

Abstract
The overall survival for patients with primary glioblastoma is very poor. Glioblastoma contains a subpopulation of glioma stem cells (GSC) that are responsible for tumour initiation, treatment resistance and recurrence. PPARα is a transcription factor involved in the control of lipid, carbohydrate and amino acid metabolism. We have recently shown that PPARα gene and protein expression is increased in glioblastoma and has independent clinical prognostic significance in multivariate analyses. In this work, we report that PPARα is overexpressed in GSC compared to foetal neural stem cells. To investigate the role of PPARα in GSC, we knocked down its expression using lentiviral transduction with short hairpin RNA (shRNA). Transduced GSC were tagged with luciferase and stereotactically xenografted into the striatum of NOD‐SCID mice. Bioluminescent and magnetic resonance imaging showed that knockdown (KD) of PPARα reduced the tumourigenicity of GSC in vivo. PPARα‐expressing control GSC xenografts formed invasive histological phenocopies of human glioblastoma, whereas PPARα KD GSC xenografts failed to establish viable intracranial tumours. PPARα KD GSC showed significantly reduced proliferative capacity and clonogenic potential in vitro with an increase in cellular senescence. In addition, PPARα KD resulted in significant downregulation of the stem cell factors c‐Myc, nestin and SOX2. This was accompanied by downregulation of the PPARα‐target genes and key regulators of fatty acid oxygenation ACOX1 and CPT1A, with no compensatory increase in glycolytic flux. These data establish the aberrant overexpression of PPARα in GSC and demonstrate that this expression functions as an important regulator of tumourigenesis, linking self‐renewal and the malignant phenotype in this aggressive cancer stem cell subpopulation. We conclude that targeting GSC PPARα expression may be a therapeutically beneficial strategy with translational potential as an adjuvant treatment. © 2018 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: PPARα, glioma stem cell, shRNA
Introduction
Gliomas form the most common group of primary central nervous system (CNS) tumours, with an incidence of 6.6 per 100 000 individuals/year 1. A total of 50% of adult gliomas are glioblastomas, which are associated with poor clinical survival 2, 3. The median survival is 15 months in the setting of a clinical trial 4, 5 and 12 months using current treatment regimens 1, 6, 7.
The peroxisome proliferator‐activated receptors (PPARs) are ligand‐activated transcription factors with diverse metabolic functions 8. PPARα activates mitochondrial and peroxisomal fatty acid oxidation and ketogenesis and inhibits glycolysis and fatty acid synthesis 9, 10, 11. Our previous work has shown that the PPARA gene and its protein product are significantly overexpressed in IDH‐wild type primary glioblastomas and that high PPARA expression functions as an independent prognostic biomarker 12. This finding has been independently cross‐validated in the Chinese Glioma Genome Atlas 13.
PPARα agonists such as fenofibrate have clinical utility in treating dyslipidaemia 14. Fenofibrate reduces glioma cell motility 15, 16 and induces cell cycle arrest and apoptosis in vitro 17, 18. Fenofibrate has also been reported to exert anti‐tumour effects by inducing ketogenesis 19 and reducing glycolytic flux 20, 21.
Stem‐like cells have been identified in glioma in vitro models 22, 23 and glioma stem cells (GSC), with the defining properties of self‐renewal, multi‐potency and in vivo tumourigenicity being isolated from human glioblastoma samples 24, 25, 26. GSC are considered responsible for tumour recurrence and treatment failure 27, 28. Karyotypically normal, untransformed (foetal) neural stem cells (NSC) share many features with patient‐derived GSC 29 and are ideal experimental controls 30. In order to improve our understanding of GSC biology, the key regulatory pathways driving the proliferation of this cancer stem cell population need to be understood. Identification of factors that distinguish NSC from transformed GSC may lead to new therapeutic agents designed to inhibit neoplastic growth with minimal toxicity to the (adult) NSC compartment 31.
Several studies to date suggest that PPARα signalling contributes to the proliferation of glioblastomas 12, 32. However, the role of PPARα expression in human GSC populations is unknown. In this study, we tested the hypothesis that PPARα expression contributes to the malignant phenotype of GSC. We used RNA interference approaches to establish the role of PPARα in maintaining the properties of GSC.
Methods
Cell culture
The human GSC (G144 and G26) and NSC (U5 and U3) cell lines (kind gifts from Dr Steve Pollard, University of Edinburgh) were cultured as monolayers in serum‐free basal media 26, 29. HEK293T (human embryonal kidney) cells (Sigma, St. Louis, MO, USA) used for producing lentiviral particles were cultured in DMEM (10% FBS and 1× non‐essential amino acids). All cell lines were cultured in 5% CO2 at 37 °C.
Protein and RNA extraction
Total protein was extracted from cell lines using Milliplex lysis buffer (Millipore, Burlington, MA, USA) and quantified using a Qubit® Protein kit and fluorometer (Life Technologies, Carlsbad, CA, USA). RNA was extracted using an RNeasy® Plus Mini Kit (Qiagen, Hilden, Germany) and the QIAcube® platform. RNA was quantified using a NanoDrop1000 spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA).
Analysis of GSC and NSC accessioned microarray data
Array data derived by Pollard et al (GSE15209) 26 was accessed from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi. Data analysis was performed using Partek Genomics Suite v.6.16.0812 (Partek, St. Louis, MO, USA) and normalised using GC‐RMA. Differentially expressed genes were analysed using an ANOVA. The false discovery rate was set at an FDR‐corrected p value of <0.05 with a 1.5‐fold expression change cut‐off.
PPARA shRNA oligonucleotide design
Human (NCBI Gene ID: 5465) PPARA (22q13) shRNA sequence primers were designed as previously described 33 using the BLOCK‐iT™ RNAi Designer (https://rnaidesigner.thermofisher.com/rnaiexpress/). Double‐stranded shRNA constructs with an upstream U6 promoter were produced using a pSilencer plasmid as the PCR template. The forward primer sequence was 5′‐CGACTCACTATAGGGCGAATTGGGT‐3′, and the reverse primer sequence contained the shRNA oligonucleotide added to the 5′ tail (see supplementary material, Table S1). Cloning of shRNA expression cassettes was carried out as previously described 33, 34, and the resulting shRNA plasmids were validated using Sanger sequencing (SourceBioscience; Nottingham, UK).
Generation of recombinant lentiviral particles and transduction
U6.shRNA and scramble (SCR) cassettes were cloned into an EGFP‐expressing lentiviral backbone (pRRL.sin.cppt.CMV.EGFP.WPRE). Viral particles were produced and titred as described previously 35. Concentrated lentiviral particles were added to G26 cells for 72 h (multiplicity of infection = 20). Stable PPARα protein knockdown (KD) was established, and a luciferase‐expressing cassette (pCignal Lenti‐TRE‐Reporter, CLS‐PCR‐1, Qiagen) was transduced into the cells using polybrene (Sigma) at 8 μg/ml before puromycin selection.
In vitro cell proliferation studies
Cells were plated at 420 cells/mm2 and cultured for 72 h. The total cell number for each replicate for each line was counted. Cells were re‐plated at 420 cells/mm2, and the experiment was repeated every 72 h for 15 days. The fold increase in cell number over day 0 was calculated using the mean value of each technical replicate for each cell line at each independent time point. Ki67 and caspase‐3 fluorescence immunocytochemistry was carried out as described previously 36 using antibodies listed in supplementary material, Supplementary materials and methods. CellTrace™ Violet proliferation studies were carried out according to the manufacturer's instructions (Thermofisher). The proliferation control and experimental samples were acquired on a Novocyte 3000 Flow Cytometer (Acea Biosciences, San Diego, CA, USA). Data were analysed using ModFit LT v3.3 software (Verity Software House, Topsham, ME, USA). Cell cycle analysis was carried out on the platforms described above using 5 μm Draq5 nuclear stain (BioLegend, San Diego, CA, USA) (15 min incubation) and cells fixed in 4% paraformaldehyde (PFA).
