

Abstract
Long noncoding RNAs (lncRNAs) can regulate target gene expression by acting in cis (locally) or in trans (non‐locally). Here, we performed genome‐wide expression analysis of Toll‐like receptor (TLR)‐stimulated human macrophages to identify pairs of cis‐acting lncRNAs and protein‐coding genes involved in innate immunity. A total of 229 gene pairs were identified, many of which were commonly regulated by signaling through multiple TLRs and were involved in the cytokine responses to infection by group B Streptococcus. We focused on elucidating the function of one lncRNA, named lnc‐MARCKS or ROCKI (Regulator of Cytokines and Inflammation), which was induced by multiple TLR stimuli and acted as a master regulator of inflammatory responses. ROCKI interacted with APEX1 (apurinic/apyrimidinic endodeoxyribonuclease 1) to form a ribonucleoprotein complex at the MARCKS promoter. In turn, ROCKI–APEX1 recruited the histone deacetylase HDAC1, which removed the H3K27ac modification from the promoter, thus reducing MARCKS transcription and subsequent Ca2+ signaling and inflammatory gene expression. Finally, genetic variants affecting ROCKI expression were linked to a reduced risk of certain inflammatory and infectious disease in humans, including inflammatory bowel disease and tuberculosis. Collectively, these data highlight the importance of cis‐acting lncRNAs in TLR signaling, innate immunity, and pathophysiological inflammation.
Keywords: cytokine production, host–pathogen interactions, innate immune system, lncRNA, TLRs
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; RNA Biology
Introduction
To date, approximately 16,000 human and 12,000 mouse genes for long noncoding RNAs (lncRNAs; defined as > 200 nucleotides) have been identified, making them the largest known class of noncoding RNAs in the mammalian genome GENCODE (https://www.gencodegenes.org). lncRNAs are versatile regulators of gene expression that interact with DNA, RNA, and proteins, and function through multiple diverse mechanisms that include acting as scaffolds, decoys, and recruiters of genetic modifiers (Cech & Steitz, 2014; Quinn & Chang, 2016; Kopp & Mendell, 2018). Of particular interest, although lncRNAs are able to regulate the expression of neighboring genes (acting in cis) and/in distantly located genes (in trans), increasing evidence suggests that the majority act in cis (Guil & Esteller, 2012). Functional classification of lncRNAs is important for developing experimental approaches to understand lncRNA functions (Kopp & Mendell, 2018).
Innate immunity is a fundamental component of anti‐pathogen defense in eukaryotes and is mediated by several families of cell surface and intracellular pattern recognition receptors, including Toll‐like receptors (TLRs), C‐type lectin receptors, RIG‐I‐like receptors, NOD‐like receptors, and AIM2‐like receptors (Mogensen, 2009). The innate immune system acts as a sensor of microbial infections to rapidly activate defense responses and to initiate long‐lasting adaptive immunity (Iwasaki & Medzhitov, 2015). The human genome encodes about 10 TLR genes, each specialized in recognizing highly conserved structural motifs produced by microbial pathogens (pathogen‐associated microbial patterns, PAMPs) (Song & Lee, 2012). TLRs can function as homodimers or heterodimers; for example, TLR2/TLR1 and TLR2/TLR6 heterodimers recognize lipopeptides from bacteria and mycoplasma; TLR4 and TLR5 homodimers recognize lipopolysaccharides (LPS) from Gram‐negative bacteria and flagellin in flagellate bacteria, respectively; and TLR3, 7, 8, and 9 homodimers respond to various forms of bacterial and viral nucleic acids (Gay & Gangloff, 2007). TLR stimulation initiates intracellular signaling cascades that lead to the production of anti‐microbial genes such as the proinflammatory cytokines interleukin (IL)‐1 and IL‐6, tumor necrosis factor‐α (TNF‐α), and type I and II interferons (IFN‐α/‐β/‐γ), which help orchestrate the innate anti‐pathogen immune response (Kawai & Akira, 2010). These proinflammatory signaling networks are tightly regulated to ensure that cells mount a robust and timely response to pathogens while concomitantly guarding against excessive and sustained inflammation, which may promote the development of chronic inflammatory diseases.
Recent microarray and RNA sequencing data have shown that immune responses alter the expression of hundreds of lncRNAs (Carpenter et al, 2013; Rapicavoli et al, 2013; Li et al, 2014; Atianand et al, 2016; Wang et al, 2017), many of which are trans‐acting regulators of protein‐coding genes (Chen et al, 2017). For example, in TLR1/2‐stimulated THP1‐derived human macrophages, the lncRNA THRIL regulates TNF‐α expression in trans by forming a complex with the ribonucleoprotein (RNP) hnRNPL that acts at the TNF‐α promoter (Li et al, 2014). In another study, lincRNA‐COX2 was found to trans‐regulate inflammatory gene expression in LPS‐stimulated bone marrow‐derived dendritic cells by interacting with hnRNP‐A/B and hnRNP‐A2/B1 (Carpenter et al, 2013). NEAT1 is another trans‐acting lncRNA that stimulates TLR3‐stimulated IL‐8 expression in HeLa cells by binding to the splicing factor SFPQ, thereby inducing SFPQ translocation away from the IL‐8 promoter (Imamura et al, 2014). While these studies confirm the importance of trans activity of lncRNAs in regulating innate immune responses, there remains a large gap in our understanding of the extent to which cis‐regulatory lncRNAs contribute to physiological and pathogenic inflammatory responses.
Here, we sought to identify global changes in coding and noncoding gene expression during the innate immune response of macrophages to TLR ligands. We identified 229 differentially expressed lncRNAs located within 5 kb of protein‐coding genes with immune‐related functions, suggesting that they may be cis‐regulatory lncRNAs with important roles in innate immunity. Consistent with this, three of the identified cis‐acting lncRNA‐coding gene pairs, lnc‐MARCKS, lnc‐BCAT1, and lnc‐BAIAP2, were shown to regulate inflammatory gene expression in macrophages stimulated with a panel of TLR ligands or infected a leading human bacterial pathogen, group B Streptococcus (GBS). We investigated the function and regulatory mechanism of lnc‐MARCKS, which we named Regulator of Cytokines and Inflammation (ROCKI), in more detail. We found that ROCKI regulated expression of its cognate protein‐coding gene myristoylated alanine‐rich protein kinase C (MARCKS) by forming an RNP complex at the MARCKS promoter with apurinic/apyrimidinic endodeoxyribonuclease 1 (APEX1). Notably, the APEX1–ROCKI complex induced recruitment of histone deacetylase HDAC1 to the promoter, which led to a reduction in the active enhancer mark H3K27ac and suppression of MARCKS transcription. In turn, ROCKI‐mediated negative regulation of MARCKS promoted inflammatory cytokine and chemokine production. Finally, we identified genetic variants of ROCKI in human monocytes that are significantly associated with a reduced risk of inflammation‐related (e.g., inflammatory bowel disease) and infection‐related (e.g., Mycobacterium tuberculosis) disease phenotypes in humans.
Collectively, our findings advance our understanding of the mechanisms by which cis‐acting lncRNAs regulate gene expression in response to bacterial and viral infections, and also suggest that ROCKI‐regulated expression of MARCKS may contribute to inflammatory diseases.
Results
TLR signaling induces genome‐wide changes in LncRNA‐ and protein‐coding genes
To understand the global changes in protein‐coding and noncoding gene expression induced by TLR activation, we analyzed the human THP1 monocyte cell line, which can be differentiated into macrophage‐like cells by treatment with phorbol‐12‐myristate‐13‐acetate (PMA). These cells are commonly used to study TLR‐mediated responses against bacterial and viral pathogens. To identify the optimal stimulation time, the cells were incubated for 2, 4, or 8 h with vehicle or one of three TLR ligands; Pam3CSK4 (Pam3), a bacterial triacyl lipoprotein mimetic and TLR1/2 ligand; Pam2CGDPKHPKSF (FSL‐1), a synthetic diacyl lipoprotein from Mycoplasma salivarium and TLR2/6 ligand; or LPS from Escherichia coli K12, a TLR4 ligand. RT–qPCR analysis revealed that expression of the proinflammatory genes IL8 and IL12B peaked at 8 h after stimulation (Fig EV1A–C). RNA‐seq analysis also identified numerous differentially expressed protein‐coding and lncRNA‐coding genes in cells stimulated for 8 h by Pam3, FSL‐1, or LPS compared with unstimulated cells (Fig 1A and B). The Circos plots in Fig 1 highlight changes in expression of two representative protein‐coding genes (NFκB, IFIT1) (Lemaitre, 2004) and two lncRNA‐coding genes (NEAT1, Lnc‐DC) (Imamura et al, 2014; Wang et al, 2014), all of which are involved in host immune responses and are dynamically regulated in response to Pam3, LPS, and FSL‐1 stimulation.
Figure EV1. Relative expression of induced genes during TLR activation.

-
A–FRepresentatives of RNA sequencing data illustrating log2fold changes of indicated genes stimulated with the three TLR ligands compared to mock control.
Source data are available online for this figure.
Figure 1. Identification of cis‐acting LncRNAs associated with innate immunity.
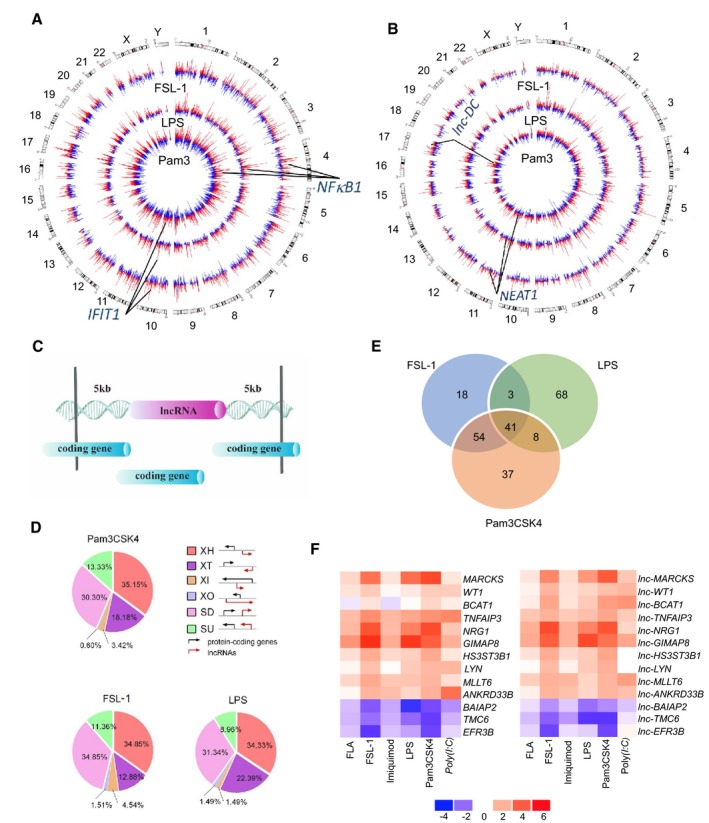
-
A, BTHP1‐derived macrophages were treated with the TLR ligands Pam3, lipopolysaccharide (LPS), or FSL‐1 and analyzed by RNA‐seq 8 h later. Circos plots show genome‐wide differential expression of protein‐coding genes (A) and non‐protein‐coding genes (B) between untreated and treated macrophages. The outermost to innermost circles show downregulated (blue) or upregulated (red) genes in cells stimulated with FSL‐1, LPS, and Pam3, respectively. Chromosomes are indicated by the numbers 1–22 and X and Y.
