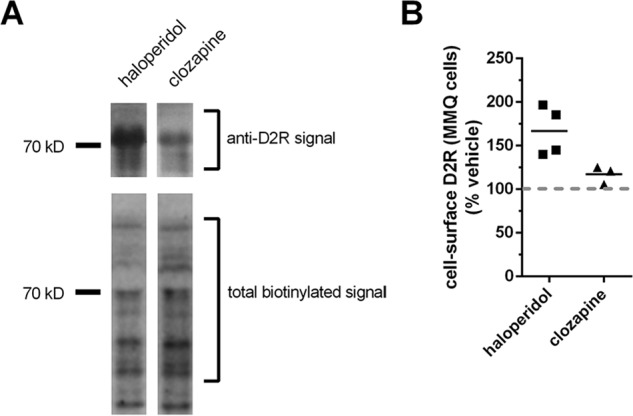
Effect of APD treatments on cell-surface levels of the D2R endogenously expressed in the pituitary-derived MMQ cell line. A, effect of APD treatment on cell-surface D2R endogenously expressed in pituitary-derived MMQ cell line. Representative lanes from Western blotting depict the effect of treatment with the indicated APDs (10 μm, 48 h) on the levels of cell-surface expression of D2R endogenously expressed in MMQ cells. Western blots of cell-surface proteins isolated by surface biotinylation of intact cells with the membrane-impermeable biotinylation reagent, EZ-LinkTM Sulfo-NHS-LC-Biotin, were probed with anti-DR antibody (top) and subsequently reprobed with HRP-conjugated streptavidin to visualize total cell-surface protein (bottom). B, quantification of cell-surface D2R in MMQ cells after treatment with the indicated APDs. Levels of endogenous cell surface D2R expression in MMQ cells visualized in A were normalized against the total cell-surface protein signal and reported as a percentage of the normalized cell-surface D2R signal in vehicle-treated cells. The signal for cell-surface D2R after treatment with haloperidol was significantly different, from both clozapine-treated (p < 0.05) and vehicle-treated (p < 0.01) cells (bar representing the mean; n = 4 for vehicle and haloperidol and 3 for clozapine; Tukey).
We also evaluated the actions of haloperidol and clozapine on the cell-surface expression of a D2R construct stably expressed in a CHO cell line. The cell line was selected and evaluated to express D2R at a density equal to or lower than that observed in mouse brain, as described previously (26, 27). In this cell line, as in MMQ cells, haloperidol treatment significantly increased cell-surface D2R expression over vehicle, and no detectable increase in expression was observed after clozapine treatment (Fig. S1).
APD treatment enhances cell-surface levels of D2R transiently expressed in HEK293T cells
Subsequently, we explored the use of a more tractable system for investigating the cellular mechanisms underlying the differential up-regulation of cell-surface D2R by haloperidol and clozapine. We found that haloperidol treatment (10 μm) produced a time-dependent enhancement of cell-surface expression of FLAG-tagged D2R transiently expressed in HEK293T cells (Fig. 2A).
Figure 2.

Effect of APD treatments on cellular levels of the D2R. A, time-dependent haloperidol-induced enhancement of cell-surface D2R expression. HEK293T cells transiently expressing the extracellular N-terminal FLAG-tagged D2R construct, FLAG-D2R, were treated with haloperidol (10 μm) for the indicated times. The levels of cell-surface D2R were then quantified by probing the intact cells with anti-FLAG antibody. The haloperidol-induced enhancement of D2R surface expression is depicted as a percentage of the signal from vehicle-treated cells. Significant enhancements of cell-surface D2R expression levels were observed at each measurement time point following haloperidol treatment, both with respect to vehicle and with respect to the previous time point (bar representing the mean; n = 6; Tukey; p < 0.001 for the 6-h treatment time point versus vehicle, and p < 0.0001 for all other comparisons). B, effect of APD treatment on the cell-surface levels of N-terminal FLAG-tagged D2SR. HEK293T cells transiently expressing the extracellular N-terminal FLAG-tagged D2SR construct, FLAG-D2SR, were treated with either haloperidol or clozapine (10 μm, 24 h). The levels of cell-surface D2SR were then quantified as in A and are reported as a percentage of the signal from vehicle-treated cells. The levels of cell-surface D2SR measured after haloperidol treatment were significantly greater than after clozapine or vehicle treatment (n = 12, Tukey, p < 0.0001). C, concentration–response curves of APD-induced enhancement of cell-surface D2R expression. HEK293T cells transiently expressing the extracellular N-terminal FLAG-tagged D2R construct, FLAG-D2R, were treated for 24 h with the indicated concentrations of haloperidol (halo), olanzapine (olanz), or clozapine (cloz). The levels of cell-surface D2R were then quantified by probing the intact cells with anti-FLAG antibody. The APD-induced enhancement of D2R surface expression was calculated as the percentage increase in cell-surface receptor levels over vehicle-treated cells and is plotted as a percentage of the response to 10 μm haloperidol (mean ± S.E. (error bars), n = 38 for 10 μm olanzapine, 6 for all other concentrations; n = 44 for 10 μm halperidol, 7 for all other concentrations; n = 50 for 10 μm clozapine, 12 for 3 and 30 μm clozapine, 6 for all others). Cell-surface levels of D2R became significantly different (Dunnett's multiple-comparison test) from vehicle after treatment with 100 nm haloperidol (p < 0.001), 1 μm olanzapine (p < 0.0001), and 3 μm clozapine (p < 0.01). Cell-surface D2R levels after treatment with 10 μm concentrations of each drug were significantly different from each other (Tukey, p < 0.0001). There was no significant difference in cell-surface D2R levels between the 3, 10, and 30 μm clozapine treatments. D, comparison of the effect of multiple APD treatments on cell-surface expression of D2R. HEK293T cells transiently expressing FLAG-D2R were treated with the indicated APDs (24 h, 30 μm for remoxipride and 10 μm for all other APDs). The levels of cell-surface D2R were then quantified as in A and are reported as a percentage of the signal of vehicle-treated cells (bar representing the median, whiskers representing the full range of data; n = 7 for amisulpride, 8 for remoxipride, 16 for tiapride, 31 for droperidol and ziprasidone, and 32 for all other drugs). Relative D2R surface expression after treatment with all APDs was significantly greater than vehicle except for the APDs clozapine and aripiprazole (Dunnett, p < 0.01). E, effect of APD treatments on total cellular expression of D2R. HEK293T cells transiently expressing FLAG-D2R were treated with the indicated APDs (10 μm, 24 h) and were then fixed and permeabilized with methanol. Total cellular D2R was then assessed by probing with anti-FLAG antibody, and levels are reported as a percentage of the signal from vehicle-treated cells (n = 6 for aripiprazole, 22 for haloperidol, 16 for all other drugs). Treatment with aripiprazole produced significantly less enhancement of total cellular receptor levels compared with the other APDs (Tukey, p < 0.005), and all APDs, except for aripiprazole, significantly enhanced total receptor levels compared with vehicle (Tukey, p < 0.0001). F, APD-mediated increase in cell-surface D2R normalized to increase in total cellular D2R. Cell-surface D2R normalized to total cellular D2R levels after clozapine treatment was significantly lower than after treatment with all other APDs or vehicle (n = 32, Dunnett, p < 0.005).
Furthermore, using this system, we showed that the differential actions of haloperidol and clozapine treatment on up-regulating cell-surface expression of D2R were also observed with the short D2 isoform (D2SR) (28) (Fig. 2B).
D2R surface expression after clozapine treatment is less than after treatment with all tested APDs except aripiprazole
We generated concentration–response curves for APD-mediated enhancement of cell-surface FLAG-D2R expression after treatment (24 h) with haloperidol, clozapine, and the second-generation APD, olanzapine (Fig. 2C). Interestingly, the APD-induced D2R cell-surface up-regulation response for clozapine plateaued at significantly lower levels compared with the other drugs at ∼30% of the haloperidol response. These data indicate that different APDs have different efficacies for up-regulating cell-surface D2R.
We then performed a wider screen that compared the cell-surface D2R up-regulation produced by a larger number of APDs. We found that a saturating concentration of clozapine produced significantly less enhancement of cell-surface D2R than that which could be produced by all tested first- and second-generation APDs, with the exception of aripiprazole (Fig. 2D). Receptor-mediated cellular responses of ligands are a function of both the fractional occupancy of receptor by the ligand and the efficacy of the ligand–receptor complex for producing the cellular response. However, because APDs, except for aripiprazole, produced D2R cell-surface up-regulation greater than the maximum that was produced by clozapine, we may conclude that the efficacy of most APDs for producing cell-surface D2R up-regulation is greater than that of clozapine.
