

Abstract
Background & Aims
Obesity promotes the development of nonalcoholic fatty liver diseases (NAFLDs), yet not all obese patients develop NAFLD. The underlying causes for this discrepancy remain elusive. LPGAT1 is an acyltransferase that catalyzes the remodeling of phosphatidylglycerol (PG), a mitochondrial phospholipid implicated in various metabolic diseases. Here, we investigated the role of LPGAT1 in regulating the onset of diet-induced obesity and its related hepatosteatosis because polymorphisms of the LPGAT1 gene promoter were strongly associated with susceptibility to obesity in Pima Indians.
Methods
Mice with whole-body knockout of LPGAT1 were generated to investigate the role of PG remodeling in NAFLD.
Results
LPGAT1 deficiency protected mice from diet-induced obesity, but led to hepatopathy, insulin resistance, and NAFLD as a consequence of oxidative stress, mitochondrial DNA depletion, and mitochondrial dysfunction.
Conclusions
This study identified an unexpected role of PG remodeling in obesity, linking mitochondrial dysfunction to NAFLD.
Keywords: LPGAT1, Cardiolipin, MEGDEL Syndrome, NAFLD, Mitochondrial Dysfunction
Abbreviations used in this paper: CL, cardiolipin; DIO, diet-induced obesity; DMEM, Dulbecco’s modified Eagle medium; EGTA, ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid; FCCP, p-trifluoromethoxy carbonyl cyanide phenylhydrazone; HFD, high-fat diet; LPGAT1, lysophosphatidylglycerol acyltransferase 1; MAM, mitochondrial-associated membrane; MEGDEL, 3-methylglutaconic aciduria with deafness, encephalopathy and Leigh-like; MGAT, monoacylglycerol acyltransferase; miR, microRNA; mRNA, messenger RNA; mtDNA, mitochondrial DNA; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PBS, phosphate-buffered saline; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PS, phosphatidylserine; ROS, reactive oxygen species; RT-PCR, reverse-transcription polymerase chain reaction; TBARS, thiobarbituric acid reactive substances; TLCL, tetra linoleoyl cardiolipin; WT, wild-type
Graphical abstract

Summary.
LPGAT1 deficiency caused hepatic insulin resistance and hepatopathy and implicated the pivotal role of phosphatidylglycerol remodeling in regulating mitochondrial function and nonalcoholic fatty liver diseases.
Obesity significantly increases the risk of nonalcoholic fatty liver disease (NAFLD), a condition that affects more than 30% of the US adult population. However, not all obese patients develop NAFLD. Although the precise molecular mechanisms underlying the discrepancy remains poorly understood, it is now widely accepted that mitochondrial dysfunction is pivotal to the pathogenesis of NAFLD and its progression to nonalcoholic steatohepatitis (NASH).1 Obese patients who developed NAFLD showed a gradual decline of the respiratory control ratio and mitochondrial coupling efficiency before the progression to NASH. Accordingly, only those obese patients who showed a loss of mitochondrial functional adaptation to the bioenergetic needs in obesity were highly prone to the development of NAFLD.1 However, the precise causes for these mitochondrial defects in NAFLD remain elusive, which has hindered ongoing efforts in developing an effective treatment of NAFLD and its dangerous progression to NASH.
Phosphatidylglycerol (PG) is a glycerophospholipid commonly recognized for its important role as a precursor for the synthesis of cardiolipin (CL), a mitochondrial signature phospholipid required for dynamic mitochondrial functions.2, 3, 4 PG deficiency in mammalian cells leads to CL deficiency, mitochondrial dysfunction, and a reduction in adenosine triphosphate production.5 Disruption of the PGS1 gene in yeast causes PG and CL deficiency and inhibition of growth on nonfermentable carbon sources.6 PG is subjected to remodeling subsequent to its de novo biosynthesis in mitochondria to incorporate appropriate acyl content for its biological functions and to prevent the harmful effect of lysophosphatidylglycerol accumulation. Consequently, defective PG remodeling is implicated in the pathogenesis of NAFLD7 and 3-methylglutaconic aciduria with deafness, encephalopathy and Leigh-like (MEGDEL) syndrome, a recessive genetic disorder of dystonia and deafness with Leigh-like syndrome.8 Patients with MEGDEL syndrome also showed hepatopathy and mitochondrial dysfunction. Defective PG remodeling also is associated with the onset of Barth syndrome, an X-linked recessive disease caused by mutations of the tafazzin gene encoding a transacylase involved in CL remodeling.9 PG and CL deficiency in Barth syndrome significantly impaired mitochondrial fatty acid oxidation, which leads to cardiomyopathy and premature death.10, 11
Our previous work showed that Lysophosphatidylglycerol Acyltransferase 1 (LPGAT1) is an acyltransferase that catalyzes the acylation of lysophosphatidylglycerol to PG, a key step involved in the PG remodeling process.3 LPGAT1 belongs to a large family of acyltransferases, which are involved in a variety of biological processes including pathways that regulate energy homeostasis, body weight, and NAFLD. LPGAT1 also was reported to regulate lipid metabolism in the liver as a putative monoacylgcyerol acyltransferase.12 Recently, a genome-wide association study linked DNA polymorphism of the LPGAT1 gene promoter to the onset of severe obesity in Pima Indians.13 LPGAT1 is expressed abundantly in a number of metabolic tissues, with highest expression in the liver.3 Moreover, LPGAT1 is implicated as a key regulator of cholesterol secretion and atherosclerosis.14 However, the metabolic function of the LPGAT1 enzyme remains elusive. In this study, we generated mice with targeted deletion of LPGAT1, and investigated the role of LPGAT1 in regulating diet-induced obesity (DIO) and its related hepatosteatosis. We show that PG remodeling by LPGAT1 plays an important role in protecting mitochondrial dysfunction associated with NAFLD.
Results
Ablation of LPGAT1 Prevents DIO, but Leads to Severe Insulin Resistance
The LPGAT1 gene promoter polymorphism recently was implicated in obesity in Pima Indians,13 but the roles of the LPGAT1 gene in lipid metabolism and energy homeostasis remain elusive. By using the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR associated protein 9 (CRISPR/Cas9)-mediated gene editing technique, we recently generated mice with a targeted deletion of the LPGAT1 gene to determine its metabolic function (Figure 1A and B). The genotype of the LPGAT1 knockout mice (LPGAT1-/-) were confirmed by both reverse-transcription polymerase chain reaction (RT-PCR) and Western blot analyses (Figure 1C and D). The LPGAT1-/- mice were born at the normal Mendelian ratio, but had significantly lower birth weight and body weight on either normal chow diet or a high-fat diet (HFD) (Figure 2A and B). However, the male LPGAT1-/- mice showed a higher percentage of body weight gain when fed a normal chow diet or a HFD (Figure 2C and D). Contrary to the findings from the genome-wide association study analysis in Pima Indians, LPGAT1-/- mice were protected from DIO, which was evidenced by a significantly lower fat mass relative to the wild-type (WT) controls (Figure 2G). Despite resistance to DIO, LPGAT1-/- mice developed glucose intolerance in response to a HFD, as indicated by the results from a glucose tolerance test (Figure 2E). The defect most likely was caused by a reduction in insulin sensitivity, as evidenced by the results from insulin tolerance tests (Figure 2F). Surprisingly, the insulin resistance was not caused by the hyperinsulinemia commonly associated with obesity. In contrast to hyperinsulinemia in WT controls, LPGAT1-/- mice showed a normal fasting serum insulin level (Figure 2H) and significantly lower levels of glucose-stimulated insulin secretion during glucose tolerance tests (Figure 2I). Likewise, the female LPGAT1 knockout mice also showed similar metabolic defects to male mice, as evidenced by significantly higher weight gain (Figure 3A and B), glucose intolerance (Figure 3C), and insulin resistance (Figure 3D).
Figure 1.