-
CSchematic showing cis‐acting lncRNAs and protein‐coding genes within 5 kb.
-
DClassification of lncRNA‐ and protein‐coding gene pairs modulated by TLR ligands according to their biotypes. Pie charts show the proportion of each pair of genes classified as XH (antisense head‐to‐head), XT (antisense tail‐to‐tail), XI (antisense inside), XO (antisense outside), SD (sense downstream), and SU (sense upstream).
-
ELncRNA‐ and protein‐coding gene pairs commonly upregulated or downregulated in THP‐1‐derived macrophages stimulated with FSL‐1, LPS, or Pam3 for 8 h.
-
FRT–qPCR validation of differentially expressed lncRNAs and mRNAs in THP1‐derived macrophages stimulated for 8 h with flagellin (FLA), FSL‐1, imiquimod, LPS, Pam3, or poly(I:C). Heat map shows the fold change in expression of 13 mRNAs (left) and lncRNAs (right).
Identification of Cis‐acting LncRNAs and protein‐coding genes associated with TLR signaling
Although lncRNAs can regulate protein‐coding gene expression by acting in cis or in trans, we focused here on identifying cis‐regulatory lncRNAs in TLR‐stimulated THP1‐derived macrophages. We performed bioinformatics analysis of the differentially expressed paired lncRNA‐coding and protein‐coding genes in Pam3‐, LPS‐, and FSL‐stimulated cells, and we selected all gene pairs located within 5 kb of each other (Fig 1C, Table EV1). Previous work has shown that most cis‐acting lncRNA genes are located within 5 kb of their corresponding regulated protein‐coding genes (Mondal et al, 2010; Orom et al, 2010; Cabili et al, 2011; Sigova et al, 2013; Luo et al, 2016).
Our analysis identified 229 candidate lncRNA‐coding and protein‐coding gene pairs, of which 116, 120, and 140 pairs were differentially expressed in cells stimulated with FSL‐1, LPS, and Pam3, respectively, and 41 gene pairs were commonly regulated by all three TLRs (Fig 1E). Among the 229 lncRNAs, 203, 25, and 4 were found to be potential cis‐regulators of 1, 2, and 3 protein‐coding genes, respectively (Table EV1). We further classified these lncRNAs into six locus biotypes based on their orientation to the protein‐coding gene (Fig 1D): XH, antisense head‐to‐head; XT, antisense tail‐to‐tail; XI, antisense inside; XO, antisense outside; SD, sense downstream; and SU, sense upstream, as described previously (Luo et al, 2016). The largest lncRNA biotype modulated by the TLRs was divergent XH, comprising 35.15, 34.85, and 34.33% of total lncRNAs differentially expressed in Pam3‐, FSL‐1‐, and LPS‐stimulated THP1‐derived macrophages, respectively (Fig 1D). These results indicate that a large number of cis‐acting lncRNA genes are non‐randomly distributed in the genome and potentially play a role in regulating innate immune responses.
Cis‐acting LncRNAs regulate expression of nearby protein‐coding genes
To narrow down our functional analyses, we analyzed the 41 gene pairs commonly regulated by Pam3, LPS, and FSL‐1 (Figs 1E and EV1D–F) and selected the 13 most differentially expressed pairs (log2‐fold change in expression of > 2 or < −2), 10 of which were upregulated and 3 were downregulated. We then validated their differential expression by RT–qPCR of FSL‐1, LPS‐ and Pam3‐treated THP1‐derived macrophages (Fig 1F). To determine whether these gene pairs were modulated by other TLRs, we also analyzed their expression in cells stimulated with Salmonella Typhimurium flagellin (FLA), poly(I:C), and imiquimod, which are TLR5, 3, and 7 ligands, respectively. Notably, the 13 gene pairs showed similar patterns of expression in response to all six TLR ligands (Fig 1F), supporting their potential importance in innate immunity.
Next, we examined the effects of the cis‐acting regulatory lncRNAs on their cognate protein‐coding genes. lncRNA expression was silenced in THP1‐derived macrophages by infection with lentiviruses encoding control or targeted shRNAs for 4 days, and relative gene expression was analyzed by RT–qPCR. We used a pool of 2–5 shRNAs per lncRNA, each targeting different gene regions, to eliminate potential off‐target effects and to enhance the knockdown (KD) efficiency. Of the 13 gene pairs examined, eight pairs were confirmed to have a cis‐regulatory relationship (Fig 2A). Macrophages depleted of lnc‐MARCKS and lnc‐BCAT1 showed significantly enhanced expression of the protein‐coding genes MARCKS, and BCAT1, respectively, compared with control shRNA‐expressing cells, which confirmed that these lncRNAs are negative regulators of the linked genes. lnc‐WT1 showed a moderate effect to negatively regulate WT1 gene (Fig 2A). In contrast, KD of lnc‐ANKRD33B, lnc‐BAIAP2, lnc‐TNFAIP3, lnc‐TMC6, and lnc‐NRG1 significantly reduced expression of the cognate protein‐coding genes, suggesting that these are positive cis‐regulatory lncRNAs (Fig 2A). lnc‐BCAT1, lnc‐WT1, lnc‐BAIAP2, and lnc‐TNFAIP3 were identified as XH lncRNAs (antisense head‐to‐head), lnc‐MARCKS as an XT lncRNA (antisense tail‐to‐tail), and lnc‐TMC6, lnc‐ANKRD33B, and lnc‐NRG1 as SD lncRNAs (sense downstream) (Figs 1D and 2A).
Figure 2. Cis‐acting LncRNAs regulate nearby genes and cytokine production in response to group B Streptococcus (GBS) infection.

-
AActivation and suppression of nearby genes by eight cis‐acting lncRNAs. RT–qPCR analysis of THP1‐derived macrophages transduced with lentiviruses expressing non‐targeting control (NC) shRNA or shRNAs targeted to the indicated lncRNAs and protein‐coding genes. Mean ± SD of n = 3. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001 by Student's t‐test.
-
B–DSilencing of cis‐regulated gene pairs affects GBS‐induced expression of IL‐6 (B and C) and IL‐1α (D). RT–qPCR analysis (B and D) or IL‐6 ELISA quantification (C) of THP1‐derived macrophages transduced with the indicated NC or targeting shRNAs for 72 h, infected with GBS for 1 h, and then treated with gentamicin for 30 min to terminate infection. RT–qPCR and ELISA were performed at 10 h post‐infection. Mean ± SD of n = 3. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001 by Student's t‐test.
Characterization of the cis‐regulatory LncRNAs
Because most of the lncRNAs identified here were annotated based on bioinformatics predictions, we performed rapid amplification of cDNA ends (RACE) to confirm their location and to determine the 5′ and 3′ end sequences of the uncharacterized lncRNAs. From these experiments, we found that lnc‐ANKRD33B and lnc‐NRG1, which were predicted to be in the sense strand overlapping the corresponding protein‐coding genes, were in fact isoforms of the protein‐coding genes. lnc‐MARCKS, lnc‐BCAT1, lnc‐BAIAP2, and lnc‐TNFAIP3 were confirmed to be bona fide transcripts, and the predicted sequences were authenticated (Fig EV2A). We identified two isoforms of lnc‐BAIAP2 and lnc‐TNFAIP3 and only one form of lnc‐MARCKS and lnc‐BCAT1 (Fig EV2A). Thus, among the 13 gene pairs functionally tested here, 6 (46%) included cis‐acting lncRNAs. This demonstrates the power of our prediction methods to identify cis‐acting lncRNAs. Interestingly, five of the six lncRNAs were antisense to their protein‐coding genes, strongly suggesting that divergent (XH and XT) biotypes are enriched among cis‐acting lncRNAs.
Figure EV2. Characterization of candidate LncRNAs and knockdown Efficiency of shRNAs targeting LncRNAs and protein‐coding genes.
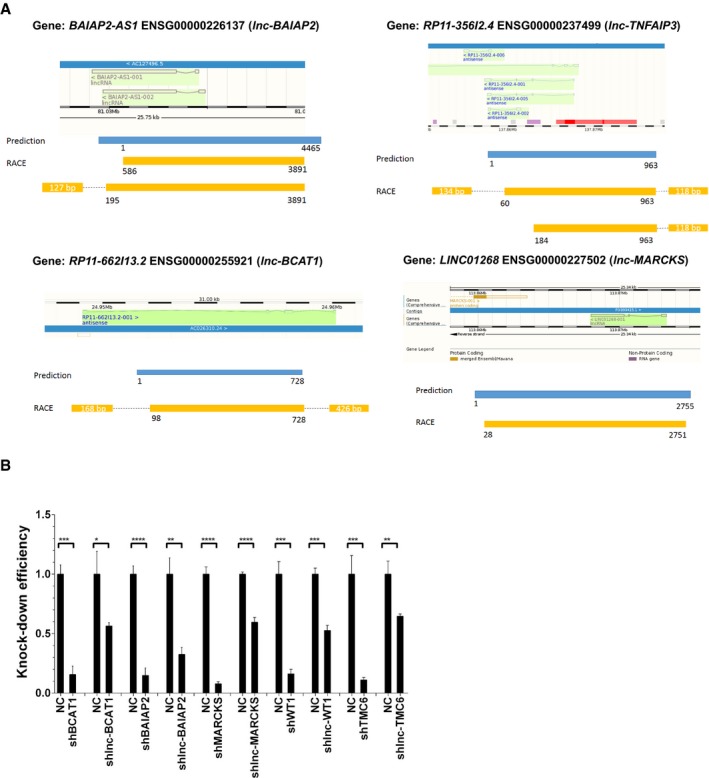
- Genetic loci of lnc‐BAIAP2, lnc‐TNFAIP3, and lnc‐BCAT1 lnc‐BAIAP2, elucidated by rapid amplification of cDNA ends (RACE).
