

Abstract
Perfectly accurate translation of mRNA into protein is not a prerequisite for life. Resulting from errors in protein synthesis, mistranslation occurs in all cells, including human cells. The human genome encodes >600 tRNA genes, providing both the raw material for genetic variation and a buffer to ensure that resulting translation errors occur at tolerable levels. On the basis of data from the 1000 Genomes Project, we highlight the unanticipated prevalence of mistranslating tRNA variants in the human population and review studies on synthetic and natural tRNA mutations that cause mistranslation or de-regulate protein synthesis. Although mitochondrial tRNA variants are well known to drive human diseases, including developmental disorders, few studies have revealed a role for human cytoplasmic tRNA mutants in disease. In the context of the unexpectedly large number of tRNA variants in the human population, the emerging literature suggests that human diseases may be affected by natural tRNA variants that cause mistranslation or de-regulate tRNA expression and nucleotide modification. This review highlights examples relevant to genetic disorders, cancer, and neurodegeneration in which cytoplasmic tRNA variants directly cause or exacerbate disease and disease-linked phenotypes in cells, animal models, and humans. In the near future, tRNAs may be recognized as useful genetic markers to predict the onset or severity of human disease.
Keywords: transfer RNA (tRNA), human genetics, cancer, neurodegeneration, protein synthesis, cytoplasmic tRNA, human genome, mistranslation, nucleotide modification
Introduction
Protein synthesis is an evolutionarily conserved process that is required by all life. In the interpretation of the genetic code, transfer RNAs (tRNAs) play a central role as they physically link amino acids to codons. Crick's adaptor hypothesis predicted the existence of tRNAs in 1955: “each amino acid would combine chemically, at a special enzyme, with a small molecule which, having a specific hydrogen-bonding surface, would combine specifically with the nucleic acid template.” (1). Just 3 years later, the first tRNAs were discovered as soluble RNAs involved in protein synthesis (2). Working with yeast, Holley et al. (3) isolated tRNAs and determined the first tRNA sequence. The first codons were mapped to amino acids using repeating polyribonucleotides as templates for protein synthesis (4, 5). The laboratories of Nirenberg et al. (6) and Khorana et al. (7) then raced to solve the complete codon catalogue using a technique that monitored the binding of radiolabeled aminoacyl-tRNAs to ribosomes separately prepared with each of the possible 64 trinucleotide codons. These efforts established the standard genetic code table for which Holley, Khorana, and Nirenberg were awarded the Nobel prize in Physiology or Medicine in 1968.
Fascinatingly, by the time the prize was awarded, exceptions to the genetic code had already been identified. In 1966, Yanofsky and co-workers (8) demonstrated tRNA mutants enabled missense suppression or amino acid mis-incorporation using a defective tryptophan synthase A gene in Escherichia coli. Early in 1968, Atkins (9) identified the first exception to triplet decoding with the discovery of frame-shifting in Salmonella typhimurium. Indeed, although the genetic code is nearly universal in the living world, several exceptions to the standard code occur in diverse organisms (10). Genome sequences and genetic and biochemical data reveal that in organisms from microbes to humans, codons can be ambiguously decoded (8, 11–13), reassigned (14, 15), or site-specifically recoded (16–18) to incorporate unexpected amino acids or amino acids beyond the standard set of 20.
Given the importance of cytosolic tRNAs to facilitate accurate synthesis of the proteome, surprisingly few examples have linked a cytosolic tRNA mutation to human disease thus far. Yet, recent examples directly connecting cytosolic tRNA mutations to disease in humans (19) and separately to neurodegeneration (20, 21) and cancer (22) in mice suggest that cytosolic tRNA variants play a greater role in disease than previously imagined. It is possible that significant changes to tRNA function are not usually tolerated in the genome or that defective tRNA alleles may be genetically buffered by multiple copies of each iso-decoder. Nevertheless, two empirical observations suggest tRNAs have a larger role in disease than previously recognized: (i) the unexpectedly large number of tRNA variants in the human population (Tables 1–3 and Table S1), and (ii) the fact that even a single nucleotide change in a single tRNA gene can cause mistranslation or stall translation leading to molecular and cellular defects (13, 21, 23). The majority of research connecting human tRNA functions to disease is focused on mutations in aminoacyl-tRNA synthetases (AARSs)4 (24), on proteins that modify nucleotides in cytosolic tRNAs (25, 26), or on the smaller pool of mitochondrial tRNAs (27–29). Two major reasons for this are a relative lack of available sequence data for cytosolic tRNAs, and a long-held assumption that excessive tRNA copy number should “buffer” potential phenotypes resulting from a single mutant.
Table 1.
Human tRNA anticodon variants
MAF is minor allele frequency. Inserted nucleotides are indicated by, e.g. _35T.
| tRNA gene | Variant | MAF (%)a | Variant counta | MAF (%)b | Variant countb | tRNA scorec | Codon identity | tRNA identity | Expression |
|
|---|---|---|---|---|---|---|---|---|---|---|
| ARMd | CHIPe | |||||||||
| Ala-AGC-2–2 | G35A | 0.02 | 1 | 84.7 | Val | Ala | + | + | ||
| Ala-AGC-6–1 | G35C | 6.55 | 328 | 6.47 | 8130 | 74.1 | Gly | Ala | + | + |
| Ala-AGC-15–1 | C36T | 0.04 | 2 | 0.03 | 42 | 56.7 | Thr | Ala | + | − |
| Ala-AGC-16–1 | G35A | 2.2 | 11 | 2.1 | 269 | 53.1 | Val | Ala | + | − |
| Ala-CGC-1–1 | G35T | 0.0008 | 1 | 79.7 | Glu | Ala | + | + | ||
| Ala-TGC-1–1 | G35A | – | 80.5 | Val | Ala | + | + | |||
| Leu-CAA-3–1 | A35C | 0.0008 | 1 | 77.3 | Trp | Leu | + | + | ||
| Ser-AGA-2–2 | G35A | 0.02 | 0.003 | 4 | 89.6 | Phe | Ser | + | + | |
| Ser-AGA-2–3 | G35A | 1.82 | 91 | 1.87 | 2347 | 89.6 | Phe | Ser | + | + |
| Ser-AGA-2–4 | G35C | 0.003 | 4 | 89.6 | Cys | Ser | + | + | ||
| Ser-AGA-2–5 | _35T | 0.0008 | 1 | 89.6 | + | + | ||||
| Ser-CGA-2–1 | _36T | 0.03 | 34 | 94 | + | − | ||||
| Ser-TGA-2–1 | G35A | 0.04 | 2 | 0.02 | 22 | 90.4 | Leu | Ser | + | + |
| Ser-TGA-3–1 | _36T | 0.0008 | 1 | 89.7 | + | + | ||||
c tRNA score was calculated using tRNA-Scan SE (128).
Table 2.
Human tRNA variants that introduce a G3:U70 base pair
MAF is minor allele frequency.
| tRNA gene | Variant | MAF (%)a | Variant counta | MAF (%)b | Variant countb | tRNA scorec | Expression |
|
|---|---|---|---|---|---|---|---|---|
| ARMd | CHIPe | |||||||
| Arg-ACG-1–3 | C70U | 0.002 | 2 | 68 | + | + | ||
| Cys-GCA-2–3 | C70U | 0.02 | 29 | 82 | + | + | ||
| Cys-GCA-1–1 | C70U | 0.02 | 1 | 0.07 | 86 | 84 | + | − |
| Cys-GCA-17–1 | C70U | 0.002 | 2 | 71 | + | + | ||
| Cys-GCA-12–1 | C70U | 0.02 | 1 | 0.01 | 16 | 72 | + | + |
| Gly-CCC-2–1 | G70U | 0.05 | 64 | 75 | + | + | ||
| Gly-GCC-2–4 | A3G | 0.08 | 4 | 0.05 | 63 | 81 | + | − |
| Gly-GCC-2–5 | A3G | 0.02 | 1 | 0.005 | 6 | 81 | + | − |
| Gly-GCC-2–1 | A3G | 0.02 | 1 | 0.02 | 20 | 81 | + | + |
| Gly-GCC-2–3 | A3G | 81 | + | + | ||||
| Gly-GCC-1–5 | A3G | 1.2 | 61 | 1.3 | 1633 | 81 | − | − |
| Gly-GCC-5–1 | A3G | 0.02 | 1 | 0.1 | 129 | 55 | − | − |
| Gly-TCC-2–6 | C70U | 0.03 | 14 | 0.09 | 111 | 74 | + | + |
| Ser-AGA-2–6 | A3G | 0.01 | 12 | 90 | + | + | ||
c tRNA score was calculated using tRNA-Scan SE (128).
Table 3.
Human tRNAArg C50T variants
| tRNA gene | SNP ID | MAF (%)a | Variant counta | MAF (%)b | Variant countb | tRNA scorec | Expression |
|
|---|---|---|---|---|---|---|---|---|
| ARMd | CHIPe | |||||||
| Arg-ACG-1–1 | rs6939540 | 2.42 | 121 | 2.2 | 2765 | 67.6 | + | + |
| Arg-ACG-1–2 | rs186104107 | 0.02 | 1 | 0.01 | 14 | 67.6 | + | + |
| Arg-TCG-6–1 | rs113170043 | 16.6 | 831 | 20.2 | 24,374 | 53.7 | + | − |
| Arg-TCT-5–1 | rs143334272 | 0.8 | 41 | 0.6 | 711 | 61.4 | + | − |
a Data are from the 1000 genomes project.
c tRNA score was calculated using tRNA-Scan SE (128).
In this review, we outline the complexity of cytosolic tRNA function and regulation in eukaryotic cells. We then summarize recent studies demonstrating examples of single nucleotide tRNA variants that elicit significant levels of amino acid mis-incorporation, which can be surprisingly well-tolerated in eukaryotic cells. Using data from the 1000 Genomes Project, we analyzed the location and frequency of naturally occurring human tRNA variants. These data reveal an abundance of mistranslating tRNAs in the human population. Finally, we summarize recent evidence linking tRNA mutations and de-regulated tRNA expression and nucleotide modification to disease in humans and model systems. Some of the studies point to the idea that tRNA mutations, which are otherwise tolerated or benign, contribute to disease in the context of other coincidental cellular defects.
tRNA function and regulation
The role of tRNA in decoding the genetic code
tRNAs are best known for their role in translation of RNA messages into proteins. tRNAs are relatively small RNA molecules, typically consisting of 76–90 nucleotides, and fold into a conserved three-dimensional structure in the shape of an upside-down L (Fig. 1A). The anticodon resides at the long end of the L-shape and binds to cognate codons in the messenger RNA (mRNA) on the ribosome. On the opposite end of the tRNA, the amino acid is ligated to the 3′-terminal adenosine residue in the acceptor stem. Accurate tRNA aminoacylation and high-fidelity decoding of codons on the ribosome are key determinants to accurate protein production.
Figure 1.