Generation of LPGAT1 knockout (KO) mice by CRISPR/Cas9 gene editing. (A) Strategy of guide RNA (gRNA) designing and LPGAT1 gene knockout. Two gRNAs were designed targeting exon 3 of the LPGAT1 gene, which resulted in a deletion of 124 bp nucleotides and termination of translation. (B) Genotyping of LPGAT1-/- and WT control mice by PCR analysis. The homozygotes of LPGAT1 knockout showed 124-bp nucleotide deletion relative to the WT control mice. (C) RT-PCR analysis of LPGAT1 mRNA expression in livers of LPGAT1-/- and the WT control mice. Data are represented as means ± SD. N = 3, ***P < .001 by t test. (D) Western blot analysis of LPGAT1 protein expression in the liver samples from LPGAT1-/- and the WT control mice.
Figure 2.

Ablation of LPGAT1 caused insulin resistance in male mice. Male LPGAT1-/- and WT control mice were fed normal chow diet (ND) or a HFD for 12 consecutive weeks, and were analyzed for the following. (A) Body weight on ND. (B) Body weight on HFD. (C) The percentage of body weight gain after birth on ND. (D) The percentage of body weight gain after birth on HFD. (E) Glucose tolerance test (GTT). (F) Insulin tolerance test (ITT). (G) Abdominal fat mass. (H) Fasting blood insulin levels. (I) Glucose-stimulated blood insulin levels. (J) Serum total cholesterol level. (K) Serum triglyceride (TAG) level. Data are represented as means ± SD (A–D, n = 10; E and F, n = 8; and G–K, n = 5–8). *P < .05, **P < .01 by t test or 1-way analysis of variance. BW, body weight; KO, knockout.
Figure 3.

LPGAT1 deficiency led to insulin resistance in female mice. Female LPGAT1-/- and WT controls mice were fed normal chow diet (ND) or a HFD for 12 consecutive weeks, and were analyzed for the following. (A) The percentage of body weight gain after birth on ND. (B) The percentage of body weight gain after birth on HFD. (C) Glucose tolerance test (GTT). (D) Insulin tolerance test (ITT). (E) Serum total cholesterol level. (F) Serum triglyceride (TAG) level. Data are represented as means ± SD (A–D, n = 8–10; and E and F, n = 5–8). *P < .05, **P < .01 by t test or 1-way analysis of variance. KO, knockout.
LPGAT1 recently was reported as one of the target genes of microRNA-30c (miR-30c), which significantly depletes the expression of LPGAT1.15 Targeted deletion of miR-30c significantly increased the plasma cholesterol level and hepatic lipid synthesis.15 Conversely, treatment of mice with miR-30c mimetics mitigated hypercholesterolemia and atherosclerosis.16 Consistent with the findings, we showed that LPGAT1 deficiency significantly decreased serum cholesterol and triglyceride levels in both male (Figure 2J and K) and female (Figure 3E and F) LPGAT1-/- mice.
LPGAT1 Deficiency Leads to Hepatopathy, Hepatosteatosis, and Hepatofibrosis
We next investigated the effect of LPGAT1 on hepatic lipid and cholesterol homeostasis because depletion of LPGAT1 messenger RNA (mRNA) by miR-30c significantly down-regulated hepatic lipid synthesis.16 Surprisingly, LPGAT1-/- mice developed spontaneous hepatosteatosis, which was exacerbated by feeding with a HFD. Accordingly, LPGAT1 deficiency significantly increased liver weight and the content of both hepatic triglyceride and cholesterol in both male (Figure 4A–C) and female (Figure 4D–F) mice. The results were corroborated further by Oil red O staining of the liver section (Figure 5B). Strikingly, LPGAT1 deficiency also caused hepatopathy, a major defect associated with MEGDEL syndrome, as evidenced by dilated hepatic venules, which were obstructed by massive accumulation of fat droplets in response to a HFD (Figure 5A, highlighted by arrows). Consistent with hepatosteatosis, LPGAT1 deficiency significantly down-regulated the expression of genes required for lipolysis, including CGI-58 and adiponutrin (Figure 5C and D), and also up-regulated the genes required for hepatic lipogenesis, including SREBP1c, FAS1, and ACC1 (Figure 5E–G), as shown by results from the RT-PCR analysis.
Figure 4.

LPGAT1 deficiency caused hepatic dyslipidemia. Male mice from Figure 2 were analyzed for changes in the following: (A) liver weight to body weight ratio, (B) liver serum triglyceride (TAG) level; and (C) liver total cholesterol level. Female mice from Figure 3 were analyzed for changes in the following: (D) liver weight to body weight ratio, (E) liver TAG level, and (F) liver total cholesterol level. Data are represented as means ± SD (A and D, n = 8–10; and B, C, E, and F, n = 5). *P < .05, **P < .01 by 1-way analysis of variance. BW, body weight.
Figure 5.

LPGAT1 deficiency aggravated diet-induced NAFLD. Male mice from Figure 2 were analyzed for (A) H&E staining of the liver section. Dilated hepatic venules are highlighted by arrows. LD, lipid droplet. Scale bar: 20 μm. (B) Analysis of hepatosteatosis by Oil red O (ORO) staining. Lipid droplets are highlighted by arrows. Scale bar: 20 μm. (C–G) Quantitative RT-PCR analysis of mRNA levels of genes related to hepatic lipid metabolism, including (C) CGI-58, (D) adiponutrin, (E) SREBP1c, (F) FAS1, and (G) ACC1. Data are represented as means ± SD (n = 4–6). *P < .05, **P < .01 by 1-way analysis of variance. KO, knockout; ND, normal chow diet.
Hepatopathy often leads to liver fibrosis before the development of NASH in patients with MEGDEL syndrome. Indeed, LPGAT1 deficiency also caused severe hepatic fibrosis in response to a HFD, as evidenced by increased expression of fibrosis markers, collagen I and III (Figure 6A and B). The results were confirmed further by Masson's trichrome staining of collagen fibers (Figure 6C, highlighted by arrows, and the area of fibrosis is quantified in Figure 6D). Again, the defects were highly reminiscent of those observed in MEGDEL syndrome, which is characterized by fibrotic staining of the dilated hepatic venules.
Figure 6.

LPGAT1 deficiency caused hepatofibrosis. Male mice from Figure 2 were analyzed for (A and B) quantitative RT-PCR analysis of mRNA levels of (A) collagen I and (B) collagen III, and (C) Masson staining of fibrosis in liver sections from LPGAT1-/- and WT controls. Fibrosis and dilation of hepatic venules are highlighted by arrows. Scale bar: 20 μm. (D) Quantification of fibrosis area in panel C by ImageJ software (1.47v; National Institutes of Health, Bethesda, MD). Data are represented as means ± SD (A and B, n = 4; and D, n = 6). **P < .01 by 1-way analysis of variance. KO, knockout; ND, normal chow diet.
Increased de novo lipogenesis plays an important role in the accumulation of triglyceride in NAFLD. To further elucidate the molecular mechanisms by which LPGAT1 deficiency caused hepatosteatosis, we isolated primary hepatocytes and analyzed the effect of LPGAT1 deficiency on the expression of several genes involved in hepatic lipid synthesis by real-time RT-PCR analysis. Consistent with findings from the liver tissue, LPGAT1 deficiency also significantly increased the expression of genes involved in lipid synthesis, including PPARα, SREBP1c, and ACC1 in primary hepatocytes under basal condition and in response to treatment with oleic acids (Figure 7A–C). Consequently, LPGAT1 deficiency also significantly increased both the number and size of lipid droplets in cultured primary hepatocytes (Figure 7D, the lipid droplet number and size are quantified in Figure 7E and F, respectively). However, LPGAT1 deficiency did not promote lipid droplet biogenesis because the total number of lipid droplets was similar between LPGAT1-/- and the WT controls in response to stimulation with oleic acid (Figure 7D and E).
Figure 7.