Colony‐forming unit assay
Cells were plated at 16 cells/mm2 and cultured for 12 days. The cells were fixed (4% PFA) and then stained with 1% crystal violet (Sigma). Calculation of colony‐forming unit (CFU) efficiency was determined as described previously 37.
Senescence‐associated β‐galactosidase staining
Cells were plated at 520 cells/mm2 and cultured for 5 days. Cells were stained for 12 h using a Senescence β‐galactosidase Staining Kit (Cell Signalling Technologies, Danvers, MA, USA). Ten high‐power fields (hpf) were examined per well and positive (cytoplasmic and nuclear blue) staining recorded as a percentage of total live cells per hpf.
Intracranial xenografting procedure, bioluminescent imagining and MRI
All animal‐handling procedures and experiments were performed in accordance with the UK Animal Scientific Procedures Act 1986 and covered by UK Home Office licenses (University of Leeds ethics committee project license:PA5C8BDBF).
KD and SCR stably transduced cells were injected into 7‐week‐old female NOD‐SCID (NOD.CB17‐Prkdcscid/NcrCrl) mice (Charles River, Wilmington, MA, USA); 30 000 cells were engrafted per animal (10 animals per cell line). Intracranial injection co‐ordinates were 1 mm rostral to bregma, 1.5 mm lateral (right) and 4 mm deep. Intracranial tumour growth was analysed every 30 days using the Xenogen IVIS Spectrum in vivo imaging system and 60 mg/kg intraperitoneal d‐luciferin (Perkin Elmer, Waltham, MA, USA). MRI data were acquired using a 7 T MRI System (AspectImaging, Watford, UK). NIfTI format images were analysed using MANGO (Mango Software, University of Texas, TX, USA). Animals that had lost ≥20% of body weight or showed persistent neurological signs were terminated by pentobarbitone overdose followed by transcardial 4% PFA perfusion. The brain was removed and fixed in 4% PFA. The experiment ran for 25 weeks.
Immunohistochemistry (IHC) and immunofluorescence (IF)
Murine brain tissue was processed on a Leica Peloris II histological platform (Leica, Wetzlar, Germany) and H&E stained using a Leica Autostainer XL platform (Leica). PPARα, Ki67 and EGFR IHC was carried out using a Leica Bond III automated immunostainer (Leica). IDH1, ATRX and GFAP IHC were carried out using a Ventana BenchMark ULTRA platform (Roche, Basel, Switzerland). Antigen retrieval techniques and antibody concentrations are detailed in supplementary material, Supplementary materials and methods and Table S4. EGFP immunofluorescence was carried out as described previously 38.
Western blotting and RT‐qPCR (reverse transcription‐quantitative PCR)
Western blotting was carried out as described previously 36 (primary antibodies are listed in supplementary material, Table S2). Extracted total RNA was reverse transcribed to cDNA for quantitative real‐time PCR using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA); qPCR was performed using a StepOne Plus Real‐Time PCR system and StepOne software v2.1 (Applied Biosystems) with Taqman® Fast Gene Expression Mastermix (Applied Biosystems), and Assay On Demand (AOD) products as listed in supplementary material, Table S3.
Lactate and glucose assays
Cells were plated at 1000 cells/mm2 and cultured for 72–96 h. Adherent cells were counted, and the culture media was collected, centrifuged at 160 × g and the supernatant kept on ice. Lactate and glucose supernatant concentrations were determined using a Cobas 8000 automated analyser (Roche) (lactate oxidase and hexokinase methods, respectively).
Statistical analysis
The normality of data distributions were tested using the Kolmogorov–Smirnov and D'Agostino and Pearson tests. A Wilcoxon matched pairs test or unpaired t‐test was used as appropriate. A Friedman test with Dunn's multiple comparison test was used for paired non‐parametric analysis of greater than two groups. A two‐way repeated‐measures ANOVA was used to compare in vitro cellular growth rates. All statistical tests were two‐tailed. Differences with p < 0.05 were considered statistically significant. Data are represented as mean ± SEM (geometric mean ± 95% CI for RT‐qPCR data). Statistical tests were performed using GraphPad Prism v5 (GraphPad Inc., San Diego, CA, USA).
Results
PPARα protein and PPARA mRNA levels were greater in GSC
PPARα protein expression was examined in three independent passages of the U3 and U5 NSC lines and G144 and G26 GSC lines. There was a significant increase in PPARα protein level in the G26 cell line compared to both U3 (p = 0.032) and U5 cell lines (p = 0.048) (Figure 1A). Immunofluorescence microscopy showed a mixed nuclear/perinuclear and cytoplasmic expression of PPARα in the GSC (Figure 1B). RT‐qPCR was performed for the U3, U5, G144 and G26 cell lines: there was a significant increase in PPARA mRNA levels in the G26 cell line compared to the U3 cell line (p = 0.039) and the U5 cell line (p = 0.049) when normalised to GAPDH or 18S expression (Figure 1C).
Figure 1.
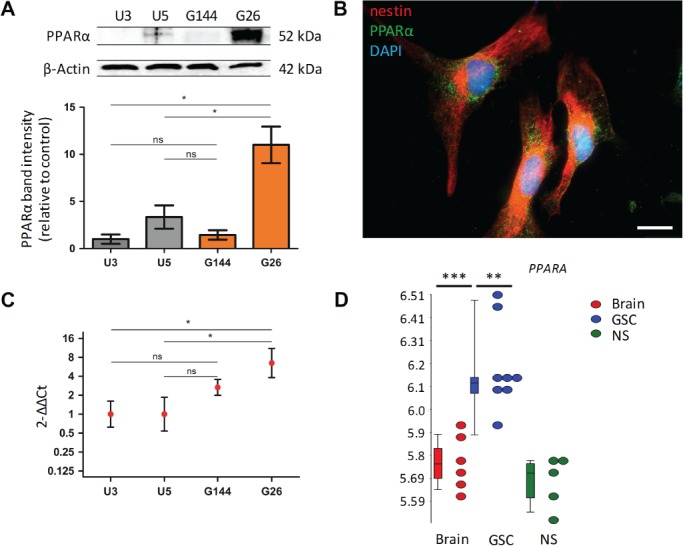
PPARα protein and PPARA gene expression are increased in GSC. (A) PPARα protein expression was examined in two NSC lines and two GSC lines at three independent passages, n = 3. Protein expression values determined using densitometric analysis, with PPARα‐integrated area density values expressed relative to the loading control β‐actin values. Expression values were calculated relative to the grouped U3 control protein homogenates. Results of equivalent statistical significance were obtained when expression values were calculated relative to the grouped U5 control protein homogenates. (B) High‐power immunofluorescence microscopy showing mixed nuclear/perinuclear and cytoplasmic expression of PPARα; ×630: oil immersion. Scale bar = 25 μm. (C) PPARA mRNA expression was examined in NSC control and GSC in vitro models by RT‐qPCR, normalised to the reference genes 18S and GAPDH (not shown). Expression values were calculated relative to the grouped U3 control samples. Results of equivalent statistical significance were obtained when expression values were calculated relative to the grouped U5 control samples. The geometric mean and 95% confidence interval are shown on a logarithmic scale (to base2). n = 3 independent experiments, all samples analysed in triplicate. (D) PPARA expression in GSC (G166, G174, G179, G144, GliNS) versus NSC versus normal adult brain tissue. In the box plots, the upper and lower ‘hinges’ correspond to the 25th and 75th percentiles, respectively. The upper/lower whisker extends to the highest/lowest value that is within 1.5× interquartile range (IQR). Data beyond the end of the whiskers are outliers. Normalised and log‐transformed mRNA gene‐level summaries are shown. The test statistic was a Friedman test with Dunn's multiple comparison test (A and C) or a one‐way ANOVA (D). Error bars show SEM. *p < 0.05, ** p < 0.01; ***p < 0.001; ns, non‐significant; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; 18S, 18 S ribosomal RNA.