- KD efficiency of shRNAs targeting the indicated lncRNAs and protein‐coding genes. RT–qPCR analysis was performed on THP‐1 cells 4 days after transduction with lentiviruses expressing non‐targeting control (NC) or targeting shRNAs. Mean ± SD of n = 3. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001 by Student's t‐test.
Cis‐acting LncRNAs regulate cytokine production in response to group B streptococcus (GBS) infection
To confirm the relevance of the identified gene pairs in the response to live bacterial infection, we used the Gram‐positive pathogen GBS as a model system to activate TLR2 signaling (Lehnardt et al, 2006; Henneke et al, 2008). THP1‐derived macrophages were infected with pooled lentiviruses encoding 2–5 shRNAs against lnc‐BAIAP2, lnc‐MARCKS, lnc‐WT1, lnc‐BCAT1, lnc‐TMC6, or their corresponding protein‐coding genes BAIAP2, MARCKS, WT1, BCAT1, or TMC6. All gene pairs were shown by RT–qPCR to be silenced with good efficiencies (Fig EV2B). Three days later, the cells were infected with GBS (multiplicity of infection = 10 bacteria/macrophage) for 1 h, after which gentamicin was added to kill extracellular bacteria, and the cells were collected for RT–qPCR analysis 10 h later. Expression of IL‐6 and IL‐1α, two potent proinflammatory genes induced by GBS infection, was significantly enhanced by KD of BAIAP2, lnc‐BAIAP2, MARCKS, WT1, and BCAT1 and significantly suppressed by KD of lnc‐MARCKS and lnc‐BCAT1 (Fig 2B–D). Although KD of lnc‐WT or lnc‐TMC6 also had modest effects on IL‐6 expression, the differences were not statistically significant (Fig 2C). These results substantiate the predicted mode of action of lnc‐BAIAP2 as a positive cis‐regulator and of lnc‐MARCKS and lnc‐BCAT1 as negative cis‐regulators of their corresponding protein‐coding genes. The data also confirm the involvement of these three gene pairs in regulating proinflammatory cytokine expression during GBS infection.
The long noncoding RNA, lnc‐MARCKS, is a positive regulator of inflammatory responses
We observed a global regulation of lnc‐MARCKS on other cytokine and chemokine expressions, such as CXCL1, IL‐24, and CCL3 (Figs 1F and 2B–D, see below Fig 6), suggesting its role as a master regulator of innate immunity. Therefore, lnc‐MARCKS was chosen for further investigation.
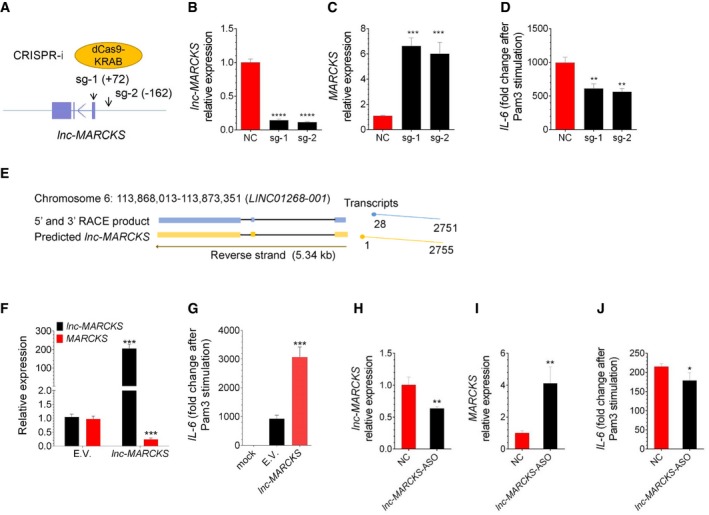
To determine the lnc‐MARCKS function by an alternative approach, THP1 cells were transduced with CRISPR inhibitor system (CRISPRi) targeting lnc‐MARCKS (Gilbert et al, 2013). Two sgRNAs were designed to target lnc‐MARCKS (Fig 3A) and > 80% inhibition of lnc‐MARCKS was achieved (Fig 3B). Consistent with the results of our functional analysis by RNAi (Fig 2B–D), CRISPRi of lnc‐MARCKS increased MARCKS expression (Fig 3C) and caused repression of IL‐6 induction in sgRNA‐expressed cells (Fig 3D), indicating that lnc‐MARCKS was a negative regulator of MARCKS and positive regulator of inflammation responses. Additionally, overexpression of an approximately 2.7‐kb transcript of lnc‐MARCKS (identified by RACE; Fig 3E) in THP1‐derived macrophages significantly downregulated basal MARCKS mRNA (Fig 3F) levels and increased IL‐6 mRNA levels upon Pam3 stimulation (Fig 3G). Further characterization showed that the lnc‐MARCKS RNA was present in THP1 macrophages at ∼6 copies per cell and mainly distributed in nucleus (Fig EV3A). A small proportion of lncRNAs undergo translation and could encode stable and functional peptides (Housman & Ulitsky, 2016; Hezroni et al, 2017). To determine whether lnc‐MARCKS lacks protein‐coding potential or not, we predicted the possible proteins in lnc‐MARCKS using SnapGene software and found one ORF (93aa) near the 3′‐end. However, the coding product cannot be aligned to any known proteins (Fig EV3B). These analyses suggested that lnc‐MARCKS did not code for small peptides. In addition, ribosome footprinting data from GWIPS shows very weak ribosomal binding on lnc‐MARCKS, which suggest that lnc‐MARCKS is a noncoding RNA (Fig EV3C) (Ingolia et al, 2009). To further confirm the noncoding potential of lnc‐MARCKS, we fused lnc‐MARCKS sequences to the reading frame of a Flag tag and transfected the vectors into 293T cells. Notably, anti‐Flag Western blot analysis failed to detect any protein expression from the constructs, confirming that lnc‐MARCKS is a bona fide noncoding RNA (Fig EV3D).
Figure 3. lnc‐MARCKS negatively regulates MARCKS expression and promotes inflammatory responses.
-
A–DRepression of lnc‐MARCKS using CRISPRi increased MARCKS expression and decreased IL‐6 induction. (A) Illustration of CRISPRi system and the relative locations of the two sgRNAs. (B‐D) lnc‐MARCKS (B), MARCKS (C), and IL‐6 (D) expressions were quantified in cells stably expressing dCas9/KRAB/BFP and the NC or sgRNAs. Mean ± SD of n = 3. **P < 0.01, ***P < 0.005, ****P < 0.0001 by Student's t‐test.
-
EThe lnc‐MARCKS gene locus. The full‐length sequence of lnc‐MARCKS was obtained by rapid amplification of cDNA ends (RACE). lnc‐MARCKS is located on human chromosome 6 and encompasses 2 exons.
-
F, GOverexpression of lnc‐MARCKS decreases MARCKS expression and enhances IL‐6 expression. RT–qPCR analysis of THP1 cells transduced with lentiviruses expressing empty vector (E.V.) or lnc‐MARCKS, treated with PMA overnight, and left unstimulated or stimulated with Pam3 for 8 h. lnc‐MARCKS and MARCKS expression (F), IL‐6 expression (G). Mean ± SD of n = 3. ***P < 0.005 by Student's t‐test.
-
H–Jlnc‐MARCKS regulates MARCKS and IL‐6 in primary macrophages. Primary macrophages were transfected with NC or lnc‐MARCKS antisense oligo to knock down lnc‐MARCKS expression. 48 h later, cells were stimulated with Pam3 for 8 h and harvested to detect lnc‐MARCKS (H), MARCKS (I), and IL‐6 expressions (J). Mean ± SD of n = 3. *P < 0.05, **P < 0.01 by Student's t‐test.
Figure EV3. Characterization of ROCKI .

- Quantification of ROCKI copy number in nucleus and cytoplasm of THP1 macrophages. Cell was fractioned to nucleus and cytoplasm sections. ROCKI DNA concentration was quantified with a standard curve and calculated to be present in THP1 macrophages at ∼6 copies per cell and was mainly distributed in the nucleus.
- ROCKI does not code any known peptides. One potential ORF near the 3′‐end region of ROCKI was predicted using SnapGene software (orange arrow), but did not align to any known peptides in human.
- Ribosomal footprinting data generated for ROCKI using GWIPS‐wiz.
- ROCKI displayed no protein‐coding potential in a eukaryotic cell expression assay. ROCKI transcripts were cloned into pFLAG‐CMV plasmid and transfected into 293FT cells for 48 h. Expression of FLAG‐fused proteins was analyzed by immunoblotting with an anti‐FLAG antibody. Left to right: E.V., empty vector; plus strand of ROCKI; minus strand of ROCKI; ROCKI with ATG plus G added to the 5′ end; ROCKI with ATG plus GG added to the 5′ end. A ZIKV coding gene (capsid protein) was probed as a protein‐coding gene control.
To further strengthen the biological significance of lnc‐MARCKS, we generated primary human macrophages and silenced lncRNA‐MARCKS expression by using an antisense oligo targeting the RNA region at 1,887–1,907 nucleotides. Consistent with the shRNA and CRISPRi results (Figs 2B–D and 3A–D), lnc‐MARCKS silencing by ASO increased MARCKS expression and repressed IL‐6 induction as compared with cells transfected with a control ASO (Fig 3H–J). Taken together, these results identify lnc‐MARCKS as a positive regulator of the inflammatory macrophage response. Therefore, we re‐named this lncRNA ROCKI (Regulator of Cytokines and Inflammation).
ROCKI functions by binding to the MARCKS distal promoter where it interacts with APEX1
To investigate the mechanism by which ROCKI cis‐regulates MARCKS expression, we first performed chromatin isolation by RNA purification (ChIRP) assays to assess ROCKI binding to the MARCKS promoter. For this, we used an antisense oligonucleotide tiling method that encompassed the entire ROCKI sequence (Li et al, 2014; Lin et al, 2014). Chromatin fractions from control or Pam3‐treated THP1‐derived macrophages were incubated with biotinylated ROCKI or control (lacZ) RNA probes and analyzed by RT–qPCR for the presence of MARCKS promoter regulatory sequences. We designed six PCR primers spanning from −1,370 to +87 relative to the +1 transcription start site. By analogy to the conserved murine MARCKS sequence, this region was expected to include a distal promoter and a core promoter (Harlan et al, 1991; Wang et al, 2002). We observed that biotinylated ROCKI was significantly enriched compared to the control probe (Fig 4A, left). Importantly, RT–qPCR analysis of the sequences pulled down with ROCKI showed specific enrichment of the −1,370/−1,140 and −1,158/−1,060 MARCKS promoter sequences, indicating that ROCKI bound to the distal, rather than the proximal, promoter (Fig 4A).
