

Abstract
Leukemic relapse is frequently accompanied by progressively aggressive clinical course. To understand the molecular mechanism of leukemic relapse, MLL/AF9-transformed mouse leukemia cells were serially transplanted in C57BL/6 mice (N = 96) by mimicking repeated recurrences, where mutations were monitored by exome sequencing (N = 42). The onset of leukemia was progressively promoted with advanced transplants, during which increasing numbers of somatic mutations were acquired (P < 0.005). Among these, mutations in Ptpn11 (p.G60R) and Braf (p.V637E) corresponded to those identified in human MLL-AML, while recurrent mutations affecting Msn (p.R295C) were observed only in mouse but not in human MLL-AML. Another mutated gene of interest was Gnb2 which was reported to be recurrently mutated in various hematological neoplasms. Gnb2 mutations (p.G77R) were significantly increased in clone size (P = 0.007) and associated with earlier leukemia onset (P = 0.011). GNB2 transcripts were significantly upregulated in human MLL-AML compared to MLL-negative AML (P < 0.05), which was supported by significantly increased Gnb2 transcript induced by MLL/AF9 overexpression (P < 0.001). In in vivo model, both mutation and overexpression of GNB2 caused leukemogenesis, and downregulation of GNB2 expression reduced proliferative potential and survival benefit, suggesting a driver role of GNB2. In conclusion, alterations of driver genes over time may play an important role in the progression of MLL-AML.
Subject terms: Cancer genetics, Acute myeloid leukaemia
Introduction
Despite advanced therapeutics, many leukemia patients become refractory to additional therapy, accounting for a major cause of leukemic deaths. Among major human acute myeloid leukemias (AMLs), MLL-rearranged AML (MLL-AML) is characteristic of poor prognosis due to refractoriness to chemotherapy and shorter period to relapse [1–3]. Multiple detailed and comprehensive studies in mouse models and human samples of MLL-AML have revealed an aberrant self-renewal of hematopoietic progenitors by constitutively activating self-renewal-related genes [4–7]. For maintenance of self-renewal potential in leukemia stem cells, chromatin modifications, including histone H3 lysine 79 (H3K79) and H3K4 methylation, play a critical role in MLL-AML [8–11]. MLL fusion proteins also activate transcription initiation by loading the TATA-binding protein (TBP) [12, 13]. According to these previous studies, epigenetic mechanisms downstream of MLL-fusion proteins are clearly involved in leukemogenesis in MLL-AML.
Recent advancements of sequencing technology and development of analytical tools allowed us to identify a whole spectrum of genetic events in various hematological neoplasms, as well as solid cancers. Multiple groups reported somatic mutations in human MLL-AML [14–17]. Among them, mutations involved in RAS pathway and receptor tyrosine kinase (RTK) genes were most frequently detected, suggesting that such signal transduction cascades might be activated in this leukemia. Even though less prevalent, mutations in the genes involved in DNA methylation and chromatin modification were also reported, indicating possible epigenetic synergy between MLL fusion genes and somatic mutations. However, it remains inconclusive whether these genetic events are associated with progression of the disease and short time to relapse. To clarify the molecular pathogenesis of progression in MLL-AML, we analyzed accumulation of genetic abnormalities in a mouse model of leukemia with human MLL/AF9 fusion gene. Then, we validated the significance of driver mutations using both in-house and publicly available human mutation and gene-expression datasets in human samples. In addition, the further analysis of clonal architecture and prognostic significance of the driver mutations allowed us to identify a novel candidate gene, Gnb2. Finally we confirmed its functional relevance by in vitro and in vivo experiments of mouse MLL/AF9 leukemia, a cytokine-dependent Ba/F3, and human hematopoietic cell lines.
Materials and Methods
Mouse model of MLL/AF9 leukemia
We exploited a mouse model of recurrent leukemia generated by mouse progenitors transduced with human MLL/AF9 as previously reported [7, 18]. Briefly, we isolated 1 × 105 granulocyte macrophage progenitor (GMP) cells from C57BL/6 mice and transduced an MLL/AF9 construct in them by a retrovirus system using a pGCDNsam-IRES-GFP vector. The GMP cells with MLL/AF9 were transplanted into nine irradiated syngeneic mice (first generation), from which bone marrow mononuclear cells (BMMNCs) including GFP-positive mouse leukemic cells were collected. Then, 1 × 106 BMMNCs per mouse were serially transplanted into 48 (second generation), 15 (third), and 24 (fourth) mice (Fig. 1a and Supplementary Figure 1). To assess disease status, we tested peripheral blood on day 14 for complete blood counts, white blood cell (WBC) phenotype (T-cell (CD4 and CD8α), B-cell (CD45R (B220)), and myeloid cell (CD11b and/or Gr-1)), and GFP positivity by flow cytometry (BD LSRFortessa X-20). In total, 96 mice were evaluated for survival periods and 57 for complete blood counts. All in vivo experiments in animals were approved by the Kyoto University Animal Care and Use Committee. To further confirm a pathogenic effect of MLL/AF9 in mouse leukemic cells, we transduced this fusion gene in Ba/F3 by the same retrovirus system as above.
Fig. 1.