PPARA gene expression was increased in whole transcriptome analysis of GSC versus NSC
Whole transcriptome expression profile data (accession number GSE15209) were analysed. Using a 1.5‐fold change cut‐off value (FDR threshold of 0.05), analysis of PPARA expression showed that this transcript was significantly increased in GSC compared to NSC and normal adult brain tissue (p = 0.006, p = 0.001, respectively) (Figure 1D). Increased expression of PPARA was noted to be within the second quintile of all overexpressed transcripts within the GSC versus NSC comparison (p = 0.006, 1.65‐fold change).
PPARα KD inhibited GSC proliferation and clonogenicity in vitro
To investigate the role of PPARα expression in GSC, we generated a stable PPARα KD GSC cell line from the G26 parent line. A control scrambled (SCR) shRNA lentiviral construct was utilised. shRNA‐mediated KD of PPARα was confirmed by western blotting 60 days after lentiviral transduction (see supplementary material, Figure S1). The addition of a luciferase cassette had no effect on shRNA PPARα KD efficiency.
PPARα KD lead to a significant decrease in the PPARα KD cell population expansion compared to the SCR shRNA cell population (p = 0.021) (population doubling time: 2.3 days versus 1.3 days for KD shRNA and SCR shRNA, respectively) (Figure 2A). There was a significant decrease in Ki67 nuclear positivity between SCR shRNA‐ versus PPARα KD shRNA‐transduced cells (30.0% versus 15.1%) (p = 0.003) (Figure 2B).
Figure 2.
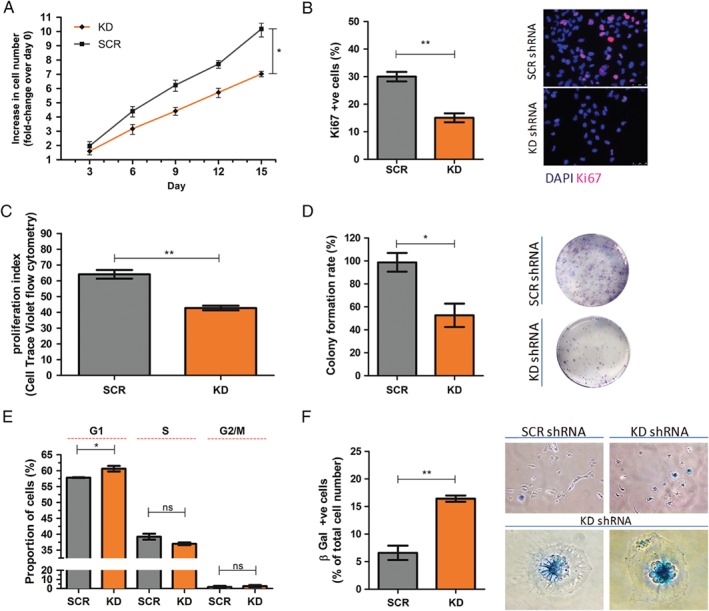
PPARα KD inhibited GSC proliferation and clonogenicity in vitro. (A) There was a significant fold decrease in proliferation in the PPARα KD GSC population compared to the SCR shRNA GSC population (population doubling time: 2.3 days versus 1.3 days, KD shRNA and SCR shRNA, respectively). The test statistic was a two‐way repeated‐measures ANOVA and Bonferroni post hoc test. Data were analysed using nonlinear regression with y = 0 (constrained). Increase (fold‐change) in cell number shown on a logarithmic scale (to base2). (B) There was a significant reduction of the Ki67 index in the PPARα KD GSC population compared to the SCR shRNA GSC population. The proportion of Ki67 nuclear positivity was quantified as the proportion of total nuclei per high‐power field (×200). Ten high‐power fields were examined per slide/technical replicate. Nuclei labelled with DAPI nuclear dye. n = 3, three technical replicates per independent experiment. Representative Ki67 IF images shown. Scale bar = 50 μm. (C) CTV cell proliferation assay; PPARα KD GSC showed a reduction in proliferation index (sum of the cells in all generations divided by the computed number of original parent cells theoretically present at the start of the experiment, where each daughter cell has half the CTV fluorescence intensity of its parental cell). Analysis was carried out using a Novocyte 3000 Flow Cytometer with 405 nm excitation laser and 445/45 nm Band Pass (BP) filter. n = 3, three technical replicates per independent experiment. (D) There was a significant increase in G1 phase cells with PPARα KD. Draq5 analysis was carried out using a Novocyte 3000 Flow Cytometer with 640 nm excitation laser and 780/60 nm BP filter. n = 3, three technical replicates per independent experiment. (E) There was a significant reduction in the number of colonies in the PPARα KD GSC population compared to the SCR shRNA cell population. Representative images of clonogenic assays are shown. (F) There was a significant increase in senescence‐associated β‐galactosidase staining in the PPARα KD GSC population compared to the SCR shRNA cell population. Representative high‐power images of β‐galactosidase staining are shown. n = 3, three technical replicates per independent passage. The test statistic was a Wilcoxon matched pair test, two‐tailed p value (B–F). Error bars show SEM. SCR, scrambled control; *p < 0.05, **p < 0.01.
A CellTrace Violet (CTV) cell proliferation assay was used to monitor cell divisions (generations) in PPARα KD shRNA‐ and SCR shRNA‐transduced cells. In keeping with the population doubling studies described above, PPARα KD shRNA‐transduced cells showed a significant reduction in proliferation index compared to SCR shRNA‐transduced cells (p = 0.002) (Figure 2C). In addition, the proportion of cells in the G1 phase of the cell cycle was shown to be significantly increased in the PPARα KD shRNA cell line compared to the SCR shRNA cell line (p = 0.034) (Figure 2D). We also studied the effect of stable PPARα KD on clonogenicity. The mean number of colonies formed by PPARα KD cells was reduced by 53.5% relative to SCR shRNA cells (p = 0.029) (Figure 2E). There was also a significant increase in β‐galactosidase (pH 6.0) positivity, a known characteristic of senescent cells, between SCR shRNA‐ versus PPARα KD shRNA‐transduced cells (6.8% versus 16.4%, p = 0.008) (Figure 2F). In conjunction with this, PPARα KD shRNA‐transduced cells were found to have aberrant cytonuclear features compared to SCR shRNA controls: the cells were notably larger and flattened with a frequent loss of the spindle morphology. Increased intracytoplasmic vacuolation and multi‐nucleation was also noted with strong perinuclear β‐galactosidase positivity (Figure 2F).
PPARα kD suppressed the tumourigenicity of GSC orthotopic xenografts
SCR and PPARα KD shRNA‐transduced G26 cells were stereotactically implanted in a NOD SCID murine model, and the effect on tumour initiation and progression was monitored. Fourteen days after xenografting, all animals showed detectable bioluminescence (BLI) signal. There was significantly less BLI signal in the PPARα KD group compared to the SCR shRNA control group at each time point during the course of the experiment (Figure 3A). Remaining animals (n = 8) were terminated after 25 weeks (Figure 3B). T2‐weighted MRI was performed 2 h antemortem. The SCR shRNA group showed evidence of right‐sided hemispheric T2‐hyperintense lesions with mass effect (Figure 3C). The PPARα KD experimental group showed no MRI signs of intracranial abnormality (Figure 3C). Twenty‐five weeks after the xenograft procedure, low power histological examination of the brains from the control SCR shRNA xenograft arm (n = 4) demonstrated extensive tumour formation (Figure 3D).