APD enhancement of surface D2R expression, relative to clozapine, is not a consequence of relative enhancement of total cellular D2R expression
We utilized a subset of five APDs to investigate whether the different efficacies of the APDs for up-regulating cell-surface expression of D2R were the result of corresponding influences on total cellular D2R. Total cellular FLAG-D2R levels were evaluated by probing cells, which were first made permeable with methanol, with anti-FLAG antibody. We found that that four of the five tested APDs, including clozapine, increased total cellular D2R to a similar extent (Fig. 2E). Hence, we may conclude that the significantly enhanced cell-surface D2R expression produced by other APDs relative to clozapine (Fig. 2D) involves post-translational mechanisms. Cell-surface D2R levels normalized against total receptor levels were lowest with clozapine, and furthermore, clozapine was the only APD for which cell-surface expression of D2R normalized to total receptor was significantly decreased (∼60% of vehicle) after drug treatment (Fig. 2F).
Unlike what was observed with clozapine, total cellular FLAG-D2R levels were significantly lower in aripiprazole-treated cells compared with all other treatments (Fig. 2E). Consequently, cell-surface levels of FLAG-D2R after aripiprazole treatment were significantly higher than with clozapine when normalized against total cell-surface receptor. These data (Fig. 2, D–F) suggest that different mechanisms account for why clozapine and aripiprazole produced similar weak enhancements of surface D2R expression, compared with other APDs (Fig. 2D); aripiprazole did not significantly increase total D2R (Fig. 2E), whereas all other APDs, including clozapine, did.
We also determined that whereas the APD-mediated promotion of D2R cell-surface expression was significantly enhanced as the total cellular expression level of D2R was increased, the differential actions of haloperidol and clozapine for up-regulating cell-surface D2R was preserved (i.e. the increase in cell-surface D2R produced by clozapine treatment as a fraction of that produced by haloperidol was not changed) (Fig. S2A).
APD-mediated up-regulation of cell-surface receptors depends upon receptor binding
Neither haloperidol nor clozapine treatment up-regulated cell-surface or total cellular levels of the mutant D2R, D2RD114A (Fig. S3, A and B), which does not bind agonists or antagonists (29). Haloperidol and clozapine do not bind β2-adrenergic receptor (β2AR) or μ-opioid receptors (MOR) with significant affinity, and neither haloperidol nor clozapine produced cell-surface up-regulation of β2AR (Fig. S4A) or MOR (Fig. S4B). Taken together, the data indicate that APD-mediated up-regulation of cell-surface receptors requires direct receptor binding.
Both haloperidol and clozapine have nanomolar affinities (KD of ∼120 and 9 nm, respectively) for the 5-HT2A serotonin receptor (5, 30). Interestingly, both haloperidol and clozapine up-regulated cell-surface levels of the 5-HT2A receptor (Fig. S4C). With 5HT2A, however, clozapine treatment produced significantly greater enhancement of cell-surface receptor expression than the maximum that could be produced by haloperidol (Fig. S4C). Thus, the experiment with the 5-HT2A receptor further illustrates that the lower efficacy of clozapine for enhancing cell-surface expression of target receptors is specific to D2R.
APDs up-regulate cell-surface D2R by binding to an intracellular D2R pool
For every APD for which we generated concentration–response curves (Figs. 2C and 3), the potency for increasing cell-surface D2R expression was observed to be lower than their previously determined affinities for D2R (16, 17, 31). In other words, the concentration required to produce ∼50% of the maximal cell-surface D2R up-regulation response was higher than the concentration that binds 50% of cell-surface D2R. The low potency with respect to binding affinity is surprising because basic pharmacological principles suggest that concentration–response curves for GPCR-mediated cellular responses should either 1) coincide with the binding curve for the receptor ligand producing the response or 2) be shifted to the left of the binding curve, due to signal amplification that can occur with G protein pathways (32). The -fold difference between affinity for D2R and potency for up-regulating D2R varied widely between the different APDs, differing from 10-fold for olanzapine and 100-fold for haloperidol (Fig. S5A, y axis). Interestingly, however, there was a correlation between the cell permeability of the different APDs, as determined previously by Dos Santos Pereira et al. (33), and the -fold difference between potency to increase cell-surface expression and affinity (Fig. S5A). In other words, the more membrane-permeable an APD, the lower the difference between the potency for increasing cell-surface D2R expression and D2R affinity. These data are consistent with the hypothesis that APD-mediated D2R cell-surface up-regulation is produced not by the APD molecules present in the extracellular solution that bind to extracellular D2R binding sites. Instead, they suggest that APD-mediated D2R cell-surface up-regulation is driven by the smaller proportion of the drug that penetrates the cell membrane and can diffuse into the cell to bind to an intracellular pool of D2R.
Figure 3.

Effect of coexpression of OCT1 on the actions of membrane-impermeable APDs in enhancing cell-surface D2R expression. A, effect of coexpression of the OCT1 transporter on the concentration–response curves for amisulpride-mediated enhancement of cell-surface D2R expression. HEK293T cells transiently expressing FLAG-D2R, either alone or co-expressing the OCT1 transporter, were treated (24 h) with the indicated concentrations of amisulpride. The amisulpride-mediated enhancement (over vehicle) of cell-surface D2R was quantified by probing the intact cells with anti-FLAG antibody and is plotted as a percentage of the response at 10 μm, the maximum amisulpride concentration that was tested (mean ± S.E. (error bars), n = 15). The percentage of maximum response with OCT1 was significantly greater at all of the sub-10 μm concentrations of amisulpride tested (t test, p < 0.0001). B, effect of coexpression of the OCT1 transporter on the concentration–response curves for tiapride-mediated enhancement of cell-surface D2R expression. Concentration–response curves for tiapride-mediated enhancement of cell-surface D2R expression were generated and plotted as in A (mean ± S.E., n = 15–16).
To test this hypothesis, we examined the effect of co-expression of the organic cation transporter, OCT1 (33), on the cell-surface D2R up-regulation actions of two APDs, amisulpride and tiapride, that have low membrane permeability. Amisulpride is a substrate for OCT1 (33), and we found that coexpression of OCT1 significantly shifted the concentration–response curve for amisulpride-mediated cell-surface D2R up-regulation to the left (Fig. 3A). Hence, we infer that amisulpride acted at an intracellular site to up-regulate cell-surface D2R, because enhancing transport of amisulpride into the cell significantly increased the potency for producing the response. Tiapride is not a substrate for OCT1, and co-expression of OCT1 had no effect on the tiapride-mediated cell-surface D2R up-regulation concentration curve (Fig. 3B). Furthermore, we may conclude that the requirement of the various APDs to cross the cell membrane to bind to an intracellular pool of D2R is why higher concentrations of drug are required to enhance cell-surface D2R than would be predicted by their affinity for D2R.
Whereas we observed a correlation between the cell permeability of different APDs and the -fold difference between potency to increase cell surface expression and affinity (Fig. S5A), we saw no such correlation between cell permeability and the efficacy of different APDs to increase cell surface expression (Fig. S5B). Together, these data indicate that whereas the cell permeability of APDs influences the potency with which they increase cell surface expression, it does not explain the different efficacies of the APDs for up-regulating cell-surface D2R. Additionally, correlation analysis using APD-D2R–binding parameters taken from Sykes et al. (16) suggests that neither affinity nor dissociation or association rates for D2R correlate with the low efficacy of clozapine for up-regulating cell-surface D2R (Fig. S5, C–E, respectively).
APD-mediated enhancement of cell-surface D2R is produced by enhancement of cell-surface insertion rates of D2R rather than via decreased cell-surface removal of the receptor
We next wanted to evaluate whether the APD-mediated enhancement of cell-surface D2R was a consequence of the enhanced rate of receptor insertion into or decreased loss of receptor from the plasma membrane. Toward this end, we utilized a previously validated pulse-chase protocol (34) that monitors the loss of a pre-biotinylated cell-surface pool of an extracellular N-terminal HaloTag-D2R construct. Cell surface–expressed HaloTag-D2R was specifically pre-biotinylated using a membrane-impermeable HaloTag-PEG-Biotin reagent (35), and then cells were treated with APDs. The relative levels of HaloTag-PEG-Biotin–labeled D2R specifically remaining at the cell surface after APD treatment were then quantified. Under these conditions, receptors that were newly inserted into the cell surface during APD treatment were not detected, and only pre-biotinylated receptors that remained at the cell surface or were internalized and reinserted during APD treatment were measured. Consequently, the relative levels of biotinylated D2R remaining at the cell surface after APD treatment are a specific measure of the loss of cell-surface D2R that occurs during APD treatment. We compared the actions of three APDs, haloperidol, aripiprazole, and clozapine, at a concentration (10 μm, 24 h) that was shown to produce a plateau in the cell-surface D2R up-regulation response for each of the APDs and thus allows for making a valid comparison between the APDs. After either haloperidol or clozapine treatment, levels of pre-biotinylated D2R remaining at the cell surface were not significantly different from vehicle or from each other (Fig. 4). Thus, we may conclude that the enhancement of cell-surface D2R produced by haloperidol treatment compared with either vehicle or clozapine was a consequence of an enhanced rate of insertion of new D2R into the plasma membrane and not due to a relatively decreased loss of receptor during APD treatment.
Figure 4.