tRNA structure. A, tRNAs fold into an L-shaped three-dimensional structure with extensive intramolecular base-pairing. This tRNAPhe structure (Protein Data Bank code 1OB5) (134) is aminoacylated with phenylalanine (AA). B, in a two-dimensional representation, tRNA resembles a cloverleaf. In both diagrams, the tRNA is colored by structural elements: acceptor stem (red), dihydrouridine (D)-arm (green), anticodon stem (cyan), anticodon (bases 34–36 in purple), variable loop (yellow), TΨC (T) arm (navy), and the conserved CCA-3′ end (white). A schematic mRNA is shown below the tRNA diagram to indicate the tRNA nucleotides that base pair with each codon position.
Codon recognition is determined by the tRNA anticodon, which base pair with tri-nucleotide codons in mRNAs during protein synthesis (Fig. 1B). The essential interaction between codon and anticodon is established not only by Watson-Crick base pairing, but also by nucleotide modifications in tRNAs (30, 31), competition between cognate and near-cognate decoding (32, 33), and wobble decoding. Generally, the first two positions of a codon form Watson-Crick pairs with the tRNA, whereas the third position is more flexible (30). In back-to-back publications, Crick hypothesized (34) what Söll et al. (35) determined experimentally that the third position of a codon can involve G:U or U:G wobble pairing with the 1st position of the anticodon at tRNA nucleotide 34. Indeed, the initial discoveries also included examples of extended wobble decoding in yeast arginine and alanine tRNAs that read codons ending in U, C, or A (35). Extended wobble decoding is facilitated by post-transcriptional tRNA modification, where adenosine residues at position 34 are modified to inosine, which pairs with U, C, or A in the third codon position (30). Additional nucleotide modifications in the anticodon loop (particularly at positions 34 and 37) also impact translation fidelity and reading-frame maintenance (36, 37). For example, in yeast, a 5-methoxycarbonylmethyl-2-thiouridine modification at anticodon base U34 represses +1 frame-shifting. Lack of the modification or hypo-modification at this site in a variety of tRNAs leads to 1.5–3.0-fold increases in ribosomal frame-shifting (38). Similarly, absence of the modified base N6-threonylcarbamoyladenosine at the anticodon adjacent position 37 also increases frame-shifting in yeast by 2-fold (39).
The standard genetic code is composed of 61 sense codons that encode 20 amino acids and 3 stop codons (UGA, UAG, and UAA) that usually signal termination of protein synthesis. Because certain tRNAs decode up to three or four different codons, the theoretical minimum number of tRNAs for an organism to encode 20 amino acids is 32 (34). Söll et al. (35) observed that the “minimum number of sRNA [tRNA] molecules required for recognition of all of the meaningful codons is relatively small, and this conclusion in turn raises the question of redundancy in the sRNA pool of a cell.” The question as to why organisms encode apparently redundant tRNA genes, which was raised the year after the code was solved, is still unanswered today. As a result of the genome sequencing revolution, we know now that nature contains examples of organisms with tRNA gene complements that are well below and vastly greater than this apparent minimal requirement. There are examples of organelles (e.g. human mitochondria with 22 tRNA genes) and even parasitic microbes (e.g. Mycoplasma mobile with 28 tRNA genes (40)) with fewer than 32 tRNA genes. Their survival depends on importing the missing tRNAs from a different cellular compartment (41) or presumably a host cell.
E. coli encodes 88 tRNA genes, whereas yeast has a small tRNAome for a eukaryote at 275 tRNA genes. Eukaryotes typically have hundreds of tRNA genes that display a general trend to increase in number and sequence diversity with the complexity of the organism (42). Unicellular protozoans encode near the theoretical minimum of tRNA genes, such as the malaria parasite Plasmodium falciparum, which has only 35 tRNA genes (43). P. falciparum was recently found to import additional tRNAs from its host (44). Some species of fish have astoundingly high tRNA gene numbers (Table S2), such as the elephant shark (Callorhinchus milii), which encodes 13,724 tRNAs (43). As exemplified in a phylogenetic comparison of yeast and human alanine tRNAs (Fig. 2), the sequence variations among tRNA iso-acceptors appear to increase with complexity as well. Yeast has 16 tRNAAla iso-acceptors, including 11 identical genes with the AGC anticodon and 5 identical genes with the TGC anticodon. In contrast, humans encode 45 tRNAAla iso-acceptors with markedly greater sequence diversity than their yeast counterparts, including examples with CGC anticodons not seen in yeast (Fig. 2).
Figure 2.
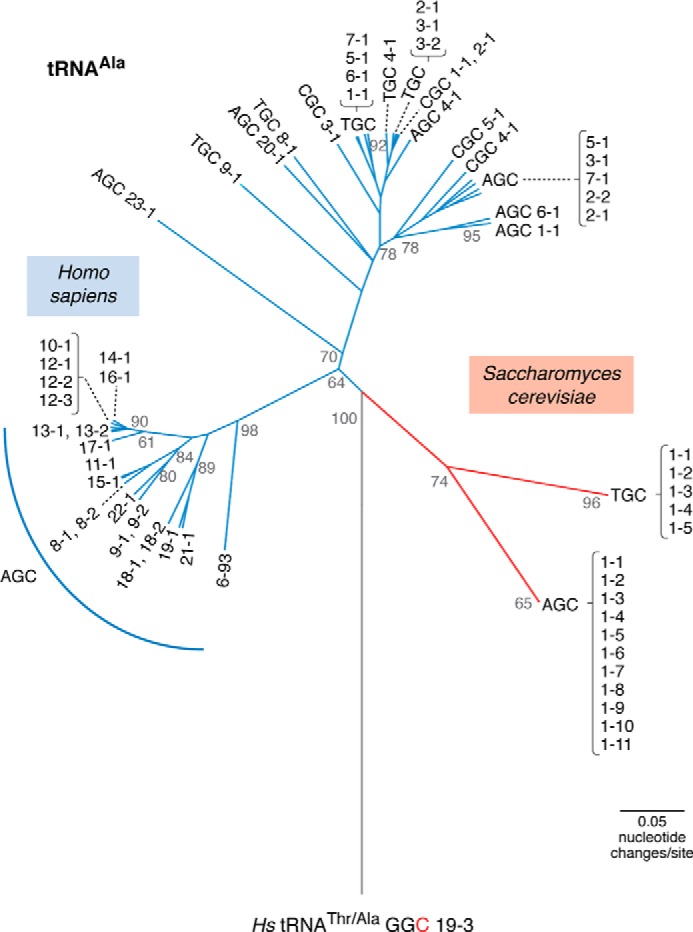
Phylogenetic relationships of human and yeast tRNAAla. The tree is based on an alignment of all known human and yeast tRNAAla iso-acceptors. The vastly expanded number and greater diversity of human tRNAAla genes compared with their yeast counterparts are evident. The tRNAs are labeled according to gene names in the genomic tRNA database (43), which include anticodon sequence followed by a numbering system where the first number indicates similar sequences, and the second number indicates a gene copy identifier. The human reference genome contains a misannotated tRNAAla, which was used to root the tree. This gene, tRNAAla-GGC-19-3, is a tRNAThr with a mutation (T36C) endowing the tRNA with an alanine anticodon. This is the only example of the alanine GGC anticodon in humans. Scale bar indicates the number of nucleotide changes per site in the tRNA sequences. The tree was calculated similarly as before (135). Briefly, a starting tree computed in MultiSeq 2.0 (136) was optimized to identify the maximum likelihood tree using PhyML 3.1 (137). Statistical branch support (out of 100) was calculated based on an approximate likelihood ratio test method (138) as implemented in PhyML.
tRNA regulation in human cells
The number of expressed tRNA genes in human cells is not well defined. Of the 610 tRNA genes in humans, the genomic tRNA database predicts 417 genes in their high-confidence set, indicating the tRNA is likely to function in protein synthesis (43). Comprehensive profiling of RNAs in human serum suggests 411 expressed tRNA genes (45). According to CHIP-seq analysis of RNA polymerase III and transcription factor occupancy, ∼350 tRNA genes are actively transcribed in a single human cell line (IMR90hTert) (46). Gogakos et al. (47) reported the expression of 288–349 tRNAs in HEK 293 cells based on two different RNA-Seq methods. Together, the data suggest 300–400 tRNA genes are expressed in any individual human cell.
The degree to which each human tRNA contributes to protein synthesis has not been determined, but evidence that cells regulate tRNA expression to control protein production is emerging. First, expression of individual tRNA genes varies between tissues (48, 49). Furthermore, the steady-state level of different tRNAs correlates with the expression of matched-codon biased mRNA transcripts (48). The observation suggests that cells can fine-tune tRNA expression profiles to match codon usage in expressed mRNAs. Indeed, the fact that efficient protein expression requires tRNA levels and decoding capacity matches the distribution of codons in mRNA is well known. Multiple E. coli strains and bioinformatic tools for codon adaptation were developed based on this principle to enhance the production of eukaryotic and other recombinant proteins in bacteria (50).
Once transcribed, tRNAs are processed via the removal and addition of nucleotides to produce a mature tRNA. Introns, 5′-leader, and 3′-trailer sequences in the original transcript are removed (51). Next, the CCA-adding enzyme elongates the pre-tRNA with the conserved CCA 3′-end. CCA-adding enzymes can append a second CCA to certain tRNAs with mismatches or excessive G:U pairs in their acceptor stems (52). The double CCA addition primes tRNAs for exonucleolytic digestion via the rapid tRNA decay pathway (53). The human CCA-adding enzyme (TRNT1) is implicated in disease. Complete loss-of-function in TRNT1 is embryonic lethal, and partial loss-of-function mutations cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (54).
tRNAs are further processed with a variety of post-transcriptional nucleotide modifications, including 2′ O-methylation of the ribose, N-acetylation and N-methylation of the nucleotide bases, as well as more complex modifications that form bases such as wybutosine (55).
In eukaryotes, tRNAHis is post-transcriptionally edited to add an extra guanine (G−1) to the 5′-end of the tRNA, a unique feature recognized and required by the cognate histidyl-tRNA synthetase (56). Depleting the tRNAHis guanylyltransferase that catalyzes G−1 addition leads to accumulation of un-aminoacylated and un-guanylated yet hyper-methylated tRNAHis in the nucleus (57). Subsequent studies point to tRNAHis m5C hypermethylation as a response to growth arrest in Saccharomyces cerevisiae, although the significance of the increase in m5C methylation is yet unclear (58). Monomethylation of the 5′-monophosphate of tRNAHis by bicoid-interacting 3 domain containing RNA methyltransferase (BCDIN3D) is thought to protect tRNAHis from degradation. BCDIN3D is overexpressed in breast cancer cells, and monomethylated tRNAHis is more abundant in breast cancer cells, yet the overall level of tRNAHis is not impacted (59). It is thought that monomethylation contributes to the formation of tRNAHis-derived fragments in breast cancer cells, which in turn regulate tumorigenic genes involved in breast cancers (60).
In fact, tRNAs are the most frequently modified noncoding RNA known, containing an average of 13 modifications per molecule (61). The combined number of expressed tRNA genes and their multiple modification states imply the existence of a large combinatorial number of tRNA microspecies in the human cell (62). Because tRNA modifications are important for translation fidelity and reading frame maintenance (34, 36, 37, 63), these microspecies have the potential to impact cellular function and disease. Modifications are also essential for regulating tRNA turnover (64) and for proper structure, folding, and stability of the tRNA (55). Indeed, many tRNA-modifying enzymes are already linked to disease (26). As described below, tRNA modification can also dynamically up- or down-regulate sets of tRNAs (65–67).
