

Abstract
DNA‐protein crosslinks (DPCs) are highly cytotoxic lesions that obstruct essential DNA transactions and whose resolution is critical for cell and organismal fitness. However, the mechanisms by which cells respond to and overcome DPCs remain incompletely understood. Recent studies unveiled a dedicated DPC repair pathway in higher eukaryotes involving the SprT‐type metalloprotease SPRTN/DVC1, which proteolytically processes DPCs during DNA replication in a ubiquitin‐regulated manner. Here, we show that chemically induced and defined enzymatic DPCs trigger potent chromatin SUMOylation responses targeting the crosslinked proteins and associated factors. Consequently, inhibiting SUMOylation compromises DPC clearance and cellular fitness. We demonstrate that ACRC/GCNA family SprT proteases interact with SUMO and establish important physiological roles of Caenorhabditis elegans GCNA‐1 and SUMOylation in promoting germ cell and embryonic survival upon DPC formation. Our findings provide first global insights into signaling responses to DPCs and reveal an evolutionarily conserved function of SUMOylation in facilitating responses to these lesions in metazoans that may complement replication‐coupled DPC resolution processes.
Keywords: DNA repair, DNA‐protein crosslinks, post‐translational modifications, proteomics, SUMO
Subject Categories: DNA Replication, Repair & Recombination; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The integrity and conservation of DNA are critical for the viability and fitness of cells and organisms. To mitigate the threat to genome stability posed by incessant genotoxic insults by endogenous and exogenous sources, cells launch a global DNA damage response (DDR), a complex network of processes that cooperatively promote efficient sensing and repair of different lesions (Jackson & Bartek, 2009; Ciccia & Elledge, 2010). While the repair systems for most types of DNA damage are now well understood, relatively little is known about how cells respond to and repair DNA‐protein crosslinks (DPCs). DPCs occur frequently and can be classified into two categories, based on their nature and origin. First, enzymatic DPCs arise as a consequence of abortive actions of DNA‐modifying enzymes, such as topoisomerases, which form covalent intermediates with DNA as part of their catalytic mechanism. Second, radiation and reactive chemicals, most prominently aldehydes, generate non‐enzymatic DPCs involving proteins residing in the vicinity of DNA (Barker et al, 2005; Ide et al, 2011; Stingele et al, 2017). In fact, formaldehyde, a potent DPC inducer and genotoxin, is generated in direct proximity to DNA as a byproduct of histone and DNA demethylation (Walport et al, 2012). Due to their large size, DPCs can form impassable roadblocks to essential DNA transactions including DNA replication and transcription and are therefore highly cytotoxic (Fu et al, 2011; Nakano et al, 2012, 2013).
The toxicity and broad diversity of DPCs that can be formed likely necessitate a flexible repertoire of cellular mechanisms for processing these lesions. Until recently, however, the pathways underlying DPC repair in eukaryotic cells remained largely elusive, due in part to the heterogeneity of DPCs and a lack of efficient approaches for the generation of defined DPCs in cells that allow for unambiguous dissection of the repair mechanisms for these lesions. Early studies suggested that repair of DPCs involves nucleotide excision repair (NER) and homologous recombination (HR), two non‐DPC‐specific repair pathways, at least in some contexts (reviewed in Ide et al, 2011). However, more recent findings in yeast, frogs, and mammals revealed the existence of a dedicated, evolutionarily conserved DPC repair mechanism involving a specialized DNA‐activated protease, known as Wss1 in yeast and SPRTN/DVC1 in higher eukaryotes, which processes DPCs proteolytically during DNA replication via an SprT‐type metalloprotease domain (Duxin et al, 2014; Stingele et al, 2014, 2015, 2016; Lopez‐Mosqueda et al, 2016; Vaz et al, 2016). Such SprT protease‐mediated trimming of proteins covalently trapped on DNA may facilitate the passage of the replication machinery and enable the subsequent, full removal of adducted peptide remnants by excision repair (Duxin et al, 2014). A critical role of SPRTN for genome stability, longevity, and health in mammals was demonstrated by recent findings that SPRTN is essential in mice, and that patients with hypomorphic SPRTN mutations, the underlying genetic determinant of Ruijs‐Aalfs syndrome, manifest with a progeroid phenotype and early‐onset cancer (Lessel et al, 2014; Maskey et al, 2014).
In human cells, the function of SPRTN in processing DPCs is centrally regulated by ubiquitin, involving a C‐terminal ubiquitin‐binding UBZ domain responsible for both its ubiquitin‐mediated recruitment to DNA damage sites and a regulatory switch controlling SPRTN proteolytic activity via DPC‐sensitive SPRTN monoubiquitylation (Centore et al, 2012; Davis et al, 2012; Mosbech et al, 2012; Lopez‐Mosqueda et al, 2016; Stingele et al, 2016; Vaz et al, 2016). By contrast, Wss1 lacks recognizable ubiquitin‐binding domains and instead contains SUMO‐interacting motifs (SIMs) that may facilitate its targeting to and processing of DPCs (Stingele et al, 2014; Balakirev et al, 2015). While no role of SUMO in regulating SPRTN function has been described, a function of the SUMO E3 ligase ZNF451 (ZATT) in facilitating the resolution of topoisomerase 2 (TOP2) cleavage complexes via tyrosyl‐DNA phosphodiesterase 2 (TDP2) that specifically removes TOP2 DNA adducts was recently reported (Schellenberg et al, 2017). However, whether this mechanism is restricted to this unique type of DNA double‐strand break‐associated DPC and precisely how signaling via ubiquitin and SUMO mechanistically underpins and regulates DPC repair processes remain unclear.
Here, we uncovered and characterized on a system‐wide level a dynamic SUMO‐dependent signaling response to DPCs in human cells, impacting the trapped proteins and associated chromatin‐bound factors to facilitate DPC clearance and cellular fitness. We demonstrate that uncharacterized SprT metalloproteases belonging to the ACRC/GCNA‐1 family interact with SUMO and that Caenorhabditis elegans GCNA‐1 promotes organismal survival upon DPC formation in conjunction with SUMOylation. Collectively, our findings provide first insights into post‐translational modification‐driven signaling responses to DPCs on a global scale and suggest a central role of SUMOylation in pathways of DPC recognition and processing that may complement DNA replication‐coupled mechanisms for resolving these lesions.
Results
Formaldehyde triggers a dynamic chromatin SUMOylation response in human cells
To explore the involvement of SUMO in cellular responses to DPCs, we first analyzed overall SUMOylation profiles of human cells exposed to the potent DPC inducer formaldehyde (McGhee & von Hippel, 1977). Strikingly, unlike a range of other genotoxic agents including ionizing radiation (IR), UV, and hydroxyurea (HU), formaldehyde elicited a prominent SUMOylation response involving both SUMO1 and SUMO2/3, which specifically impacted chromatin‐associated but not soluble proteins and correlated with the extent of DPC formation (Figs 1A–D and EV1A). This effect was apparent at formaldehyde concentrations that only modestly exceed those of human blood (100–150 μM; Luo et al, 2001) and was accompanied by formation of nuclear SUMO2/3 foci colocalizing with PML bodies in virtually all interphase cells (Figs 1B, and EV1B and C), indicating that it was not specifically coupled to DNA replication. Indeed, the DNA polymerase inhibitor aphidicolin had no effect on formaldehyde‐stimulated SUMOylation, which was also unaffected by inhibition of transcription (Fig EV1D). Like formaldehyde, treatment of cells with acetaldehyde led to increased chromatin SUMOylation (Fig EV1E). While aldehydes are potential sources of DNA interstrand crosslinks (ICLs; Langevin et al, 2011), known ICL‐inducing agents including cisplatin and mitomycin C, which unlike formaldehyde did not trigger strong DPC formation under our experimental conditions, did not markedly increase chromatin‐associated SUMO2/3 conjugates and nuclear SUMO2/3 foci (Fig 1A–C), suggesting their formation is not primarily a consequence of ICL formation. Notably, the formaldehyde‐induced increase in chromatin SUMOylation was fully reversible, declining sharply to levels comparable to those of unperturbed cells within hours after formaldehyde withdrawal (Fig 1D). Consistent with a role of formaldehyde‐induced chromatin‐associated SUMO modifications in the response to DPCs, reducing overall DPC repair capacity through depletion of the DPC protease SPRTN delayed their reversal (Fig 1E).
Figure 1. Formaldehyde triggers a dynamic chromatin SUMOylation response.
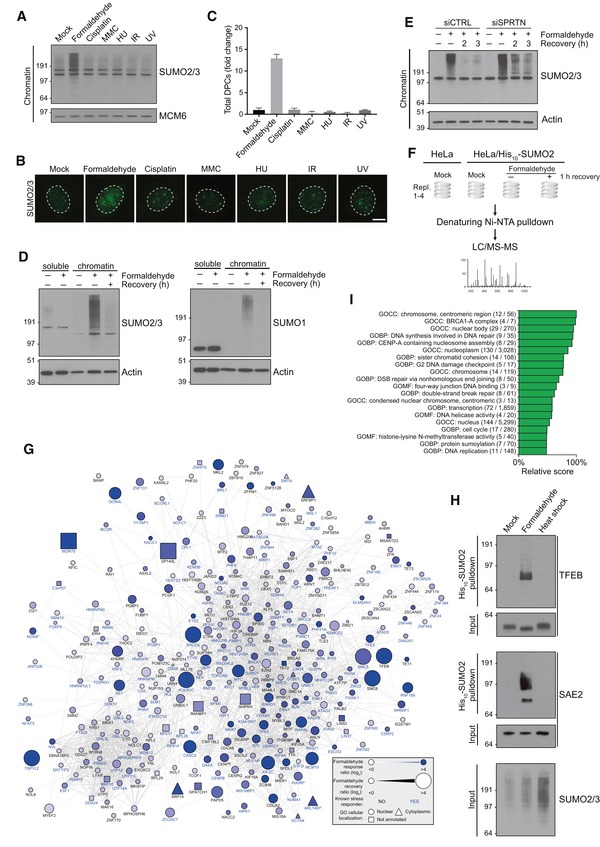
- Chromatin‐enriched fractions of HeLa cells exposed to the indicated genotoxic agents for 1 h were subjected to immunoblot analysis using SUMO2/3 antibody.
- As in (A), except that cells were preextracted in 0.2% Triton X‐100, fixed, and immunostained with SUMO2/3 antibody. Representative images are shown. Scale bar, 5 μm.
- Relative DPC levels in cells treated as in (A) were quantified using a KCl/SDS precipitation assay (mean ± SD; n = 3 independent experiments).
- HeLa cells were treated with formaldehyde for 1 h, and where indicated, propagated for an additional h in the absence of formaldehyde (recovery). Cells were then fractionated into soluble and chromatin‐enriched fractions and immunoblotted with SUMO2/3 and SUMO1 antibodies.
- As in (D), but using HeLa cells transfected with non‐targeting control (CTRL) or SPRTN siRNAs.
- Experimental set‐up for global proteomic analysis of formaldehyde‐induced SUMOylation changes.