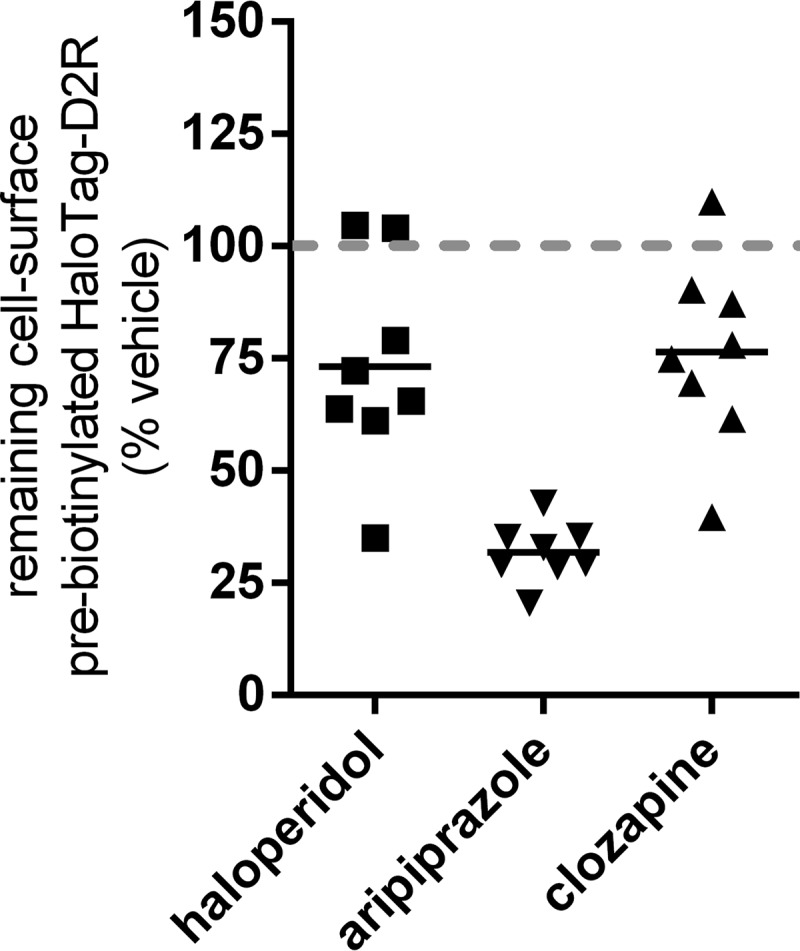
Effect of APD treatment specifically on the removal of cell-surface D2R from the plasma membrane examined via pulse-chase labeling with a membrane-impermeable biotinylation reagent. HaloTag-D2R, a D2R construct with an N-terminal extracellular HaloTag insertion, was transiently expressed in HEK293T cells, and the membrane-impermeable HaloTag-PEG-Biotin reagent was used to specifically biotinylate and pulse-label the cell-surface pool of HaloTag-D2R. After washing off unreacted HaloTag-PEG-Biotin reagent, cells were treated with the indicated APDs (10 μm, 24 h), and then the biotinylated HaloTag D2R remaining at the surface after APD treatment was quantified by probing the intact, nonpermeabilized cells with HRP-conjugated streptavidin. The levels of biotinylated receptor that remained at the surface after aripiprazole treatment were significantly lower (n = 8, Tukey) than that observed after vehicle (p < 0.0001), haloperidol (p < 0.005), or clozapine treatment (p < 0.001).
Aripiprazole treatment, however, significantly lowered the levels of pre-biotinylated D2R that remained at the cell surface (Fig. 4). Even though aripiprazole significantly increased the rate at which D2R was lost from the cell surface with respect to clozapine (∼2.5-fold; Fig. 4), the cell-surface D2R levels after aripiprazole and clozapine treatment were similar (Fig. 2D). Therefore, we can conclude that to compensate for enhanced loss of D2R during aripiprazole treatment (Fig. 4), the rate of insertion of D2R into the plasma membrane was also enhanced relative to that occurring during clozapine treatment or with vehicle. These data further emphasize that different mechanisms account for why clozapine and aripiprazole produced similar weak enhancements of surface D2R expression, compared with other APDs; lower levels of surface D2R after clozapine treatment are due to lowered rates of D2R plasma-membrane insertion, whereas lower levels of surface D2R after aripiprazole treatment are due to enhanced removal of cell-surface D2R. A simplified schematic of our interpretation of the above data can be found in the supporting material (Fig. S6)
Aripiprazole-mediated enhancement of the rate of removal of cell-surface D2R is not unexpected; aripiprazole, but neither clozapine nor haloperidol, is a partial agonist at D2R with respect to β-arrestin recruitment and will engage processes such as endocytosis that enhance the rate of internalization and down-regulation of D2R (36). Hence, these data also provide an explanation for why after aripiprazole treatment, total cellular D2R levels, although not significantly different from vehicle, were lower than that observed after treatment with other APDs (Fig. 2E).
In control experiments, we utilized the above described pulse-chase protocol involving pre-biotinylation of cell-surface HaloTag-D2R with membrane-impermeable HaloTag-PEG-Biotin to show that the differential action of haloperidol and clozapine on enhancing cell-surface D2R expression was preserved for the HaloTag-D2R construct and could be discerned via labeling cell-surface HaloTag-D2R with HaloTag-PEG-Biotin reagent after APD treatment (Fig. S7A). In another control experiment, we observed that dopamine treatment (10 μm, 45 min) significantly accelerated the removal of HaloTag-D2R from the cell surface (Fig. S7B). These control experiments indicate that the HaloTag-D2R construct 1) behaved similarly to WT D2R with respect to APD-mediated enhancement of cell-surface D2R and 2) was functional because the receptor construct responded to dopamine and underwent dopamine-induced internalization.
The differential actions of clozapine versus other APDs to promote cell-surface D2R expression are amplified in an intracellularly retained D2R mutant, D2RW160A, and a glycosylation-deficient mutant, D2RN5,N17,N23→Q
We next asked whether differential effects of APDs on protein folding could contribute to their differential enhancement of cell-surface D2R expression. Trp-160 is a tryptophan residue in the fourth transmembrane region of D2R that is highly conserved across class A GPCRs. This tryptophan participates in interactions that stabilize β2AR (37), and mutation of this residue leads to partial misfolding and endoplasmic reticulum (ER) retention of adrenergic receptors (38, 39).
Unlike what we observed with WT receptors, expression of the D2RW160A mutant construct in cells resulted in no detectable expression of the mutant at the cell surface (Fig. 5A). Notably, treatment of cells with haloperidol, fluphenazine, and droperidol produced a robust receptor-specific cell-surface signal (Fig. 5A). Clozapine treatment produced significantly less cell-surface receptor signal than all tested APDs (Fig. 5A), and the cell-surface levels were not significantly different from vehicle. Whereas total cellular D2RW160A was detected with anti-FLAG antibody, neither haloperidol nor clozapine significantly altered the total cellular levels of the mutant receptor (Fig. 5B).
Figure 5.

Effect of APD treatment on the cellular expression of trafficking-impaired D2R mutant constructs. A, effect of APD treatment on the cell-surface expression of the trafficking-deficient D2R mutant, D2RW160A. HEK293T cells transiently transfected with cDNA for the FLAG-tagged D2RW160A mutant construct, FLAG-D2RW160A, were treated with the indicated APDs (10 μm, 24 h). The relative levels of cell-surface receptor were then quantified by probing intact cells with anti-FLAG antibody and are reported as a percentage of the signal from vehicle-treated cells. The signal for cell-surface receptor after treatment with all APDs except clozapine was significantly increased (n = 8, Tukey; p < 0.01 for aripiprazole, p < 0.001 for olanzapine, p < 0.0001 for all other APDs). B, effect of APD treatment on the total cellular expression of D2R mutant, D2RW160A. HEK293T cells transiently expressing a FLAG-tagged D2RW160A mutant construct were treated with the indicated APDs (10 μm, 24 h), and total cellular D2RW160A levels are reported as a percentage of the signal from vehicle-treated cells (n = 3). C, effect of APD treatment on the surface expression of the D2R glycosylation-deficient mutant, D2RN5,N17,N23→Q. HEK293T cells transiently expressing a FLAG-tagged D2RN5,N17,N23→Q mutant construct, FLAG-D2RN5,N17,N23→Q, were treated with the indicated APDs (10 μm, 24 h), and the relative levels of cell-surface receptor were quantified as in A. The signal for cell-surface receptor after haloperidol treatment was significantly higher than in untransfected cells or cells treated with either vehicle or clozapine (n = 16, Tukey, p < 0.0001). D, effect of APD treatment on the total cellular expression of D2RN5,N17,N23→Q. HEK293T cells transiently expressing the FLAG-tagged D2RN5,N17,N23→Q mutant construct, FLAG-D2RN5,N17,N23→Q, were treated with the indicated APDs (10 μm, 24 h), and total cellular D2RN5,N17,N23→Q levels are reported as a percentage of the signal from vehicle-treated cells (n = 16). Error bars, S.E.