Figure 4. ROCKI (lnc‐MARCKS) binds to the MARCKS Promoter and Interacts with APEX1.
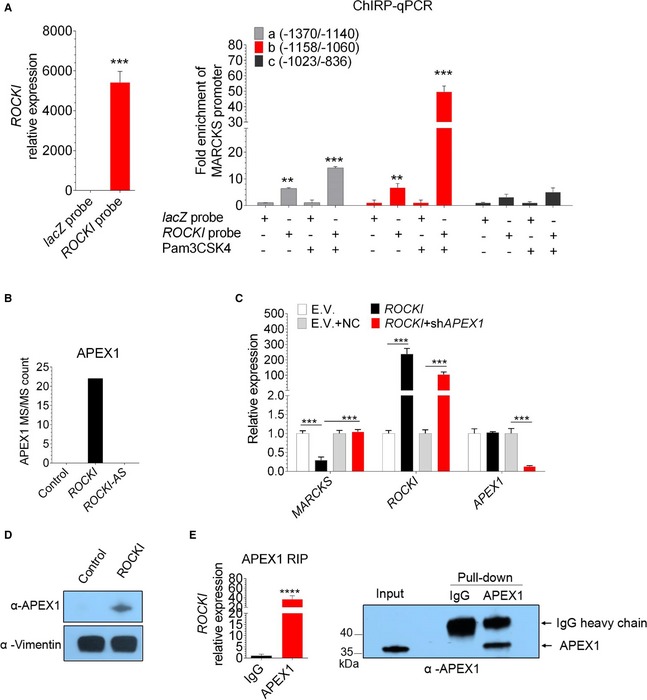
- ROCKI is recruited to the MARCKS promoter in Pam3‐stimulated THP1‐derived macrophages. ChIRP analysis of THP1‐derived macrophages treated with Pam3 for 8 h. ChIRP assays were performed using a non‐specific lacZ probe or probes specific for lnc‐MARCKS. Specificity of ROCKI probes (left), and enrichment at the MARCKS promoter (right). Probes are listed in Table EV2. Mean ± SD of n = 3. **P < 0.01, ***P < 0.005 by Student's t‐test.
- Mass spectrometry identifies APEX1 as a candidate ROCKI‐binding protein in THP1‐derived macrophages. APEX1 MS/MS count represents the ratio between the number of APEX1 peptides and all peptides detected. Mean of n = 2 samples. ROCKI and ROCKI‐AS represent ROCKI and antisense strand of ROCKI transcripts, respectively.
- ROCKI inhibition of MARCKS expression is dependent on APEX1. RT–qPCR analysis of MARCKS, ROCKI, and APEX1 expression in THP1‐derived macrophages overexpressing ROCKI and/or depleted of APEX1. Mean ± SD of n = 3. ***P < 0.001 by Student's t‐test.
- APEX1 specially binds to ROCKI. Western blot analysis of APEX1 in lacZ, or ROCKI pull‐down fractions. Vimentin, which non‐specifically binds to all RNA probes, is shown as a loading control.
- ROCKI associates with APEX1. Left: RT–qPCR analysis of ROCKI present in anti‐APEX1 antibody or control IgG immunoprecipitates from nuclear lysates of Pam3‐treated THP1‐derived macrophages. Right: APEX1 Western blot of the same immunoprecipitates. Mean ± SD of n = 4. ****P < 0.0001 by Student's t‐test.
Source data are available online for this figure.
We next asked whether ROCKI acted alone at the MARCKS promoter or might functions as part of an RNP. To identify ROCKI‐binding proteins, we in vitro‐transcribed and biotinylated ROCKI, antisense ROCKI, or a partial lacZ mRNA sequence (without protein‐coding capacity) and incubated them with nuclear proteins isolated from Pam3‐treated THP1‐derived macrophages. The associated proteins were then pulled down and analyzed by mass spectrometry. We identified the 10 most enriched ROCKI‐binding proteins as APEX1 (apurinic/apyrimidinic endodeoxyribonuclease 1), NUDT21 (NUDIX Hydrolase 21), RCC2 (regulator of chromosome condensation 2), HMGB2 (high mobility group box 2), HMGB3 (high mobility group box 3), HSD17B4 (hydroxysteroid 17‐beta dehydrogenase 4), KHSRP (KH‐type splicing regulatory protein), NCL (neuronal ceroid lipofuscinoses), CIRBP (cold‐Inducible RNA binding protein), and ELAVL1 (ELAV like RNA binding protein 1) based on the number of peptides specifically associated with ROCKI (Figs 4B and EV4A, Table EV3).
Figure EV4. Knockdown of APEX1 increases MARCKS expression.

- ROCKI‐binding proteins identified by affinity purification and mass spectrometry. The 10 most enriched candidate proteins associated with ROCKI compared with control RNA (lacZ) and antisense ROCKI (ROCKI‐AS) are shown. Mean ± SD of n = 2.
- RT–qPCR analysis of MARCKS in THP1‐derived macrophages expressing shRNAs against the candidate ROCKI‐binding genes or a NC (control) shRNA for 4 d. Mean ± SD of n = 3. **P < 0.01 by Student's t‐test.
- Knockdown efficiency of shRNAs in the experiment described in (B), as assessed by RT–qPCR analysis of the corresponding genes. Mean ± SD of n = 3. ***P < 0.005, ****P < 0.0001 by Student's t‐test.
To determine whether these candidate ROCKI‐binding proteins function in regulating MARCKS expression, we transfected THP1‐derived macrophages with shRNAs against each gene and performed RT–qPCR analysis to confirm they were efficiently knocked down (Fig EV4C). Among the 10 genes examined, silencing of APEX1 alone significantly affected MARCKS expression, inducing ~2‐fold increase in the level of MARCKS transcripts compared with the control shRNA‐transfected cells (Fig EV4B). Notably, ROCKI‐induced suppression of MARCKS inhibition was rescued by co‐transfection of THP1‐derived macrophages with shAPEX1, indicating that ROCKI‐mediated inhibition of MARCKS expression was dependent on APEX1 (Fig 4C).
We also performed pull‐down assays with in vitro‐transcribed biotinylated ROCKI or lacZ RNA followed by Western blotting with anti‐APEX1 antibody, which confirmed that APEX1 specifically interacts with ROCKI (Fig 4D). To further confirm the APEX1‐RNA interactions by an alternative experiment, we performed immunoprecipitation of endogenous APEX1 from nuclear extracts of THP1‐derived macrophages followed by RT–qPCR and the results revealed ∼40‐fold enrichment of ROCKI in the anti‐APEX1 immunoprecipitates compared to the IgG control (Fig 4E). Taken together, these results demonstrate that ROCKI and APEX1 form an RNP complex that regulates MARCKS expression.
ROCKI–APEX1 ribonucleoprotein complex facilitates HDAC1‐mediated remodeling of the MARCKS promoter
Next, we sought to understand how the ROCKI–APEX1 complex regulates MARCKS expression. We hypothesized that ROCKI may recruit APEX1 to the MARCKS promoter. To test this, we performed chromatin immunoprecipitation (ChIP) assays by immunoprecipitating endogenous APEX1 protein from nuclear lysates of THP1‐derived macrophages, eluting the associated DNA, and performing RT–qPCR analysis to probe for the presence of MARCKS promoter sequences. Indeed, APEX1 immunoprecipitates contained higher levels of the two MARCKS promoter sequences tested compared with control IgG immunoprecipitates (Fig 5A). Since APEX1 enrichment was impaired in ROCKI KD cells (Fig 5B), these data support our hypothesis that ROCKI recruits APEX1 to form an RNP at the MARCKS promoter. Importantly, ROCKI KD had no significant effect on APEX1 binding to the β‐actin promoter (ACTB2), highlighting the specific function of ROCKI–APEX1 in regulating MARCKS expression (Fig 5B).
Figure 5. ROCKI reduces H3K27ac modification of the MARCKS promoter via APEX1 binding and recruitment of HDAC1.
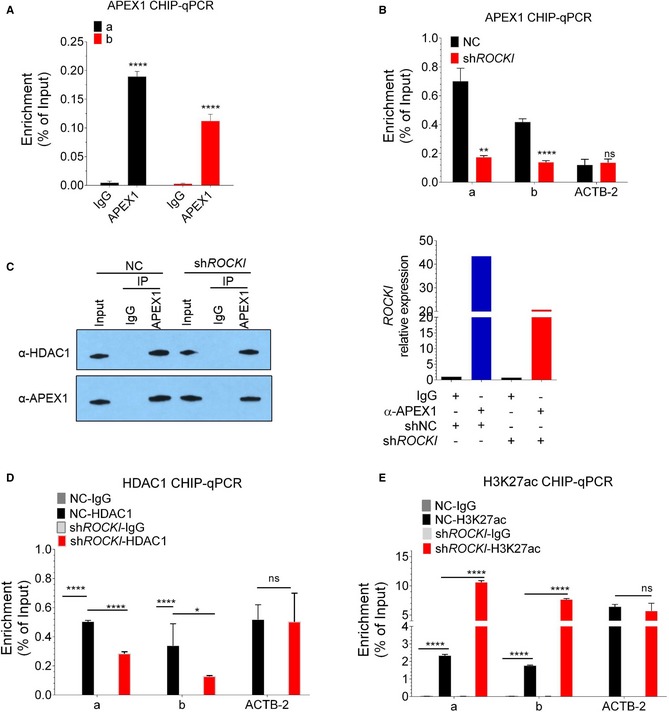
- ChIP analysis of THP1‐derived macrophages demonstrates APEX1 recruitment to the MARCKS promoter. RT–qPCR analysis of fragments of the MARCKS promoter region (a and b) or a non‐specific region (nc) co‐immunoprecipitated with anti‐APEX1 antibody. Mean ± SD of n = 4. ****P < 0.0001 by Student's t‐test.
- ChIP analysis of APEX1 recruitment to the MARCKS promoter was performed as described for (A) except the cells expressed non‐targeting control (NC) or ROCKI shRNA and treated with Pam3 for 8 h. The y‐axis shows the percentage of enrichment in normalized to input. An unrelated region of the ACTB2 promoter was amplified as a control. Mean ± SD of n = 4. **P < 0.01. ****P < 0.0001, ns, non‐significant by Student's t‐test.