Serial transplantation of mouse MLL/AF9-acute myeloid leukemia (AML) (N = 96). a Serial transplantation of MLL/AF9-transduced granulocyte-monocyte progenitor (GMP) cells in C57BL/6 mice. b Total white blood cell (WBC) counts and c GFP-positive WBC counts on day 14. Numbers of mutations identified in strain A1 (d) and B1 (e)
Whole exome sequencing and confirmatory deep sequencing
We analyzed BMMNCs obtained from the MLL/AF9 mouse model by whole exome sequencing (WES) (SureSelect XT Mouse All Exon V2). In total, we sequenced 42 mice in 8 primary AMLs (strains) from 5 donors as shown in Supplementary Figure 1. In three out of five donor mice, two strains were analyzed per mouse to compare mutation profiles between different strains from the same donors. We used DNA of mouse tails as germline control. Sequencing reads were mapped on GRCm38/mm10 by Burrows-Wheeler Aligner (BWA) [19] and candidate variants were called by Genomon pipeline [20]. Source BAM files of WES analysis were deposited under accession number PRJEB25881 in the European Nucleotide Archive. Then, we validated somatic variants by deep sequencing or Sanger sequencing of PCR amplicons as previously described [21]. To confirm relevance of somatic mutations, we translated the mutations in the mouse reference genome (GRCm38/mm10) into the corresponding protein substitutions in the human genome (GRCh38/hg38), in which driver mutations were already reported in human hematological malignancies. We also analyzed human MLL-AML samples (N = 53) by targeted sequencing as previously described [21], integrated the results with those from previously published datasets [14, 15, 22], and then compared mutations in human MLL-AML (N = 168) with those in mouse MLL-leukemia cells. Informed consent for sample collection was obtained according to a protocol approved by each Institutional Review Board in accordance with the Declaration of Helsinki.
Gene-expression analysis
mRNA transcription levels of GNB2 (NM_005273; 340 amino acids (AAs)), Gnb1, (NM_001160017; 340 AAs), and Gnb2 (NM_010312; 340 AAs) were measured by SYBR green real-time RT-PCR using a primer set indicated in Supplementary Table 1 (Roche Molecular Systems, Inc.). Expression levels of 18S rRNA were used as internal control. RNA sequencing and expression array results of GNB1 and GNB2 in human AMLs and controls were downloaded from publicly available database (BioGPS; http://biogps.org) and previously published datasets [14].
Transduction of wild type and mutant GNB2
Ba/F3 was stably transduced with Flag-wild-type and mutated GNB2 (p.G77R) (pMIG vector) as described previously [23]. Protein levels were evaluated by immunoblot. Cells were lysed, subjected to SDS-PAGE, and transferred to PVDF membrane (Millipore). Each blot was incubated with anti-Flag antibody (#1E6, Wako Pure Chemicals) or anti-Actin antibody (#I-19, Santa Cruz Biotechnology), and its signal was visualized with Immobilon Western Chemiluminescent HRP Substrate (Millipore). Phosphorylation profile in the downstream signaling pathway of GNB2 was assessed by immunoblot with anti-total AKT (#9272) and anti-pAKT (#9271) antibodies purchased from Cell Signaling. To investigate an effect of GNB2 on leukemogenesis, Ba/F3 cells transduced with wild-type GNB2, GNB2 mutant, and mock as above, were transplanted in immunodeficient mice. To assess GNB2-associated leukemic involvement, 2 × 106 cells per mouse were intravenously transplanted into NOD/SCID/γc null (NOG) mice (N = 21 in total) after 2.5 Gy irradiation. GFP positivity was measured in bone marrow cells. To further study GNB2-associated tumorigenesis, 1 × 107 cells per experiment were subcutaneously injected into nude mice (N = 30 in total) and the tumor size was measured every seven days.
Suppression of GNB2 by shRNA and depletion of Gnb2 by CRISPR-Cas9
MLL-AML cell lines (NOMO1, MOLM13, and THP1) were infected with pLKO.1 lentivirus containing shRNAs targeting GNB2 or a negative control scramble shRNA. shRNA sequences were designed according to the algorithm previously reported [24]. Targeted sequences are shown in Supplementary Table 2. Knockdown effects were measured by real-time RT-PCR as described above. To validate oncogenic potential of GNB2 and GNB1, genome-wide CRISPR-Cas9 screens using Cas9-expressing mouse MLL/AF9-AML cells were performed as previously described [25].
Statistics
Statistical analysis of means between two populations was performed using two-tailed, unpaired and/or paired Student’s t-test or nonparametric Mann–Whitney U test/Wilcoxon signed-rank test. For multiple testing, the false discovery rate (q-value) was calculated using the Benjamini–Hochberg procedure. Survival curves were assessed by Log-rank test. Correlation between the rank values of two variables was tested by Spearman rank correlation coefficient. Trends of categorical variables were assessed by the Jonckheere–Terpstra test. Statistical analyses and generation of the fishplot (https://github.com/chrisamiller/fishplot/blob/master/tests/test.R) were performed with R (https://www.r-project.org) and/or JMP9 software (SAS). A P-value and q-value of less than 0.05 was considered significant. Error bars in all graphs represent the standard error of the mean.