We then investigated whether APD treatment could mimic protein-chaperoning effects for a glycosylation-deficient D2R mutant, D2RN5,N17,N23→Q. N-Linked glycosylation of D2R is required for appropriate interaction with the chaperone protein, calnexin (40), intracellular trafficking, and plasma membrane insertion (41, 42). Treatment with haloperidol but not clozapine produced significant enhancement of cell-surface D2RN5,N17,N23→Q expression (Fig. 5C), and neither haloperidol nor clozapine treatment significantly altered the total cellular levels of D2RN5,N17,N23→Q (Fig. 5D).
Interestingly, for WT D2R, the clozapine-mediated enhancement of cell-surface D2R as a fraction of haloperidol-mediated enhancement ranged from 30 to 50%. The same clozapine versus haloperidol ratio for enhancement of cell-surface receptor levels was only 17 and 8% for the misfolding mutant, D2RW160A, and the glycosylation-deficient mutant, D2RN5,N17,N23→Q, respectively.
Cellular responses to D2R may be elicited through the activation of pertussis toxin–sensitive Gi/o G proteins (43). We explored the necessity of Gi/o G protein activation for APD-mediated enhancement of cell-surface D2R expression by inactivating these G proteins through coexpression of the catalytic subunit of pertussis toxin (Fig. S8). Differential up-regulation of cell-surface D2R by haloperidol and clozapine was conserved even after inactivating these G proteins (Fig. S8). These data indicate that activation of these G proteins is not important in the mechanisms for the differential APD-mediated enhancement of cell-surface D2R expression that we have reported here.
Treatment with haloperidol, but not clozapine, decreased cellular colocalization of D2R with an ER marker, Sec61
Because misfolded GPCRs may be retained in the ER (44, 45), we sought to determine whether the intracellular pool of D2R that is translocated to the cell surface by the various APDs originated in the ER. Consistent with earlier findings (41, 46–51), we found that a significant fraction of D2R expressed in cells is found in the ER, as evidenced by colocalization with the general ER marker, mCh-Sec61-β (Fig. 6A). Treatment with haloperidol, but not clozapine, resulted in 1) the emergence of a ring of D2R signal that coincided with the cell boundaries (Fig. 6A) and 2) significantly decreased localization between D2R and mCh-Sec61-β signals (Fig. 6B). These results confirm that APDs can differentially enhance trafficking of D2R from the ER to the plasma membrane.
Figure 6.
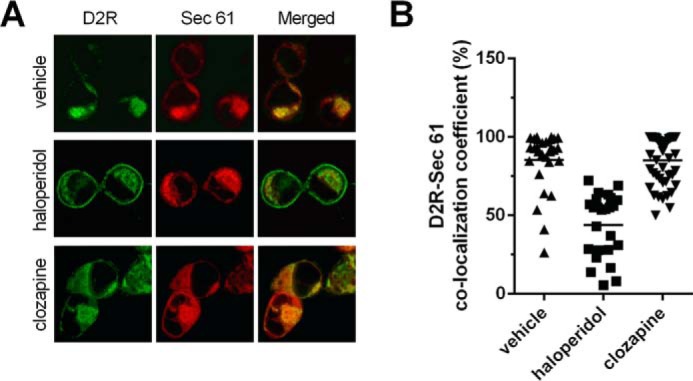
Effect of haloperidol and clozapine treatments on the cellular localization of D2R. A, confocal microscopy images of cellular distribution of D2R. HEK293T cells transiently coexpressing FLAG-D2R and mCh-Sec61-β fusion, a marker for the ER, were treated with haloperidol or clozapine (10 μm, 24 h). Representative images of D2R cellular distribution (green, left column) and Sec61 cellular distribution (red, central column) and the level of D2R-Sec61 colocalization (yellow, right column) following treatment with vehicle (top row), haloperidol (central row), and clozapine (bottom row) are depicted. Each row represents confocal microscope scans of the same field. B, quantification of the extent of colocalization between D2R and mCh-Sec61-β fusion. The graph depicts the colocalization coefficient, quantified in cells coexpressing D2R and mCh-Sec61-β fusion and defined as the number of pixels positive for both D2R and Sec61, expressed as a percentage of pixels positive for D2R, after APD treatment. The colocalization coefficient of D2R colocalized with Sec61 was significantly reduced after haloperidol treatment when compared with either vehicle or clozapine treatment (n = 25–46 cells, Tukey, p < 0.001).
Discussion
Whereas all APDs available to treat schizophrenia bind D2R at therapeutic concentrations (1–4), the cellular connections between binding D2R and suppression of psychotic symptoms are not clear. Therefore, elucidating all of the molecular and cellular actions of APDs at D2R are critical first steps toward understanding the mechanism of action of APDs. Furthermore, explaining the unique therapeutic profile of clozapine in schizophrenia requires the identification of molecular and cellular actions produced by clozapine but not by any other APD. However, until now, such unique characteristics for clozapine have not been reported. The data presented here provide novel findings relevant to the above challenges.
Here, we demonstrate that APDs bind to an intracellular pool of D2R and, even in the absence of dopamine, differentially promote trafficking of D2R from the ER to the plasma membrane. A pharmacological chaperone or pharmacoperone (44, 45) has been defined as a small molecule that enters cells and serves as a “molecular scaffold” to promote folding of proteins into appropriate tertiary structures and allows for trafficking of target proteins out of the ER. Hence, our data suggest that APDs may be acting with different efficacies as pharmacoperones at D2R.
Furthermore, an extrapolation from the concentration–response curve for haloperidol suggests that therapeutic plasma concentrations (52) of 30–40 nm will significantly enhance cell-surface D2R levels after just 24 h of treatment, signifying that the cell-surface D2R up-regulation actions of the APDs reported here may have direct clinical relevance.
APDs, except for aripiprazole, also increased total cellular levels of D2R (Fig. 2E). The APD-mediated enhancement of total cellular D2R levels could have resulted from enhanced transcription or translation or decreased degradation of the receptor. However, the ratio of cell-surface D2R up-regulation to total cellular D2R up-regulation was different between APDs (Fig. 2F), suggesting that the disparity in the APD-mediated up-regulation of cell-surface D2R was a consequence of post-translational mechanisms.
Clozapine stood out in a number of ways in our study. All APDs, except for aripiprazole, produced greater cell-surface D2R up-regulation than the maximum produced by clozapine, and thus we can conclude that they have higher efficacy than clozapine for up-regulating cell-surface D2R.
Whereas cell-surface levels of WT D2R after aripiprazole treatment were not significantly different from that after clozapine, the cell-surface D2R levels normalized against total cellular D2R were the lowest for clozapine (Fig. 2F). Furthermore, the reason that cell-surface levels of D2R after aripiprazole treatment were not significantly higher than that observed after clozapine is that aripiprazole enhances both the rate of D2R removal from and insertion into the plasma membrane, with respect to clozapine (Fig. S6). In other words, the biochemical and pharmacological reasons for why D2R cell-surface levels are low after clozapine and aripiprazole treatments are different.
The pulse-chase experiments with HaloTag-D2R also demonstrated that haloperidol treatment enhanced cell-surface expression relative to clozapine by enhancing the rate of cell-surface insertion of D2R rather than by decreasing the rate of removal (Fig. 4). It is not unreasonable then to infer, from the actions of both haloperidol and aripiprazole at HaloTag-D2R, that APDs, relative to clozapine, are enhancing the rate of cell-surface insertion of the receptor. A simple schematic illustrating this model of APD actions has been provided in the supporting material (Fig. S6).
Hence, APDs may be acting as pharmacoperones because they are binding intracellularly to D2R to enhance the rate of insertion of D2R into the plasma membrane. If so, clozapine is unique among the APDs because it has the lowest efficacy for acting as a pharmacoperone at D2R. Additional evidence for the suggestion include the observation that haloperidol, but not clozapine, decreased the retention of D2R in the ER (Fig. 6).
Even more evidence for supporting the above hypothesis follows from our observations with the intracellularly retained misfolding D2R mutant, D2RW160A, and the glycosylation-deficient mutant, D2RN5,N17,N23→Q. As described under “Results,” N-linked glycosylation of D2R is required for appropriate interaction with the chaperone protein, calnexin (40), intracellular trafficking, and plasma membrane insertion (41, 42). APDs that can act more efficiently as chemical chaperones have the potential to exert a greater role in helping to fold and transport these mutant D2R constructs to the cell surface compared with WT D2R. As expected, the differential actions of clozapine versus other APDs to promote cell-surface receptor expression were amplified in both D2RW160A and D2RN5,N17,N23→Q (Fig. 5).
Although cell membrane permeability of the various APDs contributed to the potency with which they promoted cell surface expression, it was not a predictor of the magnitude of the effect (Fig. S5, A and B, respectively). Clozapine has a relatively low affinity for D2R and a faster dissociation rate as compared with many other APDs (8, 15–17). Indeed, this fast dissociation rate has been proposed to underlie its relative lack of propensity to cause motor disorders (8). More recently, the association rate of APDs at D2R has been proposed to predict extrapyramidal side effects (16). However, correlation analysis suggests that neither affinity nor dissociation or association rates for D2R can explain the low efficacy of clozapine for up-regulating cell-surface D2R (Fig. S5, C–E, respectively). Thus, the low efficacy of clozapine for up-regulating cell-surface D2R appears to be a consequence of a novel and intrinsic property of the clozapine–D2R complex itself.