The mature and active tRNA is a substrate for amino acid ligation catalyzed by the AARS enzymes in an ATP-dependent reaction (68). Each AARS enzyme has specificity both for an amino acid and a distinct set of cognate tRNA iso-acceptors. Amino acids are ligated to the 3′-end of tRNAs, requiring the presence of a CCA 3′-tail. To ensure tRNA recognition fidelity, AARSs make essential contacts with nucleotides in their cognate tRNAs, called identity elements (69). Aminoacyl-tRNAs are then substrates for protein synthesis. The likelihood that a given aminoacyl-tRNA acts in translation depends upon many factors, including the stability of the tRNA, the number of aminoacylated-tRNAs competing for the same codon, and the expression of mRNAs containing codons read by the tRNA. tRNAs unfit for translation are degraded by the rapid tRNA decay pathway (70). Cytoplasmic tRNA levels are also regulated by export processes to other cellular compartments, including into mitochondria (71), or retrograde transport into the nucleus (72). As reviewed elsewhere (62), tRNAs perform additional functions outside of translation, either as whole tRNAs (73) or tRNA-derived fragments (74).
Phenotypes of mistranslating cells
Mistranslation occurs in all cells (75) as a result of multiple different mechanisms. Considering the small size, multitude of protein partners, and essential cellular role of tRNAs, single nucleotide changes can have a profound impact on their function and on the efficiency and fidelity of protein synthesis (12, 13, 23, 69). Proteins encoded by mRNAs containing rare codons or strongly biased codon compositions are most susceptible to the effects of tRNA variants. Loss-of-function mutations in tRNAs can cause ribosome stalling to de-regulate protein synthesis, whereas gain-of-function mutations in tRNAs can lead to mis-aminoacylation and mistranslation (12, 21).
Mistranslating tRNAs can arise from surprisingly minor changes to the nucleotide sequence. Although many tRNAs harbor major identity determinants in their anticodon, coupling aminoacylation fidelity to codon assignment, alanyl-, leucyl-, and seryl-tRNA synthetases do not recognize the anticodon nucleotides on their cognate tRNAs. Anticodon mutations in these tRNAs often elicit amino acid mis-incorporation (69). The accumulation of highly active tRNASer anticodon mutants is toxic to yeast cells, causing proteome-wide mistranslation (23). In yeast, the degree of anticodon mutant toxicity varies, depending on competition with WT tRNAs, chemical properties of the amino acids, and tRNA modifications (76).
Santos et al. (22)analyzed tRNASer variants containing Ala or Leu anticodons in murine NIH 3T3 cells grown in culture and subsequently xenografted to live mice. As determined by mass spectrometry in tumor samples recovered from the mice, the rate of mistranslation increased by ∼2-fold in the cells expressing tRNASer containing an alanine anticodon, but only marginally in cells expressing tRNASer with a leucine anticodon. Mistranslation was not toxic to the NIH 3T3 cells when grown in culture as determined by cellular viability, necrosis, and proliferation assays, indicating that increased cytosolic tRNA-dependent mistranslation was initially well-tolerated. Interestingly, expressing the mistranslating tRNAs promoted the formation of foci in vitro, suggesting a link to tumorigenesis (22). Briefly, foci formation occurs when cancer-like cells form dense clusters resembling early-stage tumors on a Petri dish (77). Mistranslating tRNAs promoted the activation of the oncogenic factors protein kinase B (Akt) and p38 when cells were treated with tumor necrosis factor-α, to a greater extent than cells expressing the WT tRNAs. Furthermore, cells mistranslating alanine codons with serine promoted angiogenesis in a chick chorioallantoic membrane assay and were highly tumorigenic when introduced in mice. Compared with the parent cells in culture, expression of the mistranslating tRNAs increased ∼8-fold in cells recovered from mouse tumors. Although mistranslating tRNA variants had undetectable cytotoxicity in cells in culture, the mutant tRNAs exacerbated or accelerated cellular pathways to cancer in a mammalian model of disease (22).
Identity element mutations are another route to mistranslating tRNAs. The phenotypic consequences of a single tRNA variant of this type are the subject of a number of recent studies. AlaRS recognizes two critical identity determinants at the 3rd base pair in the acceptor stem (G3:U70) (78), and also aminoacylates tRNAs bearing the GU pair at the 4th acceptor stem base pair (76). tRNA variants that convert nonalanine tRNAs to alanine accepting tRNAs by creating these identity elements are common in mammalian genomes (79). Some human tRNACys and tRNAThr species with G4:U69 are natural alanine acceptors, and cysteine to alanine mistranslation was detected in HEK 293 cells (79). An Animalia-specific tRNA deacylase was recently discovered that co-occurs with tRNAThr G4:U69 variants in animal genomes and de-acylates mis-charged Ala-tRNAThr (80). This enzyme may protect human cells from alanine mistranslation at threonine codons.
In our work on tRNA-dependent mistranslation, we expressed a mutant of human tRNAPro containing a G3:U70 base pair in human cells. The human tRNAPro mutant was an efficient alanine acceptor in vitro that no longer accepted proline. Our previous work in yeast demonstrated that a homologous tRNAPro mutant mistranslated multiple proline codons with alanine (13). We developed a GFP reporter (D129P) that fluoresces in response to mistranslation at the Pro-129 codon. In HEK 293 cells (12), we did not observe significant mistranslation in rich media compared with cells expressing WT tRNAPro. When the cells were starved of serum and glucose over a period of days, mistranslation accumulated to 2–5% according to the GFP reporter. Strikingly, and similar to the report of Santos et al. (22), we did not observe a loss in cellular viability or induction of the heat-shock response compared with cells expressing the WT tRNA, indicating that this level of mistranslation was well tolerated. In yeast, a mistranslating tRNA did show synthetic slow growth when coupled with deletion mutants lacking the proteome regulatory transcription factor Rpn4 (13). The above examples illustrate how phenotypes driven by a mistranslating tRNA variant can remain hidden; yet, in conditions of stress or in models of disease, an otherwise neutral tRNA variant can interact synthetically with a stressor or a second mutation to cause phenotypic defects.
Errors in protein synthesis are normally thought to be deleterious, yet mistranslation is an adaptive response in diverse organisms from bacteria (81) to yeast (13) to human cells (82). In some of these cases mistranslation provides a selective advantage. As reviewed elsewhere (83), a recurrent finding is that stress conditions reduce aminoacylation fidelity (84). For example, upon oxidative stress, methionyl-tRNA synthetase increases its mis-aminoacylation of noncognate tRNAs up to 10-fold in bacteria and mammalian cells (82, 85, 86); the additional methionine in proteins is thought to protect the proteome from oxidative damage.
tRNA variation in humans
Human tRNA variants that likely mistranslate can be readily identified in publicly available sequence data (Tables 1 and 2, S1) (43). Some of these natural variants (79) or similar variants designed in the laboratory (12, 22) do in fact cause mistranslation in human cells. In addition, tRNA variants, which simply impair tRNA folding or function, also impact the proteome. The absence of even a single tRNA gene product can alter the tRNA pool and limit the rate of protein synthesis by causing ribosome stalling (21). In both capacities, mutations in tRNA-encoding genes represent an important class of potential disease modifiers that could increase the severity of other disease-causing alleles.
To visualize the nature and extent of human tRNA variation, we analyzed data from the 1000 Genomes Project and plotted the number of unique variants at each position in an alignment of all human tRNA genes (Fig. 3, A and B). The number of unique variants were mapped on the tRNA secondary structure (Fig. 3A). We also plotted the frequency of occurrence of each of these tRNA mutations in the human population (Fig. 3C). Some tRNAs, such as those for leucine, serine, and selenocysteine, have significantly larger variable loops; variation from these regions was not included.
Figure 3.

tRNA variants observed in the 1000 Genomes Project. A, variants that occur within tRNA genes (defined by GtRNAdb (128)) were downloaded from the 1000 Genomes Project phase 3 dataset (142). Insertions and deletions were removed, as were variants with no allele frequency available. Each variant was mapped to its corresponding tRNA position, according to standardized numbering (139), using an in-house Perl script. High-confidence tRNAs were defined as tRNAs with a tRNAscan-SE score of >50 (128). For the high confidence tRNA set, unique mutations are mapped to each position in the tRNA. B, same data in A are plotted for the high-confidence set (cyan dashed line) and for all human tRNA sequences (red line). C, allele frequencies (log2 scale) of all variants that occur at each tRNA position are represented in box and whisker plots. Boxes outline quartiles of the allele frequency distribution; filled circles depict the median allele frequency; whiskers show 1.5× quartile range; and open circles depict raw data, i.e. the allele frequencies for each unique tRNA variant at the indicated position.
No position in human tRNA genes is immune to variation, yet some positions are far more variable than others (Fig. 3, A and B). The allele frequency of these variants indicates that common (>5% allele frequency) and rare variants (<5% allele frequency) are distributed across nearly all sites in the tRNA (Fig. 3C). Some sites, however, lack common variants. Although restricted to rare variants, variation is observed at position 73; this “discriminator” base is a key identity element for many AARSs. The anticodon shows variation at all three bases, albeit reduced compared with other regions of the tRNA. The data from 1000 Genomes Project suggest that across all ∼600 tRNA loci there are 25–30 unique nucleotide variants at each anticodon base, and these include both common and rare variants in the population (Fig. 3C).
Positions within the acceptor stem contain large numbers of unique variants. Many AARSs recognize acceptor stem nucleotides to ensure aminoacylation fidelity (69); thus, acceptor stem variants have the potential to elicit mistranslation or lead to a defective tRNA. Another compelling observation is that several important sites of tRNA modification display significant variation (e.g. position 37 in the anticodon loop) (Fig. 3). Consistent with this observation, mutations in tRNA-modifying enzymes that act at these positions are implicated in disease (26).
As mentioned above, most human AARSs recognize identity determinants in the tRNA anticodon except for AlaRS, LeuRS, and SerRS (69); thus, nonsynonymous anticodon variants in Ala, Leu, and Ser tRNAs are likely to mistranslate. Correspondingly, the tRNA variants that have already been shown to elicit mistranslation in human cells have mutations in the tRNA anticodon or create the identity determinants for AlaRS (12, 78, 79). Anticodon mutations in other tRNAs typically reduce or ablate aminoacylation (69). However, the degree of aminoacylation loss is not known for all anticodon positions in all tRNAs, and in some cases, efficient aminoacylation can be retained even when some identity determinants are mutated (87).
Certainly, mistranslating tRNAs can arise from a variety of mechanisms. Mutations at identity elements, which ensure cognate aminoacylation, or mutations at anti-determinants, which prevent non-cognate aminoacylation, have the potential to convert any tRNA into a mistranslator. Although such mutants likely occur in the human population, there is a paucity of biochemical data regarding human tRNA identity elements and anti-determinants, thus challenging confident identification of such variants as mistranslating tRNAs from sequence alone. Anticodon mutations in Ser, Ala, and Leu, however, will undoubtedly lead to amino acid mis-incorporation. For this reason, our discussion of specific mistranslating tRNA examples from human genomes focused on these “obvious” mistranslating tRNAs.
