

Abstract
Essentials.
The C‐terminal domain of the fibrinogen α chain (αC domain) is implicated in different severe diseases via clotting abnormalities or amyloid deposits.
Certain anomalies of the fibrinogen molecule lead to amyloid deposits in the kidney, inducing renal insufficiency.
In contrast, in Alzheimer's disease, fibrinogen is normal, but due to an inflammatory process, fibrinogen crosses into the brain and interacts with Aβ, leading to formation of pathological deposits.
Abstract
Fibrinogen, involved in coagulation, is a soluble protein composed of two sets of disulfide‐bridged Aα, Bβ, and γ‐chains. In this review, we present the clinical implications of the αC domain of the molecule in Alzheimer's disease, hereditary renal amyloidosis and a number of thrombotic and hemorrhagic disorders. In Alzheimer's disease, amyloid beta peptide (Aβ) is increased and binds to the αC domain of normal fibrinogen, triggering increased fibrin(ogen) deposition in patients’ brain parenchyma. In hereditary renal amyloidosis, fibrinogen is abnormal, with mutations located in the fibrinogen αC domain. The mutant αC domain derived from fibrinogen degradation folds incorrectly so that, in time, aggregates form, leading to amyloid deposits in the kidneys. In these patients, no thrombotic tendency has been observed. Abnormal fibrinogens with either a point mutation in the αC domain or a frameshift mutation resulting in absence of a part of the αC domain are often associated with either thrombotic events or bleeding. Mutation of an amino acid into cysteine (as in fibrinogens Dusart and Caracas V) or a frameshift mutation yielding an unpaired cysteine in the αC domain is often responsible for thrombotic events. Covalent binding of albumin to the unpaired cysteine via a disulphide bridge leads to decreased accessibility to the fibrinolytic enzymes, hence formation of poorly degradable fibrin clots, which explains the high incidence of thrombosis. In contrast, anomalies due to a frameshift mutation in the αC connector of the molecule, provoking deletion of a great part of the αC domain, are associated with bleeding.
Keywords: Alzheimer’s disease, dysfibrinogenemia, fibrinogen, fibrinogen αC domain, renal amyloidosis
Abbreviations
- αC domain
C‐terminal portions of fibrin(ogen) Aα chains, residues 220‐610
- α2AP
α2 antiplasmin
- t‐PA
tissue‐type plasminogen activator
- AEF
amyloidosis‐enhancing factor
- AD
Alzheimer’s disease
- SNP
single‐nucleotide polymorphism
- αE
extended fibrinogen α chain
- VTE
venous thromboembolism
- PE
pulmonary embolism
- BBB
blood‐brain barrier
- NSAIDs
nonsteroidal anti‐inflammatory drugs
1. INTRODUCTION
Fibrinogen is a soluble plasma glycoprotein comprising two sets of three chains, disulfide‐bridged (Aα‐Bβ‐γ)2. It consists of one central E domain containing the N terminal portions of the Aα, Bβ and γ chains, two lateral D domains connected to the E domain by coiled coils formed by parts of the three chains (Aα 50‐160, Bβ 81‐192, and γ 24‐134), and two Aα C‐terminal domains (Aα 220‐610), (αC domains) located outside the D domains (Figure 1).1, 2
Figure 1.
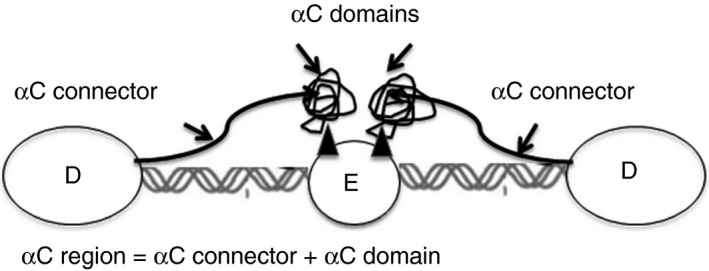
Schema of fibrinogen structure showing relationship of αC domains (αC connectors and αC compact domains) to the D and E domains
Fibrinogen is converted by thrombin into insoluble fibrin during blood clot formation. First, thrombin catalyzes the release of fibrinopeptides A and B from the Aα and Bβ chains, respectively, to form fibrin monomer. Fibrinopeptide A is released from the N‐terminal part of the Aα chain, making accessible a polymerization site “A” that interacts with the complementary “a” site located in the γ chain (T374‐E396).3 The resulting fibrin monomers interact with each other in a half‐staggered manner to produce two‐stranded protofibrils.4 Release of fibrinopeptide B, located at the N‐terminus of the fibrinogen Bβ chain, unmasks polymerization site “B” to interact with its complementary site “b” located in the C‐terminal portion of the Bβ chain, thereby generating fibrin fibers that are associated laterally.5 In parallel, FXIII activated by thrombin (FXIIIa) catalyzes formation of ε‐(γ‐glutamyl) lysyl covalent bonds between two γ chains and several α chains of adjacent fibrin molecules, and crosslinks α2‐antiplasmin (α2AP), the major plasmin inhibitor, to fibrin.6 It was further shown that factor XIII also mediates α2AP ligation to plasma fibrinogen on Aα chains prior to initiation of clotting. This process plays an important role in down‐regulating the rate of fibrinolysis.7 Using a homozygous case of dysfibrinogenemia characterized by an amino acid substitution located at the peptide bond on the Aα chain that is normally cleaved by thrombin, it was shown that clotting of fibrinogen may sometimes occur in absence of fibrinopeptide A release.8, 9, 10
Recently, it was evidenced that fibrinogen αC domain has several roles in coagulation, mediating its activity during various physiological and pathological processes. αC domain is composed of residues Aα 220‐610 consisting of a flexible, unstructured αC connector (Aα 221‐391) and a relatively compact C‐terminal portion of fibrinogen Aα chain (Aα 392‐610).11
In fibrinogen, the two αC domains interact both intramolecularly (ie, with each other) and with the central E region, preferentially through the N termini of the Bβ chains.2, 12 Initially folded on the N‐terminal part of the fibrinogen molecule, the 2 αC domains open outward after fibrinopeptide B (FPB) cleavage,1, 12 revealing novel cryptic sites for plasminogen and t‐PA binding within residues Aα 392‐610 of the αC domains13, 14 and for α2 anti plasmin binding within residues Aα 504‐610.15 Sites also become available for self‐association of the αC domains into αC polymers,16 occurring by formation of a hydrogen bond network through their N‐terminal subdomains via β‐hairpin swapping. This structure is reinforced by interaction of their C‐terminal subdomains with the αC connectors, providing the proper orientation of their reactive residues for efficient cross‐linking by factor XIIIa.16 Lateral aggregation may occur in the absence of αC regions, but their presence enhances it. Clots made from fibrinogen lacking αC domains comprise fibers that are thinner and denser, and have more branch points than normal controls16; anomalies located in this region of the molecule can induce anomalies in aggregation of the protofibrils.
After clot formation, fibrinolysis occurs. The fibrinolytic system comprises an inactive proenzyme, plasminogen, which is converted by tissue‐type plasminogen activator (t‐PA) into plasmin, which degrades fibrin. Plasminogen activation is regulated by molecular interactions between its main components, ie, by the binding of plasminogen and t‐PA to fibrin.17 Conformational changes upon conversion of fibrinogen into fibrin result in the unmasking of multiple sites that expose fibrin to the action of fibrinolytic enzymes. These include the plasminogen and t‐PA binding sites in the αC domain (Aα 392‐610) as described above,13, 14 in addition to sites found in other regions of the molecule. In 1988, Mirshahi et al18 showed, using their own monoclonal antibodies, that the Aα 148‐197 and γ 86‐302 regions were involved in t‐PA binding to fibrin. Later, Medved et al19 found that the conformational change upon conversion of fibrinogen to fibrin results in the exposure of specific epitopes involved in t‐PA binding to fibrin; these epitopes are located in Aα 148‐160 and γ 312‐324.