- Mass spectrometry‐based analysis of formaldehyde‐induced SUMOylation changes. His10‐SUMO2 conjugates from HeLa/His10‐SUMO2 cells subjected or not to formaldehyde as shown in (F) were purified on Ni‐NTA under stringent conditions and analyzed by mass spectrometry. All proteins displaying significant upregulation of SUMOylation in response to formaldehyde treatment (Table EV1) were subjected to network analysis using the STRING database, at the default interaction confidence setting of 0.4. Proteins not connected to the network were omitted.
- SUMO2 conjugates from HeLa/His10‐SUMO2 cells subjected or not to formaldehyde or heat stress for 1 h were purified as in (G) and immunoblotted with indicated antibodies.
- SUMO target proteins displaying at least 2.5‐fold upregulation of SUMOylation after formaldehyde treatment were mapped to the human proteome, which was annotated with Gene Ontology (GO) terms. Enrichment analysis was performed to find terms significantly enriched for formaldehyde‐induced SUMOylated proteins, using Fisher exact testing with multiple‐hypothesis correction to achieve a final q‐value of < 0.02. A relative score was derived from a combination of the enrichment ratio and the q‐value. A full list of all enriched terms is available (Table EV2). GOBP, GO biological processes; GOCC, GO cellular compartments; GOMF, GO molecular functions.
Source data are available online for this figure.
Figure EV1. Characterization of aldehyde‐induced chromatin SUMOylation.
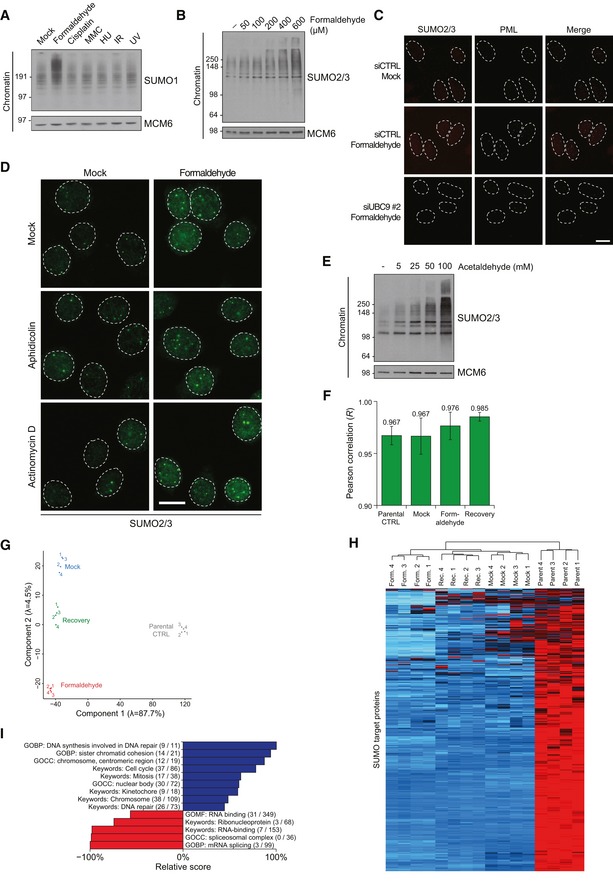
- Chromatin‐enriched fractions of HeLa cells exposed to the indicated genotoxic agents for 1 h were subjected to immunoblot analysis using antibodies to SUMO1 and MCM6 (loading control).
- Chromatin‐enriched fractions of HeLa cells exposed to the indicated doses of formaldehyde for 1 h were subjected to immunoblot analysis using SUMO2/3 and MCM6 antibodies.
- HeLa cells transfected with non‐targeting control (CTRL) or UBC9 siRNAs were left untreated or exposed to formaldehyde, preextracted and fixed 1 h later, and co‐immunostained with SUMO2/3 and PML antibodies. Representative images are shown. Scale bar, 10 μm.
- HeLa cells incubated in the presence of aphidicolin or actinomycin D to inhibit DNA replication or transcription, respectively, were left untreated or exposed to formaldehyde for 1 h. Cells were then preextracted, fixed, and immunostained with SUMO2/3 antibody. Representative images are shown. Scale bar, 10 μm.
- Chromatin‐enriched fractions of U2OS cells exposed to the indicated doses of acetaldehyde for 1 h were subjected to immunoblot analysis using antibodies to SUMO2/3 and MCM6 (loading control).
- Average Pearson correlation between same‐condition experimental replicates for global proteomic analysis of formaldehyde‐induced SUMOylation changes (Fig 1F), derived from scatter plot analysis of log2‐transformed intensity values. Error bars indicate standard deviation.
- Principal component analysis (PCA) of all experimental replicates for global proteomic analysis of formaldehyde‐induced SUMOylation changes (Fig 1F), visualizing the degree of overall variability between all experiments. Eigenvalues are displayed on the axes.
- Unsupervised hierarchical clustering analysis of Z‐scored log2‐transformed intensity values corresponding to all identified SUMO target proteins (Fig 1F; Table EV1), highlighting clusters of proteins dynamically SUMOylated in response to specific treatments. Red: < 1 standard deviations (SD); black: 0 SD; blue: 1 SD; cyan: > 2 SD.
- SUMO target proteins displaying at least 2.5‐fold upregulation of SUMOylation after formaldehyde treatment were mapped to all SUMOylated proteins identified (Fig 1F; Table EV1), which were annotated with Gene Ontology (GO) terms. Enrichment analysis was performed to find terms significantly enriched (blue) or depleted (red) for formaldehyde‐induced SUMOylated proteins, using Fisher exact testing with multiple‐hypothesis correction to achieve a final q‐value of < 0.02. A relative score was derived from a combination of the enrichment ratio and the q‐value. Selected GO terms are shown. See also Table EV2. GOBP, GO biological processes; GOCC, GO cellular compartments; GOMF, GO molecular functions.
Source data are available online for this figure.
To gain more insight into the dynamic chromatin SUMOylation response elicited by formaldehyde, we employed an unbiased proteomic approach to map the cellular proteins modified by formaldehyde‐regulated SUMOylation. To this end, SUMO2 conjugates from HeLa cells stably expressing His10‐SUMO2 that were exposed or not to formaldehyde were purified under stringent conditions (Hendriks & Vertegaal, 2016) and analyzed by mass spectrometry (MS) using label‐free quantification (Fig 1F; Cox et al, 2014). Pearson correlation showed very high reproducibility between four individual biological replicates (Fig EV1F–H), and a total of 1,041 SUMO target proteins were identified (Table EV1). Among these, 396 proteins consistently showed increased SUMOylation in response to formaldehyde exposure (FDR < 0.05; Fig 1G; Table EV1), a large majority (97.3%) of which were annotated as nuclear proteins (indicated by circles, Fig 1G). Like the overall SUMO response, the SUMOylation status of most of these factors returned to baseline levels upon brief recovery from formaldehyde treatment (indicated by small symbols, Fig 1G). Interestingly, among the 396 proteins showing formaldehyde‐stimulated SUMOylation, around 46% (182 proteins, names indicated in blue, Fig 1G) had not previously been identified across a range of similar‐sized SUMO proteomic studies as targets of SUMOylation induced by cell stresses including heat shock, proteasome inhibition, and different types of DNA damage, indicating that formaldehyde triggers a cellular stress response that is qualitatively distinct from these insults. Biochemical analysis of selected factors among this latter group of proteins confirmed their selective SUMOylation in response to formaldehyde but not heat shock (Fig 1H). GO term analysis of proteins displaying formaldehyde‐stimulated SUMOylation revealed selective enrichment of factors involved in DNA‐, chromatin‐, and cell cycle‐associated processes (Figs 1I and EV1I; Table EV2). Together, these findings suggest that formaldehyde triggers a highly dynamic chromatin SUMOylation response with a potential role in DPC repair processes.
SUMOylation directly targets defined DNMT1 DPCs
While formaldehyde is a potent inducer of DPCs, it also generates other forms of macromolecular damage, including protein–protein crosslinks. To definitively establish whether proteins covalently trapped on DNA are targeted for SUMOylation, we sought to monitor SUMO signaling in response to more defined DPCs in human cells. To this aim, we utilized the notion that DNA methyltransferases, in particular DNMT1 that maintains DNA methylation patterns in newly replicated DNA, undergo covalent crosslinking to DNA when acting on the cytosine analog 5′‐aza‐2‐deoxycytidine (5‐azadC), which can be efficiently incorporated into chromosomal DNA during replication (Fig 2A; Du et al, 2015; Maslov et al, 2012). Strikingly, treatment of cells with 5‐azadC triggered a marked DNA replication‐dependent increase in chromatin‐associated SUMO conjugates, accompanied by formation of nuclear SUMO2/3 foci, that was almost entirely abrogated by knockdown of DNMT1 (Figs 2B and C, and EV2A). By contrast, DNMT1 depletion had no impact on formaldehyde‐induced chromatin SUMOylation, as expected (Fig EV2A). These findings suggested that the 5‐azadC‐induced chromatin SUMOylation response was largely a direct consequence of DNMT1 DPC formation. Consistently, the cytotoxicity of 5‐azadC was alleviated by knockdown of DNMT1 (Fig 2D). We reasoned that crosslinked DNMT1 molecules might be direct targets of 5‐azadC‐induced SUMO modification. Indeed, 5‐azadC treatment led to strongly elevated SUMOylation of wild type DNMT1 but not a catalytically inactive mutant unable to engage in DPC formation (Fig 2E and F; Schermelleh et al, 2005), suggesting that SUMOylation might play a role in marking proteins covalently trapped on DNA for recognition by DPC‐processing factors.
Figure 2. DPCs are direct targets of SUMO‐dependent modification.
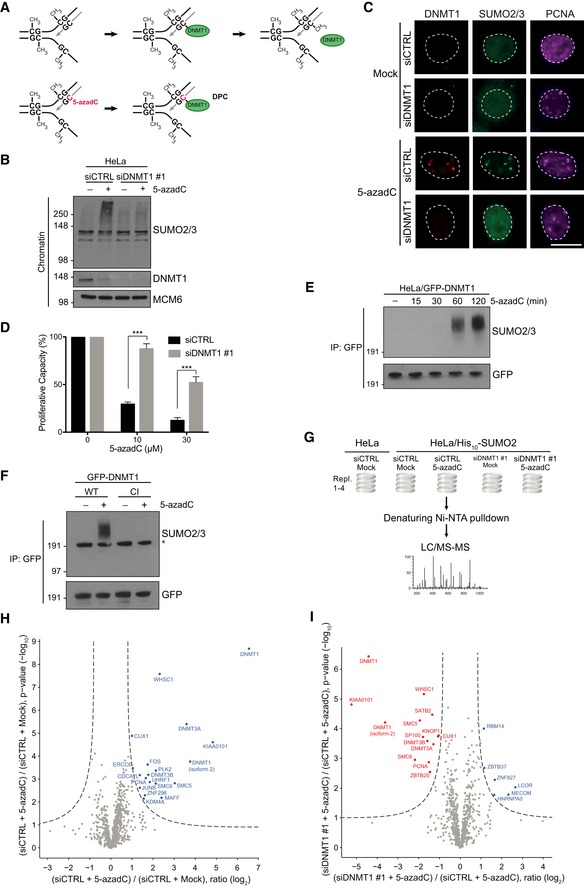
- Principle of 5‐azadC‐induced DNMT1 DPC formation. See main text for details.
