

Abstract
Background
The senescence-associated secretory phenotype (SASP) develops due to cellular senescence during conditions such as chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). However, studies comparing the degree of cellular senescence and SASP between COPD and IPF are limited. Furthermore, to the best of our knowledge, no study has examined cellular senescence and/or SASP in connective tissue disease-associated interstitial lung disease (CTD-ILD).
Methods
To compare the degree of cellular senescence among COPD, IPF, and CTD-ILD, tissue samples from surgical lung biopsies or noncancerous tissue from lobectomy specimens of patients with lung cancer were subjected to immunostaining for p16 and p21. Double-staining for p16 and phosphorylated NF-κB was performed to verify the relationship between cellular senescence and SASP.
Results
There was a greater degree of enhancement of p16 and p21 expression in patients with IPF than in those with COPD and controls. Immunostaining for p16 revealed an enhanced expression of this marker in patients with COPD compared with that in controls. No significant differences were observed in the phosphorylated NF-κB expression rate of p16-positive and p16-negative cells among patients with IPF, CTD-ILD, and COPD.
Conclusions
Epithelial cells in patients with IPF express higher levels of both cellular senescence and SASP than those in patients with COPD or controls.
Keywords: Cellular senescence, senescence-associated secretory phenotype (SASP), idiopathic pulmonary fibrosis (IPF), chronic obstructive pulmonary disease (COPD), p16
Introduction
Cellular senescence is a state characterized by the progressive cessation of cell replication (1). In senescent cells, the expression of the cell cycle inhibitors p21 and p16 is enhanced, resulting in cell cycle arrest (2,3). An increased number of senescent cells has been observed in various disorders, such as diabetes mellitus (4) and arteriosclerosis (5). Previous studies have reported an enhanced cellular senescence in lung disorders, including chronic obstructive pulmonary disease (COPD) (6) and idiopathic pulmonary fibrosis (IPF) (7,8). Smoking is considered a stimulus that induces cellular senescence in COPD (9). In IPF, ≥2 factors, including smoking (10), oxidative stress (11), TGF-β levels (12,13), and telomere shortening in alveolar epithelial cells (8,14), may be involved in the induction of cellular senescence. Although cellular senescence progresses in both COPD and IPF, it is unclear whether there is a difference in the degree of cellular senescence between these two disorders. Moreover, it is unclear whether there are any differences in the degree of cellular senescence in other interstitial lung diseases (ILDs), such as connective tissue disease-associated ILD (CTD-ILD). The present study was conducted to clarify the differences in the degree of cellular senescence among tissues obtained from patients with IPF, CTD-ILD, and COPD.
Senescent cells cause arrest of proliferation as well as exhibit the senescence-associated secretory phenotype (SASP) that is characterized by the profuse expression and secretion of various inflammatory proteins into the surrounding tissues (15). van Deursen (16) have reported that SASP contributes to malignant transformation and degenerative diseases, indicating that SASP is an important factor linking cellular senescence to COPD and ILD. A study has suggested that cellular senescence does not simultaneously occur with SASP (17); therefore, the relationship between cellular senescence and SASP warrants investigation. Because SASP develops primarily via NF-κB signaling (18-20), we investigated the relationship between cellular senescence and SASP in patients with COPD, IPF, and CTD-ILD by subjecting the tissues from such patients to immunostaining for phosphorylated NF-κB and p16.
Methods
Patients
Patients diagnosed with IPF, CTD-ILD, or COPD in pathological lung tissue at the Saitama Red Cross Hospital between January 2010 and December 2014 were enrolled in the present study. The diagnosis of IPF was established according to the 2011 ATS/ERS/JRS/ALAT guidelines (21). CTD-ILD was diagnosed by two collagen disease specialists in consultation with two respiratory medicine specialists in the Saitama Red Cross Hospital. Patients with COPD whose FEV1.0 values <70% and in whom emphysematous lesions had been histologically confirmed by a pathologist were selected for the present study. Lung tissue samples of IPF and COPD obtained from the resected lung of patients with lung cancer were considered the experimental group. Noncancerous lung tissue samples obtained from the resected lung of patients with lung cancer without a smoking history were considered to represent normal lung tissue and comprised the control group.