We investigated whether APD-mediated enhancement of cell-surface D2R levels resulted in increased levels of dopamine-mediated activation of D2R-coupled G proteins using a previously described bioluminescence resonance energy transfer (BRET)-based assay (53). Whereas we were able to generate dopamine concentration–response curves at D2R in drug-naive cells (54), we were unable to elicit dopamine responses in cells that were treated with APDs, including haloperidol and clozapine, under conditions required to produce significant up-regulation of cell-surface D2R. In other words, long-term treatment with APDs produced long-lasting antagonism, which could not be overcome with dopamine concentrations of up to 10 μm even 4 h after APD wash-off. One explanation for this result is that long-term APD treatment produces intracellular accumulation of the APDs so that the effective concentration of the APDs at D2R in the cell membrane remains high long after the APDs have been washed out from the extracellular buffer. Hence, the direct cell-biological and functional consequences of the APD-mediated enhancement of cell-surface D2R remain to be established.
Increases in brain D2R after chronic treatment with APDs have been suggested as the reason for iatrogenic psychoses and for resistance of schizophrenia patients to pharmacotherapy over time (19, 20). APD-induced up-regulation of D2R has also been implicated in TD (22), suggesting that chronic D2R up-regulation can produce permanent adverse alterations in D2R-expressing neurons. The data presented here indicate that APD-mediated D2R up-regulation may be caused, independently of dopamine antagonism, by the chaperone activity of APDs that act not only to increase total receptor expression but also to increase the relative proportion of D2R at the cell surface.
Pharmacoperones have been identified for a number of GPCRs, and their therapeutic utility has begun to be explored (55) (e.g. the use of pharmacological chaperones that act to restore plasma membrane localization of vasopressin receptor mutants as a treatment for congenital nephrogenic diabetes insipidus) (56). However, in the case of D2R, the pharmacoperone activity we observed for the APDs may in fact be deleterious. Thus, the low efficacy of clozapine as a pharmacoperone at D2R may be a hitherto unappreciated cellular process that may contribute to its unparalleled efficacy in treatment-refractory schizophrenia (5, 7, 11) and low risk for producing EPS (5–7).
An alternative possibility is that the unique efficacy of clozapine does not directly follow from the low efficacy for up-regulating cell-surface D2R. Instead, the above phenomenon that we have documented here in in vitro systems serves only as a reporter of underlying physiologically relevant cellular mechanisms that are conserved in the brain and contribute to the efficacy of clozapine. In other words, it is the underlying pharmacological and biochemical actions of clozapine at D2R, which are responsible for the low efficacy for up-regulating cell-surface D2R in vitro, that are also what contributes to the special efficacy of clozapine in the clinic. Most importantly, however, our documentation of unique cellular actions of clozapine at D2R suggests that it is not necessary to invoke the existence of a cellular target other than D2R to explain clozapine's unique therapeutic profile.
Some of the latest efforts in the discovery of novel APDs have been aimed at finding compounds that mimic the spectrum of clozapine-binding affinities at other receptors, such as D3, 5-HT2A, 5-HT2C, and 5-HT6 (57). However, close to 50 years after the discovery by Seeman et al. (2) and Snyder and co-workers (1) that D2R is the common target of all clinically available APDs, our data suggest that a new generation of APDs may yet be discovered by focusing on D2R as a central target. In addition, these data are the first to indicate that pharmacoperone activity might serve to define and differentiate between the clinical characteristics of an important class of drugs.
Experimental procedures
All reagents were purchased from Sigma-Aldrich or Thermo Fisher Scientific unless otherwise indicated.
cDNA constructs
The N-terminal extracellular FLAG epitope–tagged D2-dopamine receptor long isoform (FLAG-D2R) (58), the OCT1 transporter (59), and the ER marker, mCh-Sec61-β (60), have been described previously. The FLAG-D2SR fusion was constructed by fusing the N-terminal influenza hemagglutinin signal sequence (MKTIIALSYIFCLVFA) followed by the FLAG epitope (DYKDDDDA) and a two-amino acid linker (TV) to the N terminus of the human D2SR. The HaloTag-D2R fusion consisted of the N-terminal influenza hemagglutinin signal sequence (MKTIIALSYIFCLVFA) followed by the FLAG epitope (DYKDDDDA), a two-amino acid linker (TV), and the HaloTag protein (35) (Promega, Madison, WI), tethered to the N terminus of D2R via a three-amino acid linker (AAG). Thus, the FLAG-HaloTag-D2R construct consisted of, in order from the N to the C terminus, the influenza hemagglutinin signal sequence epitope, FLAG epitope, TV linker, HaloTag, AAG linker, and human D2R long isoform. For the FLAG-D2RW160A and the FLAG-D2RN5,N17,N23→Q mutants, the mutations refer to mutations in residues numbered as in the nontagged WT human D2R long form.
Cell culture and cDNA transfections
HEK293T cells and the prolactin-secreting pituitary-derived MMQ cells (American Type Culture Collection, ATCC, Manassas, VA) were cultured according to instructions provided by ATCC. Transfections were carried out, using Lipofectamine LTX according to the manufacturer's guidelines. Transfected DNA amounts were kept constant between groups using empty pcDNA 3.1 Zeo(+) mammalian expression vector.
Assessment of effects of APD treatment on cell-surface levels of D2R endogenously expressed in the pituitary-derived, prolactin-secreting MMQ cell line
MMQ cells were seeded in a 12-well plate at a density of 1.5 × 106 cells/well and treated with 10 μm haloperidol, clozapine, or vehicle. To increase expression of native D2R, the cells were concurrently treated with 100 nm 9-cis- and all-trans-retinoic acid (61). Following APD treatment (48 h), proteins expressed at the surface of the cultured cells were specifically biotinylated, as described previously (50), by incubation with EZ-LinkTM Sulfo-NHS-LC-Biotin reagent (Pierce, Thermo Fisher Scientific), a membrane-impermeable biotinylation reagent that reacts with primary amines of peptides projecting out into the extracellular space. The cells were treated with the EZ-LinkTM Sulfo-NHS-LC-Biotin reagent at a concentration of 1 mg/ml for 30 min at 4 °C according to the protocol provided by the manufacturer. Following incubation, excess biotinylation reagent was removed and quenched by washing twice with PBS containing glycine (100 mm, 4 °C). Cells were then lysed in lysis buffer consisting of 2% (v/v) Triton X-100 in PBS with SigmaFast protease inhibitor mixture with EDTA for 1 h at 4 °C. Cell lysates were then centrifuged (10,000 × g for 5 min at 4 °C) to remove insoluble material, and biotinylated cell-surface proteins were extracted from the cell lysate supernatant by adding 50 μl of streptavidin-agarose resin (Pierce, Thermo Fisher Scientific) and incubating with shaking for 1 h at 4 °C. Prior to the addition, the streptavidin-agarose resin was washed three times with wash buffer (150 mm NaCl, 6 mm EDTA, 50 mm Tris-HCl, 0.1% (v/v) Triton X-100, pH 7.5, 20 °C). The agarose resin and bound proteins were then separated via centrifugation at 8,000 × g for 1 min at 20 °C, and washed three times in wash buffer. 50 μl of SDS-urea sample buffer (2% (w/v) SDS, 0.01% (w/v) bromphenol blue, 8 m urea, 20 mm DTT, 50 mm Tris-HCl, pH 6.8) was then added to each sample, and bound proteins were eluted by incubation in a boiling water bath for 10 min. Proteins were resolved using SDS-PAGE and transferred to polyvinylidene fluoride membranes (Imobilon-FL, EMD Millipore, Billerica, MA) by Western blotting. The native D2R was detected on the Western blots by probing with a previously described and validated (25), affinity-purified rabbit polyclonal sera (1:500 dilution) directed against a peptide sequence corresponding to amino acids 246–305 (third intracellular loop) of mouse D2R. The antibody was previously validated by demonstrating the disappearance of signal in brain tissue from D2R knockout mice. The blots were then probed with an appropriate horseradish peroxidase (HRP)-conjugated secondary antibody. SuperSignal West Femto chemiluminescent substrate (Pierce, Thermo Fisher Scientific) was layered over the blot, and the HRP-catalyzed chemiluminescent signal was detected using a Chemidoc XRS Molecular Imager (Bio-Rad). Subsequently, total cell-surface protein in each lane of the blot was visualized by first incubating blots with 30% (v/v) hydrogen peroxide for 30 min at 37 °C to inactivate HRP conjugated to the secondary antibody and then reprobing the blots with HRP-conjugated streptavidin. The D2R signal in each lane was normalized to total cell-surface protein signal in that lane.