1.1. Implications of the αC domains in several diseases
It was shown that mutation(s) in the αC domain of fibrinogen may be responsible for severe coagulation disorders,12 and more recently this domain was also implicated in amyloidosis generation, eg, in Alzheimer's disease,20 and familial renal amyloidosis.21
Amyloidosis is caused by abnormal deposition of proteins in soft tissues, and amyloid deposits are primarily made up of protein fibers known as amyloid fibrils. These amyloid fibrils are formed when normally soluble proteins or peptides aggregate and then remain in the tissues instead of being cleared away. Amyloidosis results from a disorder of protein folding characterized by a conformational change of native globular proteins into fibrils with a β‐sheet appearance (ie, β strands connected laterally by two or three backbone hydrogen bonds, forming a twisted pleated sheet) that deposit in various organs.22 In amyloidoses, the deposits contain normal blood proteins and another factor not present in plasma, the amyloidosis‐enhancing factor (AEF), which is probably generated at the site of amyloid deposition and acts in accelerating the pre‐amyloid phase.23
1.2. Fibrinogen αC domain in Alzheimer's disease
Alzheimer's disease (AD) is a neurodegenerative disorder that involves vascular pathology20, 24; it is characterized by extraneuronal deposition of the amyloid β peptide (Aβ) in the form of plaques and by intraneuronal deposition of the microtubule‐associated Tau protein in the form of filaments. Tau interacts with microtubules by mediating microtubule assembly and stability, but in AD, Tau is hyperphosphorylated, which decreases its biological activity.25 Aβ peptide is generated from the transmembrane protein APP (amyloid precursor protein), which seems to be a dependence receptor.26, 27 Such receptors activate programmed cell death pathways in absence of their specific ligand(s) or trophic factor(s), and promote cell survival in their presence.27 Limited proteolysis of APP, first by β‐secretase and then by γ‐secretase complex produces the hydrophobic Aβ peptide,28 which aggregates to form neurotoxic, stable Aβ oligomers.29 These aggregates are evident in the initial microscopic deposition of Aβ in the form of early (diffuse) plaques in AD brains.30 Polymerization of the Aβ peptide into protease‐resistant fibrils is a significant step in AD pathogenesis.31
Interaction of Aβ peptide with fibrinogen leads to its oligomerization, and several authors have reported the crucial involvement of fibrinogen in the pathophysiology of AD—especially its association with cerebral amyloid angiopathy.32 Fibrinogen is not normally found in the brain, nevertheless it accumulates in the extravascular space in brains of AD patients.20 In these patients, deposition of the Aβ oligomers is responsible for endothelial cell damage leading to blood‐brain barrier leakage,33 attested by a diffuse pattern of staining for fibrinogen with considerable fibrinogen immunoreactivity appearing in association with Aβ deposits.34 This fibrinogen‐associated Aβ accumulates around or inside blood vessels in the brain,34 and is thought to be responsible for vascular dysfunction through provoking the degeneration of vessel wall components, affecting cerebral blood flow and worsening cognitive decline35 indeed, fibrinogen that strongly interacts with Aβ peptide was found to be deposited together with Aβ peptide at the sites of cerebral amyloid angiopathy. In addition, Aβ is a prothrombotic factor, triggering thrombin generation via FXII‐dependent activation of FXI, and hence is responsible for the chronic formation of fibrin, suggesting a new mechanism for neuronal dysfunction.36 Thus, fibrinogen does not normally cross the blood—brain barrier but, due to the cerebrovascular pathology, it does accumulate in the damaged brain vasculature and parenchyma of AD mice.37 Fibrin(ogen) deposition is due to interaction of Aβ with fibrinogen; the binding sites of fibrinogen on Aβ are located in the C‐terminus of the β‐chain (β396‐β407)32 and in the αC domain.38 Fibrin clots formed in the presence of Aβ peptide are structurally abnormal and resist degradation.39 Binding of Aβ to fibrin(ogen) renders fibrin clots more resistant to degradation via two mechanisms: (a) specific binding of Aβ to the αC domain of fibrinogen followed, upon thrombin action, by fibrin polymerization into a tight network resistant to fibrinolysis;38 and (b) Aβ overlaid on preformed clots binds to fibrin and delays lysis.39
Mounting evidence thus implicates fibrin(ogen) in AD pathogenesis. Indeed, abnormal deposition and persistence of fibrin(ogen) in AD brains resulting from Aβ‐fibrin(ogen) binding would be expected to enhance Aβ deposition and increase neuroinflammation and neurodegeneration.40 In patients, Narayan et al41 recently demonstrated that there is a significant increase in fibrinogen in brain microarray sections from AD cases compared to controls. Moreover, a novel Aβ—fibrinogen interaction inhibitor rescues both thrombosis and cognitive decline in AD mice.42
As aptly summarized by Cortes‐Canteli et al,43 AD is a multifactorial disorder with a vascular component, and increasing evidence suggests that fibrinogen and fibrin clot formation contribute to this disorder.44 Fibrin(ogen) was observed to be present in areas where neurons were degenerating, and decreasing the fibrinogen levels reduced neuronal death in AD mice. Furthermore, fibrin is also abnormally present intra‐ and extra‐vascularly in different areas of the brains of patients with AD, as well as in the brains of AD mice where it increases over time and correlates with the level of Aβ deposition. Large vessels lined with fibrin or capillaries that are completely blocked by its deposition will alter the cerebral blood flow, especially if these vascular occlusions occur chronically over the course of many years. This may play a substantial role in the cerebral hypoperfusion seen in AD patients.43, 44
1.3. Fibrinogen αC domain anomalies in renal hereditary amyloidosis
The renal hereditary amyloidoses are a rare but clinically important group of disorders that are inherited in an autosomal‐dominant fashion. Variants of the αC domain of fibrinogen cause the most common type of hereditary renal amyloidosis in Europe and, possibly, in the United States as well.45 Absence of bleeding disorders and normal clot formation indicate that the mutations do not significantly affect clotting function. Mutation induces improper folding of the mutant αC fragment derived from fibrinogen degradation so that, as shown by X‐ray fiber diffraction and electron microscopy, fibrinogen amyloid fibrils similar to other chemical types of amyloid accumulate as a β‐sheet structure; the end result is amyloid deposition in the kidneys.46
Various renal amyloidogenic mutations in fibrinogen have been described in the literature.47, 48, 49, 50, 51, 52, 53 These deposits disrupt kidney structure and cause abnormal kidney function, which tends to become progressively more abnormal as amyloid deposits accumulate with time. In these patients, renal histology was characteristic: almost complete glomerular obliteration by amyloid deposition. The disease is characterized by variable penetrance and is associated with hypertension, nephrotic syndrome, proteinuria, and renal failure. Age at onset of symptoms varies from 13 to 70 years. In all cases the clotting times of the variants responsible for renal amyloidosis was normal, except in that reported by Uemichi et al,51 where thrombin time was slightly prolonged and fibrinogen level was low.
Known amyloidogenic fibrinogen point mutations implicated in the disease are all located in the αC domain: these include R554L47; E526V,48 the mutation that most commonly causes renal amyloidosis; E540V, P552H, and T538K, mutations described by Gillmore et al49; E524K, E526K, G555F, and R554H, described by Rowczenio et al.50 Reported amyloidogenic frameshift mutations associated with the disease include: a single‐nucleotide deletion at the third base of codon 524 of the fibrinogen Aα‐chain gene (4904 del G) resulting in premature termination of the protein at codon 54851; a point deletion at position 4897 of the fibrinogen Aα‐chain gene producing a frameshift at codon 522 with truncation at codon 54852; a frameshift mutation found in a young Korean girl that is responsible for an Aα (517‐522) deletion‐insertion of a 31 amino‐acid stop.53
Biochemical analysis of amyloid fibrils from kidneys of the patient with the R554L mutation detected amino acid residues 500‐580 of fibrinogen Aα chain.47 Amyloid fibrils from patients with the E526V mutation contain a similar length peptide fragment from the variant fibrinogen Aα chain only, despite the fact that patients’ plasmas contain approximately equal amounts of normal and variant Aα chains;49 and amyloid fibrils from the patient with a single‐nucleotide deletion producing a frameshift at codon 522 are composed of a 49 amino acid fragment of the Aα chain (residues 499‐521) followed by a novel sequence created by the frameshift in the patient.51
1.4. Fibrinogen αC domain anomalies in coagulation disorders
1.4.1. Variant haplotypes located in the αC domain
Among the several haplotypes of the Aα chain associated with a single nucleotide polymorphism (SNP), only one induces an amino acid modification in the αC domain, the α‐fibrinogen T312A polymorphism. The frequency of this variant by self‐reported race is 5.1% in white patients and 13.5% in black patients.54 The α‐fibrinogen T312A variant has been shown to influence clot structure through increased factor XIII cross‐linking, since this polymorphism occurs in a region important for FXIII‐dependent cross‐linking processes, leading to the formation of fibrin clots that could predispose to clot embolization.55, 56 However, the effect of this common variant on risk of venous thromboembolism (VTE) is unclear for some authors.57
Another variant commonly encountered is Fib 420, characterized by extended α chains (αE) representing 1%‐2% of the circulating fibrinogen content. Fib 420 (αE Bβ γ)2, is a normal human variant fibrinogen with αE subunits that are 50% longer than those of the common Aα subunit due in part to an extra 236 amino acids encoded by exon VI and a variant posttranslational processing, including N‐glycosylation. Additional amino acids are located between G635 and the terminal Q847. Several lines of evidence suggest that the αE chain is less susceptible to proteolytic degradation than the common Aα chain.58
1.4.2. Dysfibrinogenemias associated with mutations in the αC domain
Fibrinogen anomalies in the αC domain often lead to coagulation disorders with highly variable clinical manifestations, from severe bleeding or thrombosis to asymptomatic (Tables 1, 2, 3, 4). Some patients presenting an αC domain anomaly suffer from a bleeding diathesis because of the formation of fibrin clots that exhibit reduced functional properties but, paradoxically, thromboembolic disorders are detected in many other patients. These latter may arise due to the formation of fibrin clots resistant to fibrinolysis by plasmin, secondary to defective t‐PA or plasminogen binding to fibrin, or else to abnormal plasminogen activation on the fibrin surface. Spontaneous abortion is another common clinical complication. The study of such cases has improved our understanding of the fibrinogen—fibrin structure, and of the mechanisms of polymerization and fibrinolysis. Characteristics of published mutations are summarized in Tables 1, 2, 3, 4, respectively, corresponding to four types of mutations reported in the literature: single amino‐acid substitution in the αC domain; 39‐amino‐acid duplication in the connector region of the αC domain; truncations affecting both the αC connector and the αC compact domain; and truncations of the αC compact domain alone.