Tissue preparation
Lung tissue from lung biopsy specimens and noncancerous lung tissue from lobectomy specimens of patients with lung cancer were used. The resected lung tissue was immersed/fixed in 10% buffered formalin for 4 h, following which it was embedded in paraffin. Among patients with IPF, lung tissues with advanced fibrosis, such as honeycomb lung, were selected, whereas among patients with COPD, lung tissues where emphysematous lesions were most prevalent were selected.
Immunohistochemistry
Immunohistochemical staining of 3-µm-thick sections of the tissue samples was performed using a fully automated immunostaining apparatus (Ventana BenchMark GX, Ultra View DAB; Roche Diagnostics International Ltd., Tokyo). The primary antibodies used were mouse monoclonal anti-p16 antibody (100 µL on each slide) (#sc-6246, Santa Cruz Biotechnology, INC, Dallas) and mouse monoclonal anti-p21 antibody (1:20, 150 µL on each slide) (#sc-1661, Santa Cruz Biotechnology, INC, Dallas). Secondary antibodies used included horseradish peroxidase-conjugated anti-mouse/rabbit antibody (#760-500, Ventana BenchMark GX); the nature and cross-reactivity of this antibody was not disclosed by the manufacturer. 3,3'-Diaminobenzidine (Ventana ultraView Universal DAB Detection Kit, Roche, Basel) was used as a chromogenic substrate for the horseradish peroxidase reaction. Nuclear counterstaining was performed using hematoxylin. Further, to examine the degree of cellular senescence, tissue samples were stained for p16 or p21. Overall, 500 epithelial cells from the honeycomb lung or alveolus obtained from each patient were assessed to determine the positive-staining rate. The pathology slides were randomly divided into five parts, and each 100 epithelial cells from the honeycomb lung or alveolus were assessed to determine the positive-staining rate. To examine the correlation between cellular senescence and SASP, tissue samples were double-stained for p16 and phosphorylated NF-κB. First, immunohistochemical staining with rabbit monoclonal anti-phosphorylated NF-κB antibody (1:50, #4808, Cell Signaling Technology, Danvers) was performed using a fully automated immunostaining apparatus. The secondary antibody (#760-500) and 3,3'-diaminobenzidine (Ventana ultraView Universal DAB Detection Kit) were used to visualize phosphorylated NF-κB antigen. Thereafter, the initial primary and secondary antibody complexes were removed by heating in a boiling water bath (100 °C for 10 min). Finally, immunohistochemical staining for mouse anti-P16 antibody (#sc-6246) was repeated using the automated immunostaining apparatus. To visualize the p16 antigen, Histo Green (#E109, AbCys, Paris) was used as the chromogenic substrate for the horseradish peroxidase reaction. Because visualizing the double-staining results is challenging, nuclear counterstaining with hematoxylin was not included in the double-staining procedure. Overall, 500 epithelial cells of the honeycomb lung or alveolus obtained from each patient were assessed to determine the phosphorylated NF-κB-positive rates of p16-positive and p16-negative cells.
Statistical analyses
Data were analyzed using the Kruskal-Wallis test, and a P value of <0.05 was considered as statistically significant. Excel Statistics Software (SSRI Co., Ltd.) was used for data analyses.
Results
Patient characteristics are summarized in Table 1. In patients with IPF, forced vital capacity and pulmonary diffusing capacity were maintained within the normal ranges, whereas median serum KL-6 level was slightly elevated [565.5 U/mL (reference range: 152–400 U/mL)]. The GAP stage was stage 1 in all patients with IPF. The percentage of younger female nonsmokers was higher among patients with CTD-ILD than among patients with IPF, and the serum KL-6 level tended to be higher in patients with CTD-ILD. The COPD group exhibited lower FEV1/FVC values than the IPF and control groups. Several cases complicated by lung cancer were noted. The IPF group included two cases of small cell carcinoma and six cases of non-small cell carcinoma. In the COPD and control groups, all cases exhibited non-small cell carcinoma.
