

Abstract
Background
Clear cell renal cell carcinoma (ccRCC) is the most common subtype of adult renal neoplasm and has a poor prognosis owing to a limited understanding of the disease mechanisms. The aim of this study was to explore and identify the key genes and signaling pathways in ccRCC.
Methods
The GSE36895 gene expression profiles were downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were then screened using software packages in R. After Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis, a protein-protein interaction (PPI) network of DEGs was constructed with Cytoscape software, and submodules were subsequently analyzed using the MCODE plug-in.
Results
Twenty-nine ccRCC samples and 23 normal samples were incorporated into this study, and a total of 468 DEGs were filtered, consisting of 180 upregulated genes and 288 downregulated genes. The upregulated DEGs were significantly enriched in the immune response, response to wounding, inflammatory response, and response to hypoxia, whereas downregulated genes were mainly enriched in ion transport, anion transport, and monovalent inorganic cation transport biological processes (BPs). According to Molecular Complex Detection analysis in PPI, C1QA, C1QB, C1QC, CCND1 and EGF had higher degrees of connectivity and could participate in the majority of important pathways, such as cytokine-cytokine receptor interactions, the chemokine signaling pathway, and the complement and coagulation cascade pathways.
Conclusions
Our study suggests that C1QA, C1QB, C1QC, CCND1 and EGF may play key roles in the progression of ccRCC, which will be useful for future studies on the underlying mechanisms of ccRCC.
Keywords: Clear cell renal cell carcinoma (ccRCC), bioinformatics analysis, microarray, differentially expressed genes (DEGs)
Introduction
Renal cell carcinoma (RCC) represents one of most common malignancies worldwide, with the second highest rates of incidence and mortality among urological tumors (1). In 2016, more than 62,700 new cases of RCC were detected, and nearly 14,240 RCC-related deaths occurred in developed countries, mainly affecting males and showing a peak in incidence at 60 to 70 years old (1,2). Clear cell RCC (ccRCC), the most common subtype of RCC, accounts for approximately 75% of all renal tumors (3). With the improvement of routine imaging technology used for diagnosis, most patients are diagnosed with a small single kidney mass. However, it is well known that ccRCC is not sensitive to chemotherapy and that radiotherapy and surgical approaches have a limited efficacy for patients with ccRCC (4). Therefore, an exploration of potential molecular mechanisms and the identification of effective biomarkers involved in the pathogenesis of ccRCC is urgently needed to improve the effectiveness of therapeutic strategies.
A recent series report showed that in addition to heredity and acquired cystic kidney disease, several other factors, such as smoking tobacco, being overweight and having hypertension, are verified to increase the risk for RCC (2,5-8). Genome association studies have reported that mutation of the VHL (von Hippel-Lindau) gene, a tumor suppressor gene that increases hypoxia inducible factor (HIF)1α and HIF 2α expression, was found in nearly 80% of ccRCCs (9). Two other genes, BAP1 and PBRM1, are also frequently altered in 15% and 50% of patients with ccRCC, respectively (10). Moreover, microRNA (miRNA), a kind of small noncoding RNA molecule that functions in the regulation of expression of targeted genes at the posttranscriptional level, plays a crucial role in ccRCC. Many miRNAs, such as miR-21 (11), miR-590-5p (12) and miR-630 (13), have been shown to act as oncogenes, while miR-584 (14), miR-92 (15) and miR-187 (16) were demonstrated to suppress the biological processes (BPs) of ccRCC. Due to the noncontinuous nature of most studies, the existing knowledge about ccRCC is inadequately understood.
Bioinformatics is a multidisciplinary field that can provide useful methods to identify and analyze the associations and molecular mechanisms among key genes and central signaling pathways (17). In this study, an original microarray dataset containing ccRCC and corresponding normal samples was downloaded from the Gene Expression Omnibus (GEO) database. Then, the differentially expressed genes (DEGs) were screened through a DEG interaction network, and hub genes were identified. Functional enrichment analyses, including Gene Ontology, Database for Annotation, Visualization and Integrated Discovery (DAVID), Kyoto Encyclopedia of Genes and Genomes (KEGG) and protein-protein interaction (PPI) network, were subsequently performed. Finally, by identifying the biological function of the hub genes and pathways, this study may offer better insight into potential molecular mechanisms for ccRCC, allowing the exploration of novel therapeutic strategies.
Methods
Microarray data
The raw gene expression data of GSE36895, contributed by Peña-Llopis et al. (18), was downloaded from the National Center of Biotechnology Information (NCBI) GEO (http://www.ncbi.nlm.nih.gov/geo/) database. This dataset comprised 29 ccRCC and 23 normal samples and was generated from patient samples using the GPL570 Affymetrix human genome U133A array (HG-U133A, Affymetrix Inc., Santa Clara CA, USA).
Data preprocessing and identification of DEGs
Briefly, the original data from the CEL files and the corresponding probe annotation information were downloaded and converted into a detectable format that was preprocessed using Bioconductor packages (http://www.bioconductor.org/) in soft R (version 3.4.1, https://www.r-project.org/). The affy package (https://bioconductor.org/packages/release/bioc/html/affy.html) was used for background correction and normalization of the data by the Robust Multi-Array Average method (19), and the missing values were inserted by the k-Nearest Neighbor method (20). After preprocessing, the limma package (http://www.bioconductor.org/packages/release/bioc/html/limma.html) was used to screen the DEGs between the ccRCC samples and the matched normal samples with a t-test (21). The adjusted P value accounted for false discovery rates (FDR) and was calculated by the Benjamini and Hochberg method (22). Hierarchical clustering was used to qualitatively analyze all of the DEGs from the microarray data and place them into two groups using the pheatmap package (23) (http://www.bioconductor.org/packages/release/bioc/html/pheatmap.html).
Subsequently, a volcano plot was generated to show up- and down-regulated genes among the DEGs. Only the DEGs with an FDR <0.05 and a |log2fold change (FC)| ≥2 were considered statistically significant.