LPGAT1 deficiency aggravated lipid accumulation in primary hepatocytes. (A–C) Quantitative RT-PCR analysis of mRNA levels of genes that promote lipogenesis, including (A) PPARα, (B) SREBP1c, and (C) ACC1 in cultured primary hepatocytes isolated from WT and LPGAT1-/- mice. (D) Confocal imaging analysis of lipid droplets in cultured primary hepatocytes isolated from LPGAT1-/- and WT control mice in response to stimulation of oleic acids. Cells were incubated with oleic acids (200 μmol/L) for 16 hours. Lipid droplets were stained by BODIPY493/503, and highlighted by arrows. Scale bar: 10 μm. (E and F) Quantification of lipid droplet (E) number and (F) size in cultured primary hepatocytes stained with BODIPY493/503 by ImageJ (National Institutes of Health, Bethesda, MD). Data are representative of at least 3 independent experiments, and are represented as means ± SD (A–C, n = 5; E and F, n = 25–30 cells). *P < .05, **P < .01 by 1-way analysis of variance. CM, completed medium; Ctrl, control; KO, knockout; LD, lipid droplet; OA, oleic acid.
To further address the issue of whether the effect of LPGAT1 deficiency on hepatosteatosis is autonomous or liver-specific, we next determined the effect of LPGAT1 deficiency on lipid droplet biogenesis in C2C12 cells, an immortalized mouse myoblast cell line. The results showed that LPGAT1 depletion did not significantly increase lipid droplet accumulation in C2C12 cells under basal conditions or in response to stimulation with oleic acids (Figure 8), further supporting a key role of LPGAT1 in hepatic lipid metabolism.
Figure 8.
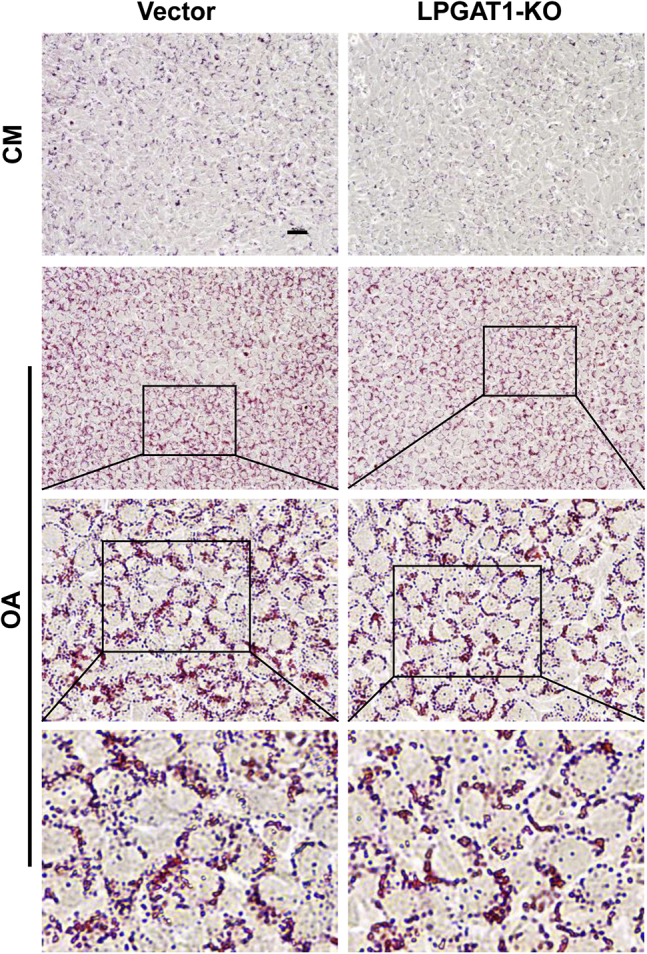
LPGAT1 deficiency did not affect lipid accumulation in C2C12 myoblast cells. The LPGAT1 gene was deleted in C2C12 cells by CRISPR/Cas9 gene editing. The LPGAT1-deficient C2C12 cells and the vector controls were treated with vehicle or 200 μmol/L oleic acid for 16 hours, stained with Oil red O, and examined for lipid droplet biogenesis by microscope imaging analysis (All-in-One Fluorescence Microscope BZ-X800; Keyence, Osaka, Japan). Images are representative of 3 independent experiments. Scale bar: 50 μm. CM, completed medium; KO, knockout; OA, oleic acid.
LPGAT1 Specifically Regulates Insulin Signaling in Hepatocytes
Mitochondrial-associated membrane (MAM) recently was identified as a major regulatory site for insulin signaling.17 To gain further insight into the molecular mechanisms underlying insulin resistance in LPGAT1-/- mice, we next analyzed the subcellular localization of LPGAT1. The results showed that LPGAT1 is localized primarily at MAM, a primary site for phospholipid remodeling (Figure 9A). Consistent with this notion, LPGAT1 deficiency significantly impaired insulin signaling in the liver, which is corroborated by a significant decrease in insulin-stimulated Protein kinase B (Akt) and Glycogen synthase kinase 3α/β (Gsk3 α/β) phosphorylation (Figure 9B, quantified in Figure 9C and D, respectively). Likewise, LPGAT1 deficiency also significantly impaired insulin signaling in cultured primary hepatocytes (Figure 9E). In contrast, LPGAT1 deficiency did not have a major effect on insulin signaling in other metabolic tissues, including skeletal muscle (Figure 9F) and heart (Figure 9G), further implicating a key role of LPGAT1 as a hepatic regulator of metabolism.
Figure 9.

LPGAT1 deficiency impaired insulin signaling. (A) Subcellular fractionation analysis of LPGAT1 stably expressed in COS-7 cells. Calnexin was blotted as an endoplasmic reticulum marker. (B–D) Western blot analysis of insulin-stimulated phosphorylation of Akt and Gsk3α/β in different metabolic tissues isolated from LPGAT1-/- and WT control mice, including (B) liver, (E) primary hepatocytes in culture, (F) skeletal muscle, and (G) heart. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as internal control for protein loading. Mice were fasted for 12 hours and then stimulated with insulin (1 U/kg body weight) for 15 minutes. Primary hepatocytes were stimulated with the indicated concentrations of insulin for 15 minutes. (C and D) Quantification of the phosphorylation level of (C) Akt and (D) Gsk3α in the liver by ImageJ (National Institutes of Health, Bethesda, MD). Data are representative of 3 independent experiments, and represented as means ± SD (n = 3). *P < .05 by 1-way analysis of variance. Ctrl, control; KO, knockout; Sk, skeletal.
In addition to MAM, LPGAT1 also is localized at the endoplasmic reticulum (ER), where it is required for ER homeostasis (Figure 9A). Remarkably, LPGAT1 deficiency also caused severe ER stress, which is exacerbated by oxidative stress by treatment of primary hepatocytes with H2O2, as evidenced by up-regulated expression of major ER stress regulators, including PERK, ATF4, BIP, ER57, GADD-34, and Xbp1 (Figure 10A–F).
Figure 10.