Results
Leukemic transformation and onset in serial transplantation
To confirm the disease progression of MLL/AF9 mouse leukemia, we evaluated 57 serially transplanted mice for peripheral blood counts and WBC phenotypes on day 14, BMMNC phenotypes, and time to leukemic evolution. By trend test (Jonckheere–Terpstra test) for the later transplant, numbers of WBC and GFP-positive WBC increased significantly (P = 8.9 × 10−6 and P = 2.1 × 10−4) (Fig. 1b, c), while hemoglobin levels and platelet counts significantly decreased (P = 3.1 × 10−4 and P = 0.0034, respectively) (Supplementary Figure 2a, b). Flow cytometry analysis of the WBC revealed that myeloid cells were significantly more dominant than T-cells and B-cells (P < 1.0 × 10−15 and P < 1.0 × 10−15) (Supplementary Figure 3). BMMNCs showed that GMP and megakaryocyte-erythroid progenitor (MEP) populations were positive for GFP (Supplementary Figure 4). Median periods to leukemia onset were 57 and 16 days in the first and the fourth transplants, respectively. Statistically, the onset of leukemia became progressively earlier in the later transplant (P = 3.6 × 10−10) (Supplementary Figure 2c). These results indicate that serial transplantation of MLL/AF9-transduced mouse leukemic cells resulted in shorter time to repopulate the bone marrow, which showed an aggressive phenotype similar to that of relapsed human MLL-AML.
Accumulation of somatic mutations in mouse MLL-AML
To identify somatic mutations acquired during serial transplantation, we analyzed eight strains originated from five donor mice with transduced MLL/AF9 (Supplementary Figure 1), where two strains per donor mice were analyzed for donor A, C, and D. In total, leukemic cells obtained from 42 recipient mice in the first (N = 8), second (N = 17), third (N = 8), and fourth (N = 9) transplants were analyzed by whole exome sequencing. The mean sequence depth for all tumor and normal samples was 126.3 × (Supplementary Figure 5). By BWA alignment and Genomon pipeline as previously described [21, 26, 27], 1084 candidate variants (the cumulative total number) were extracted, out of which we randomly selected 33 variants and 97% (32/33) of them were confirmed as somatic by deep sequencing. We applied this default filter to call the rest of mutations. As a result, 968 missense, 7 frameshift (6 deletion and 1 insertion), 67 nonsense, and 41 splice site mutations were identified in 42 mice (25.8 mutations per mouse) (Supplementary Table 3). Out of these, in the experiments of two mouse strains (strain A1 and B1), the cells obtained from different donor mice were serially transplanted four times (Supplementary Figure 1). During the serial transplantation, somatic mutations were significantly accumulated, demonstrating significant trends of increasing number of mutations in both strains (A1; P = 4.5 × 10−4 and B1; P = 0.0012, respectively) (Fig. 1d, e). To assess variation of mutations in each strain, we removed overlaps and calculated actual number of mutations; 93 and 65 mutations were identified in these strains, respectively (Fig. 2a, b), and no mutations except for one (Msn mutation) were shared between the strains (Supplementary Table 3). To further investigate the cause of the difference in acquired mutations, we compared mutational spectra between each set of two strains from the same donors (A1 vs. A2, C1 vs. C2, and D1 vs. D2). No shared mutation was identified in any set from the same donor (Supplementary Table 3), suggesting that the variation of the mutations were not derived from different donor cells but from different primary AML cells after the first transplantation.
Fig. 2.

Whole exome sequencing (WES) of mouse MLL/AF9-AML. a, b Upper panels showed 12 and 13 mice analyzed by WES in strain A1 and B1, respectively. Middle panels demonstrated landscape of somatic mutations in each strain. Green and gray indicated shared and private mutations, respectively. Driver mutations were represented in bold italic font. In lower panels, fishplots displayed clonal architecture in A1-4-7 and B1-4-5 mice according to the results of mutational landscape in their own and their ancestry transplant generations. c–e Upper panels showed each 3 mice per strain from strain C1, C2, and E1, respectively. Lower panels demonstrated landscape of somatic mutations in each strain
Candidate pathogenic mutations in mouse MLL/AF9-AML
Among whole spectrum of somatic mutations in multiple generations of mouse MLL/AF9-AML transplantation from eight different strains, we extracted candidate driver mutations according to two criteria; (1) mutations recurrently identified in the multiple independent strains or (2) mutations reported in human hematological neoplasms. Mutations of Ptpn11 (p.G60R) were found in two independent strains (A1 and C1) (Fig. 2a, c) and also in human MLL-AML samples (Fig. 3). Msn mutations (p.R295C) were also recurrent in sequenced mouse MLL/AF9-AML from 3 strains (Fig. 2) and an identical mutation was reported in mouse CALM/AF10 leukemia models [28]. In addition, Braf mutations (p.V637E) detected in all three mice of strain C2 (Fig. 2d) corresponded to those identified in human MLL-AML (BRAF p.V600E) (Fig. 3). Another mutated gene of interest was Gnb2 in Strain B1 mice (Fig. 2b) which was reported to be recurrently mutated in various human hematological neoplasms [23]. Of note is that these mutations were located at the well-conserved residues among species. If all of them were combined, these candidate driver mutations were significantly associated with earlier leukemia onset in our mouse MLL-AML model (P = 0.018) (Fig. 4a). Among them, while Msn, Ptpn11, or Braf mutation had no significant impact (Supplementary Figure 6a-c), Gnb2 mutation was a significant poor prognostic factor compared to wild type (P = 0.011) (Fig. 4b). In contrast, the number of whole mutations wasn’t significantly associated with time of leukemia onset (Supplementary Figure 2d). These results suggest that acquisition of mutations in driver genes especially Gnb2 plays a more important role in shortening of time to leukemia onset in mouse MLL/AF9-AML than mutational accumulation itself.