To assess the prevalence of likely tRNA mistranslators in the human population, we searched the Genomic tRNA database (GtRNAdb) for high-confidence Ala, Leu, and Ser tRNAs with anticodon mutations (Table 1) or mutations in the 3rd or 4th acceptor stem bp that create the G3:U70 (Table 1) or G4:U69 (Table S1) AlaRS identity element. In total, among the human Ala, Leu, and Ser iso-acceptor groups reported in GtRNAdb there are 27 unique anticodon variants. Of these, there are 14 unique nonsynonymous (Table 1) and 13 synonymous anticodon variants. Most nonsynonymous anticodon variants are rare, but three variants occur in >1% of the population. One alanine tRNA variant containing a glycine anticodon occurs in over 6% of sequenced individuals. The common occurrence of these mutations in cytosolic tRNAs is striking; analogous variants in mitochondrial tRNAs are embryonic lethal (27). Although we found a similar number of unique synonymous anticodon variants, none were found in >1% of sequenced individuals. While still encoding the “correct” amino acid, such mutants may more or less efficiently read synonymous codons for a particular amino acid, altering translation rates.
Further complicating this scenario, certain apparently synonymous anticodon variants may become mistranslators through nucleotide modification. For example, tRNAs normally containing an A34 are modified to inosine (I34) by the action of adenine deaminases acting on tRNAs (88). As noted above, I34 enables expanded wobble decoding to codons ending in U, C, or A; thus, A34 containing tRNAs are normally restricted to those amino acids with synonymous codons ending in U, C, and A. However, human genomes include examples of tRNAs bearing A34 that, if modified to I34, would lead to mistranslation (e.g. tRNA-Ser-GCT-5–1 single nucleotide polymorphism (SNP) rs550301646; tRNAAsn-ATT-1–1 and tRNA-Tyr-ATA-1–1 are in the human reference genome). In the case of tRNAAsn, A34I would incorporate Asn at Lys AAA codons. This type of phenomenon was recently examined with anticodon variants of Methanocaldococcus jannaschii tRNATyr expressed in E. coli (89, 90). In this case, a tRNATyr mutant with an AUG anticodon decoded histidine CAU and CAC codons with tyrosine at approximately equal efficiency (2–3%); the mutant tRNATyr AUG anticodon was indeed partially modified to IUG (89). Perhaps as a natural defense against mistranslation and the resulting abundant , mis-incorporation of tyrosine at glutamine CAA codons was not detected in E. coli (89).
Adding to the complexity of human tRNA variation, tRNA genes are particularly susceptible to transcription-associated mutagenesis (TAM) (91), and thus, their sequence can change more rapidly than other genes. Thornlow et al. (91) demonstrated that tRNA genes experience 7–10-fold higher rates of TAM compared with the genome-wide average. TAM occurs when DNA strands are separated during transcription, and the nontemplate strand becomes temporarily isolated and more accessible to mutagens (92). Although tRNA variation is generally selected against on a population scale, this implies that the sequence of tRNA genes within individual cells could change throughout life and that perturbations that increase tRNA expression could further increase mutation rates.
tRNA variation and disease
Like the involvement of mitochondrial tRNA variants in disease (28), recent studies have identified specific cytosolic tRNA mutants as drivers or modifiers of disease in humans and mice. In addition, tRNA mis-modification and imbalanced tRNA expression also contribute to disease. Here, we highlight examples of defective tRNA function in genetic disorders, cancers, and neurodegeneration.
tRNA mutants linked to disease
Kobayashi et al. (140) identified the first human tRNA associated with disease in 1990. The mutation, a variant of a mitochondrial tRNALeu gene, leads to the degradation of the tRNA and causes a rare disorder characterized by stroke and dementia: mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Shortly after, a mutation in mitochondrial tRNALys was found to cause another rare neurological disorder, myoclonic epilepsy and ragged-red fiber disease (93). Several other mitochondrial tRNA variants are implicated in major human diseases, including heart disease (94), hypertension (95), metabolic disease (96), and deafness (97).
Two clear examples have emerged where a cytosolic tRNA variant either contributes to or directly causes disease. One case (described under “tRNA variants in neurodegeneration”) involves a mutation in a single tRNAArg gene that causes widespread neurodegeneration in mice when associated with a second mutation in a protein-coding gene that sensitizes cells to ribosome stalling (21). The other case (detailed under “tRNA modification defects in disease”) is a single nucleotide mutation in the only functional human tRNASec gene (98, 99) that causes abdominal pain, fatigue, muscle weakness, and low plasma selenium levels in a homozygous patient (19). In this case, the tRNASec variant appears to be the primary driver of disease (19).
Imbalanced tRNA expression and disease
tRNA copy number variation
The copy number of tRNA genes varies between individuals. Iben and Maraia (100) assessed copy number variation among nuclearly encoded tRNAs from whole-genome sequencing data obtained in the 1000 Genomes Project. Their study focused on two sets of two parents and a child, from which >15-fold read coverage was obtained. Although the high similarity of tRNA iso-acceptors complicates this type of analysis, significant copy number variation in at least 11 tRNA gene loci among the six individuals was reported. Furthermore, they validated a homozygous deletion encoding on chromosome 7 in one individual. Interestingly, modification defects in this tRNA associate with type 2 diabetes in mice. The deletion is common in the human population and has no known indication of an associated phenotypic defect (101).
Tissue-specific tRNA expression
Dittmar et al. (48) demonstrated the tissue dependence of tRNA expression using a tRNA microarray that probed 42 nuclearly encoded and 21 mitochondrially encoded tRNAs from eight different tissues. They revealed that human tissues express different sets of cytosolic tRNAs. Comprehensive analysis based on RNA polymerase III occupancy of tRNA genes in mice support this finding (102).
The relevance of tissue-specific tRNA expression to disease was demonstrated by the link between tRNA abundance and cystic fibrosis (103). In this work, Kirchner et al. (103) characterized a synonymous SNP in the cystic fibrosis transmembrane conductance regulator (CFTR), which substitutes an ACT Thr codon with ACG. This synonymous mutation results in a cell type–dependent alteration of CFTR protein levels that are not explained by a change in mRNA stability or splicing. The authors discovered that is a low abundance tRNA in the cystic fibrosis model and human bronchial epithelial cell lines. The polymorphism not only reduces CFTR expression in human bronchial epithelial cells, but also impairs the folding, localization, and membrane conductance of CFTR. The findings point to a translation rate-dependent mechanism, where ribosome stalling on the ACG codon, which is read by a low abundance tRNA, causes the protein product to mis-fold and malfunction.
Phenotypic defects from synonymous codon mutations are observed in numerous other disease-relevant protein–coding genes (104). Examples include multidrug resistance 1 (MDR1) (105), estrogen receptor α (106, 107), and surfactant protein-D (108). Thus, expression of specific tRNA iso-decoders is an important consideration when synonymous mutations result in a phenotype, particularly if the protein synthesis burden is shifted to a low abundance or possibly defective tRNA. Conceivably, tRNA synonymous anticodon variants (noted above) could have a similar effect on translation rates and cellular phenotypes.
De-regulated tRNA expression
tRNA expression can change dynamically in disease. Pavon-Eternod et al. (109) demonstrated that tRNA expression increases from 3- to 10-fold in breast cancer tumors. Oncogenic factors such as RAS and C-MYC promote RNA polymerase (pol) III transcription, whereas tumor suppressors such as retinoblastoma protein (RB) and P53 inhibit pol III transcription, providing a link between common cancer mechanisms and pol III-dependent tRNA expression (110). Although cause or effect has not been established in these cases, tRNA expression changes in cancer may occur through global tRNA up-regulation to facilitate increased protein synthesis requirements in tumor cells (111).
Dysregulation of specific tRNA iso-acceptors is also implicated in cancer. Overexpression of the initiator tRNAMet promotes translation reprogramming and cell proliferation in the human breast epithelial cell lines 184A1 and MCF10A (109). This was corroborated in a comprehensive study that quantified tRNA expression profiles using tRNA microarrays and histone modification mapping across 470 patient-derived tissue samples representing various states of proliferation (112). Gingold et al. (112) demonstrated that expression is highest in the most proliferating samples and lowest in the differentiating cells.
In contrast, reduced tRNASec expression was observed in many proliferating and especially cancerous cell samples (112). tRNASec is required for the production of selenocysteine-containing proteins. Depending on the context, selenoprotein synthesis can either prevent or promote cancer (113); thus, up- or down-regulation of tRNASec may have relevance to disease. tRNA expression changes can also promote cancer through roles for tRNAs beyond protein synthesis. A recent review highlighted examples of tRNAs or tRNA-derived fragments from at least 16 iso-acceptor groups that are specifically de-regulated in cancer (110).
The tRNA expression profile in a particular cell will lead to more or less efficient translation of certain mRNAs depending on codon usage (114, 115). Differential expression of tRNAs also promotes cancer through favoring particular “translation programs.” The study of Gingold et al. (112) profiled codon usage in transcripts associated with cell cycle versus differentiation. The authors observed a dichotomy where codons with A or U in the 3rd codon position are generally more common in proliferation-associated mRNAs, and G- or C-ending codons are more common in differentiation-associated mRNA transcripts (112). The emerging view is that cells dynamically switch between “programs” of protein synthesis, in part by coordinating the transcription of tRNAs with anticodons matching the codon bias in expressed mRNAs.
Differential expression of specific tRNA iso-decoders
The expression of specific tRNA iso-decoders promotes metastasis in breast cancer model cell lines (112). The authors measured the relative abundance of different tRNA iso-decoders in cell lines selected for high rates of metastasis (MDA-LM2 and CN-LM1) and parental cell lines (MDA-231 and CN34). Two tRNAs ( and ) were highly up-regulated in both metastatic lines. These tRNAs were then overexpressed in MDA-231 cells to assess changes in the proteome resulting from their increased expression. The abundance of proteins encoded by transcripts enriched in the matching codons (GGC and GAR) increased. As measured by ribosome profiling, two such mRNAs (encoding EXOSC2 and GRIPAP1) showed higher rates of active translation in the cells overexpressing . RNAi-mediated knockdown of these mRNAs reduced in vitro invasion capacity of the cells, suggesting that EXOSC2 and GRIPAP1 are required for -promoted metastasis. Hence, the coordinated expression of tRNA iso-decoders facilitates translational reprogramming in cancer cells and is implicated in the promotion of proliferation as well as metastasis.
tRNA modification defects in disease
As mentioned previously, post-transcriptional modifications are important for tRNA function and stability. Hypo-modification can lead to rapid tRNA decay (116), and many tRNAs require anticodon modifications to ensure faithful codon recognition (34, 63). Over 50 different nucleotide modifications occur in eukaryotic tRNAs (117), and in humans, tRNAs contain an average of 13 modifications per molecule (61). Accordingly, tRNA modification defects are implicated in numerous diseases, including neurological, cardiac, respiratory, and metabolic diseases, as well as cancer and mitochondria-linked disorders (26). Most diseases that result from defects in tRNA modification are due to mutations in protein-coding genes or in mitochondrial tRNA genes, rather than cytosolic tRNA genes.