Enrichment analysis of DEGs
DAVID (24) (http://david.abcc.ncifcrf.gov/), a common functional annotation tool for bioinformatics resources, was utilized to distinguish the biological attributes, such as BP, cellular component (CC) and molecular function (MF), of important DEGs. GO term enrichment analysis was then performed, and the data were visualized. Moreover, KEGG (25) (http://www.genome.jp/kegg/) pathway enrichment analysis was performed, and the package clusterProfiler (http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html) of R was used to identify the crucial pathways that were significantly close to the PPI network. A P value of less than 0.05 was used as the cutoff criterion.
Construction of the PPI network and module analysis
Search Tool for the Retrieval of Interacting Genes (STRING, https://string-db.org/) online database (26), a biological predictive web resource including numerous proteins and known interactive functions, was employed to analyze and evaluate the interaction correlations among DEGs. A combined score of >0.4 was designated as the cutoff standard. Cytoscape software (27) (version 3.5.1) was then used to construct the PPI network according to the information from STRING. The Molecular Complex Detection plug-in (MCODE, http://apps.cytoscape.org/apps/MCODE) was used to calculate and filter central modules in the PPI network; the cutoff parameters were set as MCODE scores >3 and node numbers >4 (28). The corresponding genes in the modules may represent hub genes with significant physiological effects. A P value of <0.05 was considered significant.
Results
Screening of DEGs
A total of 76 tissues were divided into 29 ccRCC tissues, 23 normal tissues and 24 mouse samples; 52 patient samples were obtained from GSE36895. After integrated analysis from 20,483 genes in the microarray data, a total of 468 DEGs (|log2 FC| ≥2 and adjusted P value <0.05, Figure S1) were screened, including 180 upregulated genes and 288 downregulated genes in the ccRCC samples compared to normal samples (Table S1). Volcano plots (Figure 1) were generated to show the correlation between DEGs.
Figure S1.

Cluster analysis of the top 50 up- and down-regulated DEGs from tumor tissues compared with normal kidneys. Each column represents one sample, and each row represents one gene. The relative expression of a gene is described on a color scale. Red represents upregulation, and green denotes downregulation. DEG, differentially expressed gene.
Table S1. Two hundred and four DEGs were filtered from the datasets, including 52 up-regulated genes and 152 down-regulated genes in ccRCC samples compared to normal samples.
| DEGs | Gene name |
|---|---|
| Up-regulated | IGFBP3, ANGPTL4, FUT11, ENO2, HILPDA, TMEM91, CAV1, CAV2, ANGPT2, VIM, FABP5, MIR210HG, LPCAT1, NOL3, APOBEC3C, NDUFA4L2, PLA2G7, FAM49A, HK1, CDCA2, TNFAIP6, VEGFA, IRX3, PYGL, DDB2, ANXA2R, GJC1, ENTPD1, IFI16, SPAG4, ZNF395, EGLN3, CD86, SEMA5B, CXCR4, PAG1, KIAA0101, INHBB, RNASE2, COL23A1, PLOD2, LGALS1, DIRAS2, ST8SIA4, CHSY3, IGSF6, LAPTM5, ADM, LDLRAD3, DGCR5, SLFN13, RAD51AP1, CD247, LINC00887, NETO2, C1QB, CRNDE, FCER1G, STC2, TNFSF9, CD300LF, CA9, CDH13, HEY1, LHFPL2, SLFN11, CDCA7L, CCND1, LINC01094, TYROBP, CFD, TMEM45A, HIST1H2BK, SPC25, NPTX2, CCL28, RASSF2, CTSS, C10orf10, IL10RA, SCD, PGBD5, OLFML2B, GAS2L3, DTL, DDIT4, C1QC, PSMB9, C1QA, FCGR1B, KIF14, FPR3, BIRC3, MS4A7, PRKCDBP, PPP1R3C, HAPLN1, KCNJ2, HCLS1, MAP3K7CL, PRC1, LOC100132891, TOP2A, IDO1, APOLD1, CLEC5A, KISS1R, DUSP5P1, CD70, LOC101927972, C3, ALDOC, LAMP3, CD36, EVI2B, CENPK, CEP55, NKG7, TRIB3, GBP5, CSTA, PLK2, NDC80, GZMA, ERV3-2, CCR5, CPE, TGFBI, APOC1, GBP2, CST7, NUF2, ANLN, GZMB, NMB, CRTAM, SLC2A3, UBE2C, CD163, CTHRC1, TNFSF13B, GAL3ST1, FABP7, QRFPR, DLGAP5, BCL2A1, RRM2, FKBP10, TREM1, LOX, VCAN, PLVAP, TEX11, CCL5, CP, CCL4, ABCA17P, KIF20A, ADAMDEC1, PRIMA1, RAB42, CCL18, C5orf46, PTHLH, UHRF1, ENPP3, LPPR5, LYZ, C19orf33, COL21A1, SPINK13, SORCS3, NEFL, LOC102659288, CYP2J2, C10orf99, LINC01127, CXCL13, IL20RB, CCL20 |