Fig. 3.

Mutational spectrum in human MLL-AML (N = 168). A frequency of mutations (%) was shown in each affected leukemogenic pathway
Fig. 4.

Impact of driver mutations on leukemia onset and clonal expansion in MLL/AF9-AML. a, b Comparison of leukemia onset time between cases with and without driver mutations (a) or Gnb2 mutations (b). c, d Assessment of trend in variant allele frequencies of Msn (c) or Gnb2 (d) mutations
Clonal architecture in mouse MLL/AF9-AML
To further investigate clonal dynamics of the driver mutations, landscape of the mutations was demonstrated in every strain, where all the mutations were divided into either shared or private ones (Fig. 2). In strain A1 where 12 mice from 4 generations were analyzed, 36 were shared mutations and 57 were private ones (Fig. 2a). In strain B1, 1 mutation was shared and 64 were private (Fig. 2b). If all the sequenced strains were included (Fig. 2 and Supplementary Figure 7), shared and private mutations were 15.7 and 19 per strain on average, respectively. Based on these results of mutational landscape and variant allele frequency, clonal dynamics was displayed by the fishplots, showing that complex clonal architecture consisted of multiple decreased and newly emerged clones. While clones with Msn mutations were dominant in the first and second generations, new clones with Ptpn11 and Gnb2 mutations emerged in the second or third generations, and swept the initial Msn mutant clones out in the fourth generations (Fig. 2a, b). Integrating all the mutations identified in strains A1 and B1, we found that variant allele frequency significantly increased in size during serial transplantation (P = 1.7 × 10−14) (Supplementary Figure 6d). Next, we were able to conclude clonal dynamics of driver mutations. While the clone size of Gnb2 mutations significantly elevated during serial transplantation (P = 0.007) (Fig. 4d), those of Msn, Ptpn11, or Braf mutations did not significantly increase (Fig. 4c and Supplementary Figure 6e, f). Though GNB2 mutations were not identified in human MLL-AML (Fig. 3), Gnb2 mutation had a significant impact on poor prognosis in mouse MLL/AF9-AML, increased in clone size during the serial transplantation, and existed in other human hematological neoplasms [23]. Therefore, we assumed that GNB2 might be upregulated by previously unknown mechanisms as a new oncogene in human MLL-AML.
GNB2 expression in human MLL-AML
To further investigate pathological significance of GNB2, we analyzed the gene expression of GNB2 in human MLL-AML. In normal tissues, GNB2 is expressed in whole blood and bone marrow cells (Supplementary Figure 8). When compared between human MLL-rearrangement ( + ) (N = 18) and MLL-rearrangement (−) (N = 155) AML cases, mRNA levels of GNB2 extracted from RNA sequencing dataset were significantly upregulated in human MLL-rearrangement ( + ) cases (P = 0.031) (Fig. 5a). In the same cohort, we also compared GNB1 expression but identified no significant difference in contrast to the result of MLL/AF9-dependent upregulation of GNB2 (Supplementary Figure 9). To confirm this finding, we analyzed GNB2 expression by microarray in a larger cohort of AML. As a result, we again showed significantly higher expression of GNB2 in human MLL-AML (N = 38) than in non-MLL AML (N = 504) (P = 0.0091) (Fig. 5b). Finally, we assessed GNB2 expression by real-time RT-PCR in healthy donors (N = 5), human non-MLL AML cell lines (N = 4), and MLL-AML cell lines (N = 3). GNB2 expression was not significantly different between healthy donor BM cells and non-MLL AML cell lines (q = 0.67). However, MLL/AF9-positive cell lines, THP1, MOLM13, and NOMO1 significantly highly expressed this gene compared to healthy donor BM cells (q = 0.011) and non-MLL AML cell lines (q = 0.013) (Fig. 5c). To further validate these findings, we assessed Gnb1 and Gnb2 expression levels in comparison between empty- and MLL/AF9-transduced Ba/F3 cells and identified MLL/AF9-dependent upregulation of Gnb2 (P = 4.5 × 10−5) (Fig. 5d). These findings suggest the potential status of GNB2 as a candidate oncogene in human MLL-AML as well as in mouse model, and therefore we further performed confirmatory functional experiments.
Fig. 5.