Table 1.
Dysfibrinogenemia due to an amino‐acid substitution in the fibrinogen αC domain
| Name of abnormal fibrinogen | Genotype | Anomaly in the αC domain | Clinical syndrome | Reference |
|---|---|---|---|---|
| Fibrinogen Dusart | Heterozygous | Mutation of Aα 554 R to C | Thromboembolism | 59, 60, 61, 62, 63 |
| 5 other cases of Fibrinogen Dusart: | Idem | 64, 65, 66, 67, 68 | ||
| Fibrinogen Dusart Chapel Hill 1 | Heterozygous | Thromboembolism | 64 | |
| Fibrinogen Dusart German family 2 | Heterozygous | Thromboembolism | 65 | |
| Fibrinogen Dusart 3 | Heterozygous | Thromboembolism | 66 | |
| Fibrinogen Dusart 4 | Heterozygous | Venous & arterial thrombosis | 67 | |
| Fibrinogen Dusart 5 | Heterozygous | Thrombosis in portal vein | 68 | |
| Fibrinogen San Diego | Heterozygous | Mutation of Aα 554 R to H | Moderate thromboembolism | 69 |
| Fibrinogen Caracas V | Heterozygous | Mutation of Aα 532 S to C | Thromboembolism | 70 |
| Fibrinogen Bordeaux | Heterozygous | Mutation of Aα 439 R to C | Thrombosis | 71 |
| Fibrinogen Sumperk II | Double Heterozygous | Double Mutation Aα 13 G to E and Aα 314 S to C | Mild bleeding | 72 |
| Fibrinogen Caracas II | Heterozygous | Mutation of Aα 434 S to N‐glycosylated N | Asymptomatic | 73 |
| Fibrinogen Grand Lyon III | Heterozygous | Mutation of Aα 496 D to N | Asymptomatic | 74 |
| Fibrinogen Seoul II | Heterozygous | Mutation of Aα 328 Q to P | Myocardial infarct | 75 |
| Fibrinogen Sumida | Heterozygous | Mutation of Aα 472 C to S | Asymptomatic | 76 |
| Fibrinogens of several origins | Homozygous Homozygous | Mutation of Aα 519 G to R | Unknown | 77 |
| Homozygous Homozygous | Mutation of Aα 524 E to K | Unknown | 77 | |
| Unknown | Mutation of Aα 526 E to K | Unknown | 77 | |
| Unknown | Mutation of Aα 526 E to V (Christchurch IV) | Asymptomatic | 77 | |
| Unknown | Mutation of Aα 540 E to V | Unknown | 77 | |
| Unknown | Mutation of Aα 552 P to H | Unknown | 77 |
Table 2.
Dysfibrinogenemia due to an elongation of the αC domain of fibrinogen
| Name of abnormal fibrinogen | Genotype | Anomaly in the αC domain | Clinical syndrome | Reference |
|---|---|---|---|---|
| Fibrinogen Champagne Mont d'Or | Heterozygous | 39 amino acid WXXGSSGPGSTGN duplication in the connector domain starting at position 272 | Thromboembolism | 78 |
Table 3.
Dysfibrinogenemia due to a frameshift mutation in the fibrinogen αC‐connector (Aα 221‐391) resulting in a truncation affecting both the connector itself and the Aα compact domain
| Name of abnormal fibrinogen | Genotype | Anomaly in the αC domain | Clinical Syndrome | Reference |
|---|---|---|---|---|
| Fibrinogen Egyptian | Homozygous | Aα (221)Q stop | Bleeding tendency | 77 |
| Fibrinogen Bulgaria | Homozygous | Aα (229)W stop | Bleeding tendency | 77 |
| Fibrinogen Algerian | Homozygous | Aα(276)W stop | Bleeding tendency | 77 |
| Fibrinogen Chinese | Homozygous | Aα (293) frameshift‐stop | Unknown | 77 |
| Fibrinogen Iran III | Unknown | Aα (297) frameshift‐stop | Bleeding | 77 |
| Fibrinogen France VII | Homozygous | Aα (297)G stop | Unknown | 77 |
| Fibrinogen France XII | Unknown | Aα (315)W stop | Bleeding & Thrombosis | 77 |
| Fibrinogen Turkey | Homozygous | Idem | Idem | 77 |
| Fibrinogen Tunisia | Homozygous | Aα (323)G frameshift stop | Bleeding | 77 |
| Fibrinogen Germany | Homozygous | Aα (327)N frame shift stop | Bleeding | 77 |
| Fibrinogen Keokuk | Heterozygous | Lack of Aα (328‐610), Aα (328)Q stop | Asymptomatic | 79 |
| Double heterozygous: Keokuk mutation plus Aα intron 4 G‐to‐T mutation | Bleeding with severe hypofibrino‐genemia, and thrombotic episodes secondary to surgery accompanied by infusion of normal fibrinogen | |||
| Fibrinogen Otago | Homozygous | Lack of Aα (272‐610). Insertion of cytosine at position 4133 producing a frameshift which translates as 3 new amino acids Q268‐E‐P before termination at position 271 | Bleeding and miscarriages | 80 |
Table 4.