Table 1. Baseline characteristics.
| Variable | IPF | CTD-ILD | COPD | Controls |
|---|---|---|---|---|
| Cases, n | 8 | 3 | 6 | 6 |
| Male, n | 8*‡ | 0† | 6 | 2 |
| Age (years) | 68.0 | 46.0 | 73.5 | 73.0 |
| Never smoker, n | 0*‡ | 3† | 0* | 6 |
| Lung cancer coexistence, n | 8‡ | 0*† | 6 | 6 |
| KL-6 (U/mL) | 565.5 | 1,313.0 | NA | NA |
| FVC (L) | 3.41 | 2.87 | 3.00 | 2.68 |
| FVC %pred. | 107.1 | 105.5 | 89.5 | 109.7 |
| FEV1/FVC (%) | 78.1† | 86.6 | 57.8* | 77.3 |
| FEV1 (L) | 2.61† | 2.53 | 1.84 | 2.13 |
| DLco %pred. | 85.0 | 95.0 | 86.9 | 105.1 |
Data are presented as median and analyzed by the Kruskal-Wallis test. *, P<0.05 versus Control; †, P<0.05 versus COPD; ‡, P<0.05 versus CTD-ILD. KL-6, Krebs von den Lungen-6; FVC, forced vital capacity; FEV1, forced expiatory volume in 1 second; DLco, diffusing capacity of lung for carbon monoxide; NA, not applicable.
The p16-positive rate in the epithelial cells was significantly higher in the IPF group (53.2%) than in the COPD group (4.8%). Furthermore, the p16-positive rate in the epithelial cells was significantly higher in the IPF and COPD groups (53.2% and 4.8% respectively) than in the control group (0.5%) (Figures 1,2). The p21-positive rate in epithelial cells was significantly higher in the IPF group (20.7%) than in the COPD and control groups (0.8% and 0.2% respectively) (Figure 3).
Figure 1.

Low power (A) and high power (B) photomicrographs of immunohistochemical staining for p16 (brown) in lung tissues with IPF. Nuclear counterstaining was performed using hematoxylin. Original magnifications were ×40 in (A) and ×100 in (B).
Figure 2.
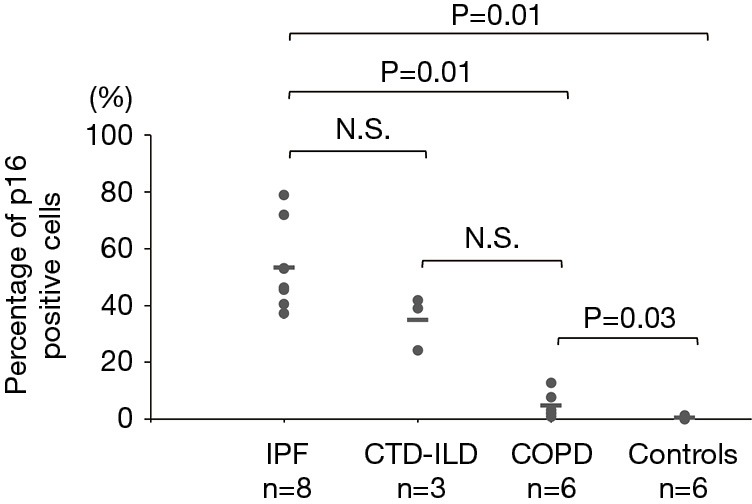
Percentage of p16-positive cells in the IPF, CTD-ILD, COPD and control groups. The horizontal bars indicate average values. The Kruskal-Wallis test was used. IPF, idiopathic pulmonary fibrosis; CTD-ILD, connective tissue disease-associated interstitial lung disease; COPD, chronic obstructive pulmonary disease; N.S., not significant.
Figure 3.
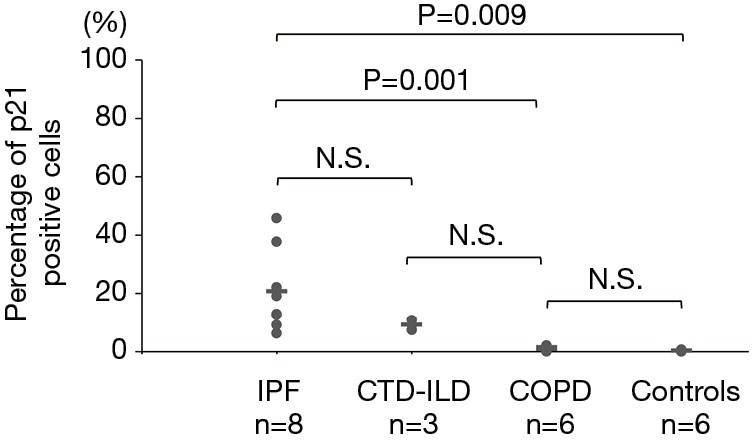
Percentage of p21-positive cells in the IPF, CTD-IP, COPD and control groups. The horizontal bars indicate average values. The Kruskal-Wallis test was used. IPF, idiopathic pulmonary fibrosis; CTD-ILD, connective tissue disease-associated interstitial lung disease; COPD, chronic obstructive pulmonary disease; N.S., not significant.