LPGAT1 deficiency caused endoplasmic reticulum stress. (A–F) Quantitative RT-PCR analysis of the mRNA level of genes involved in endoplasmic reticulum stress, including (A) PERK, (B) ATF4, (C) Bip, (D) ER57, (E) GADD-34, and (F) Xbp1, from primary hepatocytes isolated from LPGAT1-/- and the WT control mice in response to treatment of serum starvation for 4 hours or oxidative stress by H2O2 (0.5 mmol/L for 4 hours). Data are representative of 3 independent experiments, and represented as means ± SD (n = 5). **P < .01 by 1-way analysis of variance. Ctrl, control.
LPGAT1 Deficiency Leads to Mitochondrial Dysfunction in the Liver
Defective PG remodeling causes mitochondrial dysfunction, which is implicated in the pathogenesis of MEGDEL and Barth syndromes. We next questioned whether LPGAT1 deficiency also would cause mitochondrial dysfunction in the liver because mitochondrial dysfunction is implicated in the pathogenesis of NAFLD. As shown in Figure 11, LPGAT1 deficiency significantly increased the intracellular level of reactive oxygen species (ROS) in cultured primary hepatocytes, which was exacerbated further in response to oxidative stress by H2O2 (Figure 11A). Accordingly, LPGAT1 deficiency also promoted lipid peroxidation, as evidenced by increased levels of thiobarbituric acid reactive substances (TBARS), a byproduct of lipid peroxidation (Figure 11B). Moreover, oxidative stress caused depletion of mitochondrial DNA (mtDNA) copy number in cultured primary hepatocytes from LPGAT1-/- mice (Figure 11C). Consequently, LPGAT1 deficiency significantly impaired the mitochondrial respiration capacity, as evidenced by decreased mitochondrial oxygen consumption rate and blunted responses to different mitochondrial respiratory inhibitors, including oligomycin (an adenosine triphosphatase inhibitor), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP; a mitochondrial uncoupler), and rotenone (a complex І inhibitor) (Figure 11D, quantified in Figure 11E).
Figure 11.

LPGAT1 deficiency causes oxidative stress and mitochondrial dysfunction. (A) Analysis of ROS in cultured primary hepatocytes isolated from LPGAT1-/- mice and WT controls in response to H2O2 treatment (0.5 mmol/L for 4 hours). (B) TBARS assay of lipid peroxidation in the forms of malondialdehyde, a byproduct of lipid peroxidation, in the liver samples from WT and LPGAT1-/- mice. (C) Quantitative real-time PCR analysis of mtDNA copy number in cultured primary hepatocytes from LPGAT1-/- and WT control mice. (D) Seahorse XF-96 analysis of mitochondrial oxygen consumption rate (OCR) in response to treatment with the indicated mitochondrial respiratory chain inhibitors, including oligomycin, FCCP, and rotenone. (E) Quantification of OCR in panel D in response to mitochondrial respiratory chain inhibitor treatment. (F) Confocal imaging analysis of mitochondrial architecture in cultured primary hepatocytes isolated from LPGAT1-/- and WT control mice in response to H2O2 treatment (0.5 mmol/L for 4 hours) or oleic acid stimulation (200 μmol/L for 16 hours). Mitochondria were stained by MitoTracker Red. Fragmented mitochondria are highlighted by arrows. Scale bar: 10 μm. Data are representative of at least 3 independent experiments, and are represented as means ± SD (A–C, n = 4–6; D and E, n = 14). *P < .05, **P < .01, and ***P < .001 by t test or 1-way analysis of variance. Fluro, flurorescence; KO, knockout; ND, normal chow diet; prot, protein.
Oxidative stress disrupts mitochondrial dynamics, which also is implicated in the pathogenies of NAFLD and other aging-related diseases. We next determined the effect of oxidative stress on mitochondrial morphology in cultured primary hepatocytes from LPGAT1-/- mice and the WT controls by confocal imaging analysis. The results showed that LPGAT1 deficiency rendered primary hepatocytes highly sensitive to damage by oxidative stress by H2O2, which disrupted mitochondrial dynamics, leading to mitochondrial fragmentation in primary hepatocytes from LPGAT1-/- mice. Surprisingly, LPGAT1 deficiency also rendered mitochondria more sensitive to damage by lipid overload. The addition of oleic acids to the culture medium severely disrupted mitochondrial dynamics in cultured primary hepatocytes from LPGAT1-/- mice relative to the WT controls, leading to mitochondrial fragmentation (Figure 11F).
LPGAT1 Deficiency Leads to Defective PG and CL Remodeling Commonly Associated With Metabolic Diseases
We next determined the effect of LPGAT1 deficiency on the acyl profiles of PG, CL, and other phospholipids in the liver by lipidomic analysis. The results showed that LPGAT1 deficiency caused similar defects in PG remodeling as observed in MEGDEL syndrome, including a significant decrease in PG-36:2 (Figure 12A). Surprisingly, ablation of LPGAT1 also significantly depleted the content of linoleic acid (C18:2), the major fatty acyl composition of CL in metabolic tissues (Figure 12A, highlighted by a dashed box). Consequently, LPGAT1 deficiency also led to a significant decrease in tetra linoleoyl cardiolipin (TLCL) level in the liver (Figure 12B, highlighted by dashed box, and Figure 12C), a common defect associated with the etiology of NAFLD, obesity, heart failure, and other aging-related diseases.18, 19 In contrast, LPGAT1 deficiency did not significantly change the total levels of PG, CL, and other phospholipids, including phosphatidylserine (PS), phosphatidylethanolamine (PE), and phosphatidylinositol, although there was a moderate decrease in the total level of phosphatidylcholine (Figure 12D) and a slight increase in the total level of phosphatidic acid (Figure 13A and B). Interestingly, LPGAT1 deficiency also significantly changed in the acyl profiles of PS, PE, phosphatidic acid, phosphatidylcholine, and phosphatidylinositol in the liver (Figures 13 and 14). Contrary to changes in PG and CL, LPGAT1 deficiency significantly increased the linoleic acid (C18:2) content in both PS and PE (Figure 14A and B, highlighted by red lined boxes), although the biological significance of these changes remain to be elucidated.
Figure 12.

LPGAT1 deficiency changes the acyl composition of PG and CL. (A) Lipidomics analysis of the acyl compositions of PG in the liver of LPGAT1-/- and WT control mice. PG moieties enriched with linoleic acids (C18:2) are highlighted by a dashed box. (B) Lipidomics analysis of the acyl composition of CL in liver simples from LPGAT1-/- and WT control mice. TLCL is highlighted by a dashed box. (C) Analysis of total TLCL in the liver samples from LPGAT1-/- and WT control mice. (D) Lipidomics analysis of other total phospholipids, including PE, phosphatidylcholine (PC), CL, PG, phosphatidic acid (PA), phosphatidylinositol (PI), and PS in liver samples isolated from LPGAT1-/- and the WT control mice. Data are represented as means ± SD (n = 4). *P < .05 by t test. KO, knockout.
Figure 13.

LPGAT1 deficiency led to significant remodeling of phosphatidic acid (PA), phosphatidylcholine (PC), and phosphatidylinositol (PI). (A–D) Lipidomics analysis of the acyl compositions of (A) PA, (C) PC, and (D) PI in liver samples from LPGAT1-/- and the WT control mice. (B) Analysis of total PA in liver samples isolated from LPGAT1-/- and the WT control mice. Data are represented as means ± SD (n = 4). *P < .05 by t test. KO, knockout.
Figure 14.