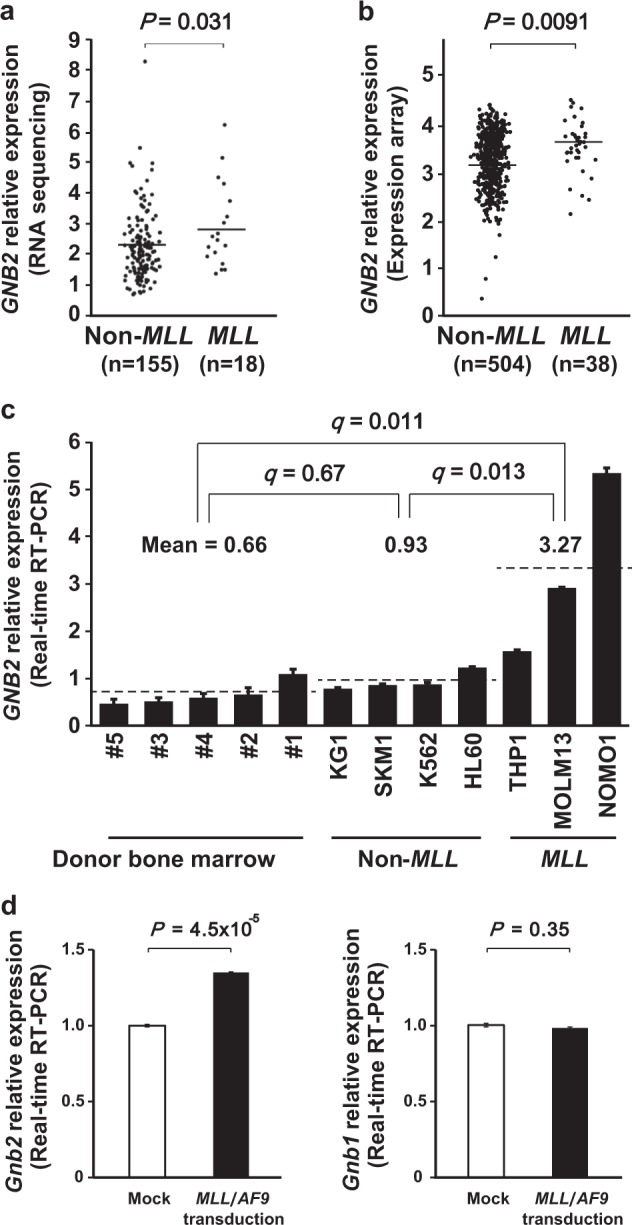
GNB2 expression in human MLL-AML. a, b GNB2 relative expression in human AML with and without MLL-fusion gene (a RNA sequencing and b expression array). c GNB2 relative expression in bone marrow mononuclear cells (BMMNCs) from healthy donors (N = 5), and AML cell lines with (N = 3) and without MLL-fusion gene (N = 4) (real-time RT-PCR). Each experiment was performed in triplicate. The Benjamini–Hochberg procedure was applied to the correction of multiple testing. d Gnb1 and Gnb2 expression levels were assessed in comparison between empty- and MLL/AF9-transduced Ba/F3 cells. Each experiment was performed in triplicate
Oncogenic potential of mutant and wild-type GNB2
We transduced wild type and mutant (p.G77R) GNB2 in an interleukin-3 (IL-3) dependent leukemic cell line Ba/F3. Overexpression of mutant GNB2 conferred cytokine-independent proliferation (Fig. 6a). To further investigate an oncogenic effect of the GNB2 mutation on cytokine independency, we assessed activation of downstream signaling pathway in Ba/F3 cells transduced with the mutation. Phosphorylation of AKT was upregulated in mutant Ba/F3 (Fig. 6b), suggesting activation of PI3K/Akt/mTOR pathway. Overexpressed wild-type GNB2 also resulted in maintaining proliferation even after IL-3 withdrawal, while mock cells cultured without IL-3 showed cell death (Fig. 6a). To further investigate tumor formation associated with GNB2, we performed in vivo experiments in which Ba/F3 cells with mock, wild-type GNB2, and mutant GNB2 were transplanted subcutaneously to nude mice (Fig. 7a–c) and intravenously to NOG mice (Fig. 7d–f). Compared between wild type vs. mutant GNB2 experiments in nude mice, tumor volumes were significantly larger in mutant than in wild type (P < 0.001 on day 14, 21, and 28) (Fig. 7a, c). Next compared between mock vs. wild-type GNB2 groups, tumor volumes were significantly larger in wild-type experiments than in mock (P < 0.01 on day 35, 42, and 49) (Fig. 7b, c). We further assessed leukemogenesis due to GNB2 alterations (both mutation and overexpression) in NOG mice. GFP-positive ratio in bone marrow cells was significantly higher in mutant experiments ( > 40%) than in wild-type ones on day 15 (P = 1.3 × 10−4) (Fig. 7d), and also significantly elevated in wild type ( > 30%) than in mock on day 28 (P = 0.0081) (Fig. 7e). Therefore, all NOG mice with mutant or wild-type GNB2 cells were proved to be affected by leukemic evolution. When compared between mutant, wild type, and mock experiments, survival period was significantly shorter in mutants than in wild type (P = 1.7 × 10−4) and also in wild type than in mock (P = 0.0057) (Fig. 7f). These findings suggest that upregulation of wild-type GNB2 had a significant leukemogenic effect, although it is less potent than that of mutant GNB2.
Fig. 6.

Cytokine independency due to GNB2. a Growth curves of Ba/F3 cells cultured without IL-3 after transduction of wild type or mutated (p.G77R) GNB2. Each experiment was performed in triplicate. b Protein levels of total Akt and phosphorylated Akt (p-Akt) were evaluated by immunoblot
Fig. 7.