Double-staining conducted for p16 and phosphorylated NF-κB revealed that the phosphorylated NF-κB-positive rates in the p16-positive cells of the IPF, CTD-ILD, and COPD groups were 25.1%, 39.7%, and 13.6% respectively (Figures 4,5); no significant differences were observed with regard to these rates among the groups. Furthermore, the phosphorylated NF-κB-positive rates in the p16-negative cells were quite low in the IPF, CTD-ILD, and COPD groups (4.7%, 7.2%, and 1.6%, respectively); no significant differences were observed among the groups (Figure 6).
Figure 4.

Double-staining for p16 (green) and phosphorylated NF-κB (brown) in a case with IPF (A, ×400) and that with controls (B, ×200) was performed. The epithelial cells with IPF were stained green and brown. IPF, idiopathic pulmonary fibrosis.
Figure 5.
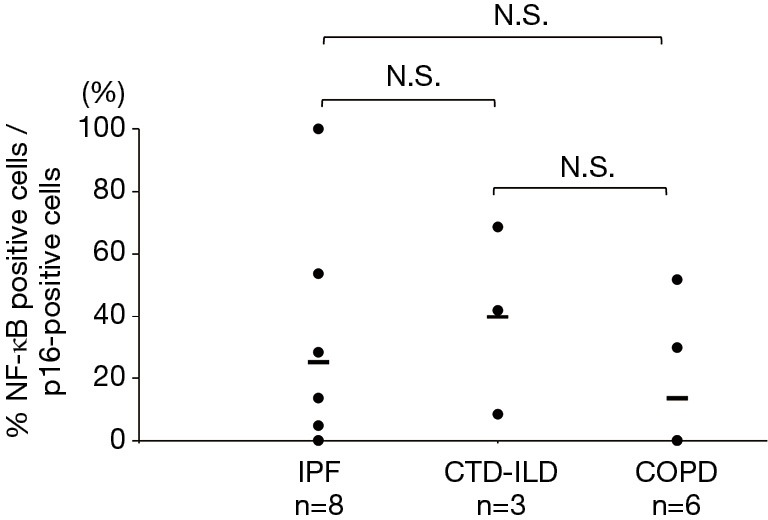
Phosphorylated NF-κB-positive rates in p16-positive cells. The horizontal bars indicate average values. The Kruskal-Wallis test was used. IPF, idiopathic pulmonary fibrosis; CTD-ILD, connective tissue disease-associated interstitial lung disease; COPD, chronic obstructive pulmonary disease; N.S., not significant.
Figure 6.
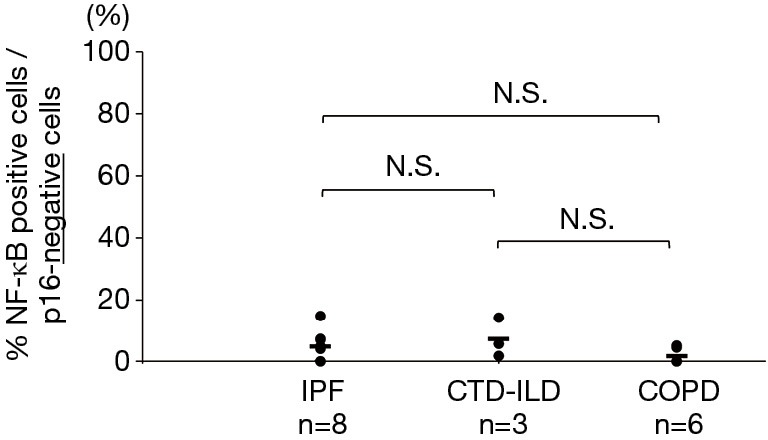
Phosphorylated NF-κB-positive rates in p16-negative cells. The horizontal bars indicate average values. The Kruskal-Wallis test was used. IPF, idiopathic pulmonary fibrosis; CTD-ILD, connective tissue disease-associated interstitial lung disease; COPD, chronic obstructive pulmonary disease; N.S., not significant.