- HeLa cells were transfected with non‐targeting control (CTRL) or DNMT1 siRNAs, exposed or not to 5‐azadC, and collected 2 h later. Chromatin‐enriched fractions were immunoblotted with antibodies to SUMO2/3, DNMT1, and MCM6 (loading control).
- Representative images of HeLa cells that were treated as in (B) and co‐immunostained with antibodies to DNMT1, SUMO2/3, and PCNA. Scale bar, 10 μm.
- Proliferative capacity of HeLa/GFP‐DNMT1 cells transfected with indicated siRNAs and exposed to the indicated 5‐azadC doses for 2 h was assayed by measuring cell proliferation with the SRB assay (mean ± SEM; n = 3 independent experiments; ***P < 0.001, Student's t‐test).
- Extracts of HeLa/GFP‐DNMT1 cells treated with 5‐azadC for the indicated times were subjected to GFP immunoprecipitation (IP) under denaturing conditions followed by immunoblotting with SUMO2/3 and GFP antibodies.
- HeLa cells were subjected to consecutive rounds of transfection with DNMT1 siRNA targeting the UTR and expression plasmid encoding WT or catalytically inactive (CI) GFP‐DNMT1. Cells were then left untreated or incubated with 5‐azadC for 2 h, and SUMOylation of GFP‐DNMT1 was analyzed as in (E). Asterisk denotes unmodified GFP‐DNMT1 recognized by the SUMO2/3 antibody due to weak cross‐reactivity.
- Experimental set‐up for global proteomic analysis of 5‐azadC‐induced SUMOylation changes.
- Mass spectrometry analysis of 5‐azadC‐induced SUMOylation changes. His10‐SUMO2 conjugates from HeLa/His10‐SUMO2 cells subjected or not to 5‐azadC as shown in (G) were purified on Ni‐NTA under stringent conditions and analyzed by mass spectrometry. Proteins displaying significantly altered SUMOylation in response to 5‐azadC treatment relative to the control condition were visualized through two‐sample t‐testing using permutation‐based FDR to achieve q‐values of < 0.05 (Table EV3).
- As in (H), except that SUMO target proteins displaying altered SUMOylation upon DNMT1 knockdown are shown. Both control (CTRL) and DNMT1 siRNA‐transfected cells were treated with 5‐azadC.
Source data are available online for this figure.
Figure EV2. Characterization of 5‐azadC‐induced DNMT1 DPC formation, SUMOylation, and resolution.

- Chromatin‐enriched fractions of HeLa cells transfected with non‐targeting control (CTRL) or DNMT1 siRNAs and treated with formaldehyde, 5‐azadC, and the DNA replication inhibitor aphidicolin as indicated for 1 h were immunoblotted with SUMO2/3, DNMT1, and MCM6 antibodies.
- Average Pearson correlation between same‐condition experimental replicates for global proteomic analysis of 5‐azadC‐induced SUMOylation changes (Fig 2G), derived from scatter plot analysis of log2‐transformed intensity values. Error bars indicate standard deviation.
- HeLa cells stably expressing GFP‐DNMT1 were transfected with indicated siRNAs, treated with 5‐azadC for 2 h, and then preextracted in stringent preextraction buffer and fixed at the indicated time points. Representative images are shown. Scale bar, 20 μm. See also Fig 3B.
- HeLa cells stably expressing GFP‐DNMT1 were transfected with indicated siRNAs, treated with 5‐azadC for 2 h, and then preextracted in stringent preextraction buffer and fixed at the indicated time points. Mean GFP signal was determined by quantitative image analysis (> 1,000 cells analyzed per condition). Data from a representative experiment are shown.
- Immunoblot analysis of U2OS cells transfected with non‐targeting control (CTRL), SPRTN, or ZNF451 siRNAs, using antibodies to SPRTN, ZNF451, and actin (loading control).
Source data are available online for this figure.
To further characterize the 5‐azadC‐induced SUMOylation response, we profiled global SUMOylation changes resulting from exposure to 5‐azadC, using a purification and MS scheme similar to that employed for mapping formaldehyde‐regulated SUMOylation processes (Fig 2G). In agreement with our biochemical observations, label‐free MS‐based quantification of four independent biological replicates with very high reproducibility revealed that 5‐azadC triggered a massive (~ 94‐fold) increase in DNMT1 SUMOylation levels and that DNMT1 itself was the major cellular substrate of 5‐azadC‐stimulated SUMOylation (Figs 2H and EV2B; Table EV3). Additional proteins were also subject to such modification, although as compared to the impact of formaldehyde‐generated non‐specific DPCs the range of proteins displaying 5‐azadC‐dependent SUMOylation was highly restricted ([Link], [Link]). These included the de novo DNA methyltransferases DNMT3A and DNMT3B, which like DNMT1 undergo direct 5‐azadC‐dependent DPC formation but play back‐up roles in replication‐coupled DNA methylation (Du et al, 2015); the DNMT1 partner protein UHRF1, which is essential for DNMT1‐dependent DNA methylation (Du et al, 2015); and PCNA and the PCNA‐interacting protein KIAA0101/PAF15, possibly reflecting the coupling of DNMT1‐mediated DNA methylation to its association with PCNA at the replication fork. Consistent with our biochemical observations (Fig 2B), the proteomic analysis further validated that the majority of 5‐azadC‐stimulated SUMOylation events were effectively suppressed by DNMT1 knockdown, implying their dependency on DNMT1 DPC formation (Fig 2I; Table EV3). Collectively, these results show that DNMT1 DPC formation triggers a prominent SUMOylation response that directly impacts the trapped proteins and associated factors.
SUMOylation is required for resolution of DNMT1‐DNA adducts
We next asked whether SUMOylation plays a role in promoting the resolution of DPCs to protect against their cytotoxicity. To explore this, we monitored the impact of inhibiting SUMOylation on the clearance of defined 5‐azadC‐induced DNMT1 DPCs, using detergent‐resistant GFP fluorescence intensity as readout for crosslinked GFP‐DNMT1 molecules in cells stably expressing this transgene (Schermelleh et al, 2005). Inhibition of 5‐azadC‐induced chromatin SUMOylation by knockdown of the SUMO E2 enzyme UBC9 led to a prominent defect in the removal of trapped GFP‐DNMT1 species following 5‐azadC treatment (Figs 3A and B, and EV2C), suggesting that SUMO‐dependent modification facilitates their efficient resolution. In support of this, UBC9 depletion exacerbated the adverse effect of 5‐azadC on cell proliferation (Fig 3C). Moreover, acute inhibition of SUMOylation by treatment with ML‐792 (SUMO‐E1i), a recently described small molecule inhibitor of the SUMO E1 enzyme SAE (He et al, 2017), abolished 5‐azadC‐induced chromatin SUMOylation and strongly impaired the timely clearance of endogenous DNMT1 DPCs (Fig 3D and E). Unlike suppression of SUMOylation, SPRTN depletion only modestly compromised cell proliferation and GFP‐DNMT1 DPC clearance following 5‐azadC treatment while knockdown of ZNF451, a SUMO E3 ligase recently implicated in resolving TOP2 DPCs (Schellenberg et al, 2017), had no effect (Figs 3F, and EV2D and E), suggesting that SUMO‐dependent modification and processing of DNMT1 DPCs can proceed via other enzymatic activities. In agreement with the notion that DNMT1 reestablishes DNA methylation patterns following DNA replication, iPOND analysis (Sirbu et al, 2012) showed that the bulk of DNMT1 species immobilized on 5‐azadC‐containing DNA as well as accompanying SUMO modifications were associated with mature but not nascent chromatin (Fig 3G). This raises the possibility that the majority of DNMT1‐DNA adducts may be out of reach for replisome‐coupled DPC‐processing pathways, consistent with the modest impact of SPRTN depletion on the clearance of these lesions. Interestingly, we noted that DNMT1 underwent 5‐azadC‐induced ubiquitylation in a SUMO‐dependent manner (Fig 3H) and that the clearance of SUMO‐modified DNMT1 molecules trapped on chromatin following treatment with 5‐azadC was impaired by inhibition of the proteasome but not DNA replication (Fig 3I and J). These observations suggest that one route of resolving SUMOylated DNMT1 DPCs may be via their ensuing ubiquitylation and proteolytic processing by the proteasome. We conclude that SUMOylation plays a critical role in facilitating the resolution of post‐replicatively formed DNMT1‐DNA adducts.
Figure 3. SUMOylation promotes resolution of DNMT1‐DNA adducts and cell fitness upon 5‐azadC treatment.
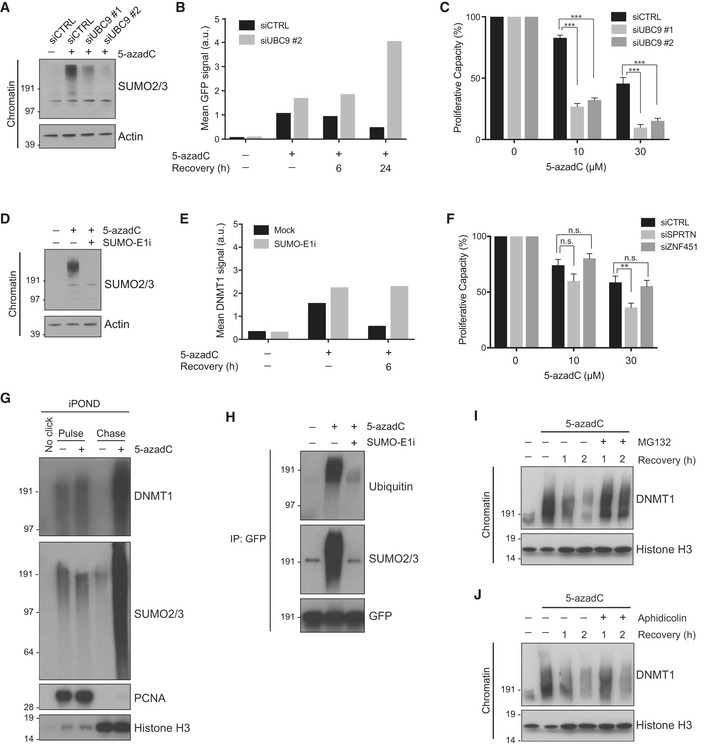
- Chromatin‐enriched fractions of HeLa cells transfected with indicated siRNAs and subsequently exposed or not to 5‐azadC for 2 h were analyzed by immunoblotting with SUMO2/3 and actin antibodies.
- HeLa cells stably expressing GFP‐DNMT1 and transfected with indicated siRNAs were left untreated or exposed to 5‐azadC for 2 h. Cells were then preextracted in stringent preextraction buffer and fixed at the indicated time points after 5‐azadC removal. Mean detergent‐resistant GFP signal was determined by quantitative image analysis (> 6,000 cells analyzed per condition). Data from a representative experiment are shown. Representative images are shown in Fig EV2C.
- Proliferative capacity of HeLa/GFP‐DNMT1 cells transfected with control or UBC9 siRNAs and exposed to the indicated 5‐azadC doses for 2 h was assayed by measuring cell proliferation with the SRB assay (mean ± SEM; n = 3 independent experiments; ***P < 0.001, Student's t‐test).
- As in (B), except that untransfected HeLa cells were exposed or not to 5‐azadC in the presence or absence of a small molecule SUMO E1 enzyme inhibitor (SUMO‐E1i).