Dysfibrinogenemia due to truncation caused by a frameshift mutation in the fibrinogen αC compact domain (Aα 392‐610)
| Name of abnormal fibrinogen | Genotype | Anomaly in the αC domain | Clinical Syndrome | Reference |
|---|---|---|---|---|
| Fibrinogen Marburg | Homozygous | Lack of Aα (464‐610) [codon Aα 461 AAA (K) to TAA (stop)] | Thromboembolism | 81 |
| Fibrinogen Milano III | Homozygous | Lack of Aα (454‐610) & 2 new C‐terminal amino acids (W452‐S453) [insertion of a thymine in exon V after the ATT triplet coding for Aα I451] | Thromboembolism | 82 |
| Fibrinogen India | Homozygous | Aα (447)T‐frameshift‐17 amino acids‐stop | Bleeding tendency | 83 |
| Fibrinogen Multinational | Heterozygous | Aα (452)G‐frameshift‐stop | Unknown | 77 |
| Fibrinogen Wilmington | Heterozygous | Cytosine deletion at nucleotide 4727 producing a frameshift at T465 followed by the additonal sequence PKMVLTVPRQWI | Bleeding | 84 |
| Fibrinogen Guarenas | Heterozygous | Nonsense mutation at G4731T that causes an Aα chain truncation at S466 | Severe bleeding in the propositus, mild in a brother, asymptomatic in others | 85 |
| Fibrinogen Lincoln | Heterozygous | Lack of Aα (479‐610) & 4 new C‐terminal amino acids resulting in a frameshift at A475, followed by H476‐C‐L‐A‐stop | Mild bleeding tendency | 86 |
| Fibrinogen San Giovanni Rotondo | Heterozygous | Single nucleotide deletion in codon A499. Appearance of a premature codon at position 518 coding for 18 new amino acids with cysteine at last position (SSTLPQLEKHSQVSSHLC) | Asymptomatic | 87 |
| Fibrinogen Nieuwegein | Homozygous | Lack of Aα 454‐610 with deletion of TG cross linking site in the αC domain | Asymptomatic | 88 |
| Fibrinogen Multinational | Unknown | Aα M476 frameshift stop | Thrombosis | 77 |
| Fibrinogen Perth | Heterozygous | Lack of Aα 494‐610 due to cytosine deletion at nucleotide 4841 & incorporation of 23 new residues (LMKLPSSTLPQLEKHSQVSSHLC) | Bleeding in some propositus, thrombosis in others | 89, 90, 91 |
| Fibrinogen Mannheim V | Heterozygous | Nucleotide deletion (C1537delA) resulting in Aα H494P mutation followed by 23 amino acids (LMKLPSSTLPQLEKHSQVSSHLC) before premature truncation after C517 | Miscarriages | 92 |
1.4.3. Mutants characterized by an amino acid substitution
The first reported case of this type was Dusart syndrome (Table 1), discovered in one of our patients who presented a severe familial thromboembolic disease and for whom we focused on thrombosis caused by abnormal fibrin structure, since clots from this patient were very tight and could not be degraded by fibrinolytic enzymes.59 The thromboembolic disease was attributed to impaired fibrin‐enhanced plasminogen activation responsible for a defect in fibrin degradability60 and to an unusual clot rigidity inducing the formation of a brittle clot, therefore resulting in a high incidence of embolism.61 Further investigation of this fibrinogen variant showed that the anomaly is due to an R554C mutation in the αC domain of fibrinogen that has not been associated with amyloid formation.62 This contrasts with the observation of Benson et al,47 who detected a fibrinogen variant with a different mutation at the same residue (R554L), but which is associated with renal amyloidosis without thrombotic disorder. Plasma Dusart fibrinogen was found to be disulfide‐linked to albumin, possibly at Aα C554; removal of the αC domain from fibrinogen Dusart by limited plasmin digestion nearly normalized fibrin polymerization.63 These observations support the conclusion that the fibrinogen αC domain plays an important role in lateral fibril association. Whether it is the presence of cysteine at Aα 554 or the albumin molecules bound to the fibrinogen at this position that causes the defective function, cannot be deduced. Since the initial discovery, five further cases of distinct families affected by Dusart syndrome have been reported; all had an impressive history of thrombosis, which was sometimes fatal.64, 65, 66, 67, 68 These six cases lend support to the concept of thromboembolic diseases due to defective fibrin lysis arising from anomalies in the αC domain of fibrinogen.
Other anomalies (n = 15) in the αC domain of the fibrinogen molecule have been described and are presented in Table 1A.69, 70, 71, 72, 73, 74, 75, 76, 77 Among these cases, four presented thrombotic disorders, another presented mild bleeding (fibrinogen Sumperk II),72 four of them were asymptomatic (Caracas II,73 Grand Lyon III,74 Sumida,76 and Christchurch IV77), and for the six others the clinical syndrome was unknown. Mutation of an αC domain amino acid to cysteine is associated with thrombotic disorders in the six fibrinogens Dusart (Aα R554C) of several origins,64, 65, 66, 67, 68 as well as in Caracas V (Aα K532C)70 and Bordeaux (Aα R439C)71; the unpaired cysteine, not being able to form a disulfide bridge, binds covalently to free ‐SH groups of albumin, resulting in formation of abnormally thin fibrin fibers that are resistant to plasmin degradation.63 Fibrinogen Sumperk II (double heterozygous mutation Aα G13E and S314C) presented only mild bleeding.72 Fibrinogen Seoul II (Aα G328P) had a myocardial infarction.75 In fibrinogen Sumida,76 the functionally important disulfide‐bridged loop Aα C442‐C472 is abolished by the Aα C472S mutation, and although the unpaired C442 binds covalently to albumin, markedly impairing lateral aggregation of protofibrils, there are no clinical manifestations—indeed, clot lysis by plasminogen and t‐PA is normal.
1.4.4. A mutant characterized by elongation of the αC domain
The patient with fibrinogen Champagne au Mont d'Or (Table 2) developed a spontaneous deep venous thrombosis complicated by pulmonary embolism (PE). But further evidence is needed to determine whether the connector prolongation predisposes to venous thrombosis by impairing fibrin degradation.78
1.4.5. Mutants characterized by a truncation in the αC connector domain
Most of the patients (Table 3) that presented a frameshift located in the αC connector (Aα 221‐391) are homozygous, with a bleeding tendency attributable to either defective factor XIIIa‐induced α‐chain crosslinks recently identified as Q223‐K508, Q223‐K539, Q237‐K418, Q237‐K508, Q237‐K539, Q237‐K556, Q366‐K539, Q563‐K539, and Q563‐K601,77, 79, 80, 93 or else to a decrease in factor XIIIa‐mediated crosslinking of PAI‐2 to several lysines, including Aα K413 and K457, which are associated with hyperfibrinolysis.94 PAI‐2 may be undetectable in normal plasma, but it is synthesized by activated monocytes in inflamed tissues,94 and aligns along fibrin strands, where it may cross‐link with fibrin(ogen).95
Only the patient with France XII dysfibrinogenemia77 and the double heterozygous Keokuk patient (Aα Q328‐stop and guanine‐to‐thymine mutation in Intron 4 of the Aα chain, inducing afibrinogenemia)79 presented both bleeding and thrombotic episodes secondary to surgery accompanied by infusion of normal fibrinogen. Heterozygosity for both mutations was required for the expression of severe hypodysfibrinogenemia and for clinical symptoms.79
Fibrinogen Otago is a homozygous dysfibrinogenemia with truncation of approximately 60% of the Aα chain (amino acids 272‐610), leading to a markedly decreased plasma fibrinogen level (0.1 g/L) that is responsible for bleeding episodes and multiple miscarriages.80
1.4.6. Mutants characterized by a truncation within the compact domain of the αC domain
This group of 14 patients with frameshift mutations leading to truncation of the αC compact domain presents a wide variety of clinical outcomes (Table 4). Severe thrombotic disorders occurred in two cases of homozygous dysfibrinogenemia, fibrinogen Marburg (lacking Aα 461‐610)81 and fibrinogen Milano III (lacking Aα 452‐610),82 as well as in one case where zygosity status is not indicated that is characterized by an Aα M476 frameshift stop.77 Patients with other mutations showed a mild bleeding tendency that may be explained by defective factor XIIIa‐induced α polymerization.77, 84, 85, 86 A variable penetrance is observed for fibrinogen Guarenas,85 since the propositus presented severe bleeding, whereas his brother, who has the same anomaly, presented only mild bleeding, and their mother, likewise affected, was asymptomatic. Patients with still other mutations were asymptomatic.87, 88 Some patients with Perth fibrinogen presented with thrombosis, and others with bleeding disorders.89, 90, 91 The patient with fibrinogen Mannheim V presented only with miscarriages.92 It is interesting to note that similar sequences with a cysteine in position 517 were found in three different abnormal dysfibrinogenemias, ie, San Giovanni Rotondo,87 Perth,89, 90, 91 and Mannheim V:92
Perth mutation Aα P495‐LMKLPSSTLPQLEKHSQVSSHL‐C517
Manheim V mutation Aα H494‐PLMKLPSSTLPQLEKHSQVSSHL‐C517
San Giovanni Rotondo mutation Aα A499‐SSTLPQLEKHSQVSSHL‐C517
Although they share an identical sequence, and the unpaired cysteine at Aα 517 generated fibrinogen‐albumin complexes in all three dysfibrogenemias, the clinical syndromes are different: the propositus with fibrinogen San Giovanni Rotondo is asymptomatic, whereas the patient with fibrinogen Mannheim V had miscarriages—and those with fibrinogen Perth present either thrombotic disorders or a bleeding tendency, as described above.