LPGAT1 deficiency significantly changed the acyl compositions of PS and PE. (A) Lipidomics analysis of the acyl compositions of PS in the liver samples from LPGAT1-/- and the WT control mice. (B) Lipidomics analysis of the acyl compositions of PE in the liver samples from LPGAT1-/- and the WT control mice. The acyl moieties enriched with linoleic acid (C18:2) are highlighted by red line boxes. Data are represented as means ± SD (n = 4). *P < .05 by t test. KO, knockout.
Discussion
Mitochondrial dysfunction plays a major role in the development of NAFLD, which is increasing because of the ongoing obesity epidemic. There is no effective treatment for this debilitating disorder owing to poor understanding of the pathogenic mechanisms and a lack of suitable drug targets. Moreover, approximately 25% of NAFLD patients are not obese and the importance of mitochondrial dysfunction in these patients remains to be determined. In this study, we identified LPGAT1 as a key regulator of mitochondrial dysfunction in NAFLD, which is supported by multiple lines of evidence. We showed that LPGAT1 deficiency rendered the mice highly susceptible to the development of severe hepatosteatosis, implicating mitochondrial dysfunction as a potential cause for NAFLD. Indeed, LPGAT1 deficiency also caused multiple defects in mitochondrial function in the liver, including oxidative stress, mtDNA depletion, mitochondrial fragmentation, and impaired oxidative phosphorylation. In contrast to a recent report that decreased LPGAT1 expression was associated with severe obesity of Pima Indians,13 LPGAT1-deficient mice are protected from DIO. Our findings are corroborated further by a recent report that mitochondrial dysfunction is the primary determining factor for susceptibility to the onset of NAFLD in obese patients.1 The LPGAT1-deficient mice were born with lower body weight, although they gained a higher percentage of weight after birth. It is interesting to investigate the causative role of LPGAT1 deficiency on body weight gain because the role of PG remodeling on embryonic development is largely unknown.
LPGAT1 catalyzes the remodeling of PG, which plays an important role in maintaining mitochondrial function.3 Defective PG remodeling causes MEGDEL syndrome, as evidenced by genetic mutation of the SERAC1 gene in human beings.8 SERAC1 is a putative enzyme required for PG remodeling. SERAC1 mutations cause hepatopathy, encephalopathy, and hypotonia. In support of LPGAT1 as a key enzyme that regulates PG remodeling, we showed in this study that LPGAT1 deficiency led to abnormal acyl compositions of PG that were highly reminiscent of the defects in MEGDEL syndromes. Consistent with the findings, LPGAT1 deficiency also caused hepatopathy and abnormal cholesterol metabolism commonly associated with MEGDEL syndrome, including dilated hepatic venules, hepatofibrosis, decreased serum cholesterol level, and accumulation of cholesterol in hepatocytes.8 Although a previous report implicated a role of LPGAT1 in triglyceride synthesis as a putative monoacylglycerol acyltransferase (MGAT),12 our data do not support the notion that hepatosteatosis in LPGAT1-/- was caused by impaired MGAT activity because LPGAT1 deficiency promoted lipogenesis in the liver and in cultured primary hepatocytes. In addition, overexpression of LPGAT1 in CV-1 in Origin Simian-7 (COS-7) cells did not promote lipid droplet formation in response to oleic acid stimulation or monoacylglycerol incubation (data not shown), which further indicated that the hepatosteatosis caused by LPGAT1 deficiency was not caused by MGAT activity. Moreover, LPGAT1 deficiency specifically promoted lipid droplet biogenesis in cultured hepatocytes, but not in C2C12 cells, whereas overexpression of MGAT2 stimulated lipid droplet biogenesis.20
PG is a precursor for the synthesis of CL, a mitochondrial signature phospholipid that plays a pivotal role in normal mitochondrial function, including mitochondrial membrane structure, respiration, mitochondrial fusion/fission, and mitophagy. The biological function of CL is determined by the composition of its 4 fatty acyl chains, which are dominated by linoleic acid (C18:2) in metabolic tissues, including liver, heart, and skeletal muscles.21 This unique CL structure, also known as TLCL, is believed to be required for mitochondrial architecture, function, and mitophagy, as evidenced by findings from research on Barth syndrome.11, 22, 23, 24 Consequently, TLCL depletion leads to mitochondrial dysfunction in Barth syndrome, including ROS production, defective oxidative phosphorylation, fatty acid oxidation, adenosine triphosphate production, and mitophagy.10, 11, 19, 23, 25 TLCL depletion also is implicated in mitochondrial dysfunction in obesity, NAFLD, and other aging-related diseases.23, 26, 27 Consistent with this notion, we showed that LPGAT1 deficiency leads to depletion of the TLCL level in the liver, further implicating an important role of PG remodeling by LPGAT1 in NAFLD.
One of the most striking features of the LPGAT1 knockout mice is severe hepatic insulin resistance in the absence of obesity and hyperinsulinemia. Although obesity is the primary cause of insulin resistance, LPGAT1 knockout mice are leaner and had a lower fasting insulin level when compared with WT control mice on a HFD. Then comes the question of how this could happen. The answer comes from the studies of the subcellular localization of LPGAT1. We showed that LPGAT1 is abundantly localized at MAM, the primary hub for insulin signaling, because both the mechanistic target of rapamycin (mTOR) and Akt, the primary downstream targets of insulin signaling, are localized at MAM. Consequently, the mechanistic target of rapamycin complex 2 (mTORC2) deficiency disrupted MAM, causing mitochondrial defects that are dependent on Akt phosphorylation in MAM.17 Disruption of MAM integrity also impaired insulin signaling in mouse and human primary hepatocytes.28 MAM earmarks the site for mitochondrial fission and mtDNA replication,29 a key process required for mitochondrial quality control by eliminating ROS-damaged mitochondria through mitophagy.30 Accordingly, we and others previously have shown that obesity and type 2 diabetes causes dilatation of MAM, leading to mitochondrial fragmentation, ROS production, and insulin resistance.21, 31 In support of a key role of LPGAT1 in regulating insulin signaling in MAM, we showed that LPGAT1 deficiency causes mitochondrial fragmentation, defective mitophagy, and severe insulin resistance. Taken together, our findings support a key role of LPGAT1 in the onset of NAFLD. More importantly, our work has identified LPGAT1 as a novel drug target for the treatment of NAFLD.
Materials and Methods
Reagents
Antibodies used in the present studies included polyclonal antibodies to phospho-Akt (Ser473, 9271), Akt (9272), phospho-glycogen synthase kinase-3α/β (phospho-Gsk-3α/β) (Ser21/9, 9331), and Gsk-3α/β (D75D3, 5676), all of which were purchased from Cell Signaling Technology (Danvers, MA). Rabbit anti–glyceraldehyde-3-phosphate dehydrogenase antibody was from Santa Cruz Biotechnology (A5441; Santa Cruz, CA). Mouse anti–β-actin antibody (A2228) was from Sigma (St. Louis, MO). Horseradish-peroxidase–conjugated donkey anti-rabbit (31460) and donkey anti-mouse (31430) IgG antibodies were purchased from GE Healthcare (Piscataway, NJ). Rat/mouse insulin enzyme-linked immunosorbent assay kit (EZRMI-13K) was purchased from Millipore (Burlington, MA). The TBARS assay kit (10009055) was purchased from Cayman Chemical (Ann Arbor, MI). LabAssay triglyceride (290-63701) and LabAssay cholesterol (294-65801) assay kits were purchased from Wako (Richmond, VA). The Multisource Genomic DNA Miniprep Kit (AP-MN-MS-GDNA) was from Axygen (Union City, CA). Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; C2920), rotenone (R8875), antimycin (A8674), oligomycin (O4876), Oil red O (O0625), and sodium oleate (O7501) were from Sigma. Hoechst 33342 (62249), BODIPY 493/503 (D3922), and MitoTracker Red CMXRos (M7512) were purchased from Invitrogen (Carlsbad, CA). Collagenase IV (17104-019) was purchased from Gibco (Carlsbad, CA).