- HeLa cells treated with 5‐azadC for 2 h in the presence or absence of SUMO‐E1i were preextracted in stringent preextraction buffer, fixed at the indicated time points after 5‐azadC removal and immunostained with DNMT1 antibody. Mean detergent‐resistant DNMT1 signal was determined by quantitative image analysis (> 6,000 cells analyzed per condition). Data from a representative experiment are shown.
- Proliferative capacity of HeLa cells transfected with SPRTN or ZNF451 siRNAs and exposed to indicated 5‐azadC doses for 2 h was assayed as in (C) (mean ± SEM; n = 3 independent experiments; **P < 0.01, n.s. not significant, Student's t‐test).
- iPOND analysis of 5‐azadC‐induced DNMT1 trapping and SUMOylation. HeLa cells were mock‐treated or preincubated with 5‐azadC for 5 min before addition of EdU for 10 min. Cells were then washed and incubated with thymidine for 0 (pulse) or 60 min (chase) before performing iPOND.
- HeLa cells stably expressing GFP‐DNMT1 were preincubated or not with SUMO‐E1i for 30 min, and where indicated, 5‐azadC was added to the medium for an additional 60 min. Cells were then processed for GFP immunoprecipitation (IP) followed by immunoblotting for the indicated proteins.
- Immunoblot analysis of chromatin‐enriched fractions of HeLa cells that were synchronized in early S phase by release from double thymidine block, pulse‐labeled with 5‐azadC for 30 min, and grown in the presence or absence of proteasome inhibitor (MG132) for the indicated times.
- As in (I), except that cells were treated or not with aphidicolin following pulse‐labeling with 5‐azadC.
Source data are available online for this figure.
The SprT protease ACRC is targeted to DPCs in a SUMO/SIM‐dependent manner
We next asked whether SprT‐type proteases, which have recently been implicated directly in DPC repair (Stingele et al, 2017), are involved in SUMO‐mediated DPC recognition and processing. Whereas the budding yeast SprT protease Wss1 recognizes SUMO conjugates via SIMs, SPRTN recruitment to DNA damage sites in human cells is strongly ubiquitin‐dependent and not known to be regulated by SUMOylation (Centore et al, 2012; Davis et al, 2012; Mosbech et al, 2012; Stingele et al, 2014; Balakirev et al, 2015). Consistently, the S phase‐specific recruitment of SPRTN to formaldehyde‐induced nuclear foci was abolished by inhibition of ubiquitylation but not SUMOylation (Fig 4A–C). Along with SPRTN, many eukaryotic species encode a second, functionally uncharacterized, SprT domain‐containing protease, generally referred to as GCNA and known in humans as ACRC (Fig 4A); indeed, GCNA proteases are more closely related to the Wss1 and SPRTN families than to any other proteases (Carmell et al, 2016). Supporting a potential involvement of ACRC in cellular responses to DPCs, we found that ectopically expressed GFP‐tagged ACRC interacted strongly with polySUMO chains and was recruited to formaldehyde‐induced foci colocalizing with SUMO in virtually all interphase cells, but did not undergo appreciable accumulation at other types of DNA damage (Figs 4B and D–F, and EV3A). Likewise, GFP‐ACRC colocalized with DNMT1 following 5‐azadC treatment (Fig 4G and H). Contrary to SPRTN, recruitment of ACRC to formaldehyde‐ and 5‐azadC‐induced DPCs was abolished by inhibition of SUMOylation but not ubiquitylation (Fig 4B, D, G and H). Sequence inspection of human ACRC revealed the presence of at least four potential SIMs in its N‐terminal portion (Fig 4A), which we surmised might underlie its SUMO‐dependent recruitment to DPCs. Indeed, while mutation of individual SIMs impaired SUMO binding to different extents, simultaneous inactivation of all four SIM motifs fully abrogated ACRC interaction with polySUMO chains and recruitment to formaldehyde‐ and 5‐azadC‐induced nuclear foci (Figs 4F–H and EV3B–D), suggesting that it recognizes SUMO conjugates at DPC sites. We conclude from these data that unlike SPRTN, the human SprT protease ACRC is targeted to DPCs in a SUMO/SIM‐dependent manner.
Figure 4. The SprT protease ACRC but not SPRTN is recruited to DPCs in a SUMO/SIM‐dependent manner.

- Domain structure of the human SprT proteases SPRTN and ACRC, showing location of SprT protease domains and additional motifs, including a ubiquitin‐binding UBZ domain in SPRTN and an N‐terminal cluster of SIMs (sequence highlighted) in ACRC. Alanine substitutions introduced to disrupt the functionality of the ACRC SIMs (SIM*) are indicated.
- Representative images of U2OS cells stably expressing GFP‐SPRTN or GFP‐ACRC that were transfected with control (CTRL) or UBC9 siRNAs, exposed to formaldehyde in the presence or absence of ubiquitin E1 enzyme (UBA1) inhibitor TAK‐243 (Ub‐E1i) as indicated and fixed one h later. Scale bar, 10 μm.
- Quantification of data in (B), showing proportion of U2OS/GFP‐SPRTN cells displaying nuclear GFP‐SPRTN foci (mean ± SEM; at least 100 cells quantified per condition per experiment; n = 3 independent experiments).
- As in (C), but showing proportion of U2OS/GFP‐ACRC cells displaying nuclear GFP‐ACRC foci (mean ± SEM; at least 100 cells quantified per condition per experiment; n = 3 independent experiments).
- U2OS cells expressing GFP‐ACRC were exposed to the indicated genotoxic agents (formaldehyde: 600 μM, 1 h; UV: 20 J/m2, 6‐h recovery; hydroxyurea (HU): 2 mM, 24 h; mitomycin C (MMC): 40 ng/ml, 24 h), preextracted and fixed, and analyzed by microscopy. Representative images are shown. Scale bar, 10 μm.
- HeLa cells transfected with plasmids encoding GFP alone or indicated GFP‐ACRC alleles were subjected to GFP IP under denaturing conditions. Beads were incubated with recombinant polySUMO22–8 chains, washed extensively, and processed for immunoblotting with SUMO2/3 and GFP antibodies.
- Representative images of U2OS cells expressing indicated GFP‐ACRC alleles that were treated with 5‐azadC in the presence or absence of SUMO inhibitor (SUMO‐E1i), fixed 2 h later, and immunostained with DNMT1 antibody. Scale bar, 10 μm.
- Quantification of data in (G), showing proportion of cells displaying GFP‐ACRC co‐localization with DNMT1 foci (mean ± SEM; at least 100 cells quantified per condition per experiment; n = 3 independent experiments).
Source data are available online for this figure.
Figure EV3. SUMO‐dependent recruitment of ectopic ACRC to sites of formaldehyde‐induced DPCs and its impact on cell fitness and DPC clearance.

- U2OS cells expressing GFP‐ACRC were exposed or not to formaldehyde, fixed 1 h later, and immunostained with SUMO2/3 antibody. Representative images are shown. Scale bar, 10 μm.
- HeLa cells transfected with plasmids encoding GFP alone or indicated GFP‐ACRC alleles were subjected to GFP IP under denaturing conditions. Beads were incubated with recombinant polySUMO23–8 chains, washed extensively, and processed for immunoblotting with SUMO2/3 and GFP antibodies.
- Representative images of U2OS cells stably expressing GFP‐ACRC SIM* that were treated or not with formaldehyde and fixed 1 h later. Scale bar, 10 μm.
- Quantification of data in (C), showing proportion of cells displaying nuclear GFP‐ACRC SIM* foci (mean ± SEM; at least 60 cells quantified per condition per experiment; n = 3 independent experiments).
- Proliferative capacity of stable U2OS/GFP‐ACRC cell lines induced or not with doxycycline (DOX) for 2 h to express the transgenes was assayed by measuring cell proliferation with the SRB assay (mean ± SEM; n = 3 independent experiments).
- U2OS/GFP‐ACRC WT cells induced or not with doxycycline (DOX) to express GFP‐ACRC were left untreated or exposed to 5‐azadC for 2 h. Cells were then preextracted in stringent preextraction buffer and fixed at the indicated time points after 5‐azadC removal, and subsequently immunostained with DNMT1 antibody. Mean detergent‐resistant DNMT1 signal was determined by quantitative image analysis (> 6,000 cells analyzed per condition). Data from a representative experiment are shown.
Source data are available online for this figure.
SUMO and the ACRC ortholog GCNA‐1 protect against DPC toxicity in Caenorhabditis elegans
The findings above suggested that ACRC might have a role in processing DPCs in a SUMO‐driven manner. However, while homologs of ACRC are encoded by many eukaryotic genomes (Carmell et al, 2016), we found that it was absent or expressed at levels below the limit of detection in a range of human primary and cancer cell lines (Bekker‐Jensen et al, 2017; our unpublished observations), in agreement with recent work suggesting that expression of ACRC orthologs is enhanced in germ and stem cells (Carmell et al, 2016). Notably, ectopic expression of ACRC in U2OS cells reduced cellular fitness in a SIM‐dependent manner and delayed the clearance of DNMT1 DPCs (Fig EV3E and F), suggesting that a potential involvement of ACRC in SUMO‐mediated responses to DPCs could be restricted to highly specific cellular settings while enforcing its expression outside this context may interfere with other DPC‐processing mechanisms. We therefore sought to examine possible physiological ACRC/GCNA protease functions in DPC responses in a whole‐organism context. To this aim, we utilized the nematode C. elegans, which contains orthologs of both ACRC and SPRTN (known as GCNA‐1 and DVC‐1, respectively; Fig 5A), the latter of which we and others have previously demonstrated plays an important role in promoting survival upon DNA damage, including DPCs, in this organism (Mosbech et al, 2012; Stingele et al, 2016). Like human ACRC, we found that GCNA‐1 interacted with polySUMO chains (Fig 5B). We then generated a gcna‐1 loss of function (lof) allele by knocking in a ~ 6.6 kb gfp‐containing selection cassette between the gcna‐1 promoter and coding sequence, simultaneously generating a transcriptional gfp reporter (Figs 5C and EV4A; Dickinson et al, 2015). Inspection of gcna‐1 promoter‐driven GFP expression confirmed that GCNA‐1 is mainly expressed in germ cells and early embryonic, proliferating cells but not in post‐mitotic tissues (Figs 5D and EV4B; Carmell et al, 2016). Interestingly, using assays probing for survival of germ and early embryonic cells, we found that gcna‐1 loss of function led to elevated formaldehyde sensitivity (Fig 5E). Likewise, gcna‐1 deficiency caused marked sensitivity to cisplatin but not UV (Figs 5F and EV4C), and GCNA‐1 and the core NER factor XPA‐1 functioned non‐epistatically in promoting survival upon cisplatin exposure (Fig EV4D). This DNA damage sensitivity profile showed striking similarities to that observed for worms lacking DVC‐1 (Stingele et al, 2016), which we independently verified were hypersensitive to formaldehyde and cisplatin (Fig 5E and F). We observed epistasis between GCNA‐1 and DVC‐1 in promoting resistance to formaldehyde but not cisplatin (Fig 5E and F), suggesting that these SprT proteases function cooperatively in response to formaldehyde‐induced DPCs but have non‐redundant roles in pathways responding to the spectrum of DNA lesions generated by cisplatin. Importantly, like gcna‐1 loss‐of‐function, an E364Q mutation in GCNA‐1 predicted to abolish the catalytic activity of its SprT protease domain (gcna‐1(E364Q)) as well as an in‐frame truncation of this domain gave rise to pronounced formaldehyde hypersensitivity (Fig 5A, C and G), suggesting that the putative protease activity of GCNA‐1 is essential for its function in mitigating the toxicity of formaldehyde‐induced DPCs.