1.4.7. Importance of the αC domain in fibrinogen assembly in and/or secretion by hepatocytes
The αC domain seems to be involved in fibrinogen assembly within and/or secretion from hepatocytes, as previously suggested by Ridgway et al80 and Jayo et al91 in the case of fibrinogen Otago (lacking amino acids 272‐610), the mother (propositus) was homozygous for the mutation and expressed very low fibrinogen level (0.06 mg/mL), whereas no circulating AαOtago chain was found in her heterozygous son, and his fibrinogen level was normal. Likewise, in fibrinogen Marburg an homozygous case of dysfibrinogenemia lacking A alpha 461‐610,81 the fibrinogen level in plasma was very low, while in her heterozygous siblings there is less than 10% of truncated Aα chain. In fibrinogens Lincoln,86 Wilmington,84 and Perth,89, 90, 91 a low level of abnormal Aα chain was found in plasma fibrinogen (ratio of truncated Aα to normal Aα chain is 0.2:1, which is considerably less than the 1:1 normally expected for heterozygotes). From all these cases, it is suggested that the αC domain is involved in assembly of the fibrinogen molecule in the hepatocyte, since the truncated chains do not compete with the normal ones during assembly of mature fibrinogen. In contrast, the Aα C442‐C472 loop which is so important in fibrinogen function has little or no effect on chain assembly and secretion, since disruption of this Aα intrachain loop (by site‐directed mutagenesis C442‐C472) did not impact fibrin(ogen) assembly nor secretion in transfected COS cells.96
1.4.8. Importance of unpaired Aα cysteine in dysfibrinogenemias
In normal fibrinogen, the αC domains are folded on the N‐terminal portion of the fibrinogen molecule and unfold upon fibrin formation, promoting lateral aggregation of protofibrils.2 As a result, anomalies in the αC domain (Tables 1 and 4) may be expected to induce anomalies in aggregation of the protofibrils. Interestingly, the mutation of Aα R554 leads to different pathologies according to whether R is mutated to L (as in hereditary renal amyloidosis, vide supra) or to C (eg, in Dusart syndrome).
In fact it appears that mutation to C of an amino acid located in the αC domain is important for thrombotic disorders. For example, in certain dysfibrinogenemias (Table 4) the deletion of amino acids Aα 465‐610 (Nieuwegein), 452‐610 (Milano III), 461‐610 (Marburg) or 467‐610 (Guarenas) results in the presence of an unpaired cysteine (C442), which in normal fibrinogen forms an intrachain disulfide bridge with Aα C472. The free ‐SH group of C442 covalently links to a free ‐SH group in albumin, which results in disturbed protofibril assembly leading to formation of a tight fibrin network and the acquisition of plasmin resistance relevant to thrombophilia. Thus it would appear that the abnormal network formation observed in such cases is caused by the covalently linked albumin rather than by absence of the carboxyl‐terminal part of the Aα chain. However, thromboembolisms were only observed in Marburg and Milano III. Other fibrinogens that bind albumin due to an anomaly in the αC domain (Mannheim, San Giovanni Rotondo, Nieuwegein) did not present any thrombotic tendency, and patients with fibrinogen Perth presented either thrombotic or hemorrhagic syndromes. With the exception of families with clear thrombotic genotype (eg, fibrinogen Dusart), in other cases the penetrance of the thrombotic phenotype may vary (eg, fibrinogen Perth), perhaps depending on the amount of albumin that becomes disulphide‐bonded to the variant.
In abnormal fibrinogens arising from a frameshift mutation in the αC connector that results in truncation of the Aα chain beginning at amino acid positions 272‐328 (Table 3), there are no unpaired cysteines available to bind albumin (Aα C442 and C472 are absent); this may explain why no thrombosis was reported in these patients.
Sauls et al97 have shown that cysteine‐fibrinogen (Hcys‐fibrinogen) obtained by in vitro incubation of H‐cyc thiolactone with purified fibrinogen shows increased resistance to fibrinolysis: H‐Cys fibrinogen has additional cysteines (seven in the Aα chain, two in the Bβ chain, three in the γ chain). Of the seven cysteine residues located in the Aα chain, three are in the αC domain, which is involved in t‐PA and plasminogen binding. Furthermore, these residues are found in the naturally occurring Aα mutations R554C in the Dusart fibrinogens62 and L532C in Caracas V70 where they are characterized by impaired fibrin‐stimulated plasminogen activation by t‐PA. It therefore seems likely that plasminogen binding in the αC domain may regulate fibrinolysis by making bound plasminogen readily available for ternary complex formation in fibrin.
2. PERSPECTIVES
From these results it appears that the αC domain of fibrin(ogen) is involved in various pathologies such as AD, renal familial (hereditary) amyloidosis, and coagulation disorders (thromboembolism or bleeding). Normal fibrin(ogen) can be found in AD plaques, whereas mutated αC domain or its fragments have been implicated in the physiopathology of renal amyloidosis and certain coagulation disorders.
Cerebral amyloid angiopathy, responsible for the vascular dysfunction seen in AD, is induced by Aβ‐fibrinogen complex; and depletion of fibrinogen lessens cerebral amyloid angiopathy.20 The blood—brain barrier (BBB) normally prevents uncontrolled entry of blood‐borne and blood‐derived products into the brain. Indeed, brain capillary endothelial cells are connected by both tight and adherens junctions, forming a continuous endothelial monolayer. This anatomical barrier only permits the passage of small circulating lipid‐soluble molecules. In AD, the BBB breakdown associated with vascular dysfunction allows influx into the brain of neurotoxic blood‐derived debris, cells, and microbial pathogens, and is associated with inflammatory and immune responses that can trigger multiple pathways of neurodegeneration.98 With the failure of several large‐scale trials of treatments designed to lower the amyloid load in the brains of AD patients, and since fibrinogen is increased in inflammatory processes, trials with nonsteroidal anti‐inflammatory drugs (NSAIDs) have begun. These studies indicate that, by decreasing fibrinogen levels, NSAIDs can attenuate the destructive process if they are started well before clinical signs develop (at least 6 months, and preferably as long as 5 years before the clinical diagnosis of AD).99
Because of the life‐threatening potential of renal insufficiency in cases of hereditary amyloidosis, double transplantation (kidney and liver) may still offer the best treatment option for eligible patients: by replacing the source of circulating amyloidogenic fibrin(ogen) with normal (non‐amyloidogenic) protein, liver transplantation prevents the formation of amyloid deposits in the transplanted kidney. It has been suggested that preemptive solitary liver transplantation early in the course of the disease might be a viable alternative, avoiding the need for hemodialysis and kidney transplantation.45 In contrast, dysfibrinogenemia associated with thrombotic or hemorrhagic disorders can be adequately managed with anticoagulant therapy or blood transfusion, respectively.
RELATIONSHIP DISCLOSURES
Shahsultan Mirshahi (S. Mirshahi) was funded by Stago to work in the INSERM laboratory (U. 965). There are no other financial conflicts of interest.
AUTHOR CONTRIBUTION
JS and CS: Researchers, analysis of several dysfibrinogenemias, discovery of Dusart Syndrome (abnormal fibrinogen with a mutation in the C‐terminal domain of the Aα chain of the molecule that leads to a severe thrombotic disorder due to defective thrombolysis); JS framed and wrote the paper. SM: Researcher, specialist in fibrin degradability, extensively involved in the conception of the paper, and in drafting and preparing the manuscript. SQM: Clinician, research on defective fibrinolytic patterns during formation of fibrin deposits, involved in literature searches and in writing the paper. RV: Research Professor, relationship between clot structure and fibrin degradability, invaluable discussions and suggestions during preparation of manuscript. LLP: Researcher, involved in critical discussions, preparation of manuscript. MM: Researcher, participated in determining which domains of the Aα chain are implicated in the binding of plasminogen activator on the fibrinogen molecule, participated in framing and writing the paper.
ACKNOWLEDGMENTS
We would like to thank Pr. Jacques Caen, who was the first to actively support our work concerning the relationship between clot structure and thrombosis. This was the starting point for all subsequent studies on Dusart Syndrome. Thank you, Pr. Caen, for giving us the opportunity to work with you.
Soria J, Mirshahi S, Mirshahi SQ, et al. Fibrinogen αC domain: Its importance in physiopathology. Res Pract Thromb Haemost. 2019;3:173–183. 10.1002/rth2.12183
Funding information
S. Mirshahi was funded by Stago to work in the INSERM laboratory (U. 965).
[Article updated on February 21, 2019 after first online publication on February 15, 2019: Author byline was corrected. “Sam Qiumas” was corrected to “Sam Qiumars Mirshahi.”]