- Left: Western blot analysis of APEX1 and HDAC1 in control rabbit IgG or anti‐APEX1 antibody immunoprecipitates from NC (control) or ROCKI‐knockdown THP1‐derived macrophages. Right: RT–qPCR analysis of ROCKI in the same immunoprecipitates.
- Knockdown of ROCKI decreased HDAC1 enrichment on the MARCKS promoter. ChIP‐qPCR analysis of HDAC1 was performed as described for (A) using an anti‐HDAC1 antibody. Mean ± SD of n = 3. *P < 0.05, ****P < 0.0001, ns, non‐significant by Student's t‐test.
- Knockdown of ROCKI enhances H3K27ac deposition on the MARCKS promoter. ChIP‐qPCR analysis of H3K27ac was performed as described for (A) using an anti‐H3K27ac antibody. Mean ± SD of n = 3. ****P < 0.0001, ns, non‐significant by Student's t‐test.
Source data are available online for this figure.
APEX1 is known to repress expression of human parathyroid hormone and renin by binding to the negative regulatory calcium‐response element in the promoters and recruiting HDAC1, which removes the activating chromatin mark H3K27ac3 and reduces gene transcription (Bhakat et al, 2003; Fuchs et al, 2003). To determine whether APEX1 might act similarly to suppress MARCKS transcription, we first performed Western blotting to probe for the presence of HDAC1 in anti‐APEX1 immunoprecipitates from nuclear lysates of control or shROCKI‐expressing THP1‐derived macrophages. These results show that HDAC1 associates with APEX1 in the nucleus and this interaction was not dependent on ROCKI expression (Fig 5C, left). RT–qPCR was performed to show the successful pull down of ROCKI in APEX1 immunoprecipitates and successful silencing in ROCKI‐knockdown cells (Fig 5C right). We next analyzed the level of HDAC1 and H3K27ac at the MARCKS promoter in these cells by performing ChIP assays with an anti‐HDAC1 or anti‐H3K27ac antibody (Fig 5D and E). As expected, HDAC1 and H3K27ac immunoprecipitates were enriched in the two MARCKS promoter sequences and the ACTB2 sequence, but only MARCKS sequence enrichment was significantly decreased in HDAC1‐CHIP (Fig 5D), while increased in H3K27ac‐CHIP (Fig 5E) by ROCKI KD. Taken together, these results show that ROCKI interacts with APEX1 to recruit HDAC1 to the MARCKS promoter, leading to a reduction in H3K27ac and repression of MARCKS transcription.
ROCKI regulates proinflammatory gene expression
To investigate how the ROCKI–APEX1–HDAC1–MARCKS axis regulates the innate inflammatory response in THP1‐derived macrophages, we performed genome‐wide RNA sequencing and analyzed differential gene expression changes in MARCKS KD‐ or ROCKI‐overexpressing cells after stimulation with Pam3 for 8 h. Genes differentially regulated by both ROCKI overexpression (800 upregulated and 1,435 downregulated) and MARCKS KD (219 upregulated and 294 downregulated) were selected based on a significant (P < 0.05) log2‐fold change of > 1 or < −0.5. We identified 128 genes that were considered to be commonly upregulated by both ROCKI and MARCKS, while 154 genes were identified to be commonly downregulated (Fig 6A and Table EV4). Notably, Gene Ontology analysis showed that the upregulated genes were significantly enriched in terms related to inflammatory responses (Fig EV5A). Among the most significantly upregulated genes were multiple cytokines, chemokines, and migration factors (Fig 6B); of these genes, we confirmed the lncRNA effects on 10 by RT–qPCR (Fig 6C).
Figure 6. ROCKI and MARCKS are Ca2+ regulators in TLR1/2 signaling.
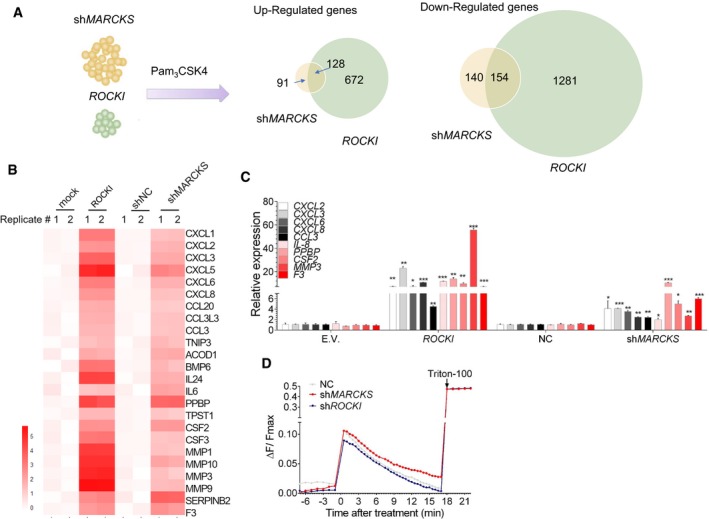
- Identification of genes regulated by both ROCKI and MARCKS by RNA‐seq of ROCKI‐overexpressing or MARCKS‐knockdown THP1‐derived macrophages stimulated with Pam3 for 8 h.
- Heatmap of representative genes commonly upregulated in ROCKI‐overexpressing and MARCKS‐knockdown THP1‐derived macrophages.
- RT–qPCR validation of inflammatory gene expression in ROCKI‐overexpressing and MARCKS‐knockdown THP1‐derived macrophages stimulated with Pam3 for 8 h. E.V., empty vector; NC, control shRNA. Mean ± SD of n = 4. *P < 0.05, **P < 0.01, ***P < 0.001 by Student's t‐test.
- MARCKS is a negative regulator of Ca2+ signaling. Fluo‐4 fluorescence of MARCKS, ROCKI, or NC (control) shRNA‐expressing THP1‐derived macrophages before and after treatment with PMA and Pam3. Triton X‐100 was added to obtain the maximal [Ca2+] signal. Fluorescence levels are expressed as the change in signal normalized to the maximal fluorescence (ΔF/F max).
Figure EV5. Target genes of MARCKS and ROCKI are enriched in immune response‐related genes.

- Gene Ontology analysis of 128 genes commonly upregulated by ROCKI overexpression and MARCKS knockdown using Gene Set Enrichment Analysis.
- Enrichment analysis of the common target genes using Thomson Reuters server (portal.genego.com).
- MARCKS and ROCKI target genes are enriched in genes downstream of histamine H1 receptor signaling. The pathway was constructed by Thomson Reuters scientists using a high quality manual curation process based on published peer‐reviewed literature. Blue and red indicate downregulated and upregulated genes, respectively.
In addition, we observed that these commonly regulated genes are highly overlapped with immune pathways, including the IL‐17 signaling pathway, IL‐1 signaling in melanoma, and histamine H1 receptor signaling in immune responses (Fig EV5C). In the histamine H1 receptor signaling, Ca2+ initiates the inflammation response by binding to PKCα (Fig EV5C). Since MARCKS protein is a well‐characterized modulator of Ca2+ signaling (Hartwig et al, 1992; Gadi et al, 2011; Rodriguez Pena et al, 2013) and APEX1 can be acetylated by calcium activation to modulate its transcriptional regulatory function (Bhakat et al, 2003, 2009), we asked whether it might play a similar role in Pam3‐stimulated THP1‐derived macrophages. To test this, we loaded cells expressing control, MARCKS, or ROCKI shRNA with the Ca2+‐sensitive fluorescent dye Fluo‐4 and examined the change in fluorescence signal after Pam3 stimulation. As shown in Fig 6D, cytoplasmic Ca2+ levels were increased by MARCKS KD and decreased by ROCKI KD compared with control cells, suggesting that the ROCKI–MARCKS axis may regulate cytokine production in TLR‐stimulated macrophages via Ca2+ signaling pathways.
Genetic links between ROCKI and inflammation‐ and infection‐related phenotypes
Finally, we used genetic variation to explore whether ROCKI function is linked to human disease phenotypes. We first sought to identify cis variants that affect ROCKI expression using quantitative trait loci (QTL) data. We obtained whole blood expression QTL (eQTL) data from the GTEx project v7 release (GTEx Consortium et al, 2017). There was a significant ROCKI eQTL for variants upstream of the promoter (Fig 7A). These variants did not show evidence for significant eQTL association with MARCKS, confirming a primary effect on ROCKI (Fig EV6A). To further determine the effects of these variants in monocytes, we analyzed CD14+ monocyte QTL datasets for H3K4me1 and H3K27ac signal from the BLUEPRINT epigenome project (Chen et al, 2016). For the same ROCKI eQTL variants, there were also significant QTLs for H3K27ac signal directly at the ROCKI promoter and H3K4me1 signal in a broader 35‐kb region spanning both MARCKS and ROCKI (Fig EV6). There was both strong co‐localization and directional consistency in the ROCKI expression, H3K27ac, and H3K4me1 QTLs (Pshared > .95), suggesting they all represent the same association signal (Fig 7A). To determine the variant responsible for the ROCKI QTL, we performed genetic fine‐mapping and calculated the posterior probability of association (PPA) that each variant was causal for the QTL signal (see Experimental Procedures). The most likely causal variant rs9387181 (PPA = 0.15) overlapped a regulatory region 6‐kb upstream of ROCKI annotated with DNase I hypersensitivity sites for monocytes and ChIP‐seq sites for immune transcription factors such as RELA and IRF4 (Fig EV6B). These data thus identify cis‐regulatory variants that affect ROCKI activity in whole blood and monocytes.
Figure 7. Genetic link between ROCKI and human disease and a model for ROCKI function.

- QTL signals for ROCKI (LINC01268) gene expression in whole blood (top, black), H3K27ac promoter signal in monocytes (middle, green), and H3K4me1 signal in monocytes (bottom, orange) in a 400‐kb window around the MARCKS/ROCKI locus. The signals were strongly co‐localized (Pshared = 0.97, Pshared = 0.98), suggesting they represent the same signal.
- Signed test statistics from UK Biobank association data for 1,463 disease‐ or medication‐related traits for rs9387181. Black crosses denote the mean ± SEM for each group. Dotted horizontal line indicates the null.
- RT–qPCR analysis of ROCKI expression in dendritic cell samples from uninfected and M. tuberculosis‐infected donors. Mean ± SD of n = 6. ***P < 0.001 by Student's t‐test.
- A cis‐regulatory model for ROCKI function in the negative regulation of MARCKS in innate immune cells. Upon TLR activation, ROCKI binds to the MARCKS promoter region and recruits APEX1, which facilitates HDAC1‐mediated removal of the H3K27ac modification, thereby inhibiting MARCKS expression.
Figure EV6. Genetic variants regulating ROCKI expression.