Measurement of surface and total cellular FLAG-tagged GPCR expression by ELISA
To evaluate the effect of APD treatment on the cell-surface expression of various GPCR constructs, we employed a previously well-characterized ELISA-based protocol that utilized antibodies to specifically label N-terminal extracellular FLAG-tagged receptors expressed on the surface of fixed nonpermeabilized cells (62). HEK293T cells were seeded in a 96-well plate (5 × 104 cells/well) and transfected with cDNA plasmids containing the indicated FLAG-tagged receptor. 24 h post-transfection, cells were treated with the specified drugs or corresponding vehicle at the indicated concentrations and for the indicated durations. After drug treatment, cells were fixed with 4% (v/v) methanol-free paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in PBS for 15 min at 4 °C and then washed twice with PBS (4 °C). Wells were blocked for 1 h at 4 °C with 5% (w/v) nonfat milk dissolved in PBS. The levels of FLAG-tagged surface receptor were then assessed by probing with HRP-conjugated mouse monoclonal anti-FLAG M2 antibody (1:5,000 dilution in 5% (w/v) nonfat milk dissolved in PBS) for 45 min at 20 °C and then washed twice in PBS (20 °C). SuperSignal West Femto chemiluminescent substrate was added to each well, and the luminescence signal was measured using a multiwell plate Glomax luminometer (Promega).
Total cell FLAG-tagged cellular receptor was evaluated in a similar manner, except that cells were fixed and permeabilized in 100% methanol for 10 min at −20 °C to allow antibody access to intracellular receptor. Subsequent washing, blocking, and antibody-probing steps were identical to those described above.
Assessment of total cellular levels of FLAG-D2RW160A by Western blotting
We were unable to detect total cellular levels of the trafficking-deficient D2R mutant construct, FLAG-D2RW160A, over background, using the ELISA method described above. A hypothesis for the inability to detect the construct is that the misfolded construct aggregates in a manner that precludes detection of the FLAG epitope by the anti-FLAG antibody in the methanol-fixed cells. Hence, the relative total cellular levels of this construct were quantified by Western blotting.
Cells were solubilized in SDS-urea sample buffer, and proteins were resolved using SDS-PAGE, and protein expression was evaluated via Western blotting. FLAG-tagged receptor was detected on Western blots by probing with HRP-conjugated anti-FLAG M2 antibody (1:5,000 dilution in 5% (w/v) nonfat milk in PBS, 1 h, 20 °C). The receptor signal in each lane was normalized against β-actin signal in that lane, which was detected by probing with mouse polyclonal anti-β-actin antibody (1:1,000 dilution in 5% (w/v) nonfat milk in PBS, 1 h, 20 °C) and an appropriate HRP-conjugated secondary. Prior to probing for β-actin, HRP conjugated to previously bound anti-FLAG M2 antibody was inactivated by incubation with 30% (v/v) hydrogen peroxide for 30 min at 37 °C.
Pulse-chase assay for specifically monitoring and comparing loss of cell-surface D2R during APD treatment utilizing the HaloTag-D2R construct
We utilized a modification of a previously validated protocol used to track endocytosis of GPCRs via pulse-chase biotinylation specifically of the cell surface–expressed GPCRs (34). HEK293T cells were seeded in 10-cm diameter culture dishes at a density of 2 × 106 cells/dish and transfected with HaloTag-D2R cDNA described above, which contains HaloTag inserted at the extracellular N terminus of D2R. HaloTag is a modified bacterial enzyme that can be specifically and covalently tagged by a wide variety of synthetic probes (35). 24 h post-transfection, cell-surface HaloTag-D2R was specifically biotinylated by incubating with the membrane-impermeable HaloTag-PEG-Biotin reagent (100 nm, 1 h, 37 °C, Promega) in the cell culture medium. Cells were then washed twice with PBS (20 °C) to remove unreacted Halotag-PEG-Biotin reagent, resuspended in Dulbecco's modified Eagle's medium via gentle pipetting, reseeded in a 96-well plate at a cell number of 160,000 cells/well, and treated with APDs as indicated. Following 24 h of APD treatment, cells were fixed in 4%(v/v) methanol-free paraformaldehyde (in PBS, 15 min, 4 °C) and then washed twice in PBS (20 °C). Wells were blocked with 3% (w/v) BSA in PBS for 45 min at 20 °C. Biotinylated HaloTag receptor remaining at the cell surface was detected using HRP-conjugated streptavidin (1:10,000 dilution in 3% (w/v) BSA dissolved in PBS). SuperSignal West Femto chemiluminescent substrate was added to each well, and the HRP-catalyzed luminescent signal was detected and quantified using the Glomax luminometer.
Confocal microscopy evaluation of the effect of APD treatment on the cellular localization of FLAG-D2R
Poly-d-lysine–coated glass coverslips were placed in the bottom of 12-well plates, and HEK293T cells were plated at a density of 1 × 105 cells/well and transfected with cDNA for constructs, FLAG-D2R, and the ER marker, mCh-Sec61-β, as indicated. 24 h post-transfection, cells were treated with APDs, again as indicated. Following APD treatment, cells were incubated with 100% methanol for 20 min at −20 °C to both fix and permeabilize the cells. Cells were washed three times in PBS (20 °C) and blocked in 5% (w/v) nonfat milk in PBS for 1 h at 20 °C under constant agitation. FLAG-D2R was detected by probing with a M2 anti-FLAG antibody (1:1,000 dilution in 5% nonfat milk in PBS, Sigma-Aldrich) overnight at 4 °C. Cells were washed two times with PBS for 15 min at 20 °C and then incubated for 1 h at 20 °C with anti-mouse Alexa Fluor 488–conjugated secondary antibody (1:5,000 dilution in 5% (w/v) nonfat milk in PBS, Molecular Probes/Thermo Fisher Scientific). Cells were imaged using a LSM 700 laser-scanning confocal microscope (Carl Zeiss, Peabody, MA). The two fluorophores, Alexa Fluor 488 (for D2R) and mCherry (mCh-Sec61-β), were excited at 488 and 555 nm, respectively, and visualized using emission spectral windows of 490–580 nm and >585 nm, respectively. We utilized the Zeiss Efficient Navigation (ZEN) software for image processing and colocalization analysis. The degree of colocalization of D2R with the ER marker, mCh-Sec61-β, was quantified using the Manders' colocalization coefficient. The fraction of D2R that colocalizes with mCh-Sec61-β was calculated by determining the fraction of the total number of D2R-positive pixels that were also positive for mCh-Sec61-β. Cutoff values for determining whether pixels were positive or negative for fluorescent signals were established by analysis of scatter plots from cells transiently expressing only one of the two colocalizing proteins, according to previously published procedures (63), and as outlined in the ZEN software manual.
Graphing and statistical analysis
Statistical analysis including “t” test and one-way analysis of variance were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, Inc., La Jolla, CA). Graphs were plotted using the same program. In the scatter plots, bars represent the mean values, and the whiskers in the box plots indicate minimum and maximum values of the indicated data.
Author contributions
J. M. S., J. C. O., J. C., J. L. C., J. R. L., J. A. J., and A. K. conceptualization; J. M. S., C. M. I., J. C. O., D. J. K., and A. K. data curation; J. M. S., J. C. O., J. R. L., and A. K. formal analysis; J. M. S. and A. K. supervision; J. M. S., C. M. I., J. C. O., J. A. C., T. J. A., D. J. K., and A. K. investigation; J. M. S., C. M. I., J. C. O., and A. K. visualization; J. M. S., J. C. O., J. C., J. L. C., and A. K. methodology; J. M. S., J. R. L., and A. K. writing-original draft; J. M. S., J. R. L., J. A. J., and A. K. writing-review and editing; A. K. resources; A. K. funding acquisition; A. K. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Nevin Lambert (Medical College of Georgia, Augusta University) for the gift of the cDNA constructs for monitoring G protein activation in cells with BRET and Dr. Jed Lampe (University of Kansas Medical Center) for the gift of OCT1 cDNA.
This work was supported in part by the Institutional Development Award (IDeA) Network for Biomedical Research Excellence from the NIGMS, National Institutes of Health, Grant P20GM103430. Abraham Kovoor is a co-founder and owns shares of Cortagenix, LLC, a neuroscience drug-discovery company. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S8.
- APD
- antipsychotic drug
- β2AR
- β2-adrenergic receptor
- BRET
- bioluminescence resonance energy transfer
- D2R
- dopamine D2 receptor(s)
- D2SR
- short D2R isoform
- ER
- endoplasmic reticulum
- HaloTag-D2R
- extracellular N-terminal HaloTag-D2R construct
- EPS
- extrapyramidal motor side effects
- GPCR
- G protein–coupled receptor
- HRP
- horseradish peroxidase
- HEK293T cells
- human embryonic kidney 293T cells
- MOR
- μ-opioid receptor(s)
- OCT1
- organic cation transporter 1
- TD
- tardive dyskinesia.