Discussion
Cellular senescence is a state wherein the cells cease to replicate (1). The expression of cyclin-dependent kinase inhibitors, such as p21 and p16, is enhanced in senescent cells, leading to permanent cell cycle arrest (2,3,22). The p21 protein arrests cell cycle progression during the G1 phase by inhibiting CDK1 or CDK2 (23,24). The expression of this protein is strictly controlled by the p53 tumor suppressor (25). The p16 protein binds to CDK4 or CDK6 to participate in the induction of cell cycle arrest (26). In addition to cell cycle arrest, senescent cells are primarily characterized by resistance to apoptosis, flattening and enlargement of the cells, changes in the expression of cellular senescence-related genes, and SASP (27). The causes of cellular senescence include the telomere shortening, oxidative stress, oncogenes, and DNA damage (28).
In patients with COPD, cellular stress and telomere shortening due to tobacco smoke induces cellular senescence (29,30), resulting in an abnormal increase in cytokine secretion from the senescent cells (SASP) and the inhibition of cellular proliferation. Cellular senescence occurs in several types of cells in COPD. Reports have shown shorter telomere length in the peripheral blood neutrophils in patients with COPD than that in normal healthy controls, with the evident progression of cellular senescence in the alveolar epithelial cells, vascular endothelial cells, fibroblasts, and stem cells (31-33).
For IPF, the hypofunction of telomerase reverse transcriptase has been reported in 15% of patients with familial interstitial pneumonia and 3% of patients with sporadic IPF (7,34). It is currently acknowledged that telomere shortening is involved in IPF development. Apoptosis and cellular senescence are induced by DNA damage due to telomere shortening; therefore, cellular senescence is considered to contribute to the onset of IPF (35). Although cellular senescence contributes to the onset of COPD and IPF, the differences in cell senescence in patients with COPD and IPF have rarely been investigated. The present study noted the cellular senescence of epithelial cells in patients with COPD and those with IPF, with a greater degree of cellular senescence being observed in those with IPF than in those with COPD. The telomere shortening was associated with poor survival in patients with IPF (36). We speculated that IPF shortens telomere length more rapidly than COPD because epithelial cell proliferation with structural modification observed in patients with IPF was more enhanced than that in those with COPD (37).
NF-κB has been shown to induce SASP (38-41). We found no significant differences in the phosphorylated NF-κB-positive rate of p16-positive and p16-negative cells between COPD and IPF, as assessed by double-staining. The similar phosphorylated NF-κB-positive rates of p16-positive and p16-negative cells in both the COPD and IPF groups indicate that SASP occurrence depends on p16 in both COPD and IPF.
Telomere shortening has been reported to occur in both IPF and CTD-ILD (42), despite differences in their etiology, indicating that cellular senescence and SASP are induced in CTD-ILD. No significant differences were observed in both the p16- and p21-positive rates in the IPF and CTD-ILD groups; however, they tended to be higher in the IPF group. We suggest that the lower degree of cell senescence in patients with CTD-ILD than that in patients with IPF is attributable to the progression of CTD-ILD that occurs at a slower rate than that of IPF as well as to the number of younger nonsmokers observed in the CTD-ILD group compared with the IPF group.
Our study has several limitations. First, the sample size in each disease group was low; however, the present series enabled us to demonstrate the differences in the degree of cellular senescence among pulmonary fibrotic lesions, emphysematous lesions, and control tissues. Second, the influence of lung cancer must be considered, because cases in all of the groups except CTD-ILD had lung cancer. Although we prepared microscopic tissue sections from the regions of pronounced fibrotic changes with no malignant cells to minimize the possible influence of lung cancer, the effects of malignancy could not be completely dismissed. Patients in both the IPF and control groups were diagnosed with lung cancer; therefore, the results in both groups might have similar cancer effects. However, we considered that the influence of lung cancer was negligible based on the comparison between both groups. It is possibly important that the expression of p16 and p21 was significantly stronger in the IPF group compared with controls. Third, the influence of smoking history must be considered. In patients with non-small cell lung carcinoma and a smoking history, there is a report that p16-positive rate was higher than in patients with non-small cell lung cancer without a smoking history (43). Nevertheless, the p16-positive rate was higher in patients with CTD-ILD without a smoking history than in those with COPD without a smoking history in the present study. Fibrotic disease might have a greater influence on the p16-positive rate than the smoking history. Moreover, it is possible that our result was influenced by the sex of the patients. In the present study, no significant difference was observed between men and women in the control group. All patients with IPF underwent lung cancer surgery; thus, our cohort primarily comprised patients with mild IPF. Although median value of pulmonary function testing was within the normal range, all patients with IPF pathologically exhibited UIP pattern; despite mild IPF, cell senescence was observed in all these patients.