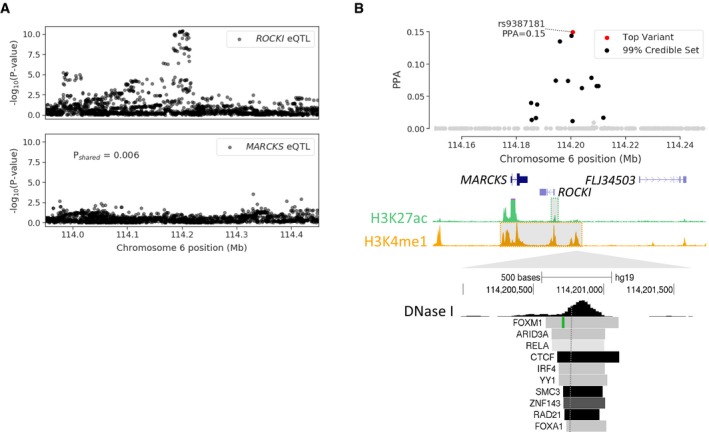
- eQTL association and Bayesian co‐localization for ROCKI (top) and MARCKS (bottom) in whole blood.
- (top) Genetic fine‐mapping of the ROCKI QTL signal and variant posterior probabilities of association (PPA), and genomic location of H3K27 and H3K4me1 QTL signals in gray. (bottom) Genomic annotations at the most likely causal variant rs9387181 6‐kb upstream of ROCKI include DNase I hypersensitivity (DHS) for monocytes and transcription factor binding ChIP‐seq for RELA, IRF4, and other factors.
We next determined the effects of the ROCKI cis‐regulatory variants on human phenotypes. We obtained association data for 1464 disease‐related phenotypes (UK Biobank) using the most probable ROCKI eQTL variant rs9387181 to represent the association signal. The ROCKI‐reducing allele of rs9387181 was broadly correlated with reduced risk of inflammatory and infection‐related, cardiovascular, and cognitive diseases and reduced use of the corresponding medications (Fig 7B). The most significant correlations for this variant within each group included reductions in flucloxacillin treatment (t = −3.3), prednisolone treatment (t = −2.8), atherosclerosis (t = −2.99), tuberculosis infection (t = −2.5), and inflammatory bowel disease (t = −2.5). These results thus provide genetic support for a link between ROCKI function in whole blood cells and isolated monocytes and the risk of inflammation‐ and infection‐related disease phenotypes. To probe further, we analyzed data from a study of gene expression in human dendritic cells infected with Mycobacterium tuberculosis (Pacis et al, 2015). Intriguingly, ROCKI expression was significantly increased in the cells infected with M. tuberculosis (Fig 7C), confirming the predicted association.
Collectively, the data presented here demonstrate a role for the lncRNA ROCKI in innate immune function during human pathogen infection. Our data support a model in which ROCKI cis‐regulates MARCKS expression via formation of an RNP with APEX1 at the MARCKS promoter, which facilitates HDAC1‐mediated removal of H3K27ac, leading to reduced MARCKS expression and suppression of Ca2+ signaling and inflammatory cytokine production (Fig 7D).
Discussion
Activation of immune cells by pathogens leads to rapid and dynamic changes in gene expression aimed at combating the infection. Previous studies have demonstrated that the expression of hundreds of lncRNAs is altered by stimulation of innate and adaptive immune cells; however, their functions remain largely unknown (Carpenter et al, 2013; Rapicavoli et al, 2013; Li et al, 2014; Atianand et al, 2016; Wang et al, 2017). Here, we analyzed the expression of coding and noncoding genes in TLR‐stimulated human macrophages to identify cis‐regulatory lncRNAs and protein‐coding genes. We identified 229 gene pairs with potential roles in immune regulation and selected 13 candidate pairs that were commonly regulated by six TLRs (TLR1/2, 2/6, 3, 4, 5, and 7) for functional validation. Eight lncRNAs were confirmed to cis‐regulate their corresponding protein‐coding genes. Of these, three (ROCKI, lnc‐BCAT1, and lnc‐WT1) were negative regulators and five (lnc‐ANKRD33B, lnc‐NRG1, lnc‐TMC6, lnc‐BAIAP2, and lnc‐TNFAIP3) were positive regulators of their cognate genes. Further analysis showed that lnc‐ANKRD33B and lnc‐NRG1 overlapped with and were assumed to be isoforms of their cognate coding genes. Interestingly, five of the remaining six lncRNAs had antisense biotypes (XH: lnc‐BCAT1, lnc‐WT1, lnc‐BAIAP2, lnc‐TNFAIP3, and XT: ROCKI). Antisense lncRNAs comprise over 90% of genic lncRNAs (lncRNA < 5 kb to a coding gene). Since > 60% of the 229 potential gene pairs identified here include antisense lncRNAs (Mancek‐Keber et al, 2012; Lee et al, 2015), we expect that further mining of our data will reveal vital information about the landscape of functional lncRNAs in TLR signaling.
Activation of TLRs regulates immune responses through a highly controlled network of gene expression. Among the four functional lncRNA‐gene pairs, MARCKS, BCAT1, and WT1 were induced by TLR ligands, acts in a negative feedback manner to inhibit cytokine productions. These genes are “brakes” in controlling excessive immune response. In the meantime, ROCKI, lnc‐BCAT1, and lnc‐WT1 were co‐induced by stimuli, acting as a negative regulator of these brake genes to maintain the proper immune responses. lnc‐BAIAP2 positively regulated BAIAP2 expression, and they were simultaneously decreased by TLR stimulus to promote immune response (Fig 2A–D). All these results confirmed that TLR pathways induce or suppress the expression of lncRNAs to turn on a lncRNA‐directed regulatory circuit, contributing to stricter and tighter regulation of TLR network. The most abundant and promising antisense lncRNAs belong to XH group and their average distance between the nascent transcription start sites is ~800 bp, a distance that can be easily co‐regulated on the genome. Local regulation of gene expression by cis‐lncRNAs may involve regulatory components in gene promoters, the process of transcription or splicing, or the sequence/structure of the RNAs. Although cis‐regulation by lncRNAs can be independent of the lncRNA transcripts themselves and could involve the RNA transcription and splicing mechanisms (Engreitz et al, 2016; Joung et al, 2017), but a number of studies showed that cis‐regulatory functions of lncRNAs were dependent on the sequence/structure of the RNAs (reviewed in Guil & Esteller, 2012 and Kopp & Mendell, 2018). For example, a divergent lncRNA Evx1 cis‐regulates adjacent coding genes based on its sequence during lineage differentiation in pluripotent cells (Luo et al, 2016). In our studies, ROCKI function was dependent upon the lncRNA sequence and ectopic expression enhanced inflammatory responses (Fig 3), representing an example of positive regulatory mechanism by lncRNA that is not a result of mere proximity of transcription.
We identified APEX1 as a ROCKI‐binding protein; interestingly, this protein has previously been reported to regulate TLR2‐dependent inflammatory responses in human keratinocytes (Lee et al, 2009). APEX1 can be acetylated by calcium activation to modulate its transcriptional regulatory function (Bhakat et al, 2003, 2009). Acetylated APEX1 binds more potently than the unacetylated protein to the parathyroid hormone promoter to repress its transcription (Bhakat et al, 2003). We observed a significant increase in ROCKI binding to the MARCKS promoter in Pam3‐stimulated THP1‐derived macrophages (Fig 4A). Further, APEX1 binding to MARCKS promoter was increased in Pam3‐stimulated THP1‐derived macrophages (Fig 5A and B) because Pam3 activates calcium, which leads to APEX1 acetylation and contributes to APEX1‐ROCKI binding to the promoter. APEX1 was confirmed to recruit HDAC1 to modify histone acylation. We did not observe an enrichment of HDAC1 in our in vitro pull‐down assay. There are two plausible explanations for this result: (i) This interaction is indirect binding, which is dependent on bridging of APEX1 as we proposed in our model Fig 7. (ii) This binding is temporal, thus hard to detect in the in vitro binding assay.
Several studies have identified a role for MARCKS in the regulation of inflammatory cytokine expression in myeloid cells. Treatment of neutrophils with LPS induces MARCKS expression and leads to its phosphorylation by protein kinase C (Aderem et al, 1988; Thelen et al, 1990; Seykora et al, 1991). Moreover, incubation of macrophages with peptides derived from the MARCKS effector domain not only blocked MARCKS phosphorylation and its disassociation from the membrane but also suppressed LPS‐induced activation of p38, JNK, and NF‐κB, thereby inhibiting inflammatory cytokine production (Lee et al, 2015). Another study proposed that MARCKS sequesters LPS to negatively regulate TLR4 signaling (Mancek‐Keber et al, 2012). Here, we found that MARCKS expression was regulated not only by TLR4 but also by TLRs 1/2, 2/6, 2, 5, and 7, suggesting that it may be a master modulator of innate immune responses. Consistent with this, MARCKS KD and ROCKI overexpression increased the expression of dozens of inflammatory factors, including chemokines and cytokines (Figs 2 and 6). Interestingly, ROCKI overexpression regulated more genes involved in cytokine and inflammation than MARCKS KD, for example, ILR2, IL1R1, CXCL16, CXCL12, and IL18BP (Fig 6). A number of genes in MAPK pathway were also induced by ROCKI overexpression, such as MAPKAPK2, MAPK10, and MAPKAPK3 (Table EV4). It remains to be determined whether ROCKI overexpression directly or indirectly affected genes which were not altered by MARCKS KD.
In addition, we observed that these commonly regulated genes highly overlapped with histamine H1 receptor signaling in immune responses (Fig EV5C). In the histamine H1 receptor signaling, Ca2+ initiates the inflammation response by binding to PKCα (Fig EV5C). Since MARCKS protein is a well‐characterized modulator of Ca2+ signaling (Hartwig et al, 1992; Gadi et al, 2011; Rodriguez Pena et al, 2013), we propose that MARCKS regulates inflammation by controlling calcium release. As shown in Fig 6D, cytoplasmic Ca2+ levels were increased by MARCKS KD and decreased by ROCKI KD compared with control cells. Thus, modulating Ca2+ signaling pathways could be a new mechanism for MARCKS to regulate inflammation responses.
Taken together, our study developed a new strategy to illustrate lncRNAs with unknown function in TLR signaling. Importantly, we observed a genetic link between ROCKI activity in blood and monocytes and the risk of inflammation and infection‐related disease phenotypes. We believe that our findings will help us understand the intact regulatory network of TLR signaling and also generate better experimental designs when investigating lncRNA transcripts in TLR signaling.