A recent example, however, provides compelling evidence of a cytosolic tRNA mutant and de-regulated nucleotide modification in human disease. A C65G mutation in was identified in a patient exhibiting abdominal pain, fatigue, muscle weakness, and low plasma selenium levels (28). Although humans encode two tRNASec genes, apparently only one is functional. A mutation in this gene has the potential to impact all 25 human selenoproteins, which are essential for normal development (118). Selenoproteins may be categorized into two groups: housekeeping and stress-related. Synthesis of housekeeping selenoproteins depends on a 5-methoxycarbonylmethyluridine (mcm5U) modification at position 34 of tRNASec, whereas further modification to 5-methoxycarbonylmethyl-2′-O-methyluridine (mcm5Um) promotes synthesis of stress-related selenoproteins (119). The tRNASec C65G variant only impaired expression of stress-related selenoproteins. This is attributed to the fact that the variant has markedly reduced levels of both the mcm5Um modification at position 34 and the N6-isopentenyl adenosine modification at position 37 (19). The finding underscores the complexity of nucleotide modification in tRNA function by showing that a mutation at one site in tRNA can impact modification at other locations in the tRNA body. In this case, a single nucleotide variant in the T-arm altered modifications in the anticodon stem loop (Fig. 1). Although the mechanism is not yet defined, presumably the C65G mutant inhibits or reduces the methyltransferase activity of the multifunction ALKBH8 gene product that catalyzes conversion of mcm5U to mcm5Um at position 34 (120).
Modifications can also drive or favor specific translation programs (65, 66). For example, melanomas harboring the V600E mutation in the proto-oncoprotein B-RAF depend on translational reprogramming controlled by up-regulation of U34 tRNA-modifying enzymes (67). Similar to the modulation of tRNA expression in metastatic breast cancer (121), the mechanism relies on coordinated regulation of both tRNAs and associated codon-biased transcripts. These modification tunable transcripts are sensitive to particular tRNA modification states (65). U34 tRNA modification promotes decoding of the “-AA” ending codons AAA, GAA, and CAA (122). Remarkably, up-regulation of U34-modifying enzymes promotes survival of melanomas dependent on hypoxia inducible factor 1α (HIF-1α) metabolism. Elevated levels of HIF-1α correlate with tumor metastasis and poor patient prognosis as well as tumor resistance to therapy (123). Indeed, the HIF-1α mRNA is enriched in AAA, GAA, and CAA codons (67). When U34-modifying enzymes ELP3, CTU1, or CTU2 were knocked down, HIF-1α protein levels decreased even though HIF-1α mRNA levels were unchanged. Thus, cancer cells are able to regulate tRNA modification enzymes to ultimately tune protein synthesis rates and protein levels in favor of oncogenesis.
tRNA variants in neurodegeneration
A common attribute of disorders linked to defective protein homeostasis is the accumulation of mistranslated or misfolded proteins in cells (20, 124). In many cell types, this problem can be counteracted through apoptosis or cell division (124). However, post-mitotic cells such as those found in the heart and brain are incapable of diluting misfolded proteins through division and lack the regenerative capacity to replace apoptotic cells readily (124). Furthermore, protein quality control decreases in post-mitotic tissues with age (125). Post-mitotic tissues may be particularly vulnerable to the consequences of tRNA variants and increasingly so with age.
Girstmair et al. (141) proposed a role for cytosolic tRNAs in Huntington's disease (HD). HD is caused by an expanded Gln repeat in the huntingtin protein, encoded by a stretch of 40–100 repeated CAG codons (126). In some cases, shorter CAG repeats appear to also cause HD, suggesting there are additional disease modifiers. Continuous translation of the repeat depletes charged , which results in more frequent frameshifting in the translation of the huntingtin gene, possibly exacerbating the disease phenotype (141). Although variants are not yet known to exacerbate HD, these findings illustrate the importance of tRNA function and abundance in pathologies of the brain.
Indeed, naturally occurring tRNA variants have the potential to deplete the abundance of a brain-specific tRNA that is essential for health. Ishimura et al. (21) uncovered a synthetic toxic effect involving a single cytosolic tRNA variant that causes widespread neurodegeneration in mice (21). Mutations in Gtpbp2 (encoding a protein that rescues stalled ribosomes) and Tr20 (encoding ) were found to co-occur in mice identified in a phenotypic screen for neurodegeneration. Mice carrying both mutations exhibit rapid neurodegeneration and die at 8–9 weeks. At 3 weeks, the mutant mice are indistinguishable from WT. The C50T mutation (n-Tr20) prevents maturation and, in combination with the loss of GTPBP2, leads to ribosome stalling. Despite many “redundant” tRNAArg iso-decoders in the cell, the lack of function of this single tRNA causes ribosome stalling. The authors measured a 3-fold increase in AGA pauses in the n-Tr20 mutant compared with a mouse containing the WT tRNA. Fascinatingly, tRNAArg C50T variants also occur in the human population, including in a TCT iso-acceptor (Table 3).
This work exemplifies the ways in which tRNA variants can exacerbate pathways to disease. Two observations from this work may have broader applicability to understanding the roles for tRNA variants in disease. First, the phenotype was tissue-specific, because expression of the n-Tr20–encoded tRNA is only observed in the central nervous system. Second, a coincident mutation in another gene sensitized cells to the loss-of-function mutation in a single tRNA gene. Together, these mutations caused disease in the animal model.
Conclusion
Humans display a remarkable array of both common and rare tRNA mutants, some with the obvious potential to mistranslate the genetic code (Tables 1 and 2 and Table S1) or create defective tRNAs (e.g. Table 3). Indeed, such tRNA variants can elicit significant levels of mistranslation in human cells and influence protein synthesis and protein homeostasis (Fig. 4). Above, we highlighted recent examples showing how tRNA variants and defective tRNA genes contribute to disease. In addition to causing disease, tRNA variants act synergistically with other disease-causing alleles by placing additional stress on protein quality control mechanisms or biasing translation programs that drive disease. Furthermore, tissue-specific tRNA expression and de-regulated tRNA expression or modification contribute to disease and phenotypic defects at the cellular level. Together, these observations suggest that cytosolic tRNA mutations may have greater importance in disease than previously recognized. We hope that the evidence provided in this review will stimulate new interest in considering cytoplasmic tRNA variants as an important factor in human genetic variation and disease.
Figure 4.

Phenotypic consequences of tRNA variation. tRNA variation can impact expression, aminoacylation, or processing and maturation of tRNAs, including nucleotide modification and tRNA folding. Alterations in the functional tRNA pool can impact mRNA translation by causing mistranslation, frame-shifting, ribosome stalling, or increased expression of codon-biased transcripts. These alterations can in turn alter the amino acid sequence, folding, and abundance of proteins across the entire proteome.
Supplementary Material
Acknowledgments
We are grateful to David Wright and Lluís Ribas de Pouplana for insightful discussions and suggestions to the manuscript.
This work was supported in part by Natural Sciences and Engineering Research Council of Canada Grants RGPIN 04282-2014 (to P. O.), 530175-2018 (to P. O.), and RGPIN-2015-04394 (to C. J. B.); Canada Foundation for Innovation Grant 229917 (to P. O.); the Ontario Research Fund 229917 (to P. O.); Canada Research Chairs 950-229917 (to P. O.); and the Ontario Centres of Excellence Grant 28922 (to P. O.). This is the first article in the “tRNAs and aminoacyl-tRNA synthetases in human disease” JBC Reviews series. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2 and supporting Refs. 1–8.
- AARS
- aminoacyl-tRNA synthetase
- SNP
- single nucleotide polymorphism
- TAM
- transcription-associated mutagenesis
- CFTR
- cystic fibrosis transmembrane conductance regulator
- mcm5U
- 5-methoxycarbonylmethyluridine
- mcm5Um
- 5-methoxycarbonylmethyl-2′-O-methyluridine
- HIF
- hypoxia-inducible factor
- GtRNAdb
- Genomic tRNA database
- pol III
- polymerase III
- HD
- Huntington's disease.
References
- 1. Crick F. H. C. (1955) On degenerate templates and the adaptor hypothesis. A Note for the RNA Tie Club, 1–17 [Google Scholar]
- 2. Hoagland M. B., Stephenson M. L., Scott J. F., Hecht L. I., and Zamecnik P. C. (1958) A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 231, 241–257 [PubMed] [Google Scholar]
- 3. Holley R. W., Everett G. A., Madison J. T., and Zamir A. (1965) Nucleotide sequences in the yeast alanine transfer ribonucleic acid. J. Biol. Chem. 240, 2122–2128 [PubMed] [Google Scholar]
- 4. Gardner R. S., Wahba A. J., Basilio C., Miller R. S., Lengyel P., and Speyer J. F. (1962) Synthetic polynucleotides and the amino acid code. VII. Proc. Natl. Acad. Sci. U.S.A. 48, 2087–2094 10.1073/pnas.48.12.2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nishimura S., Jones D. S., and Khorana H. G. (1965) Studies on polynucleotides. 48. The in vitro synthesis of a co-polypeptide containing two amino acids in alternating sequence dependent upon a DNA-like polymer containing two nucleotides in alternating sequence. J. Mol. Biol. 13, 302–324 10.1016/S0022-2836(65)80098-5 [DOI] [PubMed] [Google Scholar]
- 6. Nirenberg M., Leder P., Bernfield M., Brimacombe R., Trupin J., Rottman F., and O'Neal C. (1965) RNA codewords and protein synthesis, VII. On the general nature of the RNA code. Proc. Natl. Acad. Sci. U.S.A. 53, 1161–1168 10.1073/pnas.53.5.1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Söll D., Ohtsuka E., Jones D. S., Lohrmann R., Hayatsu H., Nishimura S., and Khorana H. G. (1965) Studies on polynucleotides, XLIX. Stimulation of the binding of aminoacyl-sRNA's to ribosomes by ribotrinucleotides and a survey of codon assignments for 20 amino acids. Proc. Natl. Acad. Sci. U.S.A. 54, 1378–1385 10.1073/pnas.54.5.1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carbon J., Berg P., and Yanofsky C. (1966) Missense suppression due to a genetically altered tRNA. Cold Spring Harb. Symp. Quant. Biol. 31, 487–497 10.1101/SQB.1966.031.01.063 [DOI] [PubMed] [Google Scholar]
- 9. Riyasaty S., and Atkins J. F. (1968) External suppression of a frameshift mutant in Salmonella. J. Mol. Biol. 34, 541–557 10.1016/0022-2836(68)90179-4 [DOI] [PubMed] [Google Scholar]