In conclusion, the present study demonstrated a difference in the degree of cellular senescence between patients with COPD and those with IPF. SASP was induced in certain p16-positive cells in both COPD and IPF. Cellular senescence and SASP were evident to the same degree in patients with CTD-ILD and those with IPF. Our findings suggest that cellular senescence and SASP are more pronounced in pulmonary fibrotic diseases than in emphysematous diseases.
Acknowledgements
None.
Ethical Statement: This study was approved by the institutional review board of the Saitama Red Cross Hospital (approval number: 20140115-2).
Footnotes
Conflicts of Interest: The authors have no conflict of interest to declare.
References
- 1.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965;37:614-36. 10.1016/0014-4827(65)90211-9 [DOI] [PubMed] [Google Scholar]
- 2.Meeker AK, Gage WR, Hicks JL, et al. Telomere length assessment in human archival tissues: combined telomere fluorescence in situ hybridization and immunostaining. Am J Pathol 2002;160:1259-68. 10.1016/S0002-9440(10)62553-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferlicot S, Youssef N, Feneux D, et al. Measurement of telomere length on tissue sections using quantitative fluorescence in situ hybridization (Q-FISH). J Pathol 2003;200:661-6. 10.1002/path.1392 [DOI] [PubMed] [Google Scholar]
- 4.Brodsky SV, Gealekman O, Chen J, et al. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ Res 2004;94:377-84. 10.1161/01.RES.0000111802.09964.EF [DOI] [PubMed] [Google Scholar]
- 5.Minamino T, Miyauchi H, Yoshida T, et al. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 2002;105:1541-4. 10.1161/01.CIR.0000013836.85741.17 [DOI] [PubMed] [Google Scholar]
- 6.Holz O, Zuhlke I, Jaksztat E, et al. Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur Respir J 2004;24:575-9. 10.1183/09031936.04.00143703 [DOI] [PubMed] [Google Scholar]
- 7.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA 2007;104:7552-7. 10.1073/pnas.0701009104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alder JK, Chen JJ, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA 2008;105:13051-6. 10.1073/pnas.0804280105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajendrasozhan S, Yang SR, Kinnula VL, et al. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008;177:861-70. 10.1164/rccm.200708-1269OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumgartner KB, Samet JM, Stidley CA, et al. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997;155:242-8. 10.1164/ajrccm.155.1.9001319 [DOI] [PubMed] [Google Scholar]
- 11.Rahman I, Skwarska E, Henry M, et al. Systemic and pulmonary oxidative stress in idiopathic pulmonary fibrosis. Free Radic Biol Med 1999;27:60-8. 10.1016/S0891-5849(99)00035-0 [DOI] [PubMed] [Google Scholar]
- 12.Waisberg DR, Barbas-Filho JV, Parra ER, et al. Abnormal expression of telomerase/apoptosis limits type II alveolar epithelial cell replication in the early remodeling of usual interstitial pneumonia/idiopathic pulmonary fibrosis. Hum Pathol 2010;41:385-91. 10.1016/j.humpath.2009.08.019 [DOI] [PubMed] [Google Scholar]
- 13.Minagawa S, Araya J, Numata T, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 2011;300:L391-401. 10.1152/ajplung.00097.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med 2008;178:729-37. 10.1164/rccm.200804-550OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campisi J, Andersen JK, Kapahi P, et al. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol 2011;21:354-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Deursen JM. The role of senescent cells in ageing. Nature 2014;509:439-46. 10.1038/nature13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blagosklonny MV. Cell cycle arrest is not senescence. Aging (Albany NY) 2011;3:94-101. 10.18632/aging.100281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chien Y, Scuoppo C, Wang X, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 2011;25:2125-36. 10.1101/gad.17276711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rovillain E, Mansfield L, Caetano C, et al. Activation of nuclear factor-kappa B signaling promotes cellular senescence. Oncogene 2011;30:2356-66. 10.1038/onc.2010.611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crescenzi E, Pacifico F, Lavorgna A, et al. NF-kappaB-dependent cytokine secretion controls Fas expression on chemotherapy-induced premature senescent tumor cells. Oncogene 2011;30:2707-17. 10.1038/onc.2011.1 [DOI] [PubMed] [Google Scholar]