| Down-regulated | CALB1, UMOD, NPHS2, HEPACAM2, LOC101929040, ATP6V1G3, DMRT2, SLC12A1, CA10, FGF1, SCNN1G, LPPR1, ATP6V0D2, RALYL, FXYD4, KNG1, TYRP1, SLC26A7, LINC01187, PTH1R, SFRP1, FAM3B, ANGPTL1, STAP1, KLK7, NPHS1, CLDN8, LOC100507537, SOST, SYNE4, CRHBP, MUC15, CWH43, HRG, ATP6V0A4, SMIM5, PIK3C2G, TMEM213, NRK, ACPP, TCEAL2, PLCXD3, FGF9, GPC5, DIO1, DDX25, LINC00645, IYD, TUBAL3, CLCNKB, TFAP2B, LOC100130278, GGT6, HS6ST2, AQP2, GRHL2, FOXI1, KCNJ1, ATP6V1C2, ENPP6, SERPINA5, SLC5A2, HYKK, PCDH9, RANBP3L, KLK6, IRX1, FLJ22763, SLC4A9, CRISP2, TMEM52B, DDN, LRRC2, XPNPEP2, C1orf168, TNNC1, ERP27, MRAP2, SLC13A2, SCN2A, LINC00551, LOC284578, ANO5, CCDC160, LOC101928303, SLC34A1, KCNJ10, SLC4A1, TCF21, ANGPTL3, SLC13A3, PACRG, RASSF10, SOWAHA, SLC30A2, HPD, ACSF2, CEL, C7, SLC7A8, PRLR, DACH1, SLC26A4, DUSP9, ADH1C, PPAPDC1A, FABP1, SLC47A2, SLC22A8, SHISA3, ALDOB, FAM169A, PLG, MTTP, TMEM45B, RBM11, CDH9, PTPRO, TSPAN8, SLIT2, DNMT3L, ERVMER34-1, ALB, SH3GL2, CRYAA, AFM, FAM151A, SOSTDC1, LOC149703, TMEM207, SLC12A3, DPEP1, BMPR1B, LOC101929480, LOC100130691, S100A2, GABRA2, CYP27B1, REEP6, RAB25, CPN2, GATA3, DAO, KCNMB2, TFCP2L1, MAN1C1, TMEM72, SMCO3, SFXN2, CNTN3, RHCG, HSD11B2, EFHD1, RNF212B, ABAT, IGSF11, PROC, SLC51B, HPGD, MAL, DCXR, CORO2B, GPR110, ESRP1, SCNN1B, UPP2, EGF, LINC00948, KCNJ13, OTOGL, ASS1, WNK4, FCAMR, ADH1B, PVALB, PCP4, GSTM3, SHISA2, TMEM178A, APELA, OGDHL, AGPAT9, SLC7A13, PTGDS, ESRRG, VTCN1, LOC727944, C16orf89, ALDH6A1, LOC389332, LOC101927244, SUCNR1, CDKN1C, CTXN3, UNC5D, CYP4F2, CHL1, G6PC, PIGR, ADH6, RGS7, SPINK1, SLC6A19, TAC1, LINC00473, CHRDL1, SLC16A10, OLFM4, TREH, ERICH4, LOC101928047, GLDC, CLSTN2, ETNPPL, FBLN5, TRIM71, ACOX2, CYP24A1, TUBB2B, FLJ35700, DCN, SLC14A1, KCNJ15, NAPSA, PRODH2, TRIM63, BEX1, SLC22A6, CHGB, NR3C2, BCHE, SLC22A12, BRE-AS1, C2orf40, LOC100505985, DEFB1, HSPA2, SCIN, FBP1, CYP8B1, LUM, HAO2, CA4, MIOX, DIRAS3, HOGA1, NDNF, LINC00113, ABCB1, CAPN3, AGR3, AZGP1, PIPOX, DNER, CKMT2, PRR15L, PCK1, TSPAN1, AGR2, PRAP1, SLC13A1, FREM2, ANXA3, MME, CYP4A11, TOX3, SLC17A1, SLC5A12, UGT3A1, CYP4F3, TINAG, GAS2, MEST, LRRC19, SLC16A9, MAL2, KL, PPP1R1A, DDC, GLYAT, AGXT2, PSAT1, LTF, MYH8, SLC27A2, FMO1, PROM1, ALDH8A1 |
DEG, differentially expressed gene; ccRCC, clear cell renal cell carcinoma.
Figure 1.
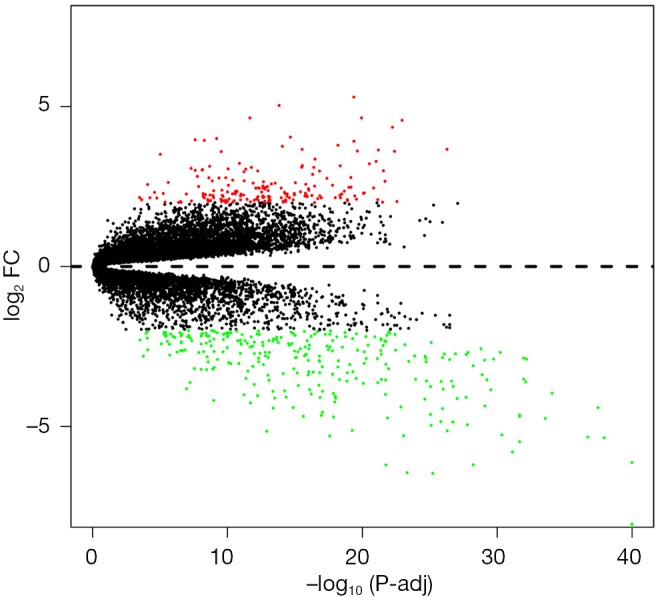
Volcano plot of differentially expressed genes. Red, green and black denote upregulated, downregulated and nonsignificant expressed genes, respectively. FC, fold change; adj, adjusted P value.
GO term enrichment analysis of DEGs
All DEGs were uploaded to the DAVID website to perform GO classification. The results of GO term analysis included the BP, CC and MF group, respectively (Table 1). As indicated in Table 1 and Figure 2, the upregulated DEGs were mainly enriched in the immune response, response to wounding, inflammatory response, response to hypoxia, and response to oxygen levels at BP, while downregulated genes were mainly enriched in ion transport, anion transport, monovalent inorganic cation transport, excretion, and cation transport. The upregulated genes were chiefly enriched in the extracellular region, extracellular space, extracellular region, Ndc80 complex, and soluble fraction; but the downregulated genes were mainly enriched in the apical part of the cell, apical plasma membrane, brush border, membrane fraction and cell fraction of CC. The upregulated DEGs were mainly enriched in cytokine activity, chemokine activity, chemokine receptor binding, protein homodimerization activity and identical protein binding; however, the downregulated DEGs were mainly enriched in anion transmembrane transporter activity, sodium ion binding, alkali metal ion binding, symporter activity and cofactor binding at MF. These results indicated that the most significant enrichment terms were immune response (Figure 2A) and ion transport (Figure 2B), which could help us understand the important DEGs involved in the pathogenesis of ccRCC.