Oncogenic potential of mutant and wild-type GNB2. Ba/F3 cells transduced with wild-type GNB2, GNB2 mutant, and mock were transplanted in immunodeficient mice. a, b, c To study GNB2-associated tumorigenesis, 1 × 107 cells per experiment were subcutaneously injected into nude mice (N = 30 in total). The tumor size was measured (a on day 23 and b on day 42) and compared between 3 groups (c). d, e, f To assess GNB2-associated leukemic involvement, 2 × 106 cells per mouse were intravenously transplanted into NOD/SCID/γc null (NOG) mice (N = 21 in total). GFP positivity was measured in bone marrow cells (d on day 15 and e on day 28). Survival time was compared between 3 groups (f)
Effect of decreased expression of wild-type GNB2
As mentioned, wild-type GNB2 expression was elevated in human MLL-AML, which is consistent with the remarkable in vivo leukemogenesis due to overexpression of wild-type GNB2. To further confirm these findings, we reduced GNB2 expression by shRNA and depleted Gnb2 by CRISPR-Cas9 system. First, knocked down wild-type GNB2 in MLL-AML cell lines; NOMO1, MOLM13, and THP1, which showed high expression of wild-type GNB2 (Fig. 5c), resulted in significantly less proliferation compared to mock experiments (Fig. 8a–c). In agreement with these findings, Gnb2 was found to be essential for mouse MLL/AF9-AML, revealed by genome-wide CRISPR-Cas9 screens [25]. sgRNAs targeting Gnb2 were significantly depleted over a 16-day incubation period, confirming a potential oncogenic function of wild-type Gnb2 in MLL-AML (P = 0.00366) (Fig. 8d, e). In contrast, sgRNAs targeting Gnb1 showed no positive impact on cell survival in the CRISPR-Cas9 screen (Supplementary Figure 10), which confirmed the results of normal GNB1 expression in human MLL-AML (Supplementary Figure 9).
Fig. 8.

Impact of decreased GNB2 expression or depleted Gnb2 genome in MLL-AML. a, b, c Growth curves (cell numbers) and GNB2 expression values of 3 human MLL-AML cell lines (a NOMO1, b MOLM13, and c THP1) treated with GNB2 shRNA and control scramble shRNA. Each experiment was performed in triplicate. d, e By genome-wide CRISPR-Cas9 screens, read counts of six sgRNAs targeting per exon (exon 1 (d) and 2 (e)) in Gnb2 were measured after a 16-day incubation period to confirm potential oncogenic function of Gnb2 in MLL/AF9-AML
Discussion
Our results from whole exome sequencing of serially transplanted mouse MLL/AF9-AML cells and confirmatory genetic and functional analyses of human MLL-AML provided insights into the molecular mechanism of leukemic progression in MLL-AML. Accumulation of somatic mutations in addition to MLL-fusion gene was common in the sequencing experiments of our mouse model and clinical samples. As well as well-known driver mutations (Ptpn11 and Braf), we found novel Msn and Gnb2 mutations in our mouse model. Out of these candidate mutations, clone size of Gnb2 mutation significantly elevated during serial transplantation and this mutation was significantly associated with earlier leukemia onset. These results suggest that GNB2 is a candidate pathogenic gene in the human disease, which was convincingly supported by high expression of this gene in human MLL-AML. The pathologic role of high expression of GNB2 was confirmed by its knockdown and knockout experiments in in vitro and in vivo disease models. Accordingly, additional mutational and transcriptome events co-occuring with MLL-fusion genes might play a significant role in the pathogenesis of progressive clinical course in MLL-AML.
As previously reported in myelodysplastic syndromes, accumulation of multiple mutations are seen at disease presentation and closely related to secondary AML evolution [21, 29, 30]. In parallel, leukemic cells of human core binding factor (CBF) leukemia frequently acquired additional mutations after initial hits of leukemogenic fusion genes [31]. CBF leukemia and MLL-AML showed similar types of genes mutated in secondary fashion, involving signal transduction (RAS pathway and RTK cascade), IDH family, and gene transcription, of which all are essential for progressive clinical phenotypes in AML. In this study of MLL/AF9-AML, we showed that somatic mutations remarkably accumulated during shortening leukemic onset and that driver mutations in the mouse sequencing reproduced human mutational profiles, including RAS pathway mutations. Of note, the presence of driver mutations were more associated with MLL/AF9-AML onset than the number of whole mutations. Analysis of complex clonal dynamics revealed distinct behaviors of clones with Msn mutations which were swept out by those with other driver mutations. Especially, clone size of mutations in Gnb2, which is involved in signal transduction, increased during the reduced clone size of Msn mutations and the shortened leukemic onset. These findings suggest that clonal dynamics of specific genetic events in MLL/AF9 AML might be associated with relapse and aggressiveness of this disease.
In general, mutations and overexpression of oncogenes provide oncogenic potential due to increased function and quantity of the coding proteins, respectively. In AML, oncogenes sometimes display activating mutations and high expression simultaneously, both of which are supposed to have a leukemogenic effect on the affected hematopoietic stem cells and/or myeloid progenitor cells. In this study of MLL/AF9-AML, we showed that a novel activating mutation of Gnb2 were clearly pathogenic in a mouse leukemia model and that high expression of GNB2 may play a significant role in human MLL-AML. Hence, our study posits the oncogenic candidacy of GNB2 in MLL-AML. GNB2 mutations were also identified in several cancers, including diffuse large B-cell lymphoma, colorectal adenocarcinoma, and lung small cell carcinoma [32–34]. After exploring and elucidating the role of GNB2, our study finds it reasonable to suggest GNB2 as an oncogene presenting its distinct phenotype of clinical progression in the synergistic manner with MLL-fusion gene.