Generation of Mice With Targeted Deletion of the LPGAT1 Gene
CRISPR/Cas9-mediated gene editing was used to generate LPGAT1 knockout mice. Briefly, guide RNAs targeting the protospacer adjacent motifs of exons of individual LPGAT1 genomic DNA were designed, and 2 protospacer adjacent motifs were chosen for the LPGAT1 gene on exon 3 (Figure 1A). The complementary oligo DNAs were synthesized and then annealed and cloned into pUC-57 under the control of T7 RNA polymerase promoter. The correct constructs were digested by the enzyme DraI, and the purified products were used as the template for the in vitro transcription using T7 high-yield RNA synthesis kit (New England Biolabs, Ipswich, MA). In parallel, the plasmid encoding Cas9, which is driven by the T7 RNA polymerase promoter, was digested by the enzyme PmeI, and the purified products were used as the templates for the in vitro transcription using the mMESSAGE mMACHINE T7 ULTRA kit (Life Technologies, Carlsbad, CA). Both guide RNAs targeting LPGAT1 and Cas9 mRNA were purified using the MEGAclear kit (Life Technologies). A mixture of guide RNAs targeting LPGAT1 (20 ng/μL) and Cas9 mRNA (200 ng/μL) were co-injected into the 1-cell fertilized embryos that were collected from the oviducts of superovulated 7- to 8-week-old B6 mice. The blastocysts were implanted into the uterus of pseudopregnant Institute of Cancer Research (ICR) mice. The peripheral blood was collected from the 4-week-old mice for flow cytometry–aided screening. We further confirmed the LPGAT1 deficiency through PCR amplification, DNA sequencing, RT-PCR, and Western blot analysis. All of the mutant mice were backcrossed with C57BL/6 mice for more than 3 generations.
Animal Care
LPGAT1-/- and age-matched WT mice (4 weeks old) were divided into 2 groups. One group was fed the HFD (D12492, 60 kcal% fat; Research Diets, Inc, New Brunswick, NJ) for 12 weeks, and the control group was fed normal chow diet (Teklad 5001 Laboratory Diet, Envigo, Huntingdon, UK). All animals were maintained in an environmentally controlled facility with a diurnal light cycle and free access to water. All experiments used littermate controls of age- and sex-matched mice, and, in accordance with the “Regulations of the People’s Republic of China on Laboratory Animal Management” and the “Administrative Measures on Quality of Laboratory Animals,” used protocols according to National Institutes of Health guidelines.
Glucose Tolerance Test and Insulin Tolerance Test
The glucose tolerance test and insulin tolerance test were performed in overnight food-deprived mice (n = 10). Glucose was delivered by oral gavage at 1.5 g/kg body weight after initial measurement of the fasting blood glucose level. Insulin was delivered by intraperitoneal injection (1 U/kg body weight; Novolin R, Novo Nordisk, Bagsværd, Denmark). Blood glucose was determined 0, 15, 30, 60, 90, and 120 minutes after the glucose or insulin load with a One Touch Ultra 2 glucometer (Lifescan, Milpitas, CA).
Primary Hepatocyte Isolation
Primary hepatocytes were isolated from male LPGAT1-/- mice and WT control mice (age, 6–10 wk). Mice were anesthetized with pentobarbital sodium, then perfused with 40 mL Krebs buffer (1 mol/L HEPES pH 7.45, 50 mmol/L ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid [EGTA] pH 7.4) from the inferior vena cava for 7 minutes, followed by 30 mL 0.2 mg/mL collagenase type IV (Sigma) in Krebs buffer (with 1 mol/L CaCl2) for 7 minutes. The perfused liver was excised, minced, and filtered through 100 mesh cell strainers (70 mm). The digestion was terminated by adding Dulbecco’s modified Eagle medium (DMEM) (Gibco) containing 10% fetal bovine serum. Hepatocytes were collected by centrifuging at 500g for 2 minutes at 4°C. Percoll (Sigma, St. Louis, MO) solution (10× phosphate-buffered saline [PBS]: Percoll = 1:9, vol/vol) was added and then centrifuged to remove the dead cells. The cell pellet was washed with DMEM twice and the hepatocytes were cultured in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin for further experiments.
Quantitative Real-Time PCR Analysis
Total RNA from LPGAT1-/- and WT mice liver tissues or primary hepatocytes were extracted using TRIzol (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. The purity and the concentration of RNA were detected by an automatic microplate spectrophotometer (OD-1000+ sepectrophotometer, Thermo Fisher Scientific, Waltham, MA). Total RNA (1 μg) was used for the preparation of complementary DNA using SuperScript II Reverse Transcriptase (18064014; Invitrogen). Quantitative real-time PCR analysis was performed using SYBR Green Master Mix (330501; Qiagen, Hilden, Germany). The relative gene expression was calculated as follows: Cycle threshold (Ct) sample = (Ct sample Gene of interest) - (Ct sample house keeping gene). Then, the relative gene expression = 2 power (Ct sample test - Ct sample control). Primer sequences used for quantitative analysis are shown in Table 1.
Table 1.
Primers Used in Real-Time PCR
| Gene | Primer sequence | |
|---|---|---|
| ND1 | Forward | 5′-TGACCCATAGCCATAATATGATTT-3′ |
| Reverse | 5′-CTCTACGTTAAACCCTGATACTAA-3′ | |
| Cyclophilin A | Forward | 5′-ACACGCCATAATGGCACTCC-3′ |
| Reverse | 5′-CAGTCTTGGCAGTGCAGAT-3′ | |
| PPARα | Forward | 5′-CCTCAGGGTACCACTACGGAGT-3′ |
| Reverse | 5′-GCCGAATAGTTCGCCGAA-3′ | |
| SREBP1c | Forward | 5′-GGAGCCATGGATTGCACATT-3′ |
| Reverse | 5′-GGCCCGGGAAGTCACTGT-3′ | |
| ACC1 | Forward | 5′-TGAGGAGG ACCGCATTTATC-3′ |
| Reverse | 5′-GAAGCTTCCTTGGTGACCAG-3′ | |
| Collagen I | Forward | 5′-GAGCGGAGAGTACTGGATCG-3′ |
| Reverse | 5′-GTTCGGGCTGATGTACCAGT-3′ | |
| Collagen III | Forward | 5′-ACCAAAAGGTGATGCTGGAC-3′ |
| Reverse | 5′-GACCTCGTGCTCCAGTTAGC-3′ | |
| Bip | Forward | 5′-TCCTATGTCGCCTTC ACT-3′ |
| Reverse | 5′-ACAGACGGGTCATTCCAC-3′ | |
| PERK | Forward | 5′-CTGTAAGAACCTGGAGCCCAAGT-3′ |
| Reverse | 5′-TCATTGGCTGTGGCATCCAT-3′ | |
| ER57 | Forward | 5′-CTGTAAGAACCTGGAGCCCAAGT-3′ |
| Reverse | 5′-TCATTGGCTGTGGCATCCAT-3′ | |
| Xpbl | Forward | 5′-AAACAGAGTAGCAGCTCAGACTGC-3′ |
| Reverse | 5′-TCCTTCTGGGTAGACCTCTGGGAG-3′ | |
| ATF4 | Forward | 5′-AGGAGTTCGCCTTGGATGCCCTG-3′ |
| Reverse | 5′-AGTGATATCCACTTCACTGCCCAG-3′ | |
| GADD34 | Forward | 5′-GGAGGAAGAGAATCAAGCCA-3′ |
| Reverse | 5′-TGGGGTCGGAGCCTGAAGAT-3′ | |
| GAPDH | Forward | 5′-AATGGTGAAGGTCGGTGTG-3′ |
| Reverse | 5′-GTGGAGTCATACTGGAACATGTAG-3′ | |
| β-actin | Forward | 5′-GGGAAATCGTGCGTGAC-3′ |
| Reverse | 5′-TTGCCAATGGTGATGACCTG-3′ |
Subcellular Fractionation
Cos-7 cells stably overexpressing FLAG-tagged LPGAT1 were homogenized with a Dounce homogenizer in 10 volumes (wt/vol) of solution consisting of 225 mmol/L mannitol, 75 mmol/L sucrose, 0.1 mmol/L EGTA, and 30 mm Tris-HCl, pH 7.4. The homogenate was first centrifuged at 600g for 10 minutes to remove cell debris and nuclear fractions. The crude mitochondrial fraction was obtained by centrifuging the supernatant at 8000g for 10 minutes. The crude mitochondrial pellet was resuspended in mitochondrial suspension buffer consisting of 250 mmol/L mannitol, 5 mmol/L HEPES, pH 7.4, and 0.5 mmol/L EGTA, and then fractionated by Percoll gradient ultracentrifuge at 95,000g for 30 minutes to isolate the pure mitochondrial fraction and MAM. The microsomal fraction was prepared from the postmitochondrial supernatant by sedimentation at 100,000g for 1 hour. The mitochondrial, MAM, and microsomal fractions were resuspended in PBS buffer and analyzed by Western blot analysis using anti-LPGAT1 (1:1000 dilution in 5% bovine serum albumin/tris buffered saline with 0.1% Tween 20 (TBST), generated by our laboratory) and anti-calnexin (NB300-518; Novus Biologicals, Centennial, CO) antibodies, which were used as an endoplasmic reticulum biomarker.