Table 1. Gene ontology analysis of up- and down-regulated DEGs associated with ccRCC.
| Expression | Category | Term | Count | P value |
|---|---|---|---|---|
| Up-regulation | GOTERM_BP_FAT | GO:0006955~immune response | 34 | 2.46E-14 |
| GOTERM_BP_FAT | GO:0009611~response to wounding | 24 | 2.39E-09 | |
| GOTERM_BP_FAT | GO:0006954~inflammatory response | 18 | 2.42E-08 | |
| GOTERM_BP_FAT | GO:0001666~response to hypoxia | 12 | 9.39E-08 | |
| GOTERM_BP_FAT | GO:0070482~response to oxygen levels | 12 | 1.58E-07 | |
| GOTERM_CC_FAT | GO:0005576~extracellular region | 51 | 8.39E-09 | |
| GOTERM_CC_FAT | GO:0005615~extracellular space | 27 | 3.05E-08 | |
| GOTERM_CC_FAT | GO:0044421~extracellular region part | 32 | 4.93E-08 | |
| GOTERM_CC_FAT | GO:0031262~Ndc80 complex | 3 | 7.25E-04 | |
| GOTERM_CC_FAT | GO:0005625~soluble fraction | 11 | 2.58E-03 | |
| GOTERM_MF_FAT | GO:0005125~cytokine activity | 11 | 1.03E-05 | |
| GOTERM_MF_FAT | GO:0008009~chemokine activity | 6 | 5.57E-05 | |
| GOTERM_MF_FAT | GO:0042379~chemokine receptor binding | 6 | 7.58E-05 | |
| GOTERM_MF_FAT | GO:0042803~protein homodimerization activity | 12 | 2.12E-04 | |
| GOTERM_MF_FAT | GO:0042802~identical protein binding | 16 | 6.18E-04 | |
| Down-regulation | GOTERM_BP_FAT | GO:0006811~ion transport | 38 | 1.42E-10 |
| GOTERM_BP_FAT | GO:0006820~anion transport | 16 | 3.03E-09 | |
| GOTERM_BP_FAT | GO:0015672~monovalent inorganic cation transport | 21 | 5.14E-08 | |
| GOTERM_BP_FAT | GO:0007588~excretion | 10 | 1.60E-07 | |
| GOTERM_BP_FAT | GO:0006812~cation transport | 26 | 6.31E-07 | |
| GOTERM_CC_FAT | GO:0045177~apical part of cell | 16 | 2.53E-07 | |
| GOTERM_CC_FAT | GO:0016324~apical plasma membrane | 14 | 2.77E-07 | |
| GOTERM_CC_FAT | GO:0005903~brush border | 8 | 9.08E-06 | |
| GOTERM_CC_FAT | GO:0005624~membrane fraction | 29 | 1.44E-04 | |
| GOTERM_CC_FAT | GO:0000267~cell fraction | 35 | 1.77E-04 | |
| GOTERM_MF_FAT | GO:0008509~anion transmembrane transporter activity | 16 | 2.63E-09 | |
| GOTERM_MF_FAT | GO:0031402~sodium ion binding | 12 | 8.54E-07 | |
| GOTERM_MF_FAT | GO:0031420~alkali metal ion binding | 16 | 9.09E-07 | |
| GOTERM_MF_FAT | GO:0015293~symporter activity | 11 | 2.60E-05 | |
| GOTERM_MF_FAT | GO:0048037~cofactor binding | 14 | 5.79E-05 |
DEG, differentially expressed gene; ccRCC, clear cell renal cell carcinoma.
Figure 2.

GO analysis and the significant DEGs in ccRCC. (A) GO analysis classified the upregulated DEGs into 1 of 3 groups: biological process, cellular component and molecular function; (B) significantly enriched GO terms of downregulated DEGs in ccRCC based on their functions. DEG, differentially expressed gene; ccRCC, clear cell renal cell carcinoma.
Signaling pathway enrichment analysis of DEGs
All DEGs were analyzed using the KEGG pathway website and the clusterProfiler package of software R, and only P values less than 0.05 were included. As shown in Table 2, the upregulated DEGs were enriched in cytokine-cytokine receptor interactions, staphylococcus aureus infection, prion disease, Chagas disease, the chemokine signaling pathway, peroxisome proliferator-activated receptors (PPAR) signaling pathway, complement and coagulation cascades, and intestinal immune network for IgA production, whereas downregulated DEGs were enriched in collecting duct acid secretion, tyrosine metabolism, aldosterone-regulated sodium reabsorption, gluconeogenesis and glycine, serine and threonine metabolism. The results of the signaling pathway enrichment analysis indicated that the most important pathways in up- and down-regulation were cytokine-cytokine receptor interaction (Figure 3A) and collecting duct acid secretion (Figure 3B).
Table 2. Signaling pathway enrichment analysis of differentially expressed genes associated with ccRCC.