As previously reported, expression of various oncogenes, which are responsible for leukemogenesis in MLL-AML, are regulated by MLL-fusion genes through histone modifications. For example, highly expressed Hox genes are associated with altered functions of histone methyltransferases and demethylases which directly or secondarily regulate methylation and acetylation status of histone H3K4, H3K36, and H3K79 [7, 9, 10, 35]. As well as such reported driver genes, GNB2 was consistently expressed to serve as an oncogene in human MLL-AML, although no somatic mutations in this gene were identified. In fact, according to ChIP sequencing of multiple hematopoietic cell lines, promoter sites of GNB2 were remarkably identified as binding regions of tri-methylated histone H3K4, which serves a major role of oncogenic potential in MLL-associated leukemia [10]. Therefore, expression of GNB2, whose activating mutation was acquired during mouse MLL/AF9 leukemia, might be epigenetically regulated by MLL fusion protein.
GNB2 encodes a β subunit of heterotrimeric G proteins, which consist of Gα, Gβ, and Gγ components that mediate signal transduction downstream of G protein-coupled receptors (GPCR). Our study and a recent paper demonstrated that GNB family proteins may also take part in non-GPCR signaling, for instance, all other types of receptor-mediated signaling, including various cytokine receptors and JAK/STAT pathways [23]. According to these concepts, RTKs and RAS pathway, which are most frequently mutated in MLL-AML, are in the downstream of the sequential signaling cascades activated by GNB2. To develop the therapeutic approach for GNB2 activation, we could further focus our attention on RTK inhibitors and RAS pathway inhibitors targeting molecules up- and downstream of this gene, suggesting that GNB2 could be a novel therapeutic target to already FDA-approved drugs.
In conclusion, this is the first comprehensive study of somatic mutations in MLL/AF9 mouse model by whole exome sequencing. Overall, we postulate that MLL-fusion genes as initial drivers may cooperate subsequently with various somatic genetic events, resulting in aggressive clinical course due to leukemic progression.
Electronic supplementary material
Acknowledgements
This work was supported by National Institutes of Health (Bethesda, MD; NIH) grants RO1HL-082983 (J.P.M.), U54 RR019391 (J.P.M.), K24 HL-077522 (J.P.M.), a grant from the AA & MDS International Foundation (Rockville, MD; J.P.M. and Cleveland, OH; H.M.), the Robert Duggan Charitable Fund (Cleveland, OH; J.P.M.), a grant from Edward P. Evans Foundation (Cleveland, OH; J.P.M), Scott Hamilton CARES grant (Cleveland, OH; H.M.), Grant-in-Aids from the Ministry of Health, Labor and Welfare of Japan and KAKENHI 23249052, 22134006, 21790907 and 15H05909 (Kyoto; S.O.), JP17km0305018, 16H05338 (Kyoto; H.M.), project for development of innovative research on cancer therapies (p-direct) (Tokyo; S.O.), the Japan Society for the Promotion of Science (JSPS) through the ‘Funding Program for World-Leading Innovative R&D on Science and Technology’, initiated by the Council for Science and Technology Policy (CSTP) (Tokyo; S.O.), JSPS Core-to-Core Program, A. Advanced Research Networks (Kyoto; H.M., S.O.), project for cancer research and therapeutics evolution (P-CREATE) from Japan Agency for Medical Research and Development JP16cm0106501 (Kyoto; S.O.), the Project for Development of Innovative Research on Cancer Therapeutics from the Japan Agency for Medical Research and Development, AMED (15cm0106056h0005 and 16cm0106501h0001) (Kyoto; S.O.), he High Performance Computing Infrastructure System Research Project (hp150232) (Kyoto; S.O.), and JP16cm0106422 (Kyoto; T.Y.), research grants from The Japan Leukemia Research Fund, The Suzuken Memorial Foundation, The UBE Foundation, Takeda Science Foundation, and The Kyoto University Foundation (Kyoto; A.Y.), KAKENHI JP18K16084 (A.K.), and research grants from The YASUDA Medical Foundation, Princess Takamatsu Cancer Research Fund, Takeda Science Foundation, The Japanese Society of Hematology, The Public Trust Fund For Clinical Cancer Research (Kyoto; H.M.). The results published here were partly based on data generated by The Cancer Genome Atlas pilot project established by the National Cancer Institute and National Human Genome Research Institute. We thank Dr. Akihiko Yokoyama for providing the MLL/AF9 vector, Maki Nakamura, Ai Takatsu, Satoko Yabuta, and Takeshi Shirahari for technical assistance, and Dr. Ravi Velaga for English editing.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Contributor Information
Seishi Ogawa, Phone: +81-75-753-9285, Email: sogawa-tky@umin.ac.jp.
Hideki Makishima, Phone: +81-75-753-9285, Email: makishima.hideki.8x@kyoto-u.ac.jp.
Electronic supplementary material
The online version of this article (10.1038/s41375-018-0253-3) contains supplementary material, which is available to authorized users.