Intracellular ROS Production Analysis
Intracellular ROS generation in primary hepatocytes were investigated using 2’,7’-dichlordehydrofluorescein-diacetate (D399; Molecular Probes, Eugene, OR) at a final concentration of 5 mmol/L. Cells were incubated with 2’,7’-dichlordehydrofluorescein-diacetate in culture medium for 30 minutes at 37°C and then resuspended in 0.5 mL PBS. The fluorescence was measured using a microplate reader (Victor3 plate reader; Perkin Elmer, Waltham, MA) set to 488-nm excitation and 525-nm emission wavelengths.
Lipid Peroxidation Assay
Lipid peroxidation was analyzed from tissue samples by measuring the production of TBARS. TBARS production was measured according to the manufacturer's instructions (TBARS assay kit, cat 10009055; Cayman Chemical). For the preparation of liver cytosol, 25 mg liver tissues were homogenized at 4°C in 250 μL RIPA lysis buffer and placed on ice for 15 minutes, and then centrifuged at 16,000g for 10 minutes. A total of 10 μL of supernatant and 10 μL of the sodium dodecyl sulfate solution were reacted with 400 μL thiobarbituric acid (TBA) buffer at 100°C for 1 hour. After centrifugation at 1600g for 10 minutes, 150 μL of each well was pipetted onto a 96-well plate. Samples were analyzed spectrophotometrically for TBARS at 535 nm in a microplate reader (Victor3 plate reader; Perkin Elmer) and normalized by tissue weight.
Oxygen Consumption Rate in Primary Hepatocytes
Primary hepatocytes were isolated and seeded in XF96 cell culture microplates (Seahorse Bioscience, Billerica, MA) at 5000 cells/well in 80 μL DMEM growth medium supplemented with 10% fetal calf serum and antibiotics, incubated at 37°C for 24 hours. Assays were initiated by removing the growth medium and replacing it with assay medium, incubated for 30 minutes in an ambient air incubator at 37°C. The mitochondria test compounds oligomycin (1.5 μmol/L), FCCP (1 μmol/L), and rotenone (1 μmol/L) were preloaded in the reagent delivery ports of A, B, and C of the O2 sensor cartridge, respectively. Oxygen consumption rate measurements then were performed according to the Seahorse Bioscience assay protocol.
Triglyceride and Cholesterol Assay
Triglyceride and cholesterol levels in serum and liver tissues were measured using the Triglyceride (290-63701; Wako) and Cholesterol (294-65801; Wako) Quantitative Assay Kits, respectively, according to the manufacturer's instructions.
Confocal Imaging Analysis
For intracellular lipid droplet analysis, primary hepatocytes were cultured in completed medium in the presence or absence of 200 μmol/L oleic acid for 16 hours. Cells then were incubated with BODIPY493/503 (5 μg/mL; Life Technologies) for 20 minutes, and analyzed under confocal microscopy (FV1200; Olympus, Shinjuku, Tokyo, Japan). To visualize mitochondria, primary hepatocytes were stained with MitoTracker Red CMXRos (50 nmol/L) for 20 minutes in a 37°C incubator, and then washed with PBS 3 times, followed by confocal imaging analysis.
mtDNA Copy Number Assay
The total DNA of primary hepatocytes was extracted using the Multisource Genomic DNA Miniprep Kit (Axygen) according to the manufacturer's instructions. Quantitative real-time PCR analysis of mtDNA copy number in hepatocytes was performed using mitochondrion-encoded reduced nicotinamide adenine dinucleotide dehydrogenase 1 as the mtDNA marker and cyclophilin A as a genomic DNA marker. The primer pairs used in the PCR analysis are shown in Table 1.
Lipidomics Analysis
For total lipids extraction, liver tissue was homogenized in a 2:1 chloroform:methanol (vol/vol) mixture and cell debris was removed by filtration. The homogenizer and collected cell debris were rinsed with fresh solvent mixture and the rinse was pooled with the previous filtrate before the addition of a 0.73% NaCl water solution, producing a final solvent system of 2:1:0.8 chloroform:methanol:water (vol/vol/vol). The lipid extracts were finally flushed with nitrogen, capped, and stored at -20°C (typically analyzed within 1 week, which is critical for CL analysis). Briefly, total lipids from LPGAT1-/- and WT mice liver tissues were analyzed by triple-quadruple mass spectrometer (Thermo Electron TSQ Quantum Ultra, Trzin, Slovenia) controlled by Xcalibur (Thermo Fisher Scientific) system software. All the mass spectrometer spectra and tandem mass spectrometer spectra were acquired automatically by a customized sequence subroutine operated under Xcalibur software.
Western Blot Analysis
The LPGAT1-/- and WT primary hepatocytes were treated with 0, 0.1, 1.0, or 10 nmol/L insulin for 15 minutes, and harvested in cell RIPA lysis buffer (20 mmol/L HEPES, 2 mmol/L EGTA, 50 mmol/L NaF, 100 mmol/L KCl, 0.2 mmol/L EDTA, 50 mmol/L β-glycerophosphate, 1.5 mmol/L Na3VO4, 10 mmol/L Na4VO7, 1 mmol/L benzamidine, 100 μL phosphatase inhibitor cocktail, 1% Triton X-100, 1.0 mmol/L phenylmethylsulfonyl fluoride), followed by centrifugation at 16,000g for 15 minutes at 4°C. The supernatant was used for Western blot analyses of total Akt, phosphor-Akt, total Gsk-3α/β, phospho-Gsk-3α/β, and glyceraldehyde-3-phosphate dehydrogenase. For analysis of insulin signaling from tissue samples, LPGAT1-/- mice and WT controls were fasted overnight, followed by intraperitoneal injection of insulin (1 U/kg body weight) or PBS, and then were euthanized 15 minutes after the injection. Tissues rapidly were dissected and frozen in liquid nitrogen. The tissue samples then were pulverized in liquid nitrogen and homogenized in the RIPA buffer with a polytron. After 30 minutes of incubation on ice, the samples were centrifuged at 16,000g for 15 minutes at 4°C. The protein concentration was determined by the Pierce BCA Protein Assay (23225; Thermo Fisher, Waltham, MA). Equal amounts of protein (30 μg) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to a polyvinylidene difluoride membrane (1620177; Bio-Rad, Hercules, CA), blocked in Tris-buffered saline with 5% milk, immunoblotted with primary antibodies (1:1000) overnight at 4°C, followed by secondary antibodies (1:5000) for 1 hour at room temperature, and developed with ECL Western Blotting Substrate (32106; Thermo Fisher).
Statistical Analysis
Statistical comparisons were performed using 2-tailed nonpaired t tests or 1-way analysis of variance to determine the difference between LPGAT1-/- and WT mice and primary hepatocytes. Values were considered statistically significant at P < .05. Data are represented as means ± SD. All authors had access to the study data and reviewed and approved the final manuscript.
Footnotes
Author Contributions: Yuguang Shi conceived the project, formulated the research, and designed the experiments; Xiaoyang Zhang, Jun Zhang, and Haoran Sun performed the main experiments and collected the data; Xiaoyang Zhang, Jun Zhang, Haoran Sun, Xueling Liu, Dan Xu, Dandan Jia, and Jia Nie analyzed the data; Jianing Wang performed the lipidomic analysis; Yue Zheng, Xianlin Han, and Feng Liu provided critical revision of the manuscript for important intellectual content; and Yuguang Shi and Jun Zhang wrote the manuscript and all authors edited it.
Conflicts of interest The authors disclose no conflicts.
Funding This work was supported in part by grants from the American Diabetes Association (1-18-IBS-329), the National Institutes of Health (R01DK076685), the Joe and Teresa Long Endowment, the Barth Syndrome Foundation, and the National Natural Science Foundation of China (31771309) (all Y.S.).