| Pathway ID | Name | Count | P value | Gene |
|---|---|---|---|---|
| Up-regulated DEGs | ||||
| hsa04060 | Cytokine-cytokine receptor interaction | 15 | 3.50E-07 | VEGFA, CXCR4, INHBB, NFSF9, CCL28, IL10RA, CD70, CCR5, TNFSF13B, CCL5, CCL4, CCL18, CXCL13, IL20RB, CCL20 |
| hsa05150 | Staphylococcus aureus infection | 6 | 4.05E-05 | C1QA, C1QB, C1QC, CDF, FPR3, C3 |
| hsa05020 | Prion diseases | 4 | 0.000659 | C1QA, C1QB, C1QC, CCL5 |
| hsa05142 | Chagas disease | 6 | 0.0011 | C1QA, C1QB, C1QC, CCL5, C3, CD247 |
| hsa04062 | Chemokine signaling pathway | 8 | 0.001257 | CXCR4, CCL28, CCR5, CCL5, CCL4, CCL18, CXCL13, CCL20 |
| hsa03320 | PPAR signaling pathway | 5 | 0.001388 | ANGPTL4, FABP5, FABP7, SCD, CD36, |
| hsa04610 | Complement and coagulation cascades | 5 | 0.0021 | C1QA, C1QB, C1QC, CFD, C3 |
| hsa04672 | Intestinal immune network for IgA production | 4 | 0.002359 | CD86, CXCR4, CCL28, TNSF13B |
| Down-regulated DEGs | ||||
| hsa04966 | Collecting duct acid secretion | 6 | 4.36E-06 | ATP6V1G3, ATP6V0D2, ATP6V0A4, CLCNKB, ATP6V1C2, SLC4A1 |
| hsa00350 | Tyrosine metabolism | 6 | 2.14E-05 | TYRP1, HPD, ADH1C, ADH1B, ADH6, DDC |
| hsa04960 | Aldosterone-regulated sodium reabsorption | 6 | 2.99E-05 | SCNN1G, FXYD4, KCNJ1, HSD11B2, SCNN1B, NR3C2 |
| hsa00010 | Glycolysis/Gluconeogenesis | 7 | 0.000119 | ADH1C, ALDOB, ADH1B, G6PC, ADH6, FBP1, PCK1 |
| hsa00260 | Glycine, serine and threonine metabolism | 5 | 0.000502 | DAO, GLDC, PIPOX, AGXT2, PSAT1 |
DEG, differentially expressed gene; ccRCC, clear cell renal cell carcinoma.
Figure 3.

Significant signaling pathway analysis of DEGs related to ccRCC performed with the KEGG pathway website and R software packages. (A) Representative dot plot of pathway enrichment analysis of upregulated DEGs; (B) pathway enrichment analysis of downregulated DEGs. Gene ratio = count/set size. DEG, differentially expressed gene; ccRCC, clear cell renal cell carcinoma; KEGG, Kyoto Encyclopedia of Genes and Genomes.
PPI network construction of DEGs and submodule analysis
To identify the interactions and key genes of the DEGs, a PPI network was constructed according to information from the STRING online database (Figure 4A). Based on the analysis of the Molecular Complex Detection (MCODE, score >7) plug-ins in the Cytoscape software, two modules were selected as submodules consisting of 66 nodes and 328 edges (Figure 4B,C). The results of GO term enrichment analysis indicated that submodule 1 was mainly associated with M phase, mitosis, nuclear division, M phase of the mitotic cell cycle and organelle fission, while submodule 2 was mainly involved in the apical plasma membrane, platelet alpha granule lumen, cytoplasmic membrane-bounded vesicle lumen, vesicle lumen and extracellular region part (Figure 4D,E).
Figure 4.

PPI network of DEGs and submodule analysis. (A) The interaction of all DEGs was examined using the STRING online database and was then filtered into the Cytoscape software to construct the PPI complex network. Red denotes upregulated genes, and green represents downregulated genes. The circles denote values of genes in log FC; (B) submodule 1 consists of 27 nodes and 173 edges; (C) submodule 2 includes 39 nodes and 155 edges; (D) the top 5 GO term enrichment analyses of module 1, which were mainly associated with M phase, mitosis, nuclear division, M phase of the mitotic cell cycle and organelle fission; (E) the top 5 GO term enrichment analyses of module 2, which were mainly linked to the apical plasma membrane, platelet alpha granule lumen, cytoplasmic membrane-bounded vesicle lumen, vesicle lumen and extracellular region. FC, fold change; PPI, protein-protein interaction; DEG, differentially expressed gene.
Pathway enrichment analysis of submodules
To investigate the potentially related pathways involved in submodules 1 and 2, the genes were uploaded into DAVID. As shown in Tables 3,4, the submodular genes were enriched in the chemokine signaling pathway, cytokine-cytokine receptor interaction, and intestinal immune network for IgA production and infection, while the submodular 2 genes were enriched in Staphylococcus aureus infection, the complement and coagulation cascades, fructose and mannose metabolism, prion diseases, aldosterone-regulated sodium reabsorption, bladder cancer, the HIF-1 (HIF) signaling pathway, viral myocarditis, systemic lupus erythematosus, glycolysis/gluconeogenesis, pancreatic cancer, pertussis, and melanoma. A network was then constructed using Cytoscape to identify the correlations between the genes and the corresponding pathways. As indicated in Figure 5, the significantly correlated genes that were involved in more than three pathways were C1QA, C1QB, C1QC, CCND1 and EGF, and the main correlated pathways were cytokine-cytokine receptor interaction, the chemokine signaling pathway, and the complement and coagulation cascade pathways.
Table 3. KEGG pathway enrichment analysis of sub-module 1.
| ID | Description | P value | P adjust | Gene ID |
|---|---|---|---|---|
| hsa04062 | Chemokine signaling pathway | 3.69E-07 | 1.36E-05 | CCL20, CCL28, CCL5, CCR5, CXCL13, CXCR4 |
| hsa04060 | Cytokine-cytokine receptor interaction | 3.40E-06 | 6.30E-05 | CCL20, CCL28, CCL5, CCR5, CXCL13, CXCR4 |
| hsa04672 | Intestinal immune network for IgA production | 0.003305 | 0.039758 | CCL28, CXCR4 |
| hsa05150 | Staphylococcus aureus infection | 0.004298 | 0.039758 | C3, FPR3 |
KEGG, Kyoto Encyclopedia of Genes and Genomes.