References
- 1.Dimartino JF, Cleary ML. Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br J Haematol. 1999;106:614–26. doi: 10.1046/j.1365-2141.1999.01439.x. [DOI] [PubMed] [Google Scholar]
- 2.Mrozek K, Bloomfield CD Chromosome aberrations, gene mutations and expression changes, and prognosis in adult acute myeloid leukemia. Hematol Am Soc Hematol Educ Prog 2006: 169–77. [DOI] [PubMed]
- 3.Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, et al. The MLL recombinome of acute leukemias in 2013. Leukemia. 2013;27:2165–76. doi: 10.1038/leu.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–7. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 5.Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–28. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- 6.Mullighan CG, Kennedy A, Zhou X, Radtke I, Phillips LA, Shurtleff SA, et al. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia. 2007;21:2000–9. doi: 10.1038/sj.leu.2404808. [DOI] [PubMed] [Google Scholar]
- 7.Zhu N, Chen M, Eng R, DeJong J, Sinha AU, Rahnamay NF, et al. MLL-AF9- and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J Clin Invest. 2016;126:997–1011. doi: 10.1172/JCI82978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–78. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem. 2006;281:18059–68. doi: 10.1074/jbc.M601903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong SH, Goode DL, Iwasaki M, Wei MC, Kuo HP, Zhu L, et al. The H3K4-Methyl epigenome regulates leukemia stem cell oncogenic potential. Cancer Cell. 2015;28:198–209. doi: 10.1016/j.ccell.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benedikt A, Baltruschat S, Scholz B, Bursen A, Arrey TN, Meyer B, et al. The leukemogenic AF4-MLL fusion protein causes P-TEFb kinase activation and altered epigenetic signatures. Leukemia. 2011;25:135–44. doi: 10.1038/leu.2010.249. [DOI] [PubMed] [Google Scholar]
- 12.Okuda H, Kanai A, Ito S, Matsui H, Yokoyama A. AF4 uses the SL1 components of RNAP1 machinery to initiate MLL fusion- and AEP-dependent transcription. Nat Commun. 2015;6:8869. doi: 10.1038/ncomms9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okuda H, Takahashi S, Takaori-Kondo A, Yokoyama A. TBP loading by AF4 through SL1 is the major rate-limiting step in MLL fusion-dependent transcription. Cell Cycle. 2016;15:2712–22. doi: 10.1080/15384101.2016.1222337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Cancer Genome Atlas Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74. [DOI] [PMC free article] [PubMed]
- 15.Lavallee VP, Baccelli I, Krosl J, Wilhelm B, Barabe F, Gendron P, et al. The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat Genet. 2015;47:1030–7. doi: 10.1038/ng.3371. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Chen W, Mysliwski M, Serio J, Ropa J, Abulwerdi FA, et al. Mutated Ptpn11 alters leukemic stem cell frequency and reduces the sensitivity of acute myeloid leukemia cells to Mcl1 inhibition. Leukemia. 2015;29:1290–300. doi: 10.1038/leu.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–22. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 19.Li H. and Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 2009;25:1754-60. [DOI] [PMC free article] [PubMed]
- 20.Laboratory of DNA Information Analysis, Human Genome Center, The Institue of Medical Science, The University of Tokyo. The last accessed date is 3/28/2016.
- 21.Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49:204–12. doi: 10.1038/ng.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grossmann V, Schnittger S, Poetzinger F, Kohlmann A, Stiel A, Eder C, et al. High incidence of RAS signalling pathway mutations in MLL-rearranged acute myeloid leukemia. Leukemia. 2013;27:1933–6. doi: 10.1038/leu.2013.90. [DOI] [PubMed] [Google Scholar]
- 23.Yoda A, Adelmant G, Tamburini J, Chapuy B, Shindoh N, Yoda Y, et al. Mutations in G protein beta subunits promote transformation and kinase inhibitor resistance. Nat Med. 2015;21:71–5. doi: 10.1038/nm.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki HI, Katsura A, Yasuda T, Ueno T, Mano H, Sugimoto K, et al. Small-RNA asymmetry is directly driven by mammalian Argonautes. Nat Struct Mol Biol. 2015;22:512–21. doi: 10.1038/nsmb.3050. [DOI] [PubMed] [Google Scholar]
- 25.Yamauchi T, Masuda T, Canver MC, Seiler M, Semba Y, Shboul M, et al. Genome-wide CRISPR-Cas9 screen identifies leukemia-specific dependence on a pre-mRNA metabolic pathway regulated by DCPS. Cancer Cell. 2018;33:386–400. doi: 10.1016/j.ccell.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47:458–68. doi: 10.1038/ng.3273. [DOI] [PubMed] [Google Scholar]
- 27.Yoshizato T, Nannya Y, Atsuta Y, Shiozawa Y, Iijima-Yamashita Y, Yoshida K, et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood. 2017;129:2347–58. doi: 10.1182/blood-2016-12-754796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutta S, Krause A, Vosberg S, Herold T, Ksienzyk B, Quintanilla-Martinez L, et al. The target cell of transformation is distinct from the leukemia stem cell in murine CALM/AF10 leukemia models. Leukemia. 2016;30:1166–76. doi: 10.1038/leu.2015.349. [DOI] [PubMed] [Google Scholar]
- 29.Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–8. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–82. doi: 10.1038/leu.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet. 2016;48:1551–6. doi: 10.1038/ng.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, et al. Genetic landscapes of relapsed and refractory diffuse large B-Cell lymphomas. Clin Cancer Res. 2016;22:2290–300. doi: 10.1158/1078-0432.CCR-15-2123. [DOI] [PubMed] [Google Scholar]
- 33.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–4. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu L, Li Q, Wong SH, Huang M, Klein BJ, Shen J, et al. ASH1L links histone H3 lysine 36 dimethylation to MLL leukemia. Cancer Discov. 2016;6:770–83. doi: 10.1158/2159-8290.CD-16-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.