Materials and Methods
Cell culture
THP1 cells were cultured as described previously (Li et al, 2014). In brief, THP1 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 50 μM β‐mercaptoethanol (Sigma). Cells were treated with 5 ng/ml phorbol‐12‐myristate‐13‐acetate (PMA) overnight to induce differentiation into macrophages, and the medium was replaced with PMA‐free medium the following day. 293FT cells were cultured in DMEM supplemented with 10% FBS. Primary macrophages were generated by stimulating peripheral blood mononuclear cell (PBMC) with M‐CSF (10 ng/ml, R&D) for 7 days in RPMI 1640 medium supplemented with 10% fetal bovine serum and 50 μM β‐mercaptoethanol.
Bacterial culture
GBS (serotype IA strain A909) was cultured overnight at 37°C without shaking in Todd‐Hewitt broth supplemented with 0.5% yeast extract. Bacteria were grown to logarithmic phase (OD = 0.4 or ~10 colony‐forming units per ml), the centrifuged, washed, and resuspended in DMEM, and serially diluted to the desired concentration. For experiments, THP1‐derived macrophages were inoculated with GBS at a multiplicity of infection of 10 bacteria per cell.
TLR ligand treatment
Human TLR agonists were purchased from Invivogen (# tlrl‐kit1hw). Aliquots of 1 × 106 THP1‐derived macrophages were seeded in 6‐well plates and incubated with Pam3 (100 ng/ml), LPS (100 ng/ml), FSL‐1 (100 ng/ml), flagellin (100 ng/ml), or imiquimod (5 μg/ml). For poly(I:C) stimulation, cells were transfected with 100 ng/ml poly(I:C) using Lipofectamine 3000 (Invitrogen) for 8 h. Cells were then washed with PBS and further processed as required for each experiment.
Interleukin‐6 (IL‐6) ELISA
In Fig 2C, the human Interleukin‐6 ELISA kit (R&D, DY20605) was used for the quantitative determination of IL‐6 expression in the supernatant of THP1 cells stimulated by GBS for 10 h. ELISA was performed following the protocol.
Library preparation and RNA‐Seq
Approximately 0.5 μg total RNA per sample was depleted of ribosomal RNA using a RiboMinus kit (Life Technologies) and then processed using NEBNext Ultra RNA Library Prep Kit for Illumina (New England BioLabs) with 15 cycles of PCR, according to the manufacturer's recommendations. The final libraries were size‐selected on 2% agarose gels to obtain products between 280 and 380 bases in length. The libraries were loaded onto an Illumina HiSeq2000 for single‐end 100 base‐pair reads with 7 base index reads.
Lentiviral‐mediated gene knockdown and ROCKI overexpression
shRNAs against protein‐coding genes were purchased from the Functional Genomics Center (sequences listed in Table EV2). shRNAs against lncRNAs were designed using the open‐access tool from the Broad Institute. Probes containing sense and antisense strands of shRNAs were annealed and cloned into pLKO.1 lentivirus vector (Addgene) following the manufacturer's instructions. ROCKI was cloned into a pFLAG‐CMV plasmid for ROCKI overexpression experiments. Primer sequences for cloning shRNAs and ROCKI are listed in Table EV2. Lentiviruses were produced in 293FT cells using the protocol described previously (Khalil et al, 2009; Li et al, 2014). Briefly, aliquots of 1 × 106 cells/well were seeded into 6‐well plates 1 day before transfection. The next day, lentiviral and packaging vectors (System Biosciences) were transfected into 293FT cells using Lipofectamine 2000 (Invitrogen) and virus‐containing supernatants were collected 2 days after transfection. THP1 cells in 12‐well plates (4 × 105/well) were transduced using 500 μl viral supernatant in the presence of 4 μg/ml polybrene, and 20 ng/ml PMA was added to induce differentiation. The transduction efficiency was enhanced by centrifuging the cell/virus mixture at 750 g for 45 min.
Primary macrophages transfection with antisense oligo (ASO)
PBMCs were seeded at 5 M/ml into 12‐well plate. After 30 min, the suspending lymphocytes were removed and the remaining monocytes were changed to the M‐CSF (10 ng/ml) containing medium. Media were changed every 2 days, and monocytes were differentiated into macrophages for 7 days. Next, transfection of 100 nM non‐targeting ASO (CGACTATACGCGCAATATGG) or ROCKI ASO (TATATTTCAACCAGGAGGCAC) was performed using RNAiMAX (Thermo Fisher Scientific) for 48 h. After stimulation with Pam3 for additional 8 h, cells were harvested in TRIzol for detection of ROCKI, MARCKS, and IL‐6 expressions.
Repression of ROCKI expression using CRISPR inhibitor (CRISPRi)
For CRISPRi repression of ROCKI, we used the system that was previously established by Gilbert (Gilbert et al, 2013) with the following modifications. Briefly, polyclonal THP1 cells expressing dCas9/KRAB/BFP fusion proteins were generated by lentivirus transduction. Cells with higher BFP expression were sorted out. Two sgRNA oligos targeting ROCKI were designed and inserted into sgRNA expressing vector (lentiGuide‐puro, Addgene 52963). Next, the vectors were packed into lentivirus and delivered into the sorted THP1‐ dCas9/KRAB/BFP cells. After 7 days of puromycin selection, cells were seeded and differentiated into macrophages, and stimulated with Pam3 for 8 h. The expressions of ROCKI, MARCKS, and IL‐6 were then determined using RT–PCR. sgRNA primers are listed in Table EV2.
RNA extraction and RT–qPCR
RNA was extracted from THP1 cells using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. RNA was treated with DNase I (Ambion) to remove contaminating DNA, and samples of ~1 μg of RNA were reverse transcribed using Superscript III or VILO Mastermix (Invitrogen). qPCR was performed using SYBR Green mix or SsoAdvanced SYBR Green mix (Bio‐Rad). The fold change in gene expression was calculated by the ΔΔCt method using glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) as an internal control. The primers used for qPCR are listed in Table EV2.
Rapid amplification of cDNA ends (RACE)
Total RNA was isolated from LPS‐treated THP1 cells and processed with a Ribo‐Zero‐rRNA Removal kit to deplete ribosomal RNA (Epicentre). RACE was performed using a SMARTer RACE kit (Clontech) according to the user manual. Briefly, ~100 ng of purified poly(A) RNA was used for each RT reaction to produce lncRNA cDNA (20 μl), and nested PCRs were performed to amplify the lncRNA cDNA ends. Primer sequences for nested PCR reactions are listed in Table EV2. RACE PCR products were cloned using Zero Blunt TOPO cloning kit (Thermo Fisher Scientific) and then sequenced.
Chromatin isolation by RNA purification (ChIRP)
ROCKI binding to the MARCKS promoter was detected by ChIRP performed using 30 probes specific for ROCKI. The probes were designed using the online probe designer version 4.2 from LGC Biosearch Technologies. Masking level was set at 2, and the minimum spacing length was set at 60 nucleotides. Oligonucleotides were biotinylated at the 3′ end. THP1 cells were incubated in medium containing 20 ng/ml PMA for 18 h and then treated with or without 100 ng/ml Pam3 for 8 h. Cells were trypsinized, washed in medium, and then washed twice with PBS. Cells were fixed in 1% glutaraldehyde in PBS at room temperature for 10 min and then quenched with 1/10th volume of 1.25 M glycine for 5 min. Fixed cells were harvested by centrifuging at 2000 RCF for 5 min and washed twice with PBS. The cells were then lysed in lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP‐40), Dounce‐homogenized 10 times on ice, and incubated for 10 min. Cell lysates were centrifuged at 2,500 g for 5 min at 4°C and the nuclear pellets were collected and sonicated (15 cycles, 30 s on, 30 s off). Isolated chromatin was analyzed for enrichment of ROCKI at the MARCKS promoter by ChIRP using the method described by Chu et al (2012). RT–qPCR primers are listed in Table EV2.
In vitro transcription and ROCKI RNA–protein pull‐down assay
ROCKI, antisense of ROCKI, or lacZ control was cloned into pBluescript KSII downstream of the T7 promoter separately (primers listed in Table EV2). The vectors were linearized by KpnI digestion, and 1 μg of linear lacZvectors was transcribed and biotinylated in vitro using a kit (Epicentre), according to the manufacturer's instructions. Biotinylated RNAs were purified, refolded, and incubated with nuclear lysates from THP1‐derived macrophages using the method described previously (Li et al, 2014). Briefly, 2 μg of diluted RNA (200 ng/μl) was incubated at 60°C for 10 min and slowly cooled to 4°C. Nuclear lysates were prepared from 2 × 108 PMA‐differentiated THP1‐derived macrophages treated with 100 ng/ml Pam3 for 8 h. Protein concentrations were measured by the DC assay (Bio‐Rad) according to the manufacturer's instructions. For the pull‐down incubations, nuclear lysates (1 mg of protein) were precleared with streptavidin beads (Invitrogen) and then incubated with 2 μg of biotinylated RNA and 40 μl of streptavidin beads for 2 h at 4°C. The beads were collected and washed three times with buffer C. RNA‐associated proteins were eluted and analyzed by mass spectrometry (Sanford Burnham Prebys Medical Discovery Institute Core) or by Western blotting, as described below.
Reverse pull‐down assays
To confirm that APEX1 binds to ROCKI, nuclear lysates (1 mg protein per sample) from 1 × 108 THP1‐derived macrophages treated with Pam3 for 8 h were precleared with protein G beads (NEB) for 2 h at 4°C and then incubated with control rabbit IgG or anti‐APEX1 antibody (4 μg per sample, Novus, NB100‐101) overnight at 4°C. Protein G beads pre‐blocked with bovine serum albumin were added to the samples for 2 h at 4°C, collected by centrifugation, washed three times with buffer C, and resuspended in 1 ml buffer C. RNA was extracted from 900 μl of each sample using TRIzol and treated with Turbo DNase to remove contaminating DNA. ROCKI levels were analyzed by RT–qPCR. The remaining 100 μl of each sample was collected, boiled for 5 min, and subjected to Western blot analysis to detect RNA‐associated APEX1.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using the method described by Deliard et al (2013) with minor modifications. Briefly, 2 × 107 THP1 macrophages were cross‐linked with 1% formaldehyde for 10 min at room temperature and incubated with glycine for 5 min. Cells were collected by centrifugation, washed twice with PBS, lysed in lysis buffer, Dounce‐homogenized 10 times on ice, and incubated for 10 min. The lysates were centrifuged at 4,000 rpm for 5 min at 4°C, and the nuclear pellets were resuspended in SDS lysis buffer and sonicated for 15 cycles (30 s on, 30 s off). DNA concentrations were quantified, and 10 μg of chromatin DNA was used for each ChIP reaction. ChIP assays were performed with 4 μg of antibody and 40 μl of protein G magnetic beads. The samples were incubated overnight at 4°C, washed, and eluted. Bead‐bound DNA was reverse cross‐linked and purified. RT–qPCR analysis was performed to detect the presence of two MARCKS promoter region sequences or an ACTB2 sequence. ACTB2 primers were purchased from Active Motif (71005). The antibodies used in these experiments include α‐HDAC1 antibody (Abcam, ab7028), α‐APEX1 (Novus, NB100‐116), and α‐H3K27ac (Abcam, ab4726).