- 10. Ling J., O'Donoghue P., and Söll D. (2015) Genetic code flexibility in microorganisms: novel mechanisms and impact on physiology. Nat. Rev. Microbiol. 13, 707–721 10.1038/nrmicro3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bezerra A. R., Simões J., Lee W., Rung J., Weil T., Gut I. G., Gut M., Bayés M., Rizzetto L., Cavalieri D., Giovannini G., Bozza S., Romani L., Kapushesky M., Moura G. R., and Santos M. A. (2013) Reversion of a fungal genetic code alteration links proteome instability with genomic and phenotypic diversification. Proc. Natl. Acad. Sci. U.S.A. 110, 11079–11084 10.1073/pnas.1302094110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lant J. T., Berg M. D., Sze D. H. W., Hoffman K. S., Akinpelu I. C., Turk M. A., Heinemann I. U., Duennwald M. L., Brandl C. J., and O'Donoghue P. (2018) Visualizing tRNA-dependent mistranslation in human cells. RNA Biol. 15, 567–575 10.1080/15476286.2017.1379645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoffman K. S., Berg M. D., Shilton B. H., Brandl C. J., and O'Donoghue P. (2017) Genetic selection for mistranslation rescues a defective co-chaperone in yeast. Nucleic Acids Res. 45, 3407–3421 10.1093/nar/gkw1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barrell B. G., Bankier A. T., and Drouin J. (1979) A different genetic code in human mitochondria. Nature 282, 189–194 10.1038/282189a0 [DOI] [PubMed] [Google Scholar]
- 15. Campbell J. H., O'Donoghue P., Campbell A. G., Schwientek P., Sczyrba A., Woyke T., Söll D., and Podar M. (2013) UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc. Natl. Acad. Sci. U.S.A. 110, 5540–5545 10.1073/pnas.1303090110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Böck A., and Stadtman T. C. (1988) Selenocysteine, a highly specific component of certain enzymes, is incorporated by a UGA-directed co-translational mechanism. Biofactors 1, 245–250 [PubMed] [Google Scholar]
- 17. Berry M. J., Martin G. W. 3rd., Tujebajeva R., Grundner-Culemann E., Mansell J. B., Morozova N., and Harney J. W. (2002) Selenocysteine insertion sequence element characterization and selenoprotein expression. Methods Enzymol. 347, 17–24 10.1016/S0076-6879(02)47004-8 [DOI] [PubMed] [Google Scholar]
- 18. Bröcker M. J., Ho J. M., Church G. M., Söll D., and O'Donoghue P. (2014) Recoding the genetic code with selenocysteine. Angew. Chem. Int. Ed. Engl. 53, 319–323 10.1002/anie.201308584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schoenmakers E., Carlson B., Agostini M., Moran C., Rajanayagam O., Bochukova E., Tobe R., Peat R., Gevers E., Muntoni F., Guicheney P., Schoenmakers N., Farooqi S., Lyons G., Hatfield D., and Chatterjee K. (2016) Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J. Clin. Invest. 126, 992–996 10.1172/JCI84747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kapur M., and Ackerman S. L. (2018) mRNA translation gone awry: translation fidelity and neurological disease. Trends Genet. 34, 218–231 10.1016/j.tig.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ishimura R., Nagy G., Dotu I., Zhou H., Yang X. L., Schimmel P., Senju S., Nishimura Y., Chuang J. H., and Ackerman S. L. (2014) Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345, 455–459 10.1126/science.1249749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Santos M., Pereira P. M., Varanda A. S., Carvalho J., Azevedo M., Mateus D. D., Mendes N., Oliveira P., Trindade F., Pinto M. T., Bordeira-Carriço R., Carneiro F., Vitorino R., Oliveira C., and Santos M. A. S. (2018) Codon misreading tRNAs promote tumor growth in mice. RNA Biol. 15, 773–786 10.1080/15476286.2018.1454244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berg M. D., Hoffman K. S., Genereaux J., Mian S., Trussler R. S., Haniford D. B., O'Donoghue P., and Brandl C. J. (2017) Evolving mistranslating tRNAs through a phenotypically ambivalent intermediate in Saccharomyces cerevisiae. Genetics 206, 1865–1879 10.1534/genetics.117.203232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meyer-Schuman R., and Antonellis A. (2017) Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum. Mol. Genet. 26, R114–R127 10.1093/hmg/ddx231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bednářová A., Hanna M., Durham I., VanCleave T., England A., Chaudhuri A., and Krishnan N. (2017) Lost in translation: defects in transfer RNA modifications and neurological disorders. Front. Mol. Neurosci. 10, 135 10.3389/fnmol.2017.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Torres A. G., Batlle E., and Ribas de Pouplana L. (2014) Role of tRNA modifications in human diseases. Trends Mol. Med. 20, 306–314 10.1016/j.molmed.2014.01.008 [DOI] [PubMed] [Google Scholar]
- 27. Kirchner S., and Ignatova Z. (2015) Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 16, 98–112 10.1038/nrg3861 [DOI] [PubMed] [Google Scholar]
- 28. Abbott J. A., Francklyn C. S., and Robey-Bond S. M. (2014) Transfer RNA and human disease. Front. Genet. 5, 158 10.3389/fgene.2014.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yasukawa T., Suzuki T., Ishii N., Ohta S., and Watanabe K. (2001) Wobble modification defect in tRNA disturbs codon-anticodon interaction in a mitochondrial disease. EMBO J. 20, 4794–4802 10.1093/emboj/20.17.4794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Agris P. F., Vendeix F. A., and Graham W. D. (2007) tRNA's wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 366, 1–13 10.1016/j.jmb.2006.11.046 [DOI] [PubMed] [Google Scholar]
- 31. Joshi K., Bhatt M. J., and Farabaugh P. J. (2018) Codon-specific effects of tRNA anticodon loop modifications on translational misreading errors in the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 46, 10331–10339 10.1093/nar/gky664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pavlov M. Y., and Ehrenberg M. (2018) Substrate-induced formation of ribosomal decoding center for accurate and rapid genetic code translation. Annu. Rev. Biophys. 47, 525–548 10.1146/annurev-biophys-060414-034148 [DOI] [PubMed] [Google Scholar]
- 33. O'Donoghue P., Prat L., Heinemann I. U., Ling J., Odoi K., Liu W. R., and Söll D. (2012) Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 586, 3931–3937 10.1016/j.febslet.2012.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crick F. H. (1966) Codon–anticodon pairing: the wobble hypothesis. J. Mol. Biol. 19, 548–555 10.1016/S0022-2836(66)80022-0 [DOI] [PubMed] [Google Scholar]
- 35. Söll D., Jones D. S., Ohtsuka E., Faulkner R. D., Lohrmann R., Hayatsu H., and Khorana H. G. (1966) Specificity of sRNA for recognition of codons as studied by the ribosomal binding technique. J. Mol. Biol. 19, 556–573 10.1016/S0022-2836(66)80023-2 [DOI] [PubMed] [Google Scholar]
- 36. Agris P. F., Narendran A., Sarachan K., Väre V. Y. P., and Eruysal E. (2017) The importance of being modified: the role of RNA modifications in translational fidelity. Enzymes 41, 1–50 10.1016/bs.enz.2017.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gustilo E. M., Vendeix F. A., and Agris P. F. (2008) tRNA's modifications bring order to gene expression. Curr. Opin. Microbiol. 11, 134–140 10.1016/j.mib.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tükenmez H., Xu H., Esberg A., and Byström A. S. (2015) The role of wobble uridine modifications in +1 translational frameshifting in eukaryotes. Nucleic Acids Res. 43, 9489–9499 10.1093/nar/gkv832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin C. A., Ellis S. R., and True H. L. (2010) The Sua5 protein is essential for normal translational regulation in yeast. Mol. Cell. Biol. 30, 354–363 10.1128/MCB.00754-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jaffe J. D., Stange-Thomann N., Smith C., DeCaprio D., Fisher S., Butler J., Calvo S., Elkins T., FitzGerald M. G., Hafez N., Kodira C. D., Major J., Wang S., Wilkinson J., Nicol R., Nusbaum C., et al. (2004) The complete genome and proteome of Mycoplasma mobile. Genome Res. 14, 1447–1461 10.1101/gr.2674004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chatterjee K., Nostramo R. T., Wan Y., and Hopper A. K. (2018) tRNA dynamics between the nucleus, cytoplasm and mitochondrial surface: location, location, location. Biochim. Biophys. Acta 1861, 373–386 10.1016/j.bbagrm.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goodenbour J. M., and Pan T. (2006) Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 34, 6137–6146 10.1093/nar/gkl725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chan P. P., and Lowe T. M. (2009) GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97 10.1093/nar/gkn787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bour T., Mahmoudi N., Kapps D., Thiberge S., Bargieri D., Ménard R., and Frugier M. (2016) Apicomplexa-specific tRip facilitates import of exogenous tRNAs into malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 113, 4717–4722 10.1073/pnas.1600476113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Umu S. U., Langseth H., Bucher-Johannessen C., Fromm B., Keller A., Meese E., Lauritzen M., Leithaug M., Lyle R., and Rounge T. B. (2018) A comprehensive profile of circulating RNAs in human serum. RNA Biol. 15, 242–250 10.1080/15476286.2017.1403003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Canella D., Praz V., Reina J. H., Cousin P., and Hernandez N. (2010) Defining the RNA polymerase III transcriptome: genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res. 20, 710–721 10.1101/gr.101337.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gogakos T., Brown M., Garzia A., Meyer C., Hafner M., and Tuschl T. (2017) Characterizing expression and processing of precursor and mature human tRNAs by hydro-tRNAseq and PAR-CLIP. Cell Rep. 20, 1463–1475 10.1016/j.celrep.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dittmar K. A., Goodenbour J. M., and Pan T. (2006) Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2, e221 10.1371/journal.pgen.0020221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Melé M., Ferreira P. G., Reverter F., DeLuca D. S., Monlong J., Sammeth M., Young T. R., Goldmann J. M., Pervouchine D. D., Sullivan T. J., Johnson R., Segrè A. V., Djebali S., Niarchou A., GTEx Consortium, et al. (2015) Human genomics. The human transcriptome across tissues and individuals. Science 348, 660–665 10.1126/science.aaa0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grote A., Hiller K., Scheer M., Münch R., Nörtemann B., Hempel D. C., and Jahn D. (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–W531 10.1093/nar/gki376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Betat H., Long Y., Jackman J. E., and Mörl M. (2014) From end to end: tRNA editing at 5′- and 3′-terminal positions. Int. J. Mol. Sci. 15, 23975–23998 10.3390/ijms151223975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilusz J. E. (2015) Controlling translation via modulation of tRNA levels: controlling translation via modulation of tRNA levels. Wiley Interdiscip. Rev. RNA 6, 453–470 10.1002/wrna.1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilusz J. E., Whipple J. M., Phizicky E. M., and Sharp P. A. (2011) tRNAs marked with CCACCA are targeted for degradation. Science 334, 817–821 10.1126/science.1213671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chakraborty P. K., Schmitz-Abe K., Kennedy E. K., Mamady H., Naas T., Durie D., Campagna D. R., Lau A., Sendamarai A. K., Wiseman D. H., May A., Jolles S., Connor P., Powell C., Heeney M. M., et al. (2014) Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood 124, 2867–2871 10.1182/blood-2014-08-591370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lorenz C., Lunse C. E., and Morl M. (2017) tRNA modifications: impact on structure and thermal adaptation. Biomolecules 7, 35 10.3390/biom7020035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Heinemann I. U., Nakamura A., O'Donoghue P., Eiler D., and Söll D. (2012) tRNAHis-guanylyltransferase establishes tRNAHis identity. Nucleic Acids Res. 40, 333–344 10.1093/nar/gkr696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gu W., Hurto R. L., Hopper A. K., Grayhack E. J., and Phizicky E. M. (2005) Depletion of Saccharomyces cerevisiae tRNA(His) guanylyltransferase Thg1p leads to uncharged tRNAHis with additional m(5)C. Mol. Cell. Biol. 25, 8191–8201 10.1128/MCB.25.18.8191-8201.