Figure 3.
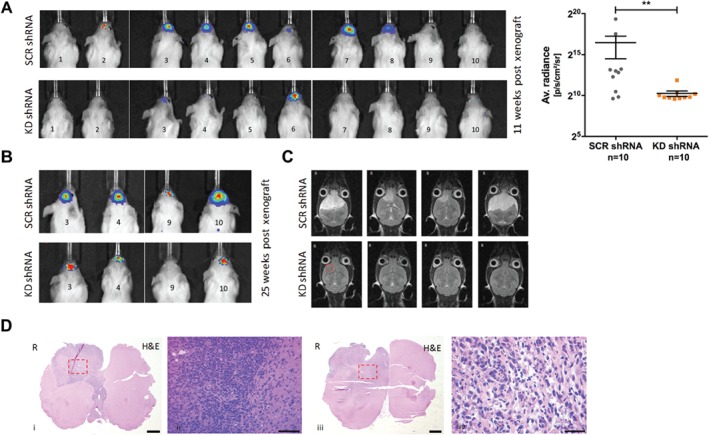
PPARα KD inhibited GSC tumourigenicity in vivo. (A) SCR‐ and KD shRNA‐transduced G26 cells were stereotactically implanted in a NOD‐SCID orthotopic murine model and the effect on tumour initiation and progression monitored. At 11 weeks after the xenografting procedure, all animals showed detectable BLI signal (p/s/cm2/sr). There was a significant decrease in BLI signal in the PPARα KD group compared to the SCR shRNA control group. (B) All remaining animals (n = 8) were terminated after 25 weeks. (C) T2‐weighted MRI was performed 2 h antemortem. The SCR shRNA group showed evidence of extensive right‐sided hemispheric T2‐hyperintense masses (axial). The PPARα KD experimental group showed no radiological evidence of any intracranial abnormality (red box denotes stereotactic injection site). (D) A total of 25 weeks after the xenograft procedure, low and power histological examination of the brains from the control SCR shRNA xenograft arm demonstrated tumour formation. All SCR shRNA xenograft experiments produced tumour masses with histological (H&E) evidence of high cellularity with pleomorphic tumour cells. Representative images shown (coronal sections). Red boxes denote areas shown at greater magnification. Scale bar = 1000 μm (i, iii); scale bar = 100 μm (ii); scale bar = 50 μm (iv). R, right hand side.
All SCR shRNA xenograft experiments produced tumour masses with histological (H&E) evidence of non‐circumscribed cellular tumours consisting of pleomorphic cells (Figure 3D) with frequent atypical mitotic figures. Ki67 IHC showed variable nuclear positivity across the tumour field (focal areas of >50% Ki67 positivity) and diffuse infiltration by Ki67‐positive cells into the adjacent host parenchyma (Figure 4A). PPARα IHC showed extensive cytoplasmic and nuclear positivity (Figure 4A). IHC performed on SCR shRNA xenografts showed the tumour cells to be negative for the expression of the IDH1R132H‐mutated protein product with strong nuclear ATRX expression and GFAP and EGFR immunopositivity (Figure 4B). EGFP expression examined by immunofluorescence recapitulated the malignant infiltration into the host parenchyma described above (Figure 4B). In contrast, the KD shRNA xenograft arm of the experiment showed no histological evidence of tumour formation (Figure 4C). Immunofluorescence microscopy of brains from the KD shRNA xenograft arm of the experiment demonstrated single cells with EGFP immunopositivity (negative for human‐specific Ki67; Figure 4C). These cells were scattered at the lateral aspect of the right anterior commissure, an area just medial to the stereotactic injection site (Figure 4D). No EGFP‐positive cells were observed in any other brain regions.
Figure 4.
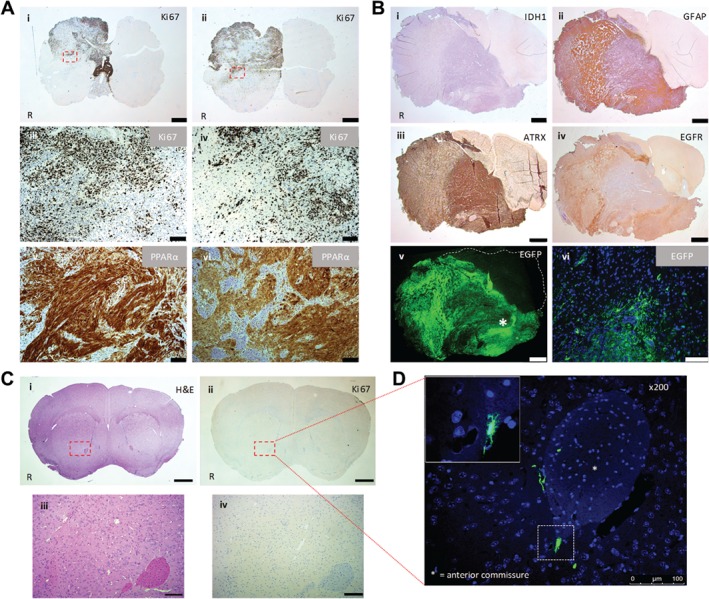
SCR shRNA xenografts: IHC and IF performed on harvested mouse brains 25 weeks after GSC implantation. (A) (i–iv) Representative Ki67 IHC. (v, vi) Representative PPARα IHC. Scale bar = 1000 μm (i, ii); scale bar = 100 μm (iii–vi). (B) SCR shRNA xenograft: IHC and IF (EGFP) performed on harvested mouse brain 25 weeks after GSC implantation. Scale bar = 1000 μm (i–v); scale bar = 50 μm (vi). (C) Representative H&E image of a coronal section and Ki67 IHC performed on a consecutive tissue section. Scale bar = 1000 μm (i, ii); 100 μm (iii, iv). (D) Immunofluorescence microscopy detection of EGFP‐positive cells. The inset in the immunofluorescent image denotes the hatched area ×400. Scale bar = 100 μm. R, right hand side. Red boxes denote areas shown at greater magnification (C and D).
PPARα shRNA KD altered the protein and gene expression of stem cell and mitogenic markers
Transduced G26 cells were examined by western blotting to assess any effects on the protein expression of key signalling mediators that occurred concomitantly with the stable KD of PPARα. The expression of c‐Myc (p = 0.029) and Cyclin D1 (p = 0.035) proteins were significantly reduced (Figure 5A). The stem cell markers nestin and SOX2 showed similarly decreased protein expression (p = 0.037, p = 0.023, respectively) (Figure 5A). The expression of the astrocytic differentiation marker GFAP was increased (p = 0.022) (Figure 5A). The PPARα transcription target EGFR showed a reduced protein expression (Figure 5A). Across multiple independent passages, no PARP cleavage was observed by western blot in the KD shRNA cell lines, establishing that the reduced proliferation rates described were not due to increased apoptosis. Indeed, no increase in active caspase 3 was observed by immunofluorescence in the KD shRNA cell line (Figure 5B).
Figure 5.