Figure 5. The ACRC ortholog GCNA‐1 and SUMOylation protect against DPC toxicity in Caenorhabditis elegans .

- Domain structure of human ACRC and its C. elegans ortholog GCNA‐1. The GCNA‐1 deletion (del) introduces a frameshift at E364, giving rise to a truncated protein containing an aberrant 22‐residue C‐terminal addition.
- HeLa cells transfected with plasmids encoding GFP alone, GFP‐ACRC, or GFP‐tagged C. elegans GCNA‐1 were subjected to GFP IP under denaturing conditions. Beads were incubated with recombinant polySUMO23–8 chains, washed extensively, and processed for immunoblotting with SUMO2/3 and GFP antibodies.
- Schematic representation of the C. elegans gcna‐1 locus, depicting mutants generated. Loss of function gcna‐1 allele (emcSi31) was created by knock‐in of a gfp selection cassette (GFP‐SEC) in the start codon (see Fig EV4A). gcna‐1 deletion (emc52), leading to a frameshift and premature stop codon in the coding sequence, and E364Q point mutant (emc53) alleles were generated by CRISPR/Cas9 targeting of exon 6.
- GFP expression driven by the gcna‐1 promoter was observed in germ cells, proliferating embryos, and young larvae but not in post‐mitotic tissues in the head (see also Fig EV4B). Scale bars, 50 μm.
- Formaldehyde survival of wild type (wt), dvc‐1, gcna‐1 loss of function (lof), and gcna‐1; dvc‐1 double mutant C. elegans (mean ± SEM; n = 4 independent experiments).
- Cisplatin survival of wild type, dvc‐1, gcna‐1, and gcna‐1; dvc‐1 double mutant C. elegans (mean ± SEM; n = 3 independent experiments).
- Formaldehyde survival of gcna‐1 deletion (del) and E364Q mutant C. elegans (mean ± SEM; n = 2 independent experiments).
- Formaldehyde survival of C. elegans grown on L4440 control (CTRL) or smo‐1 RNAi bacteria (mean ± SEM; n = 2 independent experiments).
- Formaldehyde survival of C. elegans grown on L4440 control (CTRL) or gei‐17 RNAi bacteria (mean ± SEM; n = 2 independent experiments).
- Formaldehyde survival of wild type and gcna‐1 deletion (del) mutant C. elegans grown on L4440 control (CTRL) or smo‐1 RNAi bacteria (mean ± SEM; n = 2 independent experiments).
Source data are available online for this figure.
Figure EV4. Involvement of GCNA‐1 and SUMOylation in responses to genotoxic insults in Caenorhabditis elegans .
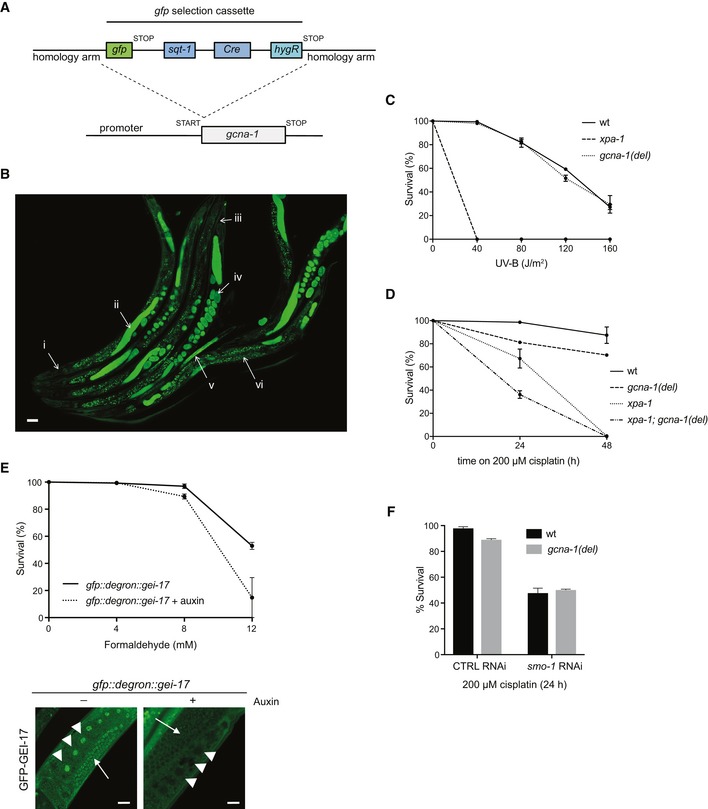
- Schematic showing the strategy for gfp selection cassette knock‐in in the start codon of C. elegans gcna‐1, simultaneously generating a transcriptional gfp reporter and gcna‐1 loss of function allele, which can be selected for by the visible “roller” phenotype produced by sqt‐1 and hygromycin resistance by hygR.
- GFP positivity in the gcna‐1 loss of function strain emcSi31 (Fig 4C) demarcates GCNA‐1 expression patterns in C. elegans (i. head: no expression; ii. GFP expression in meiotic germ cells; iii. tail: no expression; iv. GFP expression in embryos; v. GFP expression in young larvae; vi. speckles in the intestine: background fluorescence). A representative image depicting multiple animals is shown. Scale bar, 50 μm.
- UV survival of wild type (wt), gcna‐1 deletion (del), and xpa‐1 deletion C. elegans (mean ± SEM; n = 2 independent experiments).
- Cisplatin survival of wild type (wt), gcna‐1 deletion (del), xpa‐1 deletion, and xpa‐1; gcna‐1 double mutant C. elegans (mean ± SEM; n = 2 independent experiments).
- Upper panel: formaldehyde survival of gei‐17 knock‐in animals expressing GFP‐ and auxin‐responsive degron‐tagged GEI‐17 grown in the absence or presence of auxin for 24 h to induce GEI‐17 depletion (mean ± SEM; n = 2 independent experiments). Lower panel: representative images showing loss of GFP fluorescence in germ cells (arrowheads: oocytes; arrows: meiotic cells) of animals grown in the presence of auxin, demonstrating efficient auxin‐dependent depletion of GFP::degron‐tagged GEI‐17. Scale bar, 20 μm.
- Cisplatin survival of wild type and gcna‐1 deletion (del) mutant C. elegans grown on L4440 control (CTRL) or smo‐1 RNAi bacteria (mean ± SEM; n = 2 independent experiments). Due to the toxicity of smo‐1 RNAi, survival following cisplatin exposure for more than 24 h could not be assessed.
To probe for a general role of SUMO in promoting survival of germ and embryonic cells upon exposure to DPC‐inducing agents, we used RNAi to knock down SMO‐1, the single SUMO ortholog in C. elegans (Choudhury & Li, 1997). Depletion of SMO‐1 markedly enhanced formaldehyde sensitivity, an effect recapitulated by knockdown of the PIAS‐type SUMO E3 ligase, GEI‐17 (Holway et al, 2006; Figs 5H and I, and EV4E). In line with the possibility that GCNA‐1 promotes resistance to DPC‐inducing agents through a SUMO‐dependent pathway, gcna‐1 deficiency and SMO‐1 knockdown were epistatic in sensitizing worms to formaldehyde and cisplatin (Figs 5J and EV4F). Collectively, these data suggest the existence of a protective SUMO‐driven response to DPCs in C. elegans germ and embryonic cells, and implicate the GCNA‐1 SprT protease as one component involved.
Discussion
Our findings reveal an important role of dynamic SUMO‐mediated protein modifications as a means of promoting cellular responses to both enzymatic and non‐enzymatic DPCs, providing first global insights into regulatory signaling processes elicited in response to such insults. Using 5‐azadC‐induced DNMT1 DPCs as a model approach for generating defined enzymatic DPCs in human cells, we show that these lesions trigger a dynamic chromatin SUMOylation response directly impacting the crosslinked DNMT1 molecules and known DNMT1‐interacting proteins, possibly those residing in close proximity to the DPCs, in accordance with the SUMO group modification principle operating in the context of other types of DNA damage (Psakhye & Jentsch, 2012). Given that both non‐specific formaldehyde‐generated DPCs and defined enzymatic DPCs involving DNMT proteins stimulate rapid and potent chromatin SUMOylation targeting diverse and highly defined sets of factors, respectively, we speculate that SUMO modification of crosslinked proteins may be a general cellular mechanism to mark covalently trapped proteins for recognition and processing by SUMO‐targeted components of pathways promoting DPC resolution. The work described here suggests an involvement of SUMO‐binding ACRC/GCNA family SprT proteases in such processes, and we provide evidence that in C. elegans germ and early embryonic cells, GCNA‐1 has an important role in protecting against the lethality of DPC‐inducing agents including formaldehyde in a manner that involves the integrity of its protease domain and is epistatic with SUMO. However, it has been shown that GCNA proteases are predominantly expressed in germ and stem cells (Carmell et al, 2016), supported by our findings, suggesting that unlike SPRTN, the involvement of this family of SprT proteases in cellular DPC responses is, in all likelihood, highly tissue‐restricted. While we have not yet been able to experimentally verify that GCNA‐type proteins are active proteases, this seems warranted by our finding that point mutation of the highly conserved putative catalytic glutamic acid residue in the SprT domain of C. elegans GCNA‐1 sensitizes worms to formaldehyde to the same extent as gcna‐1 loss‐of‐function. Establishing the precise role of GCNA‐type SprT proteases in promoting context‐specific DPC responses in conjunction with SUMOylation will be an important task for future investigations.