Contributor Information
Jeannette Soria, Email: jeannette.soria@gmail.com.
Massoud Mirshahi, @MassoudMirshahi.
REFERENCES
- 1. Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3:1894–904. [DOI] [PubMed] [Google Scholar]
- 2. Litvinov RI, Yakovlev S, Tsurupa G, Gorkun OV, Medved L, Weisel JW. Direct evidence for specific interactions of fibrinogen αC‐domains with the central E region and with each other. Biochemistry. 2007;46:9133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Horwitz BH, Váradi A, Scheraga HA. Localization of a fibrin gamma‐chain polymerization site within segment Thr‐374 to Glu‐396 of human fibrinogen. Proc Natl Acad Sci USA. 1984;81:5980–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weisel JW, Litvinov RI. Mechanisms of fibrin polymerization and clinical implications. Blood. 2013;121:1712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Medved LV, Litvinovich SV, Ugarova TP, Lukinova NI, Kalikhevich VN, Ardemasova ZA. Localization of a fibrin polymerization site complementary to Gly‐His‐Arg sequence. FEBS Lett. 1993;320:239–42. [DOI] [PubMed] [Google Scholar]
- 6. Hethershaw EL, Cilia La Corte AL, Duval C, Ali M, Grant PJ, Ariëns RA, et al. The effect of blood coagulation factor XIII on fibrin clot structure and fibrinolysis. J Thromb Haemost. 2014;12:197–205. [DOI] [PubMed] [Google Scholar]
- 7. Mosesson MW, Siebenlist KR, Hernandez I, Lee KN, Christiansen VJ, McKee PA. Evidence that α2‐antiplasmin becomes covalently ligated to plasma fibrinogen in the circulation: a new role for plasma factor XIII in fibrinolysis regulation. J Thromb Haemost. 2008;6:1565–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soria J, Soria C, Samama M, Poirot E, Kling C. Fibrinogen Troyes‐fibrinogen Metz. Two new cases of congenital dysfibrinogenemia. Thromb Diath Haemorrh. 1972;27:619–33. [PubMed] [Google Scholar]
- 9. Soria J, Soria C, Samama M, Henschen A, Southan C. Detection of fibrinogen abnormality in dysfibrinogenemia: special report on fibrinogen Metz characterization by amino acid substitution located at the peptide bond cleaved by thrombin In: Henschen A, Graeff V, Lottspeich V, eds. Fibrinogen: Recent Biochemical and Medical Aspects. Berlin, Germany: Walter de Gruyter; 1982: pp. 129–43. [Google Scholar]
- 10. Ladure P, Ricordel Y, Soria J, Soria C, Samama M. Binding of citrate to normal fibrinogen and to Mez fibrinogen In: Henschen A, Graeff H, Lottspeich F, eds. Fibrinogen: Recent Biochemical and Medical Aspects. Berlin, Germany: Walter de Gruyter; 1982: pp. 145–52. [Google Scholar]
- 11. Medved L, Weisel JW. Recommendations for nomenclature on fibrinogen and fibrin. J Thromb Haemost. 2009;7:355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weisel JW, Medved L. The structure and function of the alpha C domains of fibrinogen. Ann N Y Acad Sci. 2001;936:312–27. [DOI] [PubMed] [Google Scholar]
- 13. Tsurupa G, Medved L. Fibrinogen alpha C domains contain cryptic plasminogen and tPA binding sites. Ann N Y Acad Sci. 2001;936:328–30. [DOI] [PubMed] [Google Scholar]
- 14. Medved L, Nieuwenhuizen W. Molecular mechanisms of initiation of fibrinolysis by fibrin. Thromb Haemost. 2003;89:409–19. [PubMed] [Google Scholar]
- 15. Tsurupa G, Yakovlev S, McKee P, Medved L. Noncovalent interaction of alpha(2)‐antiplasmin with fibrin(ogen): localization of alpha(2)‐ antiplasmin‐binding sites. Biochemistry. 2010;49:7643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsurupa G, Pechik I, Litvinov RI, Hantgan RR, Tjandra N, Weisel JW, et al. On the mechanism of αC polymer formation in fibrin. Biochemistry. 2012;51:2526–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J Biol Chem. 1982;257:2912–9. [PubMed] [Google Scholar]
- 18. Mirshahi M, Mirshahi MC, Soria J, et al. Monoclonal antibodies reacting with fibrinogen derivative inhibit the binding of tissue plasminogen activator to fragment D In: Mosesson MW, Amrani DL, Siebenlist KR, DiOrio JP, eds. Fibrinogen 3: Biochemistry, Biological Functions, Gene Regulation and Expression. Amsterdam, The Netherlands: Elsevier; 1988: pp. 147–52. [Google Scholar]
- 19. Medved L, Tsurupa G, Yakovlev S. Conformational changes upon conversion of fibrinogen into fibrin. The mechanisms of exposure of cryptic sites. Ann N Y Acad Sci. 2001;936:185–204. [DOI] [PubMed] [Google Scholar]
- 20. Cortes‐Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, et al. Fibrinogen and beta‐amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron. 2010;66:695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dember LM. Amyloidosis‐associated kidney disease. J Am Soc Nephrol. 2006;17:3458–71. [DOI] [PubMed] [Google Scholar]
- 22. Rambaran RN, Serpell LC. Amyloid fibrils: abnormal protein assembly. Prion. 2008;2:112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hol PR, Snel FW, Niewold TA, Gruys E. Amyloid‐enhancing factor (AEF) in the pathogenesis of AA‐amyloidosis in the hamster. Virchows Arch B Cell Pathol Incl Mol Pathol. 1986;52:273–81. [DOI] [PubMed] [Google Scholar]
- 24. de la Torre JC. Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004;3:184–90. [DOI] [PubMed] [Google Scholar]
- 25. Iqbal K, Liu F, Gong CX, Grundke‐Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7:656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, et al. A second cytotoxic proteolytic peptide derived from amyloid β‐protein precursor. Nature Med. 2000;6:397–404. [DOI] [PubMed] [Google Scholar]
- 27. Bredesen DE, Mehlen P, Rabizadeh S. Receptors that mediate cellular dependence. Cell Death Differ. 2005;12:1031–43. [DOI] [PubMed] [Google Scholar]
- 28. Vassar R, Bennett BD, Babu‐Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta‐secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–41. [DOI] [PubMed] [Google Scholar]
- 29. Chen YR, Glabe CG. Distinct early folding and aggregation properties of Alzheimer amyloid‐β peptides A β 40 and A β42: stable trimer or tetramer formation by A β42. J Biol Chem. 2006;281:24414–22. [DOI] [PubMed] [Google Scholar]
- 30. Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, et al. Intraneuronal αbeta 42 accumulation in human brain. Am J Pathol. 2000;156:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tjernberg LO, Callaway DJ, Tjernberg A, Hahne S, Lilliehöök C, Terenius L, et al. Molecular model of Alzheimer amyloid β‐peptide fibril formation. J Biol Chem. 1999;274:12619–25. [DOI] [PubMed] [Google Scholar]
- 32. Ahn HJ, Zamolodchikov D, Cortes‐Canteli M, Norris EH, Glickman JF, Strickland S. Alzheimer's disease peptide beta‐amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci USA. 2010;107:21812–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol. 2001;64:575–611. [DOI] [PubMed] [Google Scholar]
- 34. Ahn HJ, Chen ZL, Zamolodchikov D, Norris EH, Strickland S. Interactions of β‐amyloid peptide with fibrinogen and coagulation factor XII may contribute to Alzheimer's disease. Curr Opin Hematol. 2017;24:427–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thal DR, Capetillo‐Zarate E, Larionov S, Staufenbiel M, Zurbruegg S, Beckmann N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging. 2009;30:1936–48. [DOI] [PubMed] [Google Scholar]
- 36. Zamolodchikov D, Renné T, Strickland S. The Alzheimer's disease peptide β‐amyloid promotes thrombin generation through activation of coagulation factor XII. J Thromb Haemost. 2016;14:995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease. J Exp Med. 2007;204:1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zamolodchikov D, Berk‐Rauch HE, Oren DA, Stor DS, Singh PK, Kawasaki M, et al. Biochemical and structural analysis of the interaction between β‐amyloid and fibrinogen. Blood. 2016;128:1144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zamolodchikov D, Strickland S. Aβ delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood. 2012;119:3342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cortes‐Canteli M, Zamolodchikov D, Ahn HJ, Strickland S, Norris EH. Fibrinogen and altered hemostasis in Alzheimer's disease. J Alzheimers Dis. 2012;32:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Narayan PJ, Kim SL, Lill C, Feng S, Faull RL, Curtis MA, et al. Assessing fibrinogen extravasation into Alzheimer's disease brain using high‐content screening of brain tissue microarrays. J Neurosci Methods. 2015;247:41–9. [DOI] [PubMed] [Google Scholar]