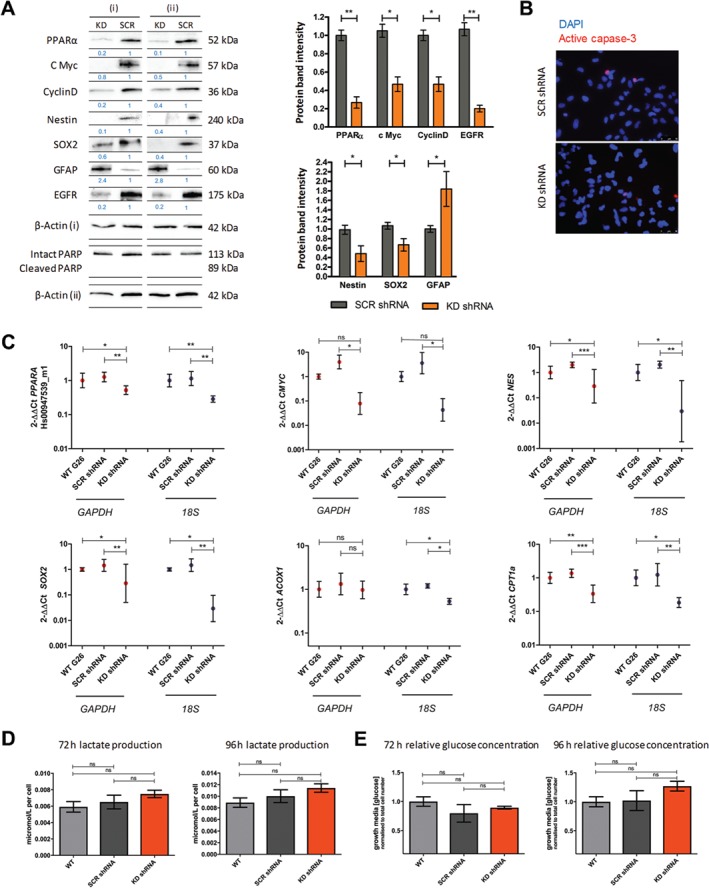
PPARα KD reduced the protein and gene expression of stemness markers in vitro with no effect on glycolytic flux. (A) Protein expression was examined at three independent passages, n = 3. Protein expression values determined using densitometric analysis, with protein‐integrated area density values expressed relative to the loading control β‐actin values. Expression values were calculated relative to the PPARα expression values. Representative western blot shown. (B) There was no significant reduction of the active caspase 3 index in the PPARα KD GSC population compared to the SCR shRNA GSC population. The proportion of active caspase 3 cellular positivity was quantified as a proportion of total nuclei per high‐power field (×200). Ten high‐power fields were examined per slide/technical replicate. Nuclei were labelled with DAPI nuclear dye. n = 3, three technical replicates per independent experiment. Representative active caspase 3 IF images shown. (C) mRNA expression was examined in the PPARα KD GSC population compared to the WT and SCR shRNA GSC population by RT‐qPCR, normalised to the reference genes 18S and GAPDH. Relative gene expression (expressed as a fold‐difference compared to control samples) was calculated using the 2−ΔΔCt method, and expression values were calculated relative to the WT control samples. The geometric mean and 95% confidence interval are shown on a logarithmic scale (to base2). n = 3 independent experiments; all samples analysed in triplicate. (D) Culture growth media lactate and glucose concentration was examined in three independent passages. Lactate/glucose concentrations were normalised to cell number at the time of media harvest. The concentration of analyte in blank control wells was subtracted from each assay output, which was then normalised to the total cell number in each well. The test statistic was a Wilcoxon matched pair test, two‐tailed P value (A) or a Friedman test with Dunn's multiple comparison test (C–E). Error bars show SEM. WT, wild type; SCR, scrambled; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; 18S, 18 S ribosomal RNA; *p < 0.05, **p < 0.01, ***p < 0.001; ns, non‐significant.
Using RT‐qPCR we found a significant reduction in PPARA mRNA levels in the KD shRNA cell lines compared to the SCR shRNA lines when normalised to GAPDH (p = 0.022) and 18S expression (p = 0.001) (Figure 5C). In keeping with the western blotting analysis of protein, there was a significant reduction in the expression of the stem cell markers NES and SOX2 in the KD shRNA cell lines compared to the SCR shRNA lines when normalised to GAPDH (p = 0.001, p = 0.002, respectively) and 18S expression (p = 0.01, p = 0.002, respectively) (Figure 5C). There was also a reduction in cMYC expression in the KD shRNA cell lines when normalised to GAPDH and 18S expression (p = 0.025, p = 0.027, respectively) (Figure 5C).
The PPARα‐regulated fatty acid oxidation enzymes ACOX1 and CPT1a were also examined by RT‐qPCR. A reduction in ACOX1 was seen when normalised to 18S expression (p = 0.027) (Figure 5C). There was a reduction in the expression of CPT1A in the KD shRNA cell lines compared to the SCR shRNA lines when normalised to GAPDH (p = 0.0002) and 18S expression (p = 0.004) (Figure 5C).
PPARα shRNA KD had no significant effect on lactate production or glucose consumption in vitro
Biochemical analysis was performed on media harvested from shRNA‐transduced cells after 72 and 96 h expansion in vitro. There was no difference in lactate production between SCR shRNA cells and KD shRNA cells after 72 or 96 h (p = 0.103; p = 0.092, respectively) (Figure 5D). There was no significant difference in relative glucose concentration in the harvested media between SCR shRNA cells and KD shRNA cells after 72 or 96 h (p = 0.172, p = 0.087, respectively) (Figure 5E).
Discussion
A key area of investigation in the search for more effective treatments for glioblastoma is the molecular manipulation of self‐renewal and proliferation pathways in GSC 39. Direct targeting of GSC may also improve the efficacy of conventional chemo‐ and radiotherapy 40. Transcription factors overexpressed in GSC could provide effective treatment targets for novel therapeutic agents. In this study, GSC were shown to express increased levels of PPARα protein and PPARA transcript when compared to NSC controls. NSC share key functional and genetic similarities to GSC and are considered an ideal experimental control in this setting 30. The analyses of PPARA expression in accessioned microarray data cross‐validated the findings derived from our in vitro models. Indeed, the increased expression of PPARA was suggested in this work to be a significant finding shared across multiple GSC cell lines. The molecular mechanisms underlying this increased expression remain to be elucidated and are an important area of future investigation.
We selected the well‐validated IDH1‐wildtype, non‐CpG island methylated G26 GSC line as a target for our lentiviral transduction work to best recapitulate a primary glioblastoma GSC subpopulation 41. Stable KD of PPARα protein expression resulted in a significantly reduced in vitro growth rate. This was confirmed using flow cytometric generational tracing, which showed a decrease in the number of cell divisions per unit time. PPARα KD additionally reduced the clonogenicity of the GSC line. These results indicate that PPARα is required for, or plays a key role in, the maintenance of GSC proliferative capacity.
Examination of the PPARα KD shRNA‐transduced cells demonstrated a significant increase in senescence‐associated β‐gal staining in vitro, indicating the induction of senescence. Cellular senescence implies a stable and long‐term loss of proliferation capacity with no loss of cellular viability or metabolic activity 42, 43, 44. Long‐term exit from the cell cycle has been suggested as a key marker of cellular senescence 42, and PPARα KD resulted in evidence of cell cycle arrest. Morphological changes consistent with a senescent phenotype were also observed 42. It is noteworthy that this indicates that molecular senescence mechanisms may remain latently functional even in aggressive GSC populations.
A defining functional characteristic of GSC is their ability to initiate and propagate histological phenocopies of human glioblastoma when xenografted intra‐cranially in immunocompromised animals 45, 46. We used orthotopic xenotransplantation to investigate the functional requirement of PPARα to maintain the tumourigenic potential of human GSC. The xenograft brains in the control SCR shRNA experimental arm showed the key histological features of a human glioblastoma. Immunophenotyping demonstrated IDH1 mutation‐negative tumour cells with strong nuclear ATRX expression 47, 48 and EGFR overexpression 49, 50, confirming an expression profile consistent with IDH‐wildtype primary glioblastoma 51. Conversely, radiological and histological examination showed that PPARα KD xenografts did not form significant tumour masses in vivo, indicating that GSC lacking PPARα expression have markedly reduced tumour‐initiating capacity. Nevertheless, the immunofluorescence examination of PPARα KD GSC‐engrafted brains demonstrated EGFP‐positive cells at the injection sites, confirming successful cell engraftment. We concluded that these EGFP‐positive cells have a significantly reduced proliferation rate but remain viable over an extended time course in vivo, in keeping with the hallmarks of senescent cells. Such scattered EGFP‐positive cells may provide sufficient signal for BLI detection in the absence of an observed tumour mass, as has been previously reported 52.