The importance of SUMOylation in promoting DPC resolution in human cell lines lacking detectable ACRC expression implies that other SUMO‐targeted DPC‐processing factors exist. The SUMO E3 ligase ZNF451/ZATT may be one such protein, as it was recently implicated in promoting efficient resolution of covalently trapped TOP2‐DNA cleavage complexes, a unique type of enzymatic DPC involving a phosphotyrosyl linkage in the context of a DNA double‐strand break that is directly reversed in a dedicated process catalyzed by the phosphodiesterase TDP2 (Schellenberg et al, 2017). However, unlike suppression of SUMOylation, we observed no adverse impact of ZNF451 depletion on cellular fitness following 5‐azadC‐induced DPCs. This suggests that ZNF451 may be primarily engaged in resolution of TOP2‐DNA cleavage complexes but largely dispensable for the reversal of other types of DPCs that are not resolved by TDP2, whereas SUMO‐dependent signaling could play a broader role in facilitating the processing of a diverse range of DPCs. Our studies highlight an interesting division of labor between ubiquitin‐ and SUMO‐dependent signaling in promoting cellular responses to these lesions. Available evidence suggests that ubiquitin‐regulated SPRTN recruitment to and processing of DPCs is tightly coupled to DNA replication (Lessel et al, 2014; Lopez‐Mosqueda et al, 2016; Stingele et al, 2016; Vaz et al, 2016). In addition, recent studies using Xenopus egg extracts revealed a parallel role of the proteasome and replication fork‐associated E3 ubiquitin ligase activity in DNA replication‐coupled DPC repair (Larsen et al, 2018). By contrast, our findings show that chromatin SUMOylation elicited in response to DPCs does not require ongoing DNA replication but is operational throughout interphase. Cells may thus possess complementary ubiquitin‐ and SUMO‐driven pathways for recognizing and resolving DPCs via mechanisms involving different protease activities and cell cycle‐dependencies. The coordinated actions of such mechanisms may be important for providing the flexibility necessary for coping with the diversity of DPC sizes, structures, and modes of DNA attachment that can be encountered. While the rapid DPC‐induced SUMO response can be triggered throughout interphase, it might be particularly relevant during conditions where DPC processing by replication‐coupled mechanisms is inefficient or even impossible. Indeed, we found that the majority of 5‐azadC‐induced DNMT1 DPCs are associated with mature chromatin but not newly synthesized DNA, raising the possibility that these lesions are effectively out of reach for the replication fork‐coupled SPRTN and proteasome pathways for DPC proteolysis. This may account for the strong SUMOylation response targeting trapped DNMT1 proteins and associated factors, and could explain the modest impact of depleting SPRTN on DNMT1 DPC resolution and cellular fitness following 5‐azadC treatment. We therefore suggest that SUMOylation may provide a means to facilitate the processing of DPCs in duplex DNA that cannot be efficiently detected and resolved by replication‐coupled DPC repair pathways. Our observations point to a role of DPC SUMOylation in stimulating subsequent proteolytic processing via the ubiquitin–proteasome system, but the precise mechanistic underpinnings remain to be established, and we do not rule out that additional SUMO‐targeted factors may also contribute to the clearance of SUMO‐modified DNMT1 DPCs and other adducted proteins. Detailed insights into these processes are of considerable biomedical relevance, given the long‐standing use of 5‐azadC for the treatment of myeloid malignancies. Our findings showing that DNMT1‐DNA adduct formation is central to its cytotoxic potential, which is markedly enhanced by SUMO inhibition, may provide new opportunities for combination therapies involving 5‐azadC and related drugs.
Collectively, our findings support an important role of SUMO‐dependent signaling in cellular DPC responses that may operate in parallel with other DPC repair mechanisms to ensure efficient DPC recognition and resolution in different contexts. Considering the widespread occurrence of SUMO group modification within the DNA damage response, whereby multiple factors residing at DNA damage sites are concurrently targeted by SUMOylation (Psakhye & Jentsch, 2012), our system‐wide analyses of DPC‐induced SUMOylation dynamics may provide a framework for the identification of additional cellular factors involved in resolution of DPCs to mitigate the cytotoxicity and disease‐promoting potential of these lesions.
Materials and Methods
Plasmids and siRNAs
Full‐length cDNAs encoding human ACRC (Invitrogen Ultimate™ ORF Collection) and C. elegans gcna‐1 (synthetic cDNA, codon‐optimized for expression in human cells) were cloned into pAcGFP1‐C1 vector (Clontech). Plasmid encoding GFP‐DNMT1 was a kind gift from Heinrich Leonhardt, Ludwig Maximilians University, Munich, Germany. Plasmid encoding GFP‐SPRTN was described previously (Mosbech et al, 2012). ACRC mutants (SIM1*: I22A, L23A, N24A, V25A; SIM2*: V76A, V77A, V78A, I79A; SIM3*: L97A, L98A, E99A, I100A; SIM4*: I121A, V122A, I123A, S124A; SIM*: SIM1‐4* combined) and catalytically inactive DNMT1 containing a C1225W point mutation were generated using the Q5 Site‐directed Mutagenesis Kit (NEB) according to the manufacturer's protocol. Plasmid DNA transfections were performed using FuGENE 6 Transfection Reagent (Promega), Novagen GeneJuice Transfection Reagent (Merck), or Lipofectamine 3000 Reagent (Invitrogen), according to the manufacturers’ protocols. siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. All siRNAs were used at a final concentration of 50 nM unless otherwise indicated. The following siRNA oligonucleotides were used: non‐targeting control (CTRL): 5′‐GGGAUACCUAGACGUUCUA‐3′; UBC9 #1: 5′‐UAGCUGUCCCAACAAAGATT‐3′; UBC9 #2: 5′‐GCUCAAGCAGAGGCCUACACGAUUU‐3′; SPRTN: 5′‐UCAAGUACCACCUGUAUUA‐3′; ZNF451: 5′‐CAGAAUUCAGGACACAAAUU‐3′; DNMT1 #1: 5′‐CGGUGCUCAUGCUUACAAC‐3′; DNMT1 #2 (targeting the 3′UTR): 5′‐CUGUAAUGAGUGGAAAUUAAGATT‐3′.
Cell culture
Human U2OS and HeLa cells were obtained from ATCC. All cell lines used in this study were cultured in DMEM containing 10% FBS and were regularly tested for mycoplasma infection. The cell lines were not authenticated. To generate U2OS cell lines constitutively expressing GFP‐ACRC WT, GFP‐ACRC SIM* and HeLa cells constitutively expressing GFP‐DNMT1, cells were transfected with the respective expression constructs and positive clones were selected by incubation in medium containing G418 (InvivoGen) for 14 days. Stable U2OS cell lines expressing GFP‐ACRC alleles in a doxycycline‐inducible manner were generated by selection in medium containing hygromycin B (Thermo Fisher Scientific) and blasticidin (InvivoGen). Clones were screened for expression of the ectopic GFP‐tagged proteins by microscopy and immunoblotting. U2OS cells stably expressing GFP‐SPRTN and HeLa cells stably expressing His10‐SUMO2 were described previously (Mosbech et al, 2012; Hendriks & Vertegaal, 2016).
Unless otherwise indicated, the following doses of drugs and genotoxic agents were used: 5‐aza‐2′‐deoxycytidine (5‐azadC; 10 μM, Sigma‐Aldrich), formaldehyde (600 μM, Merck), acetaldehyde (5 mM, Sigma), cisplatin (30 μM, Merck), mitomycin C (MMC; 15 μM, Merck), hydroxyurea (HU, Sigma‐Aldrich; 2 mM), doxycycline (1 μg/ml, Merck), MLN‐7243 (Ub‐E1i; 5 μM, Active Biochem), ultraviolet radiation (UV; 20 J/m2) and ionizing radiation (IR; 10 Gy), actinomycin D (2 μg/ml, Merck), aphidicolin (APH; 10 μM, Sigma‐Aldrich), and ML‐792 (SUMO‐E1i; 1 μM, synthesized by MedKoo Biosciences). To induce heat shock, cells were incubated at 43°C for 70 min.
Immunochemical methods, cell fractionation, and antibodies
Immunoblotting was done as previously described (Poulsen et al, 2012). To prepare cell extracts, cells were lysed in EBC buffer (50 mM Tris, pH 7.5; 150 mM NaCl; 1 mM EDTA; 0.5% NP40; 1 mM DTT) supplemented with protease and phosphatase inhibitors, incubated on ice for 10 min, and cleared by centrifugation. For immunoprecipitation, cells were lysed in EBC buffer or denaturing buffer (20 mM Tris, pH 7.5; 50 mM NaCl; 1 mM EDTA; 0.5% NP40; 0.5% SDS, 0.5% sodium deoxycholate; 1 mM DTT) supplemented with protease and phosphatase inhibitors, and lysates were sonicated and cleared by centrifugation. Lysates were incubated with GFP‐Trap agarose or magnetic beads (Chromotek) for at least 1.5 h. After extensive washing of the beads, GFP‐tagged proteins were eluted by boiling in 2× Laemmli sample buffer for 5 min and analyzed by immunoblotting. For chromatin fractionation experiments, cells were lysed in Buffer 1 (10 mM Tris, pH 8.0; 10 mM KCl; 1.5 mM MgCl2; 0.34 M sucrose; 10% glycerol; 0.1% Triton X‐100) supplemented with protease and phosphatase inhibitors and incubated on ice for 5 min. After centrifugation at 2,000 g for 5 min, the supernatant contained the soluble proteins. The pellet was washed in Buffer 1 followed by resuspension in Buffer 2 (50 mM Tris, pH 7.5; 150 mM NaCl; 1% NP40; 0.1% SDS; 1 mM MgCl2; 125 U/ml benzonase) supplemented with protease and phosphatase inhibitors. Lysates were then incubated for 15 min at 37°C and 1,000 rpm shaking. After centrifugation at 16,000 g for 10 min, the supernatant contained the chromatin‐bound proteins. Isolation of proteins associated with nascent and mature chromatin by iPOND was done as previously described (Dungrawala & Cortez, 2015).
Antibodies used in this study included: actin (clone C4, MAB1501, Merck (1:20,000 dilution), RRID:AB_2223041); DNMT1 (sc‐10221 (K‐18), Santa Cruz (1:2,000), RRID:AB_2093831); FLAG (F1804 (M2), Sigma‐Aldrich (1:1,000), RRID:AB_262044); GFP (sc‐9996 (Clone B2), Santa Cruz (1:1,000), RRID:AB_627695; 11814460001 (clones 7.1 and 13.1), Roche (1:1,000), RRID:AB_390913; 902601 (clone B34), BioLegend (1:5,000), RRID:AB_2565021; sc‐8334, Santa Cruz (1:1,000), RRID:AB_641123); MCM6 (C‐20, sc‐9843, Santa Cruz (1:500), RRID:AB_2142543); PML (sc‐5621 (H‐238), Santa Cruz (1:500), RRID:AB_2166848); SAE2 (A302‐580A, Bethyl Laboratories (1:250), RRID:AB_2034860); SPRTN (kind gift from John Rouse, University of Dundee, UK, (1:1,000)); SUMO1 (4930S, Cell Signaling Technology (1:1,000), RRID:AB_10698887); SUMO2/3 (ab3742, Abcam (1:1,000), RRID:AB_304041; ab81371 (8A2), Abcam (1:200), RRID:AB_1658424); TFEB (4240, Cell Signaling Technology (1:500), RRID:AB_11220225); ubiquitin (sc‐8017 (P4D1), Santa Cruz (1:1,000), RRID:AB_628423); and ZNF451 (SAB2108741, Sigma‐Aldrich (1:500)). Polyclonal sheep antibody to DNMT1 was raised against full‐length recombinant human DNMT1 purified from bacteria.