References
- 1. Creese I., Burt D. R., and Snyder S. H. (1976) Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192, 481–483 10.1126/science.3854 [DOI] [PubMed] [Google Scholar]
- 2. Seeman P., Lee T., Chau-Wong M., and Wong K. (1976) Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature 261, 717–719 10.1038/261717a0 [DOI] [PubMed] [Google Scholar]
- 3. Kapur S., and Mamo D. (2003) Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 1081–1090 10.1016/j.pnpbp.2003.09.004 [DOI] [PubMed] [Google Scholar]
- 4. Seeman P., Chau-Wong M., Tedesco J., and Wong K. (1975) Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc. Natl. Acad. Sci. U.S.A. 72, 4376–4380 10.1073/pnas.72.11.4376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meltzer H. Y. (2013) Update on typical and atypical antipsychotic drugs. Annu. Rev. Med. 64, 393–406 10.1146/annurev-med-050911-161504 [DOI] [PubMed] [Google Scholar]
- 6. Wenthur C. J., and Lindsley C. W. (2013) Classics in chemical neuroscience: clozapine. ACS Chem. Neurosci. 4, 1018–1025 10.1021/cn400121z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meltzer H. Y. (2012) Clozapine: balancing safety with superior antipsychotic efficacy. Clin. Schizophr. Relat. Psychoses 6, 134–144 10.3371/CSRP.6.3.5 [DOI] [PubMed] [Google Scholar]
- 8. Seeman P. (2014) Clozapine, a fast-off-D2 antipsychotic. ACS Chem. Neurosci. 5, 24–29 10.1021/cn400189s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lewis S., and Lieberman J. (2008) CATIE and CUtLASS: can we handle the truth? Br. J. Psychiatry 192, 161–163 10.1192/bjp.bp.107.037218 [DOI] [PubMed] [Google Scholar]
- 10. Kang X., and Simpson G. M. (2010) Clozapine: more side effects but still the best antipsychotic. J. Clin. Psychiatry 71, 982–983 10.4088/JCP.09com05497yel [DOI] [PubMed] [Google Scholar]
- 11. Siskind D., McCartney L., Goldschlager R., and Kisely S. (2016) Clozapine v. first- and second-generation antipsychotics in treatment-refractory schizophrenia: systematic review and meta-analysis. Br. J. Psychiatry 209, 385–392 10.1192/bjp.bp.115.177261 [DOI] [PubMed] [Google Scholar]
- 12. Leucht S., Cipriani A., Spineli L., Mavridis D., Orey D., Richter F., Samara M., Barbui C., Engel R. R., Geddes J. R., Kissling W., Stapf M. P., Lässig B., Salanti G., and Davis J. M. (2013) Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet 382, 951–962 10.1016/S0140-6736(13)60733-3 [DOI] [PubMed] [Google Scholar]
- 13. Friedman J. H. (2013) Parkinson disease psychosis: update. Behav. Neurol. 27, 469–477 10.1155/2013/645429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stahl S. M. (2013) Stahl's Essential Psychopharmacology: Neuroscientific Basis and Practical Applications, 4th Ed., p. 180, Cambridge University Press, Cambridge, UK [Google Scholar]
- 15. Kapur S., and Seeman P. (2000) Antipsychotic agents differ in how fast they come off the dopamine D2 receptors: implications for atypical antipsychotic action. J. Psychiatry Neurosci. 25, 161–166 [PMC free article] [PubMed] [Google Scholar]
- 16. Sykes D. A., Moore H., Stott L., Holliday N., Javitch J. A., Lane J. R., and Charlton S. J. (2017) Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D2 receptors. Nat. Commun. 8, 763 10.1038/s41467-017-00716-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sahlholm K., Zeberg H., Nilsson J., Ögren S. O., Fuxe K., and Århem P. (2016) The fast-off hypothesis revisited: a functional kinetic study of antipsychotic antagonism of the dopamine D2 receptor. Eur. Neuropsychopharmacol. 26, 467–476 10.1016/j.euroneuro.2016.01.001 [DOI] [PubMed] [Google Scholar]
- 18. Wisler J. W., Xiao K., Thomsen A. R. B., and Lefkowitz R. J. (2014) Recent developments in biased agonism. Curr. Opin. Cell Biol. 27, 18–24 10.1016/j.ceb.2013.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Samaha A.-N., Seeman P., Stewart J., Rajabi H., and Kapur S. (2007) “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J. Neurosci. 27, 2979–2986 10.1523/JNEUROSCI.5416-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki T., Kanahara N., Yamanaka H., Takase M., Kimura H., Watanabe H., and Iyo M. (2015) Dopamine supersensitivity psychosis as a pivotal factor in treatment-resistant schizophrenia. Psychiatry Res. 227, 278–282 10.1016/j.psychres.2015.02.021 [DOI] [PubMed] [Google Scholar]
- 21. Seeman P. (1988) Tardive dyskinesia, dopamine receptors, and neuroleptic damage to cell membranes. J. Clin. Psychopharmacol. 8, 3S–9S 10.1097/00004714-198808001-00002 [DOI] [PubMed] [Google Scholar]
- 22. Silvestri S., Seeman M. V., Negrete J. C., Houle S., Shammi C. M., Remington G. J., Kapur S., Zipursky R. B., Wilson A. A., Christensen B. K., and Seeman P. (2000) Increased dopamine D2 receptor binding after long-term treatment with antipsychotics in humans: a clinical PET study. Psychopharmacology (Berl.) 152, 174–180 10.1007/s002130000532 [DOI] [PubMed] [Google Scholar]
- 23. Judd A. M., and MacLeod R. M. (1991) Dopamine receptor and adrenoceptor agonists inhibit prolactin release from MMQ cells. Eur. J. Pharmacol. 195, 101–106 10.1016/0014-2999(91)90386-5 [DOI] [PubMed] [Google Scholar]
- 24. Ventra C., Meucci O., Grimaldi M., Scorziello A., Porcellini A., and Schettini G. (1995) Absence of D2S dopamine receptor in the prolactin-secreting MMQ pituitary clone: characterization of a wild D2L receptor coupled to native transduction mechanisms. J. Mol. Endocrinol. 14, 375–389 10.1677/jme.0.0140375 [DOI] [PubMed] [Google Scholar]
- 25. Trifilieff P., Rives M.-L., Urizar E., Piskorowski R. A., Vishwasrao H. D., Castrillon J., Schmauss C., Slättman M., Gullberg M., and Javitch J. A. (2011) Detection of antigen interactions ex vivo by proximity ligation assay: endogenous dopamine D2-adenosine A2A receptor complexes in the striatum. BioTechniques 51, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo W., Urizar E., Kralikova M., Mobarec J. C., Shi L., Filizola M., and Javitch J. A. (2008) Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. 27, 2293–2304 10.1038/emboj.2008.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klein Herenbrink C., Sykes D. A., Donthamsetti P., Canals M., Coudrat T., Shonberg J., Scammells P. J., Capuano B., Sexton P. M., Charlton S. J., Javitch J. A., Christopoulos A., and Lane J. R. (2016) The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun. 7, 10842 10.1038/ncomms10842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Monsma F. J. Jr., McVittie L. D., Gerfen C. R., Mahan L. C., and Sibley D. R. (1989) Multiple D2 dopamine receptors produced by alternative RNA splicing. Nature 342, 926–929 10.1038/342926a0 [DOI] [PubMed] [Google Scholar]
- 29. Han Y., Moreira I. S., Urizar E., Weinstein H., and Javitch J. A. (2009) Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat. Chem. Biol. 5, 688–695 10.1038/nchembio.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peprah K., Zhu X. Y., Eyunni S. V. K., Setola V., Roth B. L., and Ablordeppey S. Y. (2012) Multi-receptor drug design: haloperidol as a scaffold for the design and synthesis of atypical antipsychotic agents. Bioorg. Med. Chem. 20, 1291–1297 10.1016/j.bmc.2011.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scatton B., Cohen C., Perrault G., Oblin A., Claustre Y., Schoemaker H., Sanger D. J., Rouquier L., and Porsolt R. (2001) The preclinical pharmacologic profile of tiapride. Eur. Psychiatry 16, 29s–34s [DOI] [PubMed] [Google Scholar]
- 32. Arshavsky V. Y., and Burns M. E. (2014) Current understanding of signal amplification in phototransduction. Cell. Logist. 4, e29390 10.4161/cl.29390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dos Santos Pereira J. N., Tadjerpisheh S., Abu Abed M., Saadatmand A. R., Weksler B., Romero I. A., Couraud P.-O., Brockmöller J., and Tzvetkov M. V. (2014) The poorly membrane permeable antipsychotic drugs amisulpride and sulpiride are substrates of the organic cation transporters from the SLC22 family. AAPS J. 16, 1247–1258 10.1208/s12248-014-9649-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumagai H., Ikeda Y., Motozawa Y., Fujishiro M., Okamura T., Fujio K., Okazaki H., Nomura S., Takeda N., Harada M., Toko H., Takimoto E., Akazawa H., Morita H., Suzuki J., et al. (2015) Quantitative measurement of GPCR endocytosis via pulse-chase covalent labeling. PLoS One 10, e0129394 10.1371/journal.pone.0129394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Los G. V., Encell L. P., McDougall M. G., Hartzell D. D., Karassina N., Zimprich C., Wood M. G., Learish R., Ohana R. F., Urh M., Simpson D., Mendez J., Zimmerman K., Otto P., Vidugiris G., et al. (2008) HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382 10.1021/cb800025k [DOI] [PubMed] [Google Scholar]