Table 4. KEGG pathway enrichment analysis of sub-module 2.
| ID | Description | P value | P adjust | Gene in test set |
|---|---|---|---|---|
| hsa05150 | Staphylococcus aureus infection | 3.87E-06 | 0.000418 | C1QB, C1QC, CFD, PLG, C1QA |
| hsa04610 | Complement and coagulation cascades | 2.13E-05 | 0.001148 | C1QB, C1QC, CFD, PLG, C1QA |
| hsa00051 | Fructose and mannose metabolism | 0.000386 | 0.011718 | ALDOB, ALDOC, HK1 |
| hsa05020 | Prion diseases | 0.00046 | 0.011718 | C1QB, C1QC, C1QA |
| hsa04960 | Aldosterone-regulated sodium reabsorption | 0.000542 | 0.011718 | KCNJ1, SCNN1B, SCNN1G |
| hsa05219 | Bladder cancer | 0.000735 | 0.013238 | CCND1, EGF, VEGFA |
| hsa04066 | HIF-1 signaling pathway | 0.000866 | 0.013356 | ANGPT2, EGF, HK1, VEGFA |
| hsa05416 | Viral myocarditis | 0.002126 | 0.028696 | CAV1, CCND1, CD86 |
| hsa05322 | Systemic lupus erythematosus | 0.00258 | 0.030961 | C1QB, C1QC, CD86, C1QA |
| hsa00010 | Glycolysis, Gluconeogenesis | 0.003059 | 0.033033 | ALDOB, ALDOC, HK1 |
| hsa05212 | Pancreatic cancer | 0.00421 | 0.036303 | CCND1, EGF, VEGFA |
| hsa05133 | Pertussis | 0.00437 | 0.036303 | C1QB, C1QC, C1QA |
| hsa05218 | Melanoma | 0.00437 | 0.036303 | CCND1, EGF, FGF1 |
KEGG, Kyoto Encyclopedia of Genes and Genomes.
Figure 5.

The relationship between the genes and KEGG pathways in sub-modules 1 and 2. The red, green and yellow circles denote upregulated genes, downregulated genes and the KEGG pathway ID. The arrow represents the correlated genes that are involved in the corresponding pathway. KEGG, Kyoto Encyclopedia of Genes and Genomes.
Discussion
ccRCC, the most common subtype of RCC, is characterized by complex mechanisms with multifactorial and polygenic backgrounds. Understanding the underlying molecular mechanisms of ccRCC is important for diagnosing and treating ccRCC. Since microarrays and high-throughput sequencing can offer efficient methods for studying the human genome, they have been extensively used to explore the molecular targets for ccRCC (29). In this study, a total of 468 DEGs were screened, including 180 upregulated genes and 288 downregulated genes, in ccRCC samples and related normal kidney samples using R software packages. The results of functional annotation analysis indicated that these genes were mainly involved in the immune response, response to injury and ion transport. KEGG pathway enrichment analysis showed that the DEGs were mainly associated with cytokine-cytokine receptor interactions, collecting duct acid secretion and tyrosine metabolism. Finally, two submodules were identified in the PPI network according to information from the STRING database, and GO terms and KEGG enrichment were used to analyze the relationships between genes, pathways and biological functions.
After numerous data analyses, our results indicated that several hub genes, such as C1QA, C1QB, C1QC, CCND1 and EGF, had significant correlations with ccRCC. C1QA, C1QB, and C1QC belong to C1q, the first component of complement, which serves an important role in innate and adaptive immune responses (30). A study conducted by Hosszu suggested that C1q promoted dendritic cell (DC) maturation through binding to pathogen-associated molecular patterns and danger-associated molecular patterns, subsequently interacting with distinct cell surface molecules of DCs in the early stages of immunity (31). In patients, C1q deficiency serves as a strong susceptibility factor for systemic lupus erythematosus and rheumatoid arthritis because of autoimmune dysfunction (32). Evidence that has accumulated to date convincingly indicates that the expression of C1q in prostate epithelial cells could activate the tumor suppressor WW-domain containing oxidoreductase (WOX1), resulting in prostate cancer cell apoptosis by suppressing the synergistic effects of p53 and by destabilizing cell adhesion (33). In addition, upregulation of C1q significantly induced the death of neuroblastoma cells and breast cancer cells (33). These observations imply that C1q is important for tumor cell function, which is consistent with our findings.
The gene CCND1 encodes the cyclin D1 protein, a member of the cyclin protein family involved in regulating cell cycle progression, which dimerizes with cyclin-dependent kinase (CDK) 4 or CDK6 to regulate the G1/S phase transition of the cell cycle (34). A series of studies have demonstrated that cyclin D1 plays a vital role in tumors of the bladder (35), breast (36), and colon (37), which is in agreement with our results. Patients with bladder cancer with CCND1 defects have a doubled probability of mortality compared to patients without mutations in the gene (35). He et al. (36) observed that the expression of cyclin D1 in breast cancer tissue was higher than in normal samples, and the overexpression showed a significant correlation with tumor size, clinical stage and pathological grade. Likewise, cyclin D1 acts as an adverse factor for colorectal cancer patients, as overexpression of cyclin D1 was significantly correlated with poor overall survival and disease-free survival of the patients (37). Recent studies further demonstrated that blocking cyclin D1 expression could stop cellular proliferation and tumor growth in renal cancer cells. Although the exact mechanisms have not yet been elucidated, cyclin D1 undoubtedly plays an important role in the pathogenesis of cancer.
Epidermal growth factor (EGF), a common mitogenic factor, is now known to function by binding the EGF receptor (EGFR) generating biological effects such as cellular proliferation, differentiation, and survival (38). EGF is reported to be associated with tumor cell migration and survival due to constitutive activation of the EGFR signaling pathway (39). In addition, EGF acts as a potent inducer of upregulation of the expression of proangiogenic growth factors, such as vascular endothelial growth factor A and angiopoietin-1, participating in neoangiogenesis and the revascularization of tumor vessels (40). In an elegant study by Bracher et al. (41), researchers found that patients with melanoma had significantly increased EGF levels compared with control individuals, and EGF knockdown resulted in a reduction in primary tumor lymphangiogenesis in a mouse model, as well as impairment of melanoma cell migration. However, our data suggested that EGF expression in ccRCC samples was significantly downregulated compared with normal samples, which conflicted with these results. This discrepancy suggests that the expression of genes may differ spatially and temporally, revealing tumor heterogeneity among individuals. Furthermore, the activation of EGF-associated pathways in ccRCC might be different from that in melanoma patients. Therefore, further research is necessary to determine the potential biological relationship and mechanisms of EGF signaling in ccRCC.