KCl/SDS DPC precipitation assay
For isolation of DPCs using the KCl/SDS precipitation assay (Zhitkovich & Costa, 1992), approximately 1 × 106 cells were lysed in 1 ml Buffer A (2% SDS; 20 mM Tris, pH 7.5) followed by sonication for 25 s at an amplitude of 25%. Proteins were then precipitated by addition of 1 ml Buffer B (200 mM KCl; 20 mM Tris, pH 7.5) followed by incubation on ice for 5 min. The precipitated proteins were pelleted by centrifugation at 15,000 g for 5 min (4°C), and the supernatant was saved for quantification of soluble DNA. The pellet was washed three times by addition of 750 μl Buffer B followed by incubation at 55°C for 5 min, 30 s on ice, and centrifugation at 15,000 g at 4°C for 5 min. After washing, each pellet was resuspended in 500 μl Buffer B containing 0.04 mg/ml Proteinase K and incubated at 55°C for 3 h. After 30 s on ice and centrifugation at 15,000 g at 4°C for 5 min, the supernatant contained the crosslinked DNA. Soluble DNA and crosslinked DNA were quantified using PicoGreen, and the amount of DPCs was calculated as the ratio between crosslinked DNA and total DNA (soluble + crosslinked).
polySUMO‐binding assay
HeLa cells expressing GFP‐ACRC alleles were lysed in denaturing buffer supplemented with protease and phosphatase inhibitors, and lysates were sonicated and cleared by centrifugation. GFP‐tagged ACRC was then purified on GFP‐Trap agarose beads (Chromotek) followed by extensive washing in denaturing buffer. The beads were equilibrated in EBC buffer and incubated with recombinant polySUMO22–8 or polySUMO23–8 chains (0.2 μg/sample, Boston Biochemicals) for 2 h at 4°C with rotation. Bound material was washed five times in EBC buffer, eluted by boiling in 2× Laemmli sample buffer for 5 min, and analyzed by immunoblotting.
Immunofluorescence and high‐content image analysis
Cells were preextracted in PBS containing 0.2% Triton X‐100 for 3 min on ice or in stringent preextraction buffer (10 mM Tris–HCl, pH 7.4; 2.5 mM MgCl2; 0.5% NP‐40; 1 mM PMSF) for 8 min before fixation with 4% formaldehyde for 10 min. If cells were not preextracted, they were subjected to a permeabilization step with PBS containing 0.2% Triton X‐100 for 5 min after fixation. Coverslips were incubated with primary antibodies diluted in 1% BSA‐PBS for 1 h at room temperature followed by staining with secondary antibodies (Alexa Fluor; Life Technologies) diluted in 1% BSA‐PBS for 1 h at room temperature. For manual image acquisition, coverslips were mounted in Vectashield mounting medium (Vector Laboratories) containing nuclear stain DAPI. For automated image acquisition, DAPI staining (Thermo Fisher Scientific) was added with secondary antibodies, and coverslips were mounted in MOWIOL 4‐88 (Sigma). For manual image acquisition, we used a Leica AF6000 wide‐field microscope (Leica Microsystems) equipped with HC Plan‐Apochromat 63×/1.4 oil immersion objective, using standard settings and LAS X software (Leica Microsystems). Raw images were exported as TIFF files, and if adjustments in image contrast and brightness were applied, identical settings were used on all images of a given experiment. For automated image acquisition, an Olympus IX‐81 wide‐field microscope equipped with an MT20 Illumination system, Olympus UPLSAPO 20×/0.75 NA objective, and a digital monochrome Hamamatsu C9100 CCD (charge‐coupled device) camera was used. Automated and unbiased image analysis was carried out with the ScanR analysis software. Data were exported and processed using Spotfire (Tibco) software.
Cell proliferation assays
For analysis of cell proliferation, cells were fixed in 10% ice‐cold trichloroacetic acid (TCA) for 30 min. After washing in deionized water, cells were stained with 0.4% (w/v) sulforhodamine B (SRB) in 1% acetic acid for 15 min at room temperature. Excess dye was removed by washing with 1% acetic acid, and plates were dried at room temperature before the protein‐bound fraction was dissolved in 10 mM Tris, pH 8.0. Fluorescence was measured using microplate reader FLUOstar Omega using Omega 1.3 software (BMG Labtech).
Enrichment of SUMOylated proteins
Purification of proteins modified by His10‐SUMO2 was performed essentially as described previously (Hendriks & Vertegaal, 2016), with the following exceptions: The reduction and alkylation of peptides during the 100K MWCO filtration step were performed concomitantly by simultaneous addition of 5 mM chloroacetamide (CAA) and 5 mM tris(2‐carboxyethyl)phosphine (TCEP), as opposed to sequential incubations with CAA and dithiothreitol (DTT). Elution of StageTips was performed with 40% acetonitrile (ACN) in 0.1% formic acid, as opposed to 80% ACN in 0.1% formic acid.
Mass spectrometric analysis
Samples were analyzed on 15 cm long analytical columns, with an internal diameter of 75 μm, and packed in‐house using ReproSil‐Pur 120 C18‐AQ 1.9 μm beads (Dr. Maisch). Reversed‐phase liquid chromatography was performed using an EASY‐nLC 1200 system (Thermo). The analytical column was heated to 40°C, and elution of peptides from the column was achieved by application of gradients with stationary phase Buffer A (0.1% formic acid) and increasing amounts of mobile phase Buffer B (80% ACN in 0.1% formic acid). The primary gradient ranged from 5% buffer B to 30% buffer B over 120 min, followed by a tail‐end increase to 50% buffer B over 20 min to ensure full peptide elution, followed by a washing block of 20 min. Electrospray ionization (ESI) was achieved using a Nanospray Flex Ion Source (Thermo), with the ions analyzed using either a Q‐Exactive HF mass spectrometer (HF; Thermo) for the formaldehyde experiments or a Q‐Exactive HF‐X mass spectrometer (HFX; Thermo) for the 5‐azadC experiments. Spray voltage was set to 2 kV, capillary temperature to 275°C, and S‐Lens RF level to 50% (HF samples) or funnel RF level to 40% (HFX samples). Full scans were performed at a resolution of 60,000, with a scan range of 300–1,750 m/z, a maximum injection time of 60 ms, and an automatic gain control (AGC) target of 3,000,000 charges. Precursors were isolated with a width of 1.3 m/z, with an AGC target of 100,000 charges (HF samples) or 200,000 charges (HFX samples), and precursor fragmentation was attained using higher‐energy collision dissociation (HCD) using a normalized collision energy of 25. Only precursors with charge states 2–6 were considered, and selected precursors were excluded from repeated sequencing by setting a dynamic exclusion of 60 s. For analysis of the formaldehyde experiments on the HF instrument, two technical replicates were measured. For the first replicate, MS/MS settings included a loop count of 12, a maximum injection time of 50 ms, a resolution of 30,000, and an intensity threshold of 50,000. For the second replicate, loop count was set to 7, maximum injection time to 120 ms, resolution to 60,000, and intensity threshold to 120,000. For analysis of the 5‐azadC experiments on the HFX instrument, one technical replicate was performed, where MS/MS settings included a loop count of 9, a maximum injection time of 90 ms, a resolution of 45,000, and an intensity threshold of 120,000.
Raw data analysis
MS proteomics RAW data are available at the ProteomeXchange Consortium database via the Proteomics Identifications (PRIDE) partner repository (Vizcaino et al, 2014), under dataset ID PXD009040. All RAW files were analyzed using MaxQuant software (version 1.5.3.30; Cox & Mann, 2008; Cox et al, 2011). RAW files corresponding to the formaldehyde and 5‐azadC experiments were analyzed separately. Default MaxQuant settings were used, with exceptions outlined below. For generation of the theoretical spectral library, the HUMAN.FASTA databases were extracted from UniProt on January 6, 2016 (for formaldehyde experiments) and on February 22, 2017 (for 5‐azadC experiments). Protein N‐terminal acetylation and methionine oxidation were included as potential variable modifications (default), with a maximum allowance of three variable modifications per peptide. Label‐free quantification (LFQ) was enabled. Second peptide search was enabled (default), and matching between runs was enabled with a match time window of 2 min and an alignment time window of 40 min. Data were filtered by posterior error probability to achieve a false discovery rate of < 1% (default), at both the peptide‐spectrum match and the protein assignment levels.
Statistical analysis and data visualization
Processing of the text file output by MaxQuant was entirely performed using the freely available Perseus software (Tyanova et al, 2016). Reverse‐database hits and potential contaminant proteins were removed. LFQ intensities were log2‐transformed for further analyses. Proteins not detected in 4 out of 4 biological replicates in at least one experimental condition were removed. Scatter plot analysis, principal component analysis, Z‐scoring and subsequent generation of heatmaps, and two‐sample testing for the generation of volcano plots were all carried out in Perseus using default settings. For determination of whether a protein was a SUMO target, all His10‐SUMO‐enriched samples were individually tested versus the parental control using two‐sample testing and filtered to achieve a permutated‐based FDR of < 1% at an s0 setting of 1.5. Potential SUMO targets were moreover filtered to have at least a ratio of 1.5 over the parental control in at least one condition, in addition to having set the s0 to 1.5. For assessing differences between the differentially treated SUMO samples, non‐SUMO target proteins were removed, and all remaining proteins were investigated using two‐sample t‐testing and filtered to achieve a permutated‐based FDR of < 5% at an s0 setting of 0.5. Interaction network analysis was performed using the STRING database with default settings (interaction confidence of 0.4; Szklarczyk et al, 2017). Protein interactions were exported, and visualization was manually performed using Cytoscape (Shannon et al, 2003).
Caenorhabditis elegans strains, RNAi and sensitivity assays
All strains were cultured according to standard methods and outcrossed to N2 wild type (Bristol) at least three times. Alleles used were dvc‐1(ok260) (Mosbech et al, 2012), gei‐17(fgp1) and ieIs38 (Pelisch et al, 2017), xpa‐1(ok698) and gcna‐1(emcSi31, emc52, and emc53). Loss of function and transcriptional gfp reporter gcna‐1(emcSi31) mutant was generated by knocking in a “GFP‐SEC” selection cassette from plasmid pDD282 (kind gift from Bob Goldstein, University of North Carolina, Chapel Hill, North Carolina) in the start codon of gcna‐1, according to (Dickinson et al, 2015), by injecting pDD282 with GFP‐SEC flanked by 462 and 618 bp gcna‐1 homology arms together with plasmid pJW1219 (Ward, 2015; kind gift from Jordan Ward, University of California, San Francisco, California) expressing Cas9 and sgRNA targeting gcna‐1 (TGAAGATCTCGAAATGGTGT). gcna‐1 deletion (emc52), giving rise to a truncated protein with a predicted frameshift at E364Q that adds the 22‐amino acid sequence AGWSNSFQFLKAISTSSSSYCQ, and E364Q (emc53) mutants were generated by injection of Cas9 protein together with tracRNA and crRNA targeting gcna‐1 (TTGCTGCATGACACAGTTCA; Integrated DNA Technologies), and an ssODN (GTATGTACAACTGCTGAACGAGTACGAGATACACTGATTCATCAGCTGTGTCATGCAGCAACATGGGTGGTCGACAGGCTTC) as repair template to introduce E364Q. Mutant animals were selected by genotyping PCR. For RNAi experiments, worms were grown on RNAi bacteria (Source BioScience), for one generation (gei‐17) or 7.5 h (smo‐1) before treatment. Control RNAi was vector L4440 (kind gift from Andrew Fire, Stanford University, California). To deplete GFP::degron‐tagged GEI‐17, animals were grown for 24 h on plates containing 1 mM auxin (3‐indoleacetic acid; Sigma). Formaldehyde sensitivity was determined by allowing staged young adults to lay eggs on plates containing formaldehyde (at the indicated dose) for 24 h. Survival was determined by counting dead and living offspring. Cisplatin sensitivity was determined by allowing young staged adults, cultured for 24, 48, and 72 h on plates containing 200 μM cisplatin, to lay eggs for 24 h. Survival was scored by counting unhatched and hatched eggs. UV survival was determined as described previously (Lans et al, 2010). Independent survival assays were performed in quadruplicates or quintuplicates and replicated at least twice. For microscopy, animals were fixed on Poly‐L‐lysine hydrobromide (Sigma) slides with 4% paraformaldehyde in PBS, mounted using Vectashield (Vector Laboratories), and imaged using an LSM700 confocal microscope (Carl Zeiss).