References
- 1.Koliaki C., Szendroedi J., Kaul K., Jelenik T., Nowotny P., Jankowiak F., Herder C., Carstensen M., Krausch M., Knoefel W.T., Schlensak M., Roden M. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Chen D., Zhang X.-Y., Shi Y. Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem J. 2006;398:169–176. doi: 10.1042/BJ20060303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y., Cao J., Shi Y. Identification and characterization of a gene encoding human LPGAT1, an endoplasmic reticulum-associated lysophosphatidylglycerol acyltransferase. J Biol Chem. 2004;279:55866–55874. doi: 10.1074/jbc.M406710200. [DOI] [PubMed] [Google Scholar]
- 4.Nie J., Hao X., Chen D., Han X., Chang Z., Shi Y. A novel function of the human CLS1 in phosphatidylglycerol synthesis and remodeling. Biochim Biophys Acta. 2010;1801:438–445. doi: 10.1016/j.bbalip.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawasaki K., Kuge O., Chang S.C., Heacock P.N., Rho M., Suzuki K., Nishijima M., Dowhan W. Isolation of a Chinese hamster ovary (CHO) cDNA encoding phosphatidylglycerophosphate (PGP) synthase, expression of which corrects the mitochondrial abnormalities of a PGP synthase-defective mutant of CHO-K1 cells. J Biol Chem. 1999;274:1828–1834. doi: 10.1074/jbc.274.3.1828. [DOI] [PubMed] [Google Scholar]
- 6.Zhong Q., Gohil V.M., Ma L., Greenberg M.L. Absence of cardiolipin results in temperature sensitivity, respiratory defects, and mitochondrial DNA instability independent of pet56. J Biol Chem. 2004;279:32294–32300. doi: 10.1074/jbc.M403275200. [DOI] [PubMed] [Google Scholar]
- 7.Gorden D.L., Ivanova P.T., Myers D.S., McIntyre J.O., VanSaun M.N., Wright J.K., Matrisian L.M., Brown H.A. Increased diacylglycerols characterize hepatic lipid changes in progression of human nonalcoholic fatty liver disease; comparison to a murine model. PLoS One. 2011;6:e22775. doi: 10.1371/journal.pone.0022775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wortmann S.B., Vaz F.M., Gardeitchik T., Vissers L.E., Renkema G.H., Schuurs-Hoeijmakers J.H., Kulik W., Lammens M., Christin C., Kluijtmans L.A., Rodenburg R.J., Nijtmans L.G., Grunewald A., Klein C., Gerhold J.M., Kozicz T., van Hasselt P.M., Harakalova M., Kloosterman W., Baric I., Pronicka E., Ucar S.K., Naess K., Singhal K.K., Krumina Z., Gilissen C., van Bokhoven H., Veltman J.A., Smeitink J.A., Lefeber D.J., Spelbrink J.N., Wevers R.A., Morava E., de Brouwer A.P. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. 2012;44:797–802. doi: 10.1038/ng.2325. [DOI] [PubMed] [Google Scholar]
- 9.Claypool S.M., Koehler C.M. The complexity of cardiolipin in health and disease. Trends Biochem Sci. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y.W., Claypool S.M. Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front Genet. 2015;6:3. doi: 10.3389/fgene.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu P., Shi Y. Regulation of autophagy by mitochondrial phospholipids in health and diseases. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:114–129. doi: 10.1016/j.bbalip.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hiramine Y., Emoto H., Takasuga S., Hiramatsu R. Novel acyl-coenzyme A:monoacylglycerol acyltransferase plays an important role in hepatic triacylglycerol secretion. J Lipid Res. 2010;51:1424–1431. doi: 10.1194/jlr.M002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Traurig M.T., Orczewska J.I., Ortiz D.J., Bian L., Marinelarena A.M., Kobes S., Malhotra A., Hanson R.L., Mason C.C., Knowler W.C., Bogardus C., Baier L.J. Evidence for a role of LPGAT1 in influencing BMI and percent body fat in Native Americans. Obesity (Silver Spring) 2013;21:193–202. doi: 10.1002/oby.20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vickers K.C., Moore K.J. Small RNA overcomes the challenges of therapeutic targeting of microsomal triglyceride transfer protein. Circ Res. 2013;113:1189–1191. doi: 10.1161/CIRCRESAHA.113.302732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soh J., Iqbal J., Queiroz J., Fernandez-Hernando C., Hussain M.M. MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat Med. 2013;19:892–900. doi: 10.1038/nm.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irani S., Pan X., Peck B.C., Iqbal J., Sethupathy P., Hussain M.M. MicroRNA-30c mimic mitigates hypercholesterolemia and atherosclerosis in mice. J Biol Chem. 2016;291:18397–18409. doi: 10.1074/jbc.M116.728451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Betz C., Stracka D., Prescianotto-Baschong C., Frieden M., Demaurex N., Hall M.N. Feature article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A. 2013;110:12526–12534. doi: 10.1073/pnas.1302455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zorzano A., Liesa M., Sebastian D., Segales J., Palacin M. Mitochondrial fusion proteins: dual regulators of morphology and metabolism. Semin Cell Dev Biol. 2010;21:566–574. doi: 10.1016/j.semcdb.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Li J., Romestaing C., Han X., Li Y., Hao X., Wu Y., Sun C., Liu X., Jefferson L.S., Xiong J., Lanoue K.F., Chang Z., Lynch C.J., Wang H., Shi Y. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12:154–165. doi: 10.1016/j.cmet.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J., Xu D., Nie J., Cao J., Zhai Y., Tong D., Shi Y. Monoacylglycerol acyltransferase-2 is a tetrameric enzyme that selectively heterodimerizes with diacylglycerol acyltransferase-1. J Biol Chem. 2014;289:10909–10918. doi: 10.1074/jbc.M113.530022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J., Liu X., Wang H., Zhang W., Chan D.C., Shi Y. Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression. Proc Natl Acad Sci U S A. 2012;109:6975–6980. doi: 10.1073/pnas.1120043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlame M., Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006;580:5450–5455. doi: 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 23.Shi Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J Biomed Res. 2010;24:6–15. doi: 10.1016/S1674-8301(10)60003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu P., Liu X., Zhang J., Wang H.G., Ye J.M., Shi Y. Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy. 2015;11:643–652. doi: 10.1080/15548627.2015.1023984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han X., Yang J., Yang K., Zhao Z., Abendschein D.R., Gross R.W. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry. 2007;46:6417–6428. doi: 10.1021/bi7004015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paradies G., Petrosillo G., Paradies V., Ruggiero F.M. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic Biol Med. 2010;48:1286–1295. doi: 10.1016/j.freeradbiomed.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 27.Shigenaga M.K., Hagen T.M., Ames B.N. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tubbs E., Theurey P., Vial G., Bendridi N., Bravard A., Chauvin M.A., Ji-Cao J., Zoulim F., Bartosch B., Ovize M., Vidal H., Rieusset J. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes. 2014;63:3279–3294. doi: 10.2337/db13-1751. [DOI] [PubMed] [Google Scholar]
- 29.Lewis S.C., Uchiyama L.F., Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science. 2016;353:aaf5549. doi: 10.1126/science.aaf5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Twig G., Elorza A., Molina A.J., Mohamed H., Wikstrom J.D., Walzer G., Stiles L., Haigh S.E., Katz S., Las G., Alroy J., Wu M., Py B.F., Yuan J., Deeney J.T., Corkey B.E., Shirihai O.S. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tubbs E., Chanon S., Robert M., Bendridi N., Bidaux G., Chauvin M.A., Ji-Cao J., Durand C., Gauvrit-Ramette D., Vidal H., Lefai E., Rieusset J. Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes. 2018;67:636–650. doi: 10.2337/db17-0316. [DOI] [PubMed] [Google Scholar]