It has been shown that both PPARα pharmacological antagonism and siRNA‐mediated PPARα KD reduce the expression of c‐Myc, cyclin D1 and CDK4 in renal cell carcinoma (RCC) in vitro models 53. The PPARα agonist Wy‐14643 has also been shown to decrease the expression of the let‐7C miRNA in wild‐type mice, with no similar repression seen in PPARα‐null animals 54. let‐7C miRNA targets and represses c‐Myc expression 54. c‐Myc plays a role in the initiation and proliferation of glial brain tumours, and there is evidence of deregulation of the c‐Myc pathway in glioblastoma 55, 56, 57. The full transcriptional functions of c‐Myc remain to be elucidated 58, but the induction of cyclin D1 59 and the repression of p21WAF1/CIP1 expression have been previously reported 60, 61. We investigated a putative PPARα/c‐Myc interaction in our PPARα KD in vitro model: c‐Myc protein expression was found to be decreased in shRNA‐mediated PPARα KD GSC. This was accompanied by a significant decrease in cyclin D1 expression and a concomitant G1 phase cell cycle arrest.
PPARα has also been reported to play a role in EGFR phosphorylation and activation 62, 63. PPARα‐LXRα/RXRα heterodimers positively regulate EGFR promotor activity, and a putative PPARα DNA response element has been described upstream of the EGFR promoter 63. We have previously reported that EGFR mRNA expression significantly correlates with high PPARA mRNA expression in the TCGA primary glioblastoma dataset 12. In keeping with these findings in surgical tumour specimens, PPARα KD in GSC was found to significantly reduce the protein expression of EGFR in vitro. EGFR activation and subsequent receptor dimerisation promote cellular proliferation via activation of the MAPK and PI3K‐Akt pathways 64, and this reduction of EGFR expression may be an additional factor in the decreased expression of c‐Myc, which is an immediate early‐response gene downstream of many ligand–membrane receptor complexes 58.
PPARα KD also resulted in reduced expression of nestin and SOX2 proteins with an increase in GFAP protein expression. GFAP is a commonly used astrocyte maturation marker 65, 66, 67. GSC populations are known to upregulate GFAP along with other astrocyte differentiation markers (AQP4 and ALDH1A1) following the induction of a differentiated and cell cycle‐arrested state 26, 68. The altered expression of this differentiation marker was therefore in keeping with a reduction in GSC proliferative capacity and a senescent (post‐mitotic) state. Whether this PPARα KD‐driven cellular state is reversible or represents terminal differentiation warrants further investigation (Figure 6) 69.
Figure 6.
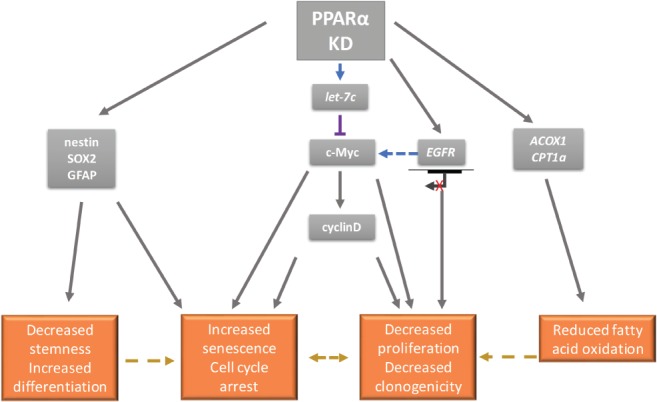
Schematic model of the effects of PPARα KD in a GSC in vitro model. PPARα KD exerts an anti‐proliferative effect on GSC via an altered stemness phenotype, increased rates of cellular senescence and putative changes in the metabolic flux of the GSC population. It is hypothesised that this may be attributed to downstream changes in key molecular mediators of malignant transformation such as c‐Myc, EGFR and cyclin D1.
PPARα drives the transcription of key fatty acid oxidation (FAO) enzymes, including carnitine palmitoyltransferase 1 alpha (CPT1α; CPT1A) and acyl‐coenzyme A oxidase 1 (ACOX1) 8. Both murine sub‐ventricular zone NSC and human GSC have been reported as being dependent on FAO 70, 71. In this study, PPARα KD reduced the gene expression of CPT1A and ACOX1, with a concomitant reduction in proliferation and clonogenic potential. PPARα antagonism in RCC models decreases FAO and enhances glycolysis 53. We assayed in vitro lactate and glucose concentrations and showed that a compensatory increase in glycolysis (pyruvate to lactate conversion; the Warburg effect 72) did not occur in GSC. This may be due to the reduction in c‐Myc expression, which has been associated with decreased glycolytic rates 73, 74, 75. In addition, we propose that FAO‐dependent GSC have only a small requirement for glucose oxidation 70, 76, and PPARα KD, through effects on FAO enzyme expression, may deplete GSC populations of their prime FAO bioenergetic source with no compensatory glycolytic flux, resulting in the anti‐proliferative phenotype described. Interestingly, the unique metabolic requirements of GSC compared to the aberrantly differentiated cells of the tumour mass 40 may explain the paradox of increased PPARA expression in mediating prolonged clinical survival 12 versus KD of PPARα in GSC inhibiting tumour growth. We hypothesise that high PPARA exerts an inhibitory effect on glioblastoma glycolysis 77, an effect not seen in the GSC population. The differing roles of molecular mediators of malignancy in disparate GSC and tumour mass cell populations is a key area for future investigation and has crucial implications when designing adjuvant treatment strategies to inhibit tumour recurrence.
In summary, our study establishes the expression of PPARα in GSC. The stable KD of PPARα in GSC completely abolished intracranial tumour formation. This was associated with the induction of cellular senescence in vitro, driven by the reduced expression of mitogenic and stemness factors. These data provide evidence of the role of PPARα in GSC as an important molecular regulator, linking proliferation and self‐renewal with a critical role in maintaining the malignant phenotype. Targeting PPARα in GSC populations may therefore have translational potential as a novel adjuvant therapeutic approach to abrogate the contribution of GSC to the poor overall clinical survival for glioblastoma patients.
Author contributions statement
HRH, HS, CLK‐C, KCK, JU, AW, and KMK designed the study. HRH, HS, CLK‐C, TB, KMH, JR, KCK, LSB, AH, OC‐L, WGS, HB, RAK, FM, and TB performed the data collection, analysis and interpretation. HRH, HS, CLK‐C, KCK, and AW produced the manuscript. All authors approved final manuscript.
SUPPLEMENTARY MATERIAL ONLINE.