Quantification and statistical analysis
Statistical analysis of data was performed using GraphPad Prism (version 7). Information about statistical tests is provided in the figure legends and methods description. No samples were excluded from the analysis, and no statistical method was used to predetermine sample size. For all experiments, samples were not randomized and the investigators were not blinded to the group allocation during experiments and outcome assessment. Unless otherwise stated, all experiments were repeated at least three times with similar outcomes.
Author contributions
Conceptualization: PS and NM; Methodology: NB, LA, PS, IAH, KT, JCYL, HL, MLN, and NM; Investigation: NB, LA, PS, IAH, KT, and JCYL; Writing–Original Draft: NM; Writing–Review & Editing: NM; Supervision: HL, MLN, and NM; Project Administration: NM; Funding Acquisition: LA, PS, IAH, HL, and NM.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Heinrich Leonhardt, John Rouse, Bob Goldstein, Jordan Ward, and Andrew Fire for providing reagents, members of the Mailand laboratory for helpful discussions, and Katrine Weischenfeldt and the Protein Imaging Platform at the Novo Nordisk Foundation Center for Protein Research for technical support. This work was supported by grants from The Novo Nordisk Foundation (grants no. NNF14CC0001, NNF15OC0016926, and NNF18OC0030752), The Lundbeck Foundation, European Research Council [ERC, grant agreement no. 616236 (DDRegulation)], European Union's Horizon 2020 research and innovation program (Marie Skłodowska‐Curie grant agreement no. 798560), European Molecular Biology Organization (grant agreement ALTF 503‐2016), Danish National Research Foundation (grant no. DNRF115), the Danish Cancer Society, the Dutch Cancer Society, and CancerGenomiCs.nl from the Netherlands Organization for Scientific Research.
The EMBO Journal (2019) 38: e101496
Data availability
The mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009040.
References
- Balakirev MY, Mullally JE, Favier A, Assard N, Sulpice E, Lindsey DF, Rulina AV , Gidrol X, Wilkinson KD (2015) Wss1 metalloprotease partners with Cdc48/Doa1 in processing genotoxic SUMO conjugates. Elife 4: e06763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker S, Weinfeld M, Murray D (2005) DNA‐protein crosslinks: their induction, repair, and biological consequences. Mutat Res 589: 111–135 [DOI] [PubMed] [Google Scholar]
- Bekker‐Jensen DB, Kelstrup CD, Batth TS, Larsen SC, Haldrup C, Bramsen JB , Sorensen KD, Hoyer S, Orntoft TF, Andersen CL, Nielsen ML, Olsen JV (2017) An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst 4: 587–599 e584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmell MA, Dokshin GA, Skaletsky H, Hu YC, van Wolfswinkel JC, Igarashi KJ, Bellott DW, Nefedov M, Reddien PW, Enders GC, Uversky VN, Mello CC, Page DC (2016) A widely employed germ cell marker is an ancient disordered protein with reproductive functions in diverse eukaryotes. Elife 5: e19993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centore RC, Yazinski SA, Tse A, Zou L (2012) Spartan/C1orf124, a reader of PCNA ubiquitylation and a regulator of UV‐induced DNA damage response. Mol Cell 46: 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury BK, Li SS (1997) Identification and characterization of the SMT3 cDNA and gene from nematode Caenorhabditis elegans . Biochem Biophys Res Commun 234: 788–791 [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EJ, Lachaud C, Appleton P, Macartney TJ, Nathke I, Rouse J (2012) DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat Struct Mol Biol 19: 1093–1100 [DOI] [PubMed] [Google Scholar]
- Dickinson DJ, Pani AM, Heppert JK, Higgins CD, Goldstein B (2015) Streamlined genome engineering with a self‐excising drug selection cassette. Genetics 200: 1035–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Johnson LM, Jacobsen SE, Patel DJ (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol 16: 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Cortez D (2015) Purification of proteins on newly synthesized DNA using iPOND. Methods Mol Biol 1228: 123–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, Walter JC (2014) Repair of a DNA‐protein crosslink by replication‐coupled proteolysis. Cell 159: 346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, Walter JC (2011) Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 146: 931–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Riceberg J, Soucy T, Koenig E, Minissale J, Gallery M, Bernard H, Yang X, Liao H, Rabino C, Shah P, Xega K, Yan ZH, Sintchak M, Bradley J, Xu H, Duffey M, England D, Mizutani H, Hu Z et al (2017) Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat Chem Biol 13: 1164–1171 [DOI] [PubMed] [Google Scholar]
- Hendriks IA, Vertegaal AC (2016) Label‐free identification and quantification of SUMO target proteins. Methods Mol Biol 1475: 171–193 [DOI] [PubMed] [Google Scholar]
- Holway AH, Kim SH, La Volpe A, Michael WM (2006) Checkpoint silencing during the DNA damage response in Caenorhabditis elegans embryos. J Cell Biol 172: 999–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide H, Shoulkamy MI, Nakano T, Miyamoto‐Matsubara M, Salem AM (2011) Repair and biochemical effects of DNA‐protein crosslinks. Mutat Res 711: 113–122 [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461: 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ (2011) Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475: 53–58 [DOI] [PubMed] [Google Scholar]
- Lans H, Marteijn JA, Schumacher B, Hoeijmakers JH, Jansen G, Vermeulen W (2010) Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet 6: e1000941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Raschle M, Walter JC, Duxin JP (2018) Replication‐coupled DNA‐protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts. Mol Cell 73: 574–588.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic‐Terzic I, Lopez‐Mosqueda J, Philipp M, Sim JC, Smith KR, Oehler J, Cabrera E, Freire R, Pope K, Nahid A, Norris F, Leventer RJ, Delatycki MB, Barbi G, von Ameln S, Hogel J et al (2014) Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet 46: 1239–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Mosqueda J, Maddi K, Prgomet S, Kalayil S, Marinovic‐Terzic I, Terzic J, Dikic I (2016) SPRTN is a mammalian DNA‐binding metalloprotease that resolves DNA‐protein crosslinks. Elife 5: e21491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Li H, Zhang Y, Ang CY (2001) Determination of formaldehyde in blood plasma by high‐performance liquid chromatography with fluorescence detection. J Chromatogr B Biomed Sci Appl 753: 253–257 [DOI] [PubMed] [Google Scholar]
- Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, Machida YJ (2014) Spartan deficiency causes genomic instability and progeroid phenotypes. Nat Commun 5: 5744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov AY, Lee M, Gundry M, Gravina S, Strogonova N, Tazearslan C, Bendebury A, Suh Y, Vijg J (2012) 5‐aza‐2′‐deoxycytidine‐induced genome rearrangements are mediated by DNMT1. Oncogene 31: 5172–5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee JD, von Hippel PH (1977) Formaldehyde as a probe of DNA structure. r. Mechanism of the initial reaction of Formaldehyde with DNA. Biochemistry 16: 3276–3293 [DOI] [PubMed] [Google Scholar]
- Mosbech A, Gibbs‐Seymour I, Kagias K, Thorslund T, Beli P, Povlsen L, Nielsen SV, Smedegaard S, Sedgwick G, Lukas C, Hartmann‐Petersen R, Lukas J, Choudhary C, Pocock R, Bekker‐Jensen S, Mailand N (2012) DVC1 (C1orf124) is a DNA damage‐targeting p97 adaptor that promotes ubiquitin‐dependent responses to replication blocks. Nat Struct Mol Biol 19: 1084–1092 [DOI] [PubMed] [Google Scholar]
- Nakano T, Ouchi R, Kawazoe J, Pack SP, Makino K, Ide H (2012) T7 RNA polymerases backed up by covalently trapped proteins catalyze highly error prone transcription. J Biol Chem 287: 6562–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Miyamoto‐Matsubara M, Shoulkamy MI, Salem AM, Pack SP, Ishimi Y, Ide H (2013) Translocation and stability of replicative DNA helicases upon encountering DNA‐protein cross‐links. J Biol Chem 288: 4649–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch F, Tammsalu T, Wang B, Jaffray EG, Gartner A, Hay RT (2017) A SUMO‐dependent protein network regulates chromosome congression during oocyte meiosis. Mol Cell 65: 66–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen M, Lukas C, Lukas J, Bekker‐Jensen S, Mailand N (2012) Human RNF169 is a negative regulator of the ubiquitin‐dependent response to DNA double‐strand breaks. J Cell Biol 197: 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psakhye I, Jentsch S (2012) Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151: 807–820 [DOI] [PubMed] [Google Scholar]
- Schellenberg MJ, Lieberman JA, Herrero‐Ruiz A, Butler LR, Williams JG, Munoz‐Cabello AM, Mueller GA, London RE, Cortes‐Ledesma F, Williams RS (2017) ZATT (ZNF451)‐mediated resolution of topoisomerase 2 DNA‐protein cross‐links. Science 357: 1412–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermelleh L, Spada F, Easwaran HP, Zolghadr K, Margot JB, Cardoso MC, Leonhardt H (2005) Trapped in action: direct visualization of DNA methyltransferase activity in living cells. Nat Methods 2: 751–756 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Cortez D (2012) Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat Protoc 7: 594–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Schwarz MS, Bloemeke N, Wolf PG, Jentsch S (2014) A DNA‐dependent protease involved in DNA‐protein crosslink repair. Cell 158: 327–338 [DOI] [PubMed] [Google Scholar]
- Stingele J, Habermann B, Jentsch S (2015) DNA‐protein crosslink repair: proteases as DNA repair enzymes. Trends Biochem Sci 40: 67–71 [DOI] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjaer S, Tainer JA, Skehel JM, Groll M, Boulton SJ (2016) Mechanism and regulation of DNA‐protein crosslink repair by the DNA‐dependent metalloprotease SPRTN. Mol Cell 64: 688–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Boulton SJ (2017) Mechanisms of DNA‐protein crosslink repair. Nat Rev Mol Cell Biol 18: 563–573 [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C (2017) The STRING database in 2017: quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Res 45: D362–D368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN , Vendrell I, Fischer R, Torrecilla I, Drobnitzky N, Freire R, Amor DJ, Lockhart PJ, Kessler BM, McKenna GW, Gileadi O, Ramadan K (2016) Metalloprotease SPRTN/DVC1 orchestrates replication‐coupled DNA‐protein crosslink repair. Mol Cell 64: 704–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez‐Bartolome S et al (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32: 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walport LJ, Hopkinson RJ, Schofield CJ (2012) Mechanisms of human histone and nucleic acid demethylases. Curr Opin Chem Biol 16: 525–534 [DOI] [PubMed] [Google Scholar]
- Ward JD (2015) Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co‐conversion and inactivation of NHEJ repair. Genetics 199: 363–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitkovich A, Costa M (1992) A simple, sensitive assay to detect DNA‐protein crosslinks in intact cells and in vivo . Carcinogenesis 13: 1485–1489 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
The mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009040.