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Preston M. A., D'Silva S., Kon Y., and Phizicky E. M. (2013) tRNAHis 5-methylcytidine levels increase in response to several growth arrest conditions in Saccharomyces cerevisiae. RNA 19, 243–256 10.1261/rna.035808.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Martinez A., Yamashita S., Nagaike T., Sakaguchi Y., Suzuki T., and Tomita K. (2017) Human BCDIN3D monomethylates cytoplasmic histidine transfer RNA. Nucleic Acids Res. 45, 5423–5436 10.1093/nar/gkx051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tomita K., and Liu Y. (2018) Human BCDIN3D is a cytoplasmic tRNA(His)-specific 5′-monophosphate methyltransferase. Front. Genet. 9, 305 10.3389/fgene.2018.00305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Saikia M., Fu Y., Pavon-Eternod M., He C., and Pan T. (2010) Genome-wide analysis of N1-methyl-adenosine modification in human tRNAs. RNA 16, 1317–1327 10.1261/rna.2057810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schimmel P. (2018) The emerging complexity of the tRNA world: mammalian tRNAs beyond protein synthesis. Nat. Rev. Mol. Cell Biol. 19, 45–58 10.1038/nrm.2017.77 [DOI] [PubMed] [Google Scholar]
- 63. Towns W. L., and Begley T. J. (2012) Transfer RNA methyltransferases and their corresponding modifications in budding yeast and humans: activities, predications, and potential roles in human health. DNA Cell Biol. 31, 434–454 10.1089/dna.2011.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hopper A. K. (2013) Transfer RNA post-transcriptional processing, turnover, and subcellular dynamics in the yeast Saccharomyces cerevisiae. Genetics 194, 43–67 10.1534/genetics.112.147470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dedon P. C., and Begley T. J. (2014) A system of RNA modifications and biased codon use controls cellular stress response at the level of translation. Chem. Res. Toxicol. 27, 330–337 10.1021/tx400438d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Endres L., Dedon P. C., and Begley T. J. (2015) Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 12, 603–614 10.1080/15476286.2015.1031947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rapino F., Delaunay S., Rambow F., Zhou Z., Tharun L., De Tullio P., Sin O., Shostak K., Schmitz S., Piepers J., Ghesquière B., Karim L., Charloteaux B., Jamart D., Florin A., et al. (2018) Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 558, 605–609 10.1038/s41586-018-0243-7 [DOI] [PubMed] [Google Scholar]
- 68. Ibba M., and Soll D. (2000) Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 69, 617–650 10.1146/annurev.biochem.69.1.617 [DOI] [PubMed] [Google Scholar]
- 69. Giegé R., Sissler M., and Florentz C. (1998) Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 26, 5017–5035 10.1093/nar/26.22.5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Payea M. J., Sloma M. F., Kon Y., Young D. L., Guy M. P., Zhang X., De Zoysa T., Fields S., Mathews D. H., and Phizicky E. M. (2018) Widespread temperature sensitivity and tRNA decay due to mutations in a yeast tRNA. RNA 24, 410–422 10.1261/rna.064642.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kessler A. C., Silveira d'Almeida G., and Alfonzo J. D. (2018) The role of intracellular compartmentalization on tRNA processing and modification. RNA Biol. 15, 554–566 10.1080/15476286.2017.1371402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hopper A. K., Pai D. A., and Engelke D. R. (2010) Cellular dynamics of tRNAs and their genes. FEBS Lett. 584, 310–317 10.1016/j.febslet.2009.11.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kwon N. H., Lee M. R., Kong J., Park S. K., Hwang B. J., Kim B. G., Lee E.-S., Moon H.-G., and Kim S. (2018) Transfer-RNA-mediated enhancement of ribosomal proteins S6 kinases signaling for cell proliferation. RNA Biol. 15, 635–648 10.1080/15476286.2017.1356563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lee Y. S., Shibata Y., Malhotra A., and Dutta A. (2009) A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 23, 2639–2649 10.1101/gad.1837609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schwartz M. H., and Pan T. (2017) Function and origin of mistranslation in distinct cellular contexts. Crit. Rev. Biochem. Mol. Biol. 52, 205–219 10.1080/10409238.2016.1274284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zimmerman S. M., Kon Y., Hauke A. C., Ruiz B. Y., Fields S., and Phizicky E. M. (2018) Conditional accumulation of toxic tRNAs to cause amino acid misincorporation. Nucleic Acids Res. 46, 7831–7843 10.1093/nar/gky623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Alvarez A., Barisone G. A., and Diaz E. (2014) Focus formation: a cell-based assay to determine the oncogenic potential of a gene. J. Vis. Exp. 94, e51742 10.3791/51742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hou Y.-M., Francklyn C., and Schimmel P. (1989) Molecular dissection of a transfer RNA and the basis for its identity. Trends Biochem. Sci. 14, 233–237 10.1016/0968-0004(89)90033-9 [DOI] [PubMed] [Google Scholar]
- 79. Sun L., Gomes A. C., He W., Zhou H., Wang X., Pan D. W., Schimmel P., Pan T., and Yang X.-L. (2016) Evolutionary gain of alanine mischarging to noncognate tRNAs with a G4:U69 base pair. J. Am. Chem. Soc. 138, 12948–12955 10.1021/jacs.6b07121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kuncha S. K., Mazeed M., Singh R., Kattula B., Routh S. B., and Sankaranarayanan R. (2018) A chiral selectivity relaxed paralog of DTD for proofreading tRNA mischarging in Animalia. Nat. Commun. 9, 511 10.1038/s41467-017-02204-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pezo V., Metzgar D., Hendrickson T. L., Waas W. F., Hazebrouck S., Döring V., Marlière P., Schimmel P., and De Crécy-Lagard V. (2004) Artificially ambiguous genetic code confers growth yield advantage. Proc. Natl. Acad. Sci. U.S.A. 101, 8593–8597 10.1073/pnas.0402893101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Netzer N., Goodenbour J. M., David A., Dittmar K. A., Jones R. B., Schneider J. R., Boone D., Eves E. M., Rosner M. R., Gibbs J. S., Embry A., Dolan B., Das S., Hickman H. D., Berglund P., et al. (2009) Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 462, 522–526 10.1038/nature08576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Moghal A., Mohler K., and Ibba M. (2014) Mistranslation of the genetic code. FEBS Lett. 588, 4305–4310 10.1016/j.febslet.2014.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wu J., Fan Y., and Ling J. (2014) Mechanism of oxidant-induced mistranslation by threonyl-tRNA synthetase. Nucleic Acids Res. 42, 6523–6531 10.1093/nar/gku271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lee J. Y., Kim D. G., Kim B. G., Yang W. S., Hong J., Kang T., Oh Y. S., Kim K. R., Han B. W., Hwang B. J., Kang B. S., Kang M. S., Kim M. H., Kwon N. H., and Kim S. (2014) Promiscuous methionyl-tRNA synthetase mediates adaptive mistranslation to protect cells against oxidative stress. J. Cell Sci. 127, 4234–4245 10.1242/jcs.152470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gomes A. C., Kordala A. J., Strack R., Wang X., Geslain R., Delaney K., Clark W. C., Keenan R., and Pan T. (2016) A dual fluorescent reporter for the investigation of methionine mistranslation in live cells. RNA 22, 467–476 10.1261/rna.054163.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fechter P., Rudinger-Thirion J., Tukalo M., and Giegé R. (2001) Major tyrosine identity determinants in Methanococcus jannaschii and Saccharomyces cerevisiae tRNA(Tyr) are conserved but expressed differently. Eur. J. Biochem. 268, 761–767 10.1046/j.1432-1327.2001.01931.x [DOI] [PubMed] [Google Scholar]
- 88. Torres A. G., Piñeyro D., Rodríguez-Escribà M., Camacho N., Reina O., Saint-Léger A., Filonava L., Batlle E., and Ribas de Pouplana L. (2015) Inosine modifications in human tRNAs are incorporated at the precursor tRNA level. Nucleic Acids Res. 43, 5145–5157 10.1093/nar/gkv277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Biddle W., Schmitt M. A., and Fisk J. D. (2016) Modification of orthogonal tRNAs: unexpected consequences for sense codon reassignment. Nucleic Acids Res. 44, 10042–10050 10.1093/nar/gkw948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Biddle W., Schmitt M. A., and Fisk J. D. (2015) Evaluating sense codon reassignment with a simple fluorescence screen. Biochemistry 54, 7355–7364 10.1021/acs.biochem.5b00870 [DOI] [PubMed] [Google Scholar]
- 91. Thornlow B. P., Hough J., Roger J. M., Gong H., Lowe T. M., and Corbett-Detig R. B. (2018) Transfer RNA genes experience exceptionally elevated mutation rates. Proc. Natl. Acad. Sci. U.S.A. 115, 8996–9001 10.1073/pnas.1801240115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jinks-Robertson S., and Bhagwat A. S. (2014) Transcription-associated mutagenesis. Annu. Rev. Genet. 48, 341–359 10.1146/annurev-genet-120213-092015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shoffner J. M., Lott M. T., Lezza A. M., Seibel P., Ballinger S. W., and Wallace D. C. (1990) Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 61, 931–937 10.1016/0092-8674(90)90059-N [DOI] [PubMed] [Google Scholar]
- 94. Jia Z., Wang X., Qin Y., Xue L., Jiang P., Meng Y., Shi S., Wang Y., Qin Mo J., and Guan M.-X. (2013) Coronary heart disease is associated with a mutation in mitochondrial tRNA. Hum. Mol. Genet. 22, 4064–4073 10.1093/hmg/ddt256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li H., Geng J., Yu H., Tang X., Yang X., and Xue L. (2018) Mitochondrial tRNAThr 15909A>G mutation associated with hypertension in a Chinese Han pedigree. Biochem. Biophys. Res. Commun. 495, 574–581 10.1016/j.bbrc.2017.11.061 [DOI] [PubMed] [Google Scholar]
- 96. Ding Y., Xia B.-H., Zhang C.-J., and Zhuo G.-C. (2018) Mitochondrial tRNALeu(UUR) C3275T, tRNAGln T4363C and tRNALys A8343G mutations may be associated with PCOS and metabolic syndrome. Gene 642, 299–306 10.1016/j.gene.2017.11.049 [DOI] [PubMed] [Google Scholar]
- 97. Meng F., He Z., Tang X., Zheng J., Jin X., Zhu Y., Ren X., Zhou M., Wang M., Gong S., Mo J. Q., Shu Q., and Guan M.-X. (2018) Contribution of the tRNAIle 4317A→G mutation to the phenotypic manifestation of the deafness-associated mitochondrial 12S rRNA 1555A→G mutation. J. Biol. Chem. 293, 3321–3334 10.1074/jbc.RA117.000530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. O'Neill V. A., Eden F. C., Pratt K., and Hatfield D. L. (1985) A human opal suppressor tRNA gene and pseudogene. J. Biol. Chem. 260, 2501–2508 [PubMed] [Google Scholar]