- 21.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788-824. 10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herbig U, Jobling WA, Chen BP, et al. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell 2004;14:501-13. 10.1016/S1097-2765(04)00256-4 [DOI] [PubMed] [Google Scholar]
- 23.Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res 2005;65:3980-5. 10.1158/0008-5472.CAN-04-3995 [DOI] [PubMed] [Google Scholar]
- 24.Deng C, Zhang P, Harper JW, et al. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995;82:675-84. 10.1016/0092-8674(95)90039-X [DOI] [PubMed] [Google Scholar]
- 25.Hong H, Takahashi K, Ichisaka T, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009;460:1132-5. 10.1038/nature08235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Villacañas O, Perez JJ, Rubio-Martinez J. Structural analysis of the inhibition of Cdk4 and Cdk6 by p16(INK4a) through molecular dynamics simulations. J Biomol Struct Dyn 2002;20:347-58. 10.1080/07391102.2002.10506853 [DOI] [PubMed] [Google Scholar]
- 27.Kuilman T, Michaloglou C, Mooi WJ, et al. The essence of senescence. Genes Dev 2010;24:2463-79. 10.1101/gad.1971610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007;8:729-40. 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- 29.Tsuji T, Aoshiba K, Nagai A. Cigarette smoke induces senescence in alveolar epithelial cells. Am J Respir Cell Mol Biol 2004;31:643-9. 10.1165/rcmb.2003-0290OC [DOI] [PubMed] [Google Scholar]
- 30.Morlá M, Busquets X, Pons J, et al. Telomere shortening in smokers with and without COPD. Eur Respir J 2006;27:525-8. 10.1183/09031936.06.00087005 [DOI] [PubMed] [Google Scholar]
- 31.Jin M, Lee EC, Ra SW, et al. Relationship of absolute telomere length with quality of life, exacerbations, and mortality in COPD. Chest 2018;154:266-73. 10.1016/j.chest.2018.05.022 [DOI] [PubMed] [Google Scholar]
- 32.Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med 2006;174:886-93. 10.1164/rccm.200509-1374OC [DOI] [PubMed] [Google Scholar]
- 33.Müller KC, Welker L, Paasch K, et al. Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir Res 2006;7:32. 10.1186/1465-9921-7-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 2007;356:1317-26. 10.1056/NEJMoa066157 [DOI] [PubMed] [Google Scholar]
- 35.Zhou Y, Peng H, Sun H, et al. Chitinase 3-like 1 suppresses injury and promotes fibroproliferative responses in Mammalian lung fibrosis. Sci Transl Med 2014;6:240ra76. 10.1126/scitranslmed.3007096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snetselaar R, van Batenburg AA, van Oosterhout MFM, et al. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS One 2017;12:e0189467. 10.1371/journal.pone.0189467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savale L, Chaouat A, Bastuji-Garin S, et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2009;179:566-71. 10.1164/rccm.200809-1398OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008;133:1019-31. 10.1016/j.cell.2008.03.039 [DOI] [PubMed] [Google Scholar]
- 39.Orjalo AV, Bhaumik D, Gengler BK, et al. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci USA 2009;106:17031-6. 10.1073/pnas.0905299106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Acosta JC, O'Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008;133:1006-18. 10.1016/j.cell.2008.03.038 [DOI] [PubMed] [Google Scholar]
- 41.Hardy K, Mansfield L, Mackay A, et al. Transcriptional networks and cellular senescence in human mammary fibroblasts. Mol Biol Cell 2005;16:943-53. 10.1091/mbc.e04-05-0392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snetselaar R, van Moorsel CHM, Kazemier KM, et al. Telomere length in interstitial lung diseases. Chest 2015;148:1011-8. 10.1378/chest.14-3078 [DOI] [PubMed] [Google Scholar]
- 43.Zhang B, Zhu W, Yang P, et al. Cigarette smoking and P16INK4α gene promoter hypermethylation in non-small cell lung carcinoma patients: a meta-analysis. PLoS One 2011;6:e28882. 10.1371/journal.pone.0028882 [DOI] [PMC free article] [PubMed] [Google Scholar]