Supplementary materials and methods
Figure S1. Western blot analysis of shRNA KD on the expression of PPARα protein
Figure S2. Generational tracing in KD‐ and SCR shRNA‐transduced cell lines
Table S1. PPARA shRNA primer sequences
Table S2. Details of primary antibodies used for western blotting
Table S3. Primer sets used for RT‐qPCR assays
Table S4. Details of primary antibodies and antigen retrieval used for immunohistochemistry
Supporting information
Supplementary materials and methods
Figure S1. Western blot analysis of shRNA KD on the expression of PPARα protein
Figure S2. Generational tracing in KD‐ and SCR shRNA‐transduced cell lines
Table S1. PPARA shRNA primer sequences
Table S2. Details of primary antibodies used for western blotting
Table S3. Primer sets used for RT‐qPCR assays
Table S4. Details of primary antibodies and antigen retrieval used for immunohistochemistry
Acknowledgements
HRH is supported by the Pathological Society and Jean Shanks Foundation Pathological Research Training Fellowship. HLS and JBU are supported by a Biotechnology and Biological Sciences Research Council (BBSRC) grant (BB/M017532). CLK‐C is funded by the Functional Neurosurgery Fund, North Bristol NHS Trust, Bristol. WG Singleton is jointly funded by the Medical Research Council (MRC) and the Brain Tumour Charity. The authors are grateful to Dr Steve Pollard, University of Edinburgh, for providing the U3, U5, G144 and G26 cell lines and Ms Sarah Chir, North Bristol NHS Trust, for her assistance with tissue sectioning and immunohistochemistry.
No conflicts of interest were declared.
References
- 1. Ostrom QT, Gittleman H, Xu J, et al CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2009–2013. Neuro Oncol 2016; 18(Suppl 5): v1–v75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rouse C, Gittleman H, Ostrom QT, et al Years of potential life lost for brain and CNS tumors relative to other cancers in adults in the United States, 2010. Neuro Oncol 2016; 18: 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haynes HR, Camelo‐Piragua S, Kurian KM. Prognostic and predictive biomarkers in adult and pediatric gliomas: toward personalized treatment. Front Oncol 2014; 4: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chinot OL, Wick W, Mason W, et al Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. N Engl J Med 2014; 370: 709–722. [DOI] [PubMed] [Google Scholar]
- 5. Gilbert MR, Dignam JJ, Armstrong TS, et al A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 2014; 370: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gramatzki D, Dehler S, Rushing EJ, et al Glioblastoma in the Canton of Zurich, Switzerland revisited: 2005 to 2009. Cancer 2016; 122: 2206–2215. [DOI] [PubMed] [Google Scholar]
- 7. Stupp R, Mason WP, van den Bent MJ, et al Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–996. [DOI] [PubMed] [Google Scholar]
- 8. Grabacka M, Pierzchalska M, Reiss K. Peroxisome proliferator activated receptor α ligands as anticancer drugs targeting mitochondrial metabolism. Curr Pharm Biotechnol 2013; 14: 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frederiksen KS, Wulff EM, Sauerberg P, et al Prediction of PPAR‐alpha ligand‐mediated physiological changes using gene expression profiles. J Lipid Res 2004; 45: 592–601. [DOI] [PubMed] [Google Scholar]
- 10. Ribet C, Montastier E, Valle C, et al Peroxisome proliferator‐activated receptor‐alpha control of lipid and glucose metabolism in human white adipocytes. Endocrinology 2010; 151: 123–133. [DOI] [PubMed] [Google Scholar]
- 11. Muoio DM, Way JM, Tanner CJ, et al Peroxisome proliferator‐activated receptor‐alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002; 51: 901–909. [DOI] [PubMed] [Google Scholar]
- 12. Haynes HR, White P, Hares KM, et al The transcription factor PPARalpha is overexpressed and is associated with a favourable prognosis in IDH‐wildtype primary glioblastoma. Histopathology 2016; 70: 1030–1043. [DOI] [PubMed] [Google Scholar]
- 13. Shi Y, Tao T, Liu N, et al PPARα, a predictor of patient survival in glioma, inhibits cell growth through the E2F1/miR‐19a feedback loop. Oncotarget 2016; 7: 84623–84633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang F, Jaber LA, Berlie HD, et al Evolution of peroxisome proliferator‐activated receptor agonists. Ann Pharmacother 2007; 41: 973–983. [DOI] [PubMed] [Google Scholar]
- 15. Drukala J, Urbanska K, Wilk A, et al ROS accumulation and IGF‐IR inhibition contribute to fenofibrate/PPARalpha‐mediated inhibition of glioma cell motility in vitro. Mol Cancer 2010; 9: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Panigrahy D, Kaipainen A, Huang S, et al PPAR agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci U S A 2008; 105: 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han D‐F, Zhang J‐X, Wei W‐J, et al Fenofibrate induces G0/G 1 phase arrest by modulating the PPARα/FoxO1/p27(kip) pathway in human glioblastoma cells. Tumour Biol 2015; 36: 3823–3829. [DOI] [PubMed] [Google Scholar]
- 18. Wilk A, Urbanska K, Grabacka M, et al Fenofibrate‐induced nuclear translocation of FoxO3A triggers Bim‐mediated apoptosis in glioblastoma cells in vitro. Cell Cycle 2012; 11: 2660–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grabacka MM, Wilk A, Antonczyk A, et al Fenofibrate induces ketone body production in melanoma and glioblastoma cells. Front Endocrinol (Lausanne) 2016; 7: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilk A, Wyczechowska D, Zapata A, et al Molecular mechanisms of fenofibrate‐induced metabolic catastrophe and glioblastoma cell death. Mol Cell Biol 2015; 35: 182–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han D, Wei W, Chen X, et al NF‐κB/RelA‐PKM2 mediates inhibition of glycolysis by fenofibrate in glioblastoma cells. Oncotarget 2015; 6: 26119–26128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ignatova TN, Kukekov VG, Laywell ED, et al Human cortical glial tumors contain neural stem‐like cells expressing astroglial and neuronal markers in vitro. Glia 2002; 39: 193–206. [DOI] [PubMed] [Google Scholar]
- 23. Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem‐like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A 2004; 101: 781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galli R, Binda E, Orfanelli U, et al Isolation and characterization of tumorigenic, stem‐like neural precursors from human glioblastoma. Cancer Res 2004; 64: 7011–7021. [DOI] [PubMed] [Google Scholar]
- 25. Singh SK, Hawkins C, Clarke ID, et al Identification of human brain tumour initiating cells. Nature 2004; 432: 396–401. [DOI] [PubMed] [Google Scholar]
- 26. Pollard SM, Yoshikawa K, Clarke ID, et al Glioma stem cell lines expanded in adherent culture have tumor‐specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009; 4: 568–580. [DOI] [PubMed] [Google Scholar]
- 27. Chen J, Li Y, Yu T‐S, et al A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012; 488: 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piccirillo SGM, Spiteri I, Sottoriva A, et al Contributions to drug resistance in glioblastoma derived from malignant cells in the sub‐ependymal zone. Cancer Res 2014; 75: 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okawa S, Gagrica S, Blin C, et al Proteome and secretome characterization of glioblastoma‐derived neural stem cells. Stem Cells 2017; 35: 967–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stangeland B, Mughal AA, Grieg Z, et al Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget 2015; 6: 26192–26215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Auffinger B, Spencer D, Pytel P, et al The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev Neurother 2015; 15: 741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Binello E, Mormone E, Emdad L, et al Characterization of fenofibrate‐mediated anti‐proliferative pro‐apoptotic effects on high‐grade gliomas and anti‐invasive effects on glioma stem cells. J Neurooncol 2014; 117: 225–234. [DOI] [PubMed] [Google Scholar]
- 33. Harper SQ, Davidson BL. Plasmid‐based RNA interference: construction of small‐hairpin rna expression vectors. Methods Mol Biol 2005; 309: 219–235. [DOI] [PubMed] [Google Scholar]