- 36. Allen J. A., Yost J. M., Setola V., Chen X., Sassano M. F., Chen M., Peterson S., Yadav P. N., Huang X. P., Feng B., Jensen N. H., Che X., Bai X., Frye S. V., Wetsel W. C., Caron M. G., Javitch J. A., Roth B. L., and Jin J. (2011) Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U.S.A. 108, 18488–18493 10.1073/pnas.1104807108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanson M. A., Cherezov V., Griffith M. T., Roth C. B., Jaakola V.-P., Chien E. Y. T., Velasquez J., Kuhn P., and Stevens R. C. (2008) A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 16, 897–905 10.1016/j.str.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lan T.-H., Liu Q., Li C., Wu G., and Lambert N. A. (2012) Sensitive and high resolution localization and tracking of membrane proteins in live cells with BRET. Traffic 13, 1450–1456 10.1111/j.1600-0854.2012.01401.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kobayashi H., Ogawa K., Yao R., Lichtarge O., and Bouvier M. (2009) Functional rescue of β-adrenoceptor dimerization and trafficking by pharmacological chaperones. Traffic 10, 1019–1033 10.1111/j.1600-0854.2009.00932.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Free R. B., Hazelwood L. A., Cabrera D. M., Spalding H. N., Namkung Y., Rankin M. L., and Sibley D. R. (2007) D1 and D2 dopamine receptor expression is regulated by direct interaction with the chaperone protein calnexin. J. Biol. Chem. 282, 21285–21300 10.1074/jbc.M701555200 [DOI] [PubMed] [Google Scholar]
- 41. Fishburn C. S., Elazar Z., and Fuchs S. (1995) Differential glycosylation and intracellular trafficking for the long and short isoforms of the D2 dopamine receptor. J. Biol. Chem. 270, 29819–29824 10.1074/jbc.270.50.29819 [DOI] [PubMed] [Google Scholar]
- 42. Min C., Zheng M., Zhang X., Guo S., Kwon K.-J., Shin C. Y., Kim H.-S., Cheon S. H., and Kim K.-M. (2015) N-Linked glycosylation on the N-terminus of the dopamine D2 and D3 receptors determines receptor association with specific microdomains in the plasma membrane. Biochim. Biophys. Acta 1853, 41–51 10.1016/j.bbamcr.2014.09.024 [DOI] [PubMed] [Google Scholar]
- 43. Beaulieu J.-M., Espinoza S., and Gainetdinov R. R. (2015) Dopamine receptors: IUPHAR Review 13. Br. J. Pharmacol. 172, 1–23 10.1111/bph.12906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tao Y.-X., and Conn P. M. (2014) Chaperoning G protein-coupled receptors: from cell biology to therapeutics. Endocr. Rev. 35, 602–647 10.1210/er.2013-1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ulloa-Aguirre A., and Michael Conn P. (2011) Pharmacoperones: a new therapeutic approach for diseases caused by misfolded G protein-coupled receptors. Recent Pat. Endocr. Metab. Immune Drug Discov. 5, 13–24 10.2174/187221411794351851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kovoor A., Seyffarth P., Ebert J., Barghshoon S., Chen C.-K., Schwarz S., Axelrod J. D., Cheyette B. N. R., Simon M. I., Lester H. A., and Schwarz J. (2005) D2 dopamine receptors colocalize regulator of G-protein signaling 9–2 (RGS9–2) via the RGS9 DEP domain, and RGS9 knock-out mice develop dyskinesias associated with dopamine pathways. J. Neurosci. 25, 2157–2165 10.1523/JNEUROSCI.2840-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kubale V., Blagotinšek K., Nøhr J., Eidne K. A., and Vrecl M. (2016) The conserved arginine cluster in the insert of the third cytoplasmic loop of the long form of the D2 dopamine receptor (D2L-R) acts as an intracellular retention signal. Int. J. Mol. Sci. 17, 1152 10.3390/ijms17071152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Prou D., Gu W. J., Le Crom S., Vincent J. D., Salamero J., and Vernier P. (2001) Intracellular retention of the two isoforms of the D2 dopamine receptor promotes endoplasmic reticulum disruption. J. Cell Sci. 114, 3517–3527 [DOI] [PubMed] [Google Scholar]
- 49. Sedaghat K., Nantel M.-F., Ginsberg S., Lalonde V., and Tiberi M. (2006) Molecular characterization of dopamine D2 receptor isoforms tagged with green fluorescent protein. Mol. Biotechnol. 34, 1–14 10.1385/MB:34:1:1 [DOI] [PubMed] [Google Scholar]
- 50. Sharma M., Celver J., Octeau J. C., and Kovoor A. (2013) Plasma membrane compartmentalization of D2 dopamine receptors. J. Biol. Chem. 288, 12554–12568 10.1074/jbc.M112.443945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takeuchi Y., and Fukunaga K. (2003) Differential subcellular localization of two dopamine D2 receptor isoforms in transfected NG108–15 cells. J. Neurochem. 85, 1064–1074 10.1046/j.1471-4159.2003.01763.x [DOI] [PubMed] [Google Scholar]
- 52. de Oliveira I. R., de Sena E. P., Pereira E. L., Miranda A. M., de Oliveira N. F., Ribeiro M. G., de Castro-e-Silva E., Dardennes R. M., Samuel-Lajeunesse B., and Marcilio C. (1996) Haloperidol blood levels and clinical outcome: a meta-analysis of studies relevant to testing the therapeutic window hypothesis. J. Clin. Pharm. Ther. 21, 229–236 10.1111/j.1365-2710.1996.tb01143.x [DOI] [PubMed] [Google Scholar]
- 53. Masuho I., Martemyanov K. A., and Lambert N. A. (2015) Monitoring G protein activation in cells with BRET. Methods Mol. Biol. 1335, 107–113 10.1007/978-1-4939-2914-6_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Octeau J. C., Schrader J. M., Masuho I., Sharma M., Aiudi C., Chen C.-K., Kovoor A., and Celver J. (2014) G protein β5 is targeted to D2-dopamine receptor-containing biochemical compartments and blocks dopamine-dependent receptor internalization. PLoS One 9, e105791 10.1371/journal.pone.0105791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mouillac B., and Mendre C. (2017) Biased agonist pharmacochaperones: small molecules in the toolbox for selectively modulating GPCR activity. In Topics in Medicinal Chemistry, pp. 1–18, Springer, Berlin [Google Scholar]
- 56. Bernier V., Morello J.-P., Zarruk A., Debrand N., Salahpour A., Lonergan M., Arthus M.-F., Laperrière A., Brouard R., Bouvier M., and Bichet D. G. (2006) Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 17, 232–243 [DOI] [PubMed] [Google Scholar]
- 57. Garzya V., Forbes I. T., Gribble A. D., Hadley M. S., Lightfoot A. P., Payne A. H., Smith A. B., Douglas S. E., Cooper D. G., Stansfield I. G., Meeson M., Dodds E. E., Jones D. N. C., Wood M., Reavill C., et al. (2007) Studies towards the identification of a new generation of atypical antipsychotic agents. Bioorg. Med. Chem. Lett. 17, 400–405 10.1016/j.bmcl.2006.10.036 [DOI] [PubMed] [Google Scholar]
- 58. Celver J., Sharma M., and Kovoor A. (2012) D2-dopamine receptors target regulator of G protein signaling 9-2 to detergent-resistant membrane fractions. J. Neurochem. 120, 56–69 10.1111/j.1471-4159.2011.07559.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boxberger K. H., Hagenbuch B., and Lampe J. N. (2014) Common drugs inhibit human organic cation transporter 1 (OCT1)-mediated neurotransmitter uptake. Drug Metab. Dispos. Biol. Fate Chem. 42, 990–995 10.1124/dmd.113.055095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zurek N., Sparks L., and Voeltz G. (2011) Reticulon short hairpin transmembrane domains are used to shape ER tubules. Traffic 12, 28–41 10.1111/j.1600-0854.2010.01134.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Samad T. A., Krezel W., Chambon P., and Borrelli E. (1997) Regulation of dopaminergic pathways by retinoids: activation of the D2 receptor promoter by members of the retinoic acid receptor-retinoid X receptor family. Proc. Natl. Acad. Sci. U.S.A. 94, 14349–14354 10.1073/pnas.94.26.14349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Celver J., Sharma M., and Kovoor A. (2010) RGS9–2 mediates specific inhibition of agonist-induced internalization of D2-dopamine receptors. J. Neurochem. 114, 739–749 10.1111/j.1471-4159.2010.06805.x [DOI] [PubMed] [Google Scholar]
- 63. Dunn K. W., Kamocka M. M., and McDonald J. H. (2011) A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 300, C723–C742 10.1152/ajpcell.00462.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.