In addition to the genes discussed above, the pathways that were found to be enriched by subnetwork modules suggested that the processes involved in ccRCC were also mainly linked with the cytokine-cytokine receptor interaction pathway, chemokine signaling pathway, and complement and coagulation cascade pathways. The pathways of cytokine-cytokine receptor interaction and chemokine signaling are crucial in the regulation of the immunological and inflammatory response (42,43). Emerging evidence indicates that inhibition of the expression of C-X-C chemokine receptor type 4 (CXCR4), one of the major immunological receptors in the cytokine-cytokine receptor interaction pathway, blocked perineural invasion of prostate cancer in vitro and in vivo (44). Moreover, activation of the chemokine signaling pathway could inhibit proliferation and invasion of prostate adenocarcinoma cells via upregulation of C-C chemokine receptor type 5 (CCR5) expression (45). The complement and coagulation cascade pathways were crucially involved in activating the progression of the systemic inflammatory response after traumatic organ failure (46). The interaction of elevated inflammation levels and complement effectors predisposes patients to thrombosis by modifying the phospholipid membranes of vascular endothelial cells (47). ccRCC creates a tumor microenvironment that promotes the inflammatory response, promoting the formation of tumor thrombi as well as metastasis of tumor cells due to their immunogenic characteristics. In the present study, C1q family members of the complement system, such as C1QA, C1QB, and C1QC, were associated with the inflammatory response. Thus, the complement and coagulation cascade pathways might be viewed as indispensable in different phases of ccRCC.
Conclusions
The present study, performed with GEO analysis, showed that genes such as C1QA, C1QB, C1QC, CCND1 and EGF, cytokine-cytokine receptor interactions, the chemokine signaling pathway, and the complement and coagulation cascade pathways, might be importantly associated with the pathogenesis of ccRCC. This study provided new insight for the understanding of molecular mechanisms in ccRCC. However, further experiments are required to confirm and validate these predicted results.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7-30. 10.3322/caac.21332 [DOI] [PubMed] [Google Scholar]
- 2.Capitanio U, Montorsi F. Renal cancer. Lancet 2016;387:894-906. 10.1016/S0140-6736(15)00046-X [DOI] [PubMed] [Google Scholar]
- 3.Srinivasan R, Ricketts CJ, Sourbier C, et al. New strategies in renal cell carcinoma: targeting the genetic and metabolic basis of disease. Clin Cancer Res 2015;21:10-7. 10.1158/1078-0432.CCR-13-2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Escudier B, Eisen T, Porta C, et al. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23:vii65-71. 10.1093/annonc/mds227 [DOI] [PubMed] [Google Scholar]
- 5.Theis RP, Grieb SM, Burr D, et al. Smoking, environmental tobacco smoke, and risk of renal cell cancer: a population-based case-control study. BMC Cancer 2008;8:387. 10.1186/1471-2407-8-387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frew IJ, Moch H. A clearer view of the molecular complexity of clear cell renal cell carcinoma. Annu Rev Pathol 2015;10:263-89. 10.1146/annurev-pathol-012414-040306 [DOI] [PubMed] [Google Scholar]
- 7.Gati A, Kouidhi S, Marrakchi R, et al. Obesity and renal cancer: Role of adipokines in the tumor-immune system conflict. Oncoimmunology 2014;3:e27810. 10.4161/onci.27810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deckers IA, Brandt PA, Engeland M, et al. Polymorphisms in genes of the renin-angiotensin-aldosterone system and renal cell cancer risk: Interplay with hypertension and intakes of sodium, potassium and fluid. Int J Cancer 2015;136:1104-16. 10.1002/ijc.29060 [DOI] [PubMed] [Google Scholar]
- 9.Moore LE, Nickerson ML, Brennan P, et al. Von Hippel-Lindau (VHL) inactivation in sporadic clear cell renal cancer: associations with germline VHL polymorphisms and etiologic risk factors. PLoS Genet 2011;7:e1002312. 10.1371/journal.pgen.1002312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joseph RW, Kapur P, Serie DJ, et al. Clear cell renal cell carcinoma subtypes identified by BAP1 and PBRM1 expression. J Urol 2016;195:180-7. 10.1016/j.juro.2015.07.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osanto S, Qin Y, Buermans HP, et al. Genome-wide microRNA expression analysis of clear cell renal cell carcinoma by next generation deep sequencing. PLoS One 2012;7:e38298. 10.1371/journal.pone.0038298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao X, Tang C, Xiao S, et al. Enhancement of proliferation and invasion by MicroRNA-590-5p via targeting PBRM1 in clear cell renal carcinoma cells. Oncol Res 2013;20:537-44. 10.3727/096504013X13775486749335 [DOI] [PubMed] [Google Scholar]
- 13.Zhao JJ, Chen PJ, Duan RQ, et al. Up-regulation of miR-630 in clear cell renal cell carcinoma is associated with lower overall survival. Int J Clin Exp Pathol 2014;7:3318-23. [PMC free article] [PubMed] [Google Scholar]
- 14.Ueno K, Hirata H, Shahryari V, et al. Tumour suppressor microRNA-584 directly targets oncogene Rock-1 and decreases invasion ability in human clear cell renal cell carcinoma. Br J Cancer 2011;104:308. 10.1038/sj.bjc.6606028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valera VA, Walter BA, Linehan WM, et al. Regulatory effects of microRNA-92 (miR-92) on VHL gene expression and the hypoxic activation of miR-210 in clear cell renal cell carcinoma. J Cancer 2011;2:515. 10.7150/jca.2.515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao J, Lei T, Xu C, et al. MicroRNA-187, down-regulated in clear cell renal cell carcinoma and associated with lower survival, inhibits cell growth and migration though targeting B7-H3. Biochem Biophys Res Commun 2013;438:439-44. 10.1016/j.bbrc.2013.07.095 [DOI] [PubMed] [Google Scholar]