- 99. Santesmasses D., Mariotti M., and Guigó R. (2017) Computational identification of the selenocysteine tRNA (tRNASec) in genomes. PLoS Comput. Biol. 13, e1005383 10.1371/journal.pcbi.1005383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Iben J. R., and Maraia R. J. (2014) tRNA gene copy number variation in humans. Gene 536, 376–384 10.1016/j.gene.2013.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wei F.-Y., Suzuki T., Watanabe S., Kimura S., Kaitsuka T., Fujimura A., Matsui H., Atta M., Michiue H., Fontecave M., Yamagata K., Suzuki T., and Tomizawa K. (2011) Deficit of tRNALys modification by Cdkal1 causes the development of type 2 diabetes in mice. J. Clin. Invest. 121, 3598–3608 10.1172/JCI58056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kutter C., Brown G. D., Gonçalves A., Wilson M. D., Watt S., Brazma A., White R. J., and Odom D. T. (2011) Pol III binding in six mammals shows conservation among amino acid isotypes despite divergence among tRNA genes. Nat. Genet. 43, 948–955 10.1038/ng.906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kirchner S., Cai Z., Rauscher R., Kastelic N., Anding M., Czech A., Kleizen B., Ostedgaard L. S., Braakman I., Sheppard D. N., and Ignatova Z. (2017) Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol. 15, e2000779 10.1371/journal.pbio.2000779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Rauscher R., and Ignatova Z. (2018) Timing during translation matters: synonymous mutations in human pathologies influence protein folding and function. Biochem. Soc. Trans. 46, 937–944 10.1042/BST20170422 [DOI] [PubMed] [Google Scholar]
- 105. Kimchi-Sarfaty C., Oh J. M., Kim I. W., Sauna Z. E., Calcagno A. M., Ambudkar S. V., and Gottesman M. M. (2007) A “Silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528 10.1126/science.1135308 [DOI] [PubMed] [Google Scholar]
- 106. Fernández-Calero T., Cabrera-Cabrera F., Ehrlich R., and Marín M. (2016) Silent polymorphisms: can the tRNA population explain changes in protein properties? Life 6, 9 10.3390/life6010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Horjales S., Cota G., Señorale-Pose M., Rovira C., Román E., Artagaveytia N., Ehrlich R., and Marín M. (2007) Translational machinery and protein folding: evidence of conformational variants of the estrogen receptor α. Arch. Biochem. Biophys. 467, 139–143 10.1016/j.abb.2007.07.029 [DOI] [PubMed] [Google Scholar]
- 108. Foreman M. G., Kong X., DeMeo D. L., Pillai S. G., Hersh C. P., Bakke P., Gulsvik A., Lomas D. A., Litonjua A. A., Shapiro S. D., Tal-Singer R., and Silverman E. K. (2011) Polymorphisms in surfactant protein-D are associated with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 44, 316–322 10.1165/rcmb.2009-0360OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Pavon-Eternod M., Gomes S., Rosner M. R., and Pan T. (2013) Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA 19, 461–466 10.1261/rna.037507.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Huang S. Q., Sun B., Xiong Z. P., Shu Y., Zhou H. H., Zhang W., Xiong J., and Li Q. (2018) The dysregulation of tRNAs and tRNA derivatives in cancer. J. Exp. Clin. Cancer Res. 37, 101 10.1186/s13046-018-0745-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mahlab S., Tuller T., and Linial M. (2012) Conservation of the relative tRNA composition in healthy and cancerous tissues. RNA 18, 640–652 10.1261/rna.030775.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gingold H., Tehler D., Christoffersen N. R., Nielsen M. M., Asmar F., Kooistra S. M., Christophersen N. S., Christensen L. L., Borre M., Sørensen K. D., Andersen L. D., Andersen C. L., Hulleman E., Wurdinger T., Ralfkiær E., et al. (2014) A dual program for translation regulation in cellular proliferation and differentiation. Cell 158, 1281–1292 10.1016/j.cell.2014.08.011 [DOI] [PubMed] [Google Scholar]
- 113. Hatfield D. L., Yoo M.-H., Carlson B. A., and Gladyshev V. N. (2009) Selenoproteins that function in cancer prevention and promotion. Biochim. Biophys. Acta 1790, 1541–1545 10.1016/j.bbagen.2009.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Quax T. E., Claassens N. J., Söll D., and van der Oost J. (2015) Codon bias as a means to fine-tune gene expression. Mol. Cell 59, 149–161 10.1016/j.molcel.2015.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Frumkin I., Lajoie M. J., Gregg C. J., Hornung G., Church G. M., and Pilpel Y. (2018) Codon usage of highly expressed genes affects proteome-wide translation efficiency. Proc. Natl. Acad. Sci. U.S.A. 115, E4940–E4949 10.1073/pnas.1719375115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Alexandrov A., Chernyakov I., Gu W., Hiley S. L., Hughes T. R., Grayhack E. J., and Phizicky E. M. (2006) Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21, 87–96 10.1016/j.molcel.2005.10.036 [DOI] [PubMed] [Google Scholar]
- 117. Boccaletto P., Machnicka M. A., Purta E., Piatkowski P., Baginski B., Wirecki T. K., de Crécy-Lagard V., Ross R., Limbach P. A., Kotter A., Helm M., and Bujnicki J. M. (2018) MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307 10.1093/nar/gkx1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Schweizer U., and Fradejas-Villar N. (2016) Why 21? The significance of selenoproteins for human health revealed by inborn errors of metabolism. FASEB J. 30, 3669–3681 10.1096/fj.201600424 [DOI] [PubMed] [Google Scholar]
- 119. Carlson B., Yoo M.-H., Tsuji P., Gladyshev V. N., and Hatfield D. L. (2009) Mouse models targeting selenocysteine tRNA expression for elucidating the role of selenoproteins in health and development. Molecules 14, 3509–3527 10.3390/molecules14093509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Songe-Møller L., van den Born E., Leihne V., Vågbo C. B., Kristoffersen T., Krokan H. E., Kirpekar F., Falnes P. Ø., and Klungland A. (2010) Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol. Cell. Biol. 30, 1814–1827 10.1128/MCB.01602-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Goodarzi H., Nguyen H. C. B., Zhang S., Dill B. D., Molina H., and Tavazoie S. F. (2016) Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 165, 1416–1427 10.1016/j.cell.2016.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Nedialkova D. D., and Leidel S. A. (2015) Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161, 1606–1618 10.1016/j.cell.2015.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Masoud G. N., and Li W. (2015) HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 5, 378–389 10.1016/j.apsb.2015.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Josefson R., Andersson R., and Nyström T. (2017) How and why do toxic conformers of aberrant proteins accumulate during ageing? Essays Biochem. 61, 317–324 10.1042/EBC20160085 [DOI] [PubMed] [Google Scholar]
- 125. de Toda I. M., Vida C., Ortega E., and De La Fuente M. (2016) Hsp70 basal levels, a tissue marker of the rate of aging and longevity in mice. Exp Gerontol. 84, 21–28 10.1016/j.exger.2016.08.013 [DOI] [PubMed] [Google Scholar]
- 126. Bates G. P., Dorsey R., Gusella J. F., Hayden M. R., Kay C., Leavitt B. R., Nance M., Ross C. A., Scahill R. I., Wetzel R., Wild E. J., and Tabrizi S. J. (2015) Huntington disease. Nat. Rev. Dis. Primers 1, 15005 [DOI] [PubMed] [Google Scholar]
- 127. Parisien M., Wang X., and Pan T. (2013) Diversity of human tRNA genes from the 1000-genomes project. RNA Biol. 10, 1853–1867 10.4161/rna.27361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Chan P. P., and Lowe T. M. (2016) GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189 10.1093/nar/gkv1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Casper J., Zweig A. S., Villarreal C., Tyner C., Speir M. L., Rosenbloom K. R., Raney B. J., Lee C. M., Lee B. T., Karolchik D., Hinrichs A. S., Haeussler M., Guruvadoo L., Navarro Gonzalez J., et al. (2018) The UCSC genome browser database: 2018 update. Nucleic Acids Res. 46, D762–D769 10.1093/nar/gkx1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hrabeta-Robinson E., Marcus E., Cozen A. E., Phizicky E. M., and Lowe T. M. (2017) High-throughput small RNA sequencing enhanced by AlkB-facilitated RNA de-methylation (ARM-Seq). Methods Mol. Biol. 1562, 231–243 10.1007/978-1-4939-6807-7_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gerstein M. B., Kundaje A., Hariharan M., Landt S. G., Yan K. K., Cheng C., Mu X. J., Khurana E., Rozowsky J., Alexander R., Min R., Alves P., Abyzov A., Addleman N., Bhardwaj N., et al. (2012) Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100 10.1038/nature11245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang J., Zhuang J., Iyer S., Lin X., Whitfield T. W., Greven M. C., Pierce B. G., Dong X., Kundaje A., Cheng Y., Rando O. J., Birney E., Myers R. M., Noble W. S., Snyder M., and Weng Z. (2012) Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22, 1798–1812 10.1101/gr.139105.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wang J., Zhuang J., Iyer S., Lin X. Y., Greven M. C., Kim B. H., Moore J., Pierce B. G., Dong X., Virgil D., Birney E., Hung J. H., and Weng Z. (2013) Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 41, D171–D176 10.1093/nar/gks1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Parmeggiani A., Krab I. M., Watanabe T., Nielsen R. C., Dahlberg C., Nyborg J., and Nissen P. (2006) Enacyloxin IIa pinpoints a binding pocket of elongation factor Tu for development of novel antibiotics. J. Biol. Chem. 281, 2893–2900 10.1074/jbc.M505951200 [DOI] [PubMed] [Google Scholar]
- 135. Hohn M. J., Park H. S., O'Donoghue P., Schnitzbauer M., and Söll D. (2006) Emergence of the universal genetic code imprinted in an RNA record. Proc. Natl. Acad. Sci. U.S.A. 103, 18095–18100 10.1073/pnas.0608762103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Roberts E., Eargle J., Wright D., and Luthey-Schulten Z. (2006) MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics 7, 382 10.1186/1471-2105-7-382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., and Gascuel O. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 138. Shimodaira H., and Hasegawa M. (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114–1116 10.1093/oxfordjournals.molbev.a026201 [DOI] [Google Scholar]
- 139. Sprinzl M., and Vassilenko K. S. (2005) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 33, D139–D140 10.1093/nar/gki012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kobayashi Y., Momoi M. Y., Tominaga K., Momoi T., Nihei K., Yanagisawa M., Kagawa Y., and Ohta S. (1990) A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem. Biophys. Res. Commun. 173, 816–822 10.1016/S0006-291X(05)80860-5 [DOI] [PubMed] [Google Scholar]
- 141. Girstmair H., Saffert P., Rode S., Czech A., Holland G., Bannert N., and Ignatova Z. (2013) Depletion of cognate charged transfer RNA causes translational frameshifting within the expanded CAG stretch in huntingtin. Cell Rep. 3, 148–159 10.1016/j.celrep.2012.12.019 [DOI] [PubMed] [Google Scholar]
- 142. 1000 Genomes Project Consortium, Auton A., Brooks L. D., Durbin R. M., Garrison E. P., Kang H. M., Korbel J. O., Marchini J. L., McCarthy S., McVean G. A., and Abecasis G. R. (2015) A global reference for human genetic variation. Nature. 526, 68–74 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.