- 42. Ahn HJ, Glickman JF, Poon KL, Zamolodchikov D, Jno‐Charles OC, Norris EH, et al. A novel Aβ‐fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer's disease mice. J Exp Med. 2014;211:1049–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cortes‐Canteli M, Mattei L, Richards AT, Norris EH, Strickland S. Fibrin deposited in the Alzheimer's disease brain promotes neuronal degeneration. Neurobiol Aging. 2015;36:608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cortes‐Canteli M, Strickland S. Fibrinogen, a possible key player in Alzheimer's disease. J Thromb Haemost. 2009;7(suppl 1):146–115. [DOI] [PubMed] [Google Scholar]
- 45. Stangou AJ, Banner NR, Hendry BM, Rela M, Portmann B, Wendon J, et al. Hereditary fibrinogen A alpha‐chain amyloidosis: phenotypic characterization of a systemic disease and the role of liver transplantation. Blood. 2010;115:2998–3007. [DOI] [PubMed] [Google Scholar]
- 46. Serpell LC, Benson M, Liepnieks JJ, Fraser PE. Structural analyses of fibrinogen amyloid fibrils. Amyloid. 2007;14:199–203. [DOI] [PubMed] [Google Scholar]
- 47. Benson MD, Liepnieks J, Uemichi T, Wheeler G, Correa R. Hereditary renal amyloidosis associated with a mutant fibrinogen alpha‐chain. Nat Genet. 1993;3:252–5. [DOI] [PubMed] [Google Scholar]
- 48. Uemichi T, Liepnieks JJ, Benson MD. Hereditary renal amyloidosis with a novel variant fibrinogen. J Clin Invest. 1994;93:731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gillmore JD, Lachmann HJ, Rowczenio D, Gilbertson JA, Zeng CH, Liu ZH, et al. Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen A‐chain amyloidosis. J Am Soc Nephrol. 2009;20:444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rowczenio D, Stensland M, de Souza GA, Strøm EH, Gilbertson JA, Taylor G, et al. Renal amyloidosis associated with 5 novel variants in the fibrinogen A alpha chain protein. Kidney Int Rep. 2017;2:461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Uemichi T, Liepnieks JJ, Yamada T, Gertz MA, Bang N, Benson MDA. Frame shift mutation in the fibrinogen A alpha chain gene in a kindred with renal amyloidosis. Blood. 1996;87:4197–203. [PubMed] [Google Scholar]
- 52. Hamidi Asl L, Liepnieks JJ, Uemichi T, Rebibou J‐M, Justrabo E, Droz D, et al. Renal amyloidosis with a frame shift mutation in fibrinogen a‐alpha chain gene producing a novel amyloid protein. Blood. 1997;90:4799–805. [PubMed] [Google Scholar]
- 53. Kang HG, Bybee A, Ha IS, Park MS, Gilbertson JA, Cheong HI, et al. Hereditary amyloidosis in early childhood associated with a novel insertion‐deletion (indel) in the fibrinogen A chain gene. Kidney Int. 2005;68:1994–8. [DOI] [PubMed] [Google Scholar]
- 54. Carter AM, Catto AJ, Kohler HP, Ariëns RAS, Stickland MH, Grant PJ. Fibrinogen Thr312Ala polymorphism and venous thromboembolism. Blood. 2000;96:1177–9. [PubMed] [Google Scholar]
- 55. Li JF, Lin Y, Yang YH, Gan HL, Liang Y, Lie J, et al. Fibrinogen Aα Thr312Ala polymorphism specifically contributes to chronic thromboembolic pulmonary hypertension by increasing fibrin resistance. PLoS One. 2013;8:e69635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Standeven KF, Grant PJ, Carter AM, Scheiner T, Weisel JW, Ariëns RA. Functional analysis of the fibrinogen Aα Thr312Ala polymorphism: effects on fibrin structure and function. Circulation. 2003;107:2326–30. [DOI] [PubMed] [Google Scholar]
- 57. Rasmussen‐Torvik LJ, Cushman M, Tsai MY, Zhang Y, Heckbert SR, Rosamond WD, et al. The association of alpha‐fibrinogen Thr312Ala polymorphism and venous thromboembolism in the LITE study. Thromb Res. 2007;121:173–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fu Y, Grieninger G. Fib420: a normal human variant of fibrinogen with two extended Aα chains. Proc Natl Acad Sci USA. 1994;91:2625–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Soria J, Soria C, Caen JP. A new type of dysfibrinogenemia with defective fibrin lysis ‐ Dusard syndrome. Possible relation to Thrombosis. Br J Haematol. 1983;53:573–86. [DOI] [PubMed] [Google Scholar]
- 60. Lijnen HR, Soria J, Soria C, Collen D, Caen JP. Dysfibrinogenemia (fibrinogen Dusard) associated with impaired fibrin‐enhanced plasminogen activation. Thromb Haemost. 1984;51:108–9. [PubMed] [Google Scholar]
- 61. Collet JP, Soria J, Mirshahi M, Hirsch M, Dagonnet FB, Caen J, et al. Dusart syndrome: a new concept of the relationship between fibrin clot architecture and fibrin clot degradability: hypofibrinolysis related to an abnormal clot structure. Blood. 1993;82:2462–9. [PubMed] [Google Scholar]
- 62. Koopman J, Haverkate F, Grimbergen J, Lord ST, Mosesson MW, DiOrio JP, et al. Molecular basis for fibrinogen Dusart (A alpha 554 Arg–>Cys) and its association with abnormal fibrin polymerization and thrombophilia. J Clin Invest. 1993;91:1637–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mosesson MW, Siebenlist KR, Hainfeld JF, Wall JS, Soria J, Soria C, et al. The relationship between the fibrinogen D domain self‐association/cross‐linking site (gammaXL) and the fibrinogen Dusart abnormality (Aalpha R554C‐albumin): clues to thrombophilia in the “Dusart syndrome”. J Clin Invest. 1996;97:2342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wada Y, Lord ST. A correlation between thrombotic disease and a specific fibrinogen abnormality (A alpha 554 Arg–>Cys) in two unrelated kindred, Dusart and Chapel Hill III. Blood. 1994;84:3709–14. [PubMed] [Google Scholar]
- 65. Meyer M, Kutscher G, Binnewies T, et al. Mutation spectrum in fibrinogen genes: molecular analysis in 11 German families with dysfibrinogenemia [abstract]. Thromb Haemost. 2000;96:1191–3. [Google Scholar]
- 66. Tarumi T, Martincic D, Thomas A, Janco R, Hudson M, Baxter P, et al. Familial thrombophilia associated with fibrinogen Paris V: Dusart syndrome. Blood. 2000;96:1191–3. [PubMed] [Google Scholar]
- 67. Ramanathan R, Gram J, Feddersen S, Nybo M, Larsen A, Sidelmann JJ. Dusart syndrome in a Scandinavian family characterized by arterial and venous thrombosis at young age. Scand J Clin Lab Invest. 2013;73:585–90. [DOI] [PubMed] [Google Scholar]
- 68. Shen YM, Trang V, Sarode R, Brennan S. Fibrinogen Dusart presenting as recurrent thromboses in the hepatic portal system. Blood Coagul Fibrinolysis. 2014;25:392–4. [DOI] [PubMed] [Google Scholar]
- 69. Morris TA, Marsh JJ, Chiles PG, Magaña MM, Liang NC, Soler X, et al. High prevalence of dysfibrinogenemia among patients with chronic thromboembolic pulmonary hypertension. Blood. 2009;114:1929–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Marchi R, Lundberg U, Grimmberger J, Koopman J, Torres A, de Bosch NB, et al. Fibrinogen Caracas V, an abnormal fibrinogen with an A alpha‐ 532 Ser‐Cys substitution associated with thrombosis. Thromb Haemost. 2000;84:263–70. [PubMed] [Google Scholar]
- 71. Hanss M, Vergnes C, Rugeri L, Ffrench P, De Mazancourt P. A new electrophoretic variant of fibrinogen associated with venous thromboembolism, fibrinogen Bordeaux A alpha Arg 439–>Cys. J Thromb Haemost. 2008;6:1422–4. [DOI] [PubMed] [Google Scholar]
- 72. Kotlín R, Suttnar J, Cápová I, Hrachovinová I, Urbánková M, Dyr JE. Fibrinogen Šumperk II : dysfibrinogenemia in an individual with two coding mutations. Am J Hematol. 2012;87:555–7. [DOI] [PubMed] [Google Scholar]
- 73. Woodhead JL, Nagaswami C, Matsuda M, Arocha‐Piñango CL, Weisel JW. The ultrastructure of fibrinogen Caracas II molecules, fibers, and clots. J Biol Chem. 1996;271:4946–53. [DOI] [PubMed] [Google Scholar]
- 74. Hanss M, Chevreaud C, French P, Négrier C, de Mazancourt P. Clinical and biological features of 3 cases of hypofibrinogenemia associated with three different mutations (gamma Ala341Thr, Bbeta Tyr326Cys and Aalpha Asp496Asn). Thromb Haemost. 2007;98:689–91. [PubMed] [Google Scholar]
- 75. Park R, Doh HJ, An SS, Choi JR, Chung KH, Song KS. A novel fibrinogen variant (fibrinogen Seoul II; Aalpha Gln328Pro) characterized by impaired fibrin alpha‐chain cross‐linking. Blood. 2006;108:1919–24. [DOI] [PubMed] [Google Scholar]
- 76. Ikeda M, Arai S, Mukai S, Takezawa Y, Terasawa F, Okumura N. Novel heterozygous dysfibrinogenemia, Sumida (AαC472S), showed markedly impaired lateral aggregation of protofibrils and mildly lower functional fibrinogen levels. Thromb Res. 2015;135:710–7. [DOI] [PubMed] [Google Scholar]
- 77. Groupe français d’étude sur l’hémostase et la thrombose, avileble in GFHT, Human fibrinogen data base, fibrinogen variantes A‐alpha chain , 2018.