- 17.Loscalzo J, Barabasi AL. Systems biology and the future of medicine. Wiley Interdiscip Rev Syst Biol Med 2011;3:619-27. 10.1002/wsbm.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 2012;44:751-9. 10.1038/ng.2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautier L, Cope L, Bolstad BM, et al. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004;20:307-15. 10.1093/bioinformatics/btg405 [DOI] [PubMed] [Google Scholar]
- 20.Keller JM, Gray MR, Givens JA. A fuzzy k-nearest neighbor algorithm. IEEE Trans Syst Man Cybern 1985;4:580-5. 10.1109/TSMC.1985.6313426 [DOI] [Google Scholar]
- 21.Diboun I, Wernisch L, Orengo CA, Koltzenburg M. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genomics 2006;7:252. 10.1186/1471-2164-7-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 1995;57:289-300. [Google Scholar]
- 23.Huang da W , Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44-57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 24.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27-30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bar-Joseph Z, Gifford DK, Jaakkola TS. Fast optimal leaf ordering for hierarchical clustering. Bioinformatics 2001;17 Suppl 1:S22-9. 10.1093/bioinformatics/17.suppl_1.S22 [DOI] [PubMed] [Google Scholar]
- 26.Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447-52. 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498-504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang B, Li C, Zhao J. Identification of key pathways and genes in colorectal cancer using bioinformatics analysis. Med Oncol 2016;33:111. 10.1007/s12032-016-0829-6 [DOI] [PubMed] [Google Scholar]
- 29.King HC, Sinha AA. Gene expression profile analysis by DNA microarrays: promise and pitfalls. JAMA 2001;286:2280-8. 10.1001/jama.286.18.2280 [DOI] [PubMed] [Google Scholar]
- 30.Kishore U, Reid KB. C1q: structure, function, and receptors. Immunopharmacology 2000;49:159-70. 10.1016/S0162-3109(00)80301-X [DOI] [PubMed] [Google Scholar]
- 31.Hosszu KK, Santiago-Schwarz F, Peerschke EI, et al. Evidence that a C1q/C1qR system regulates monocyte-derived dendritic cell differentiation at the interface of innate and acquired immunity. Innate Immun 2010;16:115-27. 10.1177/1753425909339815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghebrehiwet B, Hosszu KK, Valentino A, et al. Monocyte Expressed Macromolecular C1 and C1q Receptors as Molecular Sensors of Danger: Implications in SLE. Front Immunol 2014;5:278. 10.3389/fimmu.2014.00278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong Q, Sze CI, Lin SR, et al. Complement C1q activates tumor suppressor WWOX to induce apoptosis in prostate cancer cells. PLoS One 2009;4:e5755. 10.1371/journal.pone.0005755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witzel II, Koh LF, Perkins ND. Regulation of cyclin D1 gene expression. Biochem Soc Trans 2010;38:217-22. 10.1042/BST0380217 [DOI] [PubMed] [Google Scholar]
- 35.Seiler R, Thalmann GN, Rotzer D, et al. CCND1/CyclinD1 status in metastasizing bladder cancer: a prognosticator and predictor of chemotherapeutic response. Mod Pathol 2014;27:87-95. 10.1038/modpathol.2013.125 [DOI] [PubMed] [Google Scholar]
- 36.He Y, Liu Z, Qiao C, et al. Expression and significance of Wnt signaling components and their target genes in breast carcinoma. Mol Med Rep 2014;9:137-43. 10.3892/mmr.2013.1774 [DOI] [PubMed] [Google Scholar]
- 37.Li Y, Wei J, Xu C, et al. Prognostic significance of cyclin D1 expression in colorectal cancer: a meta-analysis of observational studies. PLoS One 2014;9:e94508. 10.1371/journal.pone.0094508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeng F, Harris RC. Epidermal growth factor, from gene organization to bedside. Semin Cell Dev Biol 2014;28:2-11. 10.1016/j.semcdb.2014.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 2011;30:3477-88. 10.1038/onc.2011.160 [DOI] [PubMed] [Google Scholar]
- 40.Luangdilok S, Box C, Harrington K, et al. MAPK and PI3K signalling differentially regulate angiogenic and lymphangiogenic cytokine secretion in squamous cell carcinoma of the head and neck. Eur J Cancer 2011;47:520-9. 10.1016/j.ejca.2010.10.009 [DOI] [PubMed] [Google Scholar]
- 41.Bracher A, Cardona AS, Tauber S, et al. Epidermal growth factor facilitates melanoma lymph node metastasis by influencing tumor lymphangiogenesis. J Invest Dermatol 2013;133:230-8. 10.1038/jid.2012.272 [DOI] [PubMed] [Google Scholar]
- 42.Leonard WJ, Lin JX. Cytokine receptor signaling pathways. J Allergy Clin Immunol 2000;105:877-88. 10.1067/mai.2000.106899 [DOI] [PubMed] [Google Scholar]
- 43.Haque N, Barjis I, Haque N, et al. Modeling of chemokine signaling pathway. Available online: https://dl.acm.org/citation.cfm?id=2349533&dl=ACM&coll=DL
- 44.Zhang S, Qi L, Li M, et al. Chemokine CXCL12 and its receptor CXCR4 expression are associated with perineural invasion of prostate cancer. J Exp Clin Cancer Res 2008;27:62. 10.1186/1756-9966-27-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vaday GG, Peehl DM, Kadam PA, et al. Expression of CCL5 (RANTES) and CCR5 in prostate cancer. Prostate 2006;66:124-34. 10.1002/pros.20306 [DOI] [PubMed] [Google Scholar]
- 46.Amara U, Rittirsch D, Flierl M, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol 2008;632:71-9. 10.1007/978-0-387-78952-1_6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Markiewski MM, Nilsson B, Ekdahl KN, et al. Complement and coagulation: strangers or partners in crime? Trends Immunol 2007;28:184-92. 10.1016/j.it.2007.02.006 [DOI] [PubMed] [Google Scholar]