- 34. Hartfield EM, Rinaldi F, Glover CP, et al Connexin 36 expression regulates neuronal differentiation from neural progenitor cells. PLoS One 2011; 6: e14746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scott H, Howarth J, Lee YB, et al MiR‐3120 is a mirror microRNA that targets heat shock cognate protein 70 and auxilin messenger RNAs and regulates clathrin vesicle uncoating. J Biol Chem 2012; 287: 14726–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Redondo J, Hares K, Wilkins A, et al Reductions in kinesin expression are associated with nitric oxide‐induced axonal damage. J Neurosci Res 2015; 93: 882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Redondo J, Sarkar P, Kemp K, et al Reduced cellularity of bone marrow in multiple sclerosis with decreased MSC expansion potential and premature ageing in vitro. Mult Scler 2018; 24: 919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kemp KC, Hares K, Redondo J, et al Bone marrow transplantation stimulates neural repair in Friedreich's ataxia mice. Ann Neurol 2018; 83: 779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seymour T, Nowak A, Kakulas F. Targeting aggressive cancer stem cells in glioblastoma. Front Oncol 2015; 5: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Spencer DA, Auffinger BM, Murphy JP, et al Hitting a moving target: glioma stem cells demand new approaches in glioblastoma therapy. Curr Cancer Drug Targets 2017; 17: 236–254. [DOI] [PubMed] [Google Scholar]
- 41. Stricker SH, Feber A, Engström PG, et al Widespread resetting of DNA methylation in glioblastoma‐initiating cells suppresses malignant cellular behavior in a lineage‐dependent manner. Genes Dev 2013; 27: 654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kuilman T, Michaloglou C, Mooi WJ, et al The essence of senescence. Genes Dev 2010; 24: 2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dimri GP, Lee X, Basile G, et al A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92: 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Debacq‐Chainiaux F, Erusalimsky JD, Campisi J, et al Protocols to detect senescence‐associated beta‐galactosidase (SA‐βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009; 4: 1798–1806. [DOI] [PubMed] [Google Scholar]
- 45. Stylli SS, Luwor RB, Ware TMB, et al Mouse models of glioma. J Clin Neurosci 2015; 22: 619–626. [DOI] [PubMed] [Google Scholar]
- 46. Lee J, Kotliarova S, Kotliarov Y, et al Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum‐cultured cell lines. Cancer Cell 2006; 9: 391–403. [DOI] [PubMed] [Google Scholar]
- 47. Reuss DE, Sahm F, Schrimpf D, et al ATRX and IDH1‐R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol 2015; 129: 133–146. [DOI] [PubMed] [Google Scholar]
- 48. Jiao Y, Killela PJ, Reitman ZJ, et al Frequent ATRX, CIC, and FUBP1 mutations refine the classification of malignant gliomas. Oncotarget 2012; 3: 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Verhaak RGW, Hoadley KA, Purdom E, et al Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010; 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yan H, Parsons DW, Jin G, et al IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360: 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Louis DN, Perry A, Reifenberger G, et al The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016; 131: 803–820. [DOI] [PubMed] [Google Scholar]
- 52. Kaur H, Ali SZ, Huey L, et al The transcriptional modulator HMGA2 promotes stemness and tumorigenicity in glioblastoma. Cancer Lett 2016; 377: 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abu Aboud O, Wettersten HI, Weiss RH. Inhibition of PPARα induces cell cycle arrest and apoptosis, and synergizes with glycolysis inhibition in kidney cancer cells. PLoS One 2013; 8: e71115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shah YM, Morimura K, Yang Q, et al Peroxisome proliferator‐activated receptor alpha regulates a microrna‐mediated signaling cascade responsible for hepatocellular proliferation. Mol Cell Biol 2007; 27: 4238–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jensen NA, Pedersen KM, Lihme F, et al Astroglial c‐Myc overexpression predisposes mice to primary malignant gliomas. J Biol Chem 2003; 278: 8300–8308. [DOI] [PubMed] [Google Scholar]
- 56. Wang J, Wang H, Li Z, et al c‐Myc is required for maintenance of glioma cancer stem cells. PLoS One 2008; 3: e3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Annibali D, Whitfield JR, Favuzzi E, et al Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat Commun 2014; 5: 4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dang CV. MYC on the path to cancer. Cell 2012; 149: 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Daksis JI, Lu RY, Facchini LM, et al Myc induces cyclin D1 expression in the absence of de novo protein synthesis and links mitogen‐stimulated signal transduction to the cell cycle. Oncogene 1994; 9: 3635–3645. [PubMed] [Google Scholar]
- 60. Gartel AL, Ye X, Goufman E, et al Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci U S A 2001; 98: 4510–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mitchell KO, El‐Deiry WS. Overexpression of c‐Myc inhibits p21WAF1/CIP1 expression and induces S‐phase entry in 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA)‐sensitive human cancer cells. Cell Growth Differ 1999; 10: 223–230. [PubMed] [Google Scholar]
- 62. Gardner OS, Dewar BJ, Earp HS, et al Dependence of peroxisome proliferator‐activated receptor ligand‐induced mitogen‐activated protein kinase signaling on epidermal growth factor receptor transactivation. J Biol Chem 2003; 278: 46261–46269. [DOI] [PubMed] [Google Scholar]
- 63. Mahankali M, Farkaly T, Bedi S, et al Phosphatidic Acid (PA) can displace PPARα/LXRα binding to the EGFR promoter causing its transrepression in luminal cancer cells. Sci Rep 2015; 5: 15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Patel R, Leung HY. Targeting the EGFR‐family for therapy: biological challenges and clinical perspective. Curr Pharm Des 2012; 18: 2672–2679. [DOI] [PubMed] [Google Scholar]
- 65. Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol 2011; 93: 421–443. [DOI] [PubMed] [Google Scholar]
- 66. Sun Y, Pollard S, Conti L, et al Long‐term tripotent differentiation capacity of human neural stem (NS) cells in adherent culture. Mol Cell Neurosci 2008; 38: 245–258. [DOI] [PubMed] [Google Scholar]
- 67. Imura T, Kornblum HI, Sofroniew MV. The predominant neural stem cell isolated from postnatal and adult forebrain but not early embryonic forebrain expresses GFAP. J Neurosci 2003; 23: 2824–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Carén H, Stricker SH, Bulstrode H, et al Glioblastoma stem cells respond to differentiation cues but fail to undergo commitment and terminal cell‐cycle arrest. Stem Cell Rep 2015; 5: 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carén H, Beck S, Pollard SM. Differentiation therapy for glioblastoma – too many obstacles? Mol Cell Oncol 2016; 3: e1124174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lin H, Patel S, Affleck VS, et al Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol 2017; 19: 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stoll EA, Makin R, Sweet IR, et al Neural stem cells in the adult subventricular zone oxidize fatty acids to produce energy and support neurogenic activity. Stem Cells 2015; 33: 2306–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 2016; 41: 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ortega ÁD, Sánchez‐Aragó M, Giner‐Sánchez D, et al Glucose avidity of carcinomas. Cancer Lett 2009; 276: 125–135. [DOI] [PubMed] [Google Scholar]
- 74. Shim H, Dolde C, Lewis BC, et al c‐Myc transactivation of LDH‐A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A 1997; 94: 6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kim J‐W, Zeller KI, Wang Y, et al Evaluation of Myc E‐Box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol 2004; 24: 5923–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vlashi E, Lagadec C, Vergnes L, et al Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A 2011; 108: 16062–16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen W, Cloughesy T, Kamdar N, et al Imaging proliferation in brain tumors with 18F‐FLT PET: comparison with 18F‐FDG. J Nucl Med 2005; 46: 945–952. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Western blot analysis of shRNA KD on the expression of PPARα protein
Figure S2. Generational tracing in KD‐ and SCR shRNA‐transduced cell lines
Table S1. PPARA shRNA primer sequences
Table S2. Details of primary antibodies used for western blotting
Table S3. Primer sets used for RT‐qPCR assays
Table S4. Details of primary antibodies and antigen retrieval used for immunohistochemistry