- 78. Hanss MM, French PO, Mornex JF, Chabuet M, Biot F, De Mazancourt P, et al. Two novel fibrinogen variants found in patients with pulmonary embolism and their families. J Thromb Haemost. 2003;1:1251–7. [DOI] [PubMed] [Google Scholar]
- 79. Lefebvre P, Velasco PT, Dear A, Lounes KC, Lord ST, Brennan SO, et al. Severe hypodysfibrinogenemia in compound heterozygotes of the fibrinogen AalphaIVS4 + 1G>T mutation and an AalphaGln328 truncation (fibrinogen Keokuk). Blood. 2004;103:2571–6. [DOI] [PubMed] [Google Scholar]
- 80. Ridgway H, Brennan SO, Faed JM, George PM. Fibrinogen Otago: a major alpha chain truncation associated with severe hypofibrinogenaemia and recurrent miscarriages. Br J Haematol. 1997;98:632–9. [DOI] [PubMed] [Google Scholar]
- 81. Koopman J, Haverkate F, Grimbergen J, Egbring R, Lord ST. Fibrinogen Marburg: a homozygous case of dysfibrinogenemia, lacking amino acids A alpha 461‐610 (Lys 461 AAA–>stop TAA). Blood. 1992;80:1972–9. [PubMed] [Google Scholar]
- 82. Furlan M, Steinmann C, Jungo M, Bögli C, Baudo F, Redaelli R, et al. Frameshift mutation in Exon V of the A alpha‐chain gene leading to truncated A alpha‐chains in the homozygous dysfibrinogen Milano III. J Biol Chem. 1994;269:33129–34. [PubMed] [Google Scholar]
- 83. Sumitha E, Jayandharan GR, Arora N, Abraham A, David S, Devi GS, et al. Molecular basis of quantitative fibrinogen disorders in 27 patients from India. Haemophilia. 2013;19:611–8. [DOI] [PubMed] [Google Scholar]
- 84. Brennan SO, Mosesson MW, Lowen R, Frantz C. Dysfibrinogenemia (fibrinogen Wilmington) due to a novel Aalpha chain truncation causing decreased plasma expression and impaired fibrin polymerisation. Thromb Haemost. 2006;96:88–9. [DOI] [PubMed] [Google Scholar]
- 85. Marchi R, Carvajal Z, Meyer M, Soria J, Ruiz‐Saez A, Arocha‐Piñango CL, et al. Fibrinogen Guarenas, an abnormal fibrinogen with an Aalpha‐chain truncation due to a nonsense mutation at Aalpha 467 Glu (GAA)–>stop (TAA). Thromb Res. 2006;118:637–50. [DOI] [PubMed] [Google Scholar]
- 86. Ridgway HJ, Brennan SO, Gibbons S, George PM. Fibrinogen Lincoln: a new truncated alpha chain variant with delayed clotting. Br J Haematol. 1996;93:177–84. [DOI] [PubMed] [Google Scholar]
- 87. Margaglione M, Vecchione G, Santacroce R, D'Angelo F, Casetta B, Papa ML, et al. A frameshift mutation in the human fibrinogen Aalpha‐chain gene (Aalpha(499)Ala frameshift stop) leading to dysfibrinogen San Giovanni Rotondo. Thromb Haemost. 2001;86:1483–8. [PubMed] [Google Scholar]
- 88. Collen A, Maas A, Kooistra T, Lupu F, Grimbergen J, Haas FJLM, et al. Aberrant fibrin formation and cross‐linking of fibrinogen Nieuwegein, a variant with a shortened Aalpha‐chain, alters endothelial capillary tube formation. Blood. 2001;97:973–80. [DOI] [PubMed] [Google Scholar]
- 89. Homer VM, Mullin JL, Brennan SO, Barr A, George PM. Novel Aα chain truncation (fibrinogen Perth) resulting in low expression and impaired fibrinogen polymerization. J Thromb Haemost. 2003;1:1245–50. [DOI] [PubMed] [Google Scholar]
- 90. Westbury SK, Duval C, Philippou H, Brown R, Lee KR, Murden SL, et al. Partial deletion of the αC‐domain in the Fibrinogen Perth variant is associated with thrombosis, increased clot strength and delayed fibrinolysis. Thromb Haemost. 2013;110:1135–44. [DOI] [PubMed] [Google Scholar]
- 91. Jayo A, Arnold E, González‐Manchón C, Green D, Lord ST. Hypodysfibrinogenemia causing mild bleeding and thrombotic complications in a compound heterozygote of AalphaIVS4+1G>T mutation and Aalpha4841delC (Aα Perth). Thromb Haemost. 2009;101:770–2. [PMC free article] [PubMed] [Google Scholar]
- 92. Dempfle CE, George PM, Borggrefe M, Neumaier M, Brennan SO. Demonstration of heterodimeric fibrinogen molecules partially conjugated with albumin in a novel dysfibrinogen: fibrinogen Mannheim V. Thromb Haemost. 2009;102:29–34. [DOI] [PubMed] [Google Scholar]
- 93. Wang W. Identification of respective lysine donor and glutamine acceptor sites involved in factor XIIIa‐catalyzed fibrin α chain cross‐linking. J Biol Chem. 2011;286:44952–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ritchie H, Lawrie LC, Mosesson MW, Booth NA. Characterization of crosslinking sites in fibrinogen for plasminogen activator inhibitor 2 (PAI‐2). Ann N Y Acad Sci. 2001;936:215–8. [DOI] [PubMed] [Google Scholar]
- 95. Kruithof EK, Baker MS, Bunn CL. Biological and clinical aspects of plasminogen activator inhibitor type 2. Blood. 1995;86:4007–24. [PubMed] [Google Scholar]
- 96. Zhang JZ, Redman C. Fibrinogen assembly and secretion. Role of intrachain disulfide loops. J Biol Chem. 1996;271:30083–8. [DOI] [PubMed] [Google Scholar]
- 97. Sauls DL, Lockhart E, Warren ME, Lenkowski A, Wilhelm SE, Hoffman M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: a potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry. 2006;45:2480–7. [DOI] [PubMed] [Google Scholar]
- 98. Sweeney MD, Sagare AP. Zlokovic BV blood‐brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. McGeer PL, Guo JP, Lee M, Kennedy K, McGeer EG. Alzheimer's disease can be spared by nonsteroidal anti‐inflammatory drugs. J Alzheimer Dis. 2018;62:1219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]