Co‐immunoprecipitation assay
To determine whether APEX1 and HDAC1 interact, APEX1 was immunoprecipitated as described for the reverse RNA pull‐down assay. The beads were resuspended in 1 ml buffer C, and 900 μl was subjected to Western blot analysis of HDAC1 protein in the immunoprecipitates. The remaining 100 μl of sample was collected for RT–qPCR analysis of ROCKI.
Western blotting
Protein lysates were separated on 4–20% SDS/PAGE gels and transferred to PVDF membranes. Western blotting was performed using Fast Western Blot kits (Thermo Scientific Pierce) with the following antibodies: mouse anti‐FLAG M2 antibody (Sigma, F1804), rabbit anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; Cell Signaling Technology, 2118), rabbit anti‐APEX1 (NOVUS, NB100‐101), mouse anti‐HDAC1 (NOVUS, NB100‐56340SS), rabbit anti‐H3K27ac (Cell Signaling Technology, 9733S), and mouse anti‐vimentin (Abcam, ab8978).
Calcium flux assay
To detect changes in intracellular Ca2+ concentration in THP1‐derived macrophages, 4 × 105 cells expressing control, MARCKS, or ROCKI shRNA were preincubated with the Ca2+ indicator Fluo‐4 (Thermo Fisher, F14201) at 6 μM for 30 min, washed with Hanks’ Balanced Salt Solution, and aliquoted into a 96‐well plate at 4 × 104/well. Fluorescence signals were detected before and every 30 s after stimulation with PMA (50 ng/ml) and Pam3 (500 ng/ml). The maximal [Ca2+] response was determined by adding 0.1% Triton X‐100 to the cells at the end of the detection period. The change in fluorescence was normalized to the maximal fluorescence signal and is expressed as ΔF/F max.
MARCKS and ROCKI QTL analysis
Publicly available whole blood eQTL summary statistics were downloaded from the Genotype‐Tissue Expression (Consortium et al, 2017) project v7 release (Consortium et al, 2017). We extracted all variants that were tested for association with ROCKI and MARCKS expression for further analysis. We also downloaded CD14+ monocyte QTL summary statistic datasets for gene expression, H3K4me1, and H3K27ac from the BLUEPRINT epigenome project (Chen et al, 2016). The monocyte eQTL dataset could not be used in these analyses because it used an earlier release (GENCODE v15) for transcript annotation and ROCKI was not among the genes tested in this study. We extracted QTLs for H3K27ac and H3K4me1 signal in a genomic region around MARCKS and ROCKI.
For each dataset, we calculated Bayes factors for each variant in a 1 MB window around the signal following the approach of Wakefield (Wakefield, 2009). We then used a Bayesian co‐localization method to determine whether the whole blood eQTL association for ROCKI and monocyte H3K27ac QTL association upstream of the ROCKI promoter and eQTL association for MARCKS showed evidence of a shared causal variant (Giambartolomei et al, 2014). We considered signals with a shared probability (Pshared) > 0.9.
We then performed fine‐mapping to determine the causal variant for the shared signal using ROCKI eQTL and H3K27ac QTL data. For each variant in a 1 MB window around the signal and in common to both datasets, we multiplied Bayes factors for each signal and calculated a posterior probability of association (PPA) for each variant as the Bayes factor divided by the sum of Bayes factors across all variants. We then intersected candidate casual variants with DNase I hypersensitivity sites (DHS) and transcription factor ChIP‐seq sites from the ENCODE project (Maurano et al, 2012).
UK Biobank GWAS data analysis for phenotypic associations
Publicly available UK Biobank GWAS summary statistics for 2,419 phenotypes were downloaded (https://sites.google.com/broadinstitute.org/ukbbgwasresults). Phenotypes that did not appear to be related to diseases or disease treatments were removed, and we then extracted summary statistics for the most significantly associated ROCKI eQTL variant (rs9387181). For each phenotype, we aligned summary results to the ROCKI‐reducing allele of rs9387181. We then categorized 1,464 disease‐ and medication‐related phenotypes into 22 groups, removing phenotypes that did not fit in a clear group. We then plotted the signed test statistic of phenotypes within each group, and the average signed test statistic within each group.
Statistical analysis
Differences between groups were analyzed using Student's t‐test (paired, two‐sided). Data are presented as the mean ± standard deviation (SD). N = 3. Differences were considered statistically significant when P < 0.05.
Author contributions
QZ, T‐CC, VSP, and TMR designed experiments. VSP performed initial studies to generate gene expression data in response to TLR activation. QZ and T‐CC performed experiments to identify cis‐regulation and mechanistic studies. YQ analyzed cis‐regulatory ncRNAs and coding gene expression. SKT analyzed immune mechanisms. JC and KJG analyzed lincRNA and coding genes genetic variation in diseases. AD and TRG analyzed RNA‐seq data after TLR ligand stimulation. ZL and JD contributed to TLR stimulation experiments. SG analyzed RNA‐seq data and pathways analysis in ROCKI overexpression and MARCKS KD. C‐MT and VN participated in GBS studies. KU participated in analyzing ncRNAs relevance in TB. QZ, T‐CC, and TMR wrote the manuscript with input from authors.
Conflict of interest
TMR is a founder of ViRx Pharmaceuticals and a member of its scientific advisory board. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Howard Chang and members of the Rana Lab for helpful discussions and advice. We are grateful to Xiayu Stacy Huang and Alexey Eroshkin of The Genomics and Informatics and Data Management core facilities at Sanford Burnham Prebys Medical Discovery Institute for help in data analysis. We thank Steve Head and the staff of the Next Generation Sequencing core facility at The Scripps Research Institute for help with the HT‐seq. This work was supported in part by NIH grants to TMR (CA177322, DP1DA039562, AI125103, DA046171).
The EMBO Journal (2019) 38: e100041
Data availability
Data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE109084 (related to Fig 1) and GSE108680 (related to Fig 6).
References
- Aderem AA, Albert KA, Keum MM, Wang JK, Greengard P, Cohn ZA (1988) Stimulus‐dependent myristoylation of a major substrate for protein kinase C. Nature 332: 362–364 [DOI] [PubMed] [Google Scholar]
- Atianand MK, Hu W, Satpathy AT, Shen Y, Ricci EP, Alvarez‐Dominguez JR, Bhatta A, Schattgen SA, McGowan JD, Blin J, Braun JE, Gandhi P, Moore MJ, Chang HY, Lodish HF, Caffrey DR, Fitzgerald KA (2016) A long noncoding RNA lincRNA‐EPS acts as a transcriptional brake to restrain inflammation. Cell 165: 1672–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S (2003) Role of acetylated human AP‐endonuclease (APE1/Ref‐1) in regulation of the parathyroid hormone gene. EMBO J 22: 6299–6309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakat KK, Mantha AK, Mitra S (2009) Transcriptional regulatory functions of mammalian AP‐endonuclease (APE1/Ref‐1), an essential multifunctional protein. Antioxid Redox Signal 11: 621–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon‐Vega B, Regev A, Rinn JL (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry‐Bezy M, Lawrence JB, O'Neill LA, Moore MJ, Caffrey DR, Fitzgerald KA (2013) A long noncoding RNA mediates both activation and repression of immune response genes. Science 341: 789–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech TR, Steitz JA (2014) The noncoding RNA revolution‐trashing old rules to forge new ones. Cell 157: 77–94 [DOI] [PubMed] [Google Scholar]
- Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido‐Martín D, Watt S, Yan Y, Kundu K, Ecker S, Datta A, Richardson D, Burden F, Mead D, Mann AL, Fernandez JM, Rowlston S, Wilder SP, Farrow S, Shao X et al (2016) Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167: 1398–1414.e1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Satpathy AT, Chang HY (2017) Gene regulation in the immune system by long noncoding RNAs. Nat Immunol 18: 962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Quinn J, Chang HY (2012) Chromatin isolation by RNA purification (ChIRP). J Vis Exp 10.3791/3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT, Laboratory DA, Coordinating Center ‐Analysis Working G, Statistical Methods groups‐Analysis Working G, Enhancing Gg, Fund NIHC, Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida, Biospecimen Collection Source Site N, Biospecimen Collection Source Site R, Biospecimen Core Resource V, Brain Bank Repository‐University of Miami Brain Endowment B, Leidos Biomedical‐Project M, Study E, Genome Browser Data I, Visualization EBI, Genome Browser Data I, Visualization‐Ucsc Genomics Institute UoCSC, Lead a, Laboratory DA, Coordinating C, management NIHp, Biospecimen c, Pathology, e QTLmwg , Battle A, Brown CD, Engelhardt BE, Montgomery SB (2017) Genetic effects on gene expression across human tissues. Nature 550: 204–213 29022597 [Google Scholar]
- Deliard S, Zhao J, Xia Q, Grant SF (2013) Generation of high quality chromatin immunoprecipitation DNA template for high‐throughput sequencing (ChIP‐seq). J Vis Exp 10.3791/50286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES (2016) Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539: 452–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S, Philippe J, Corvol P, Pinet F (2003) Implication of Ref‐1 in the repression of renin gene transcription by intracellular calcium. J Hypertens 21: 327–335 [DOI] [PubMed] [Google Scholar]
- Gadi D, Wagenknecht‐Wiesner A, Holowka D, Baird B (2011) Sequestration of phosphoinositides by mutated MARCKS effector domain inhibits stimulated Ca(2+) mobilization and degranulation in mast cells. Mol Biol Cell 22: 4908–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay NJ, Gangloff M (2007) Structure and function of Toll receptors and their ligands. Annu Rev Biochem 76: 141–165 [DOI] [PubMed] [Google Scholar]
- Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10: e1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern‐Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS (2013) CRISPR‐mediated modular RNA‐guided regulation of transcription in eukaryotes. Cell 154: 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guil S, Esteller M (2012) Cis‐acting noncoding RNAs: friends and foes. Nat Struct Mol Biol 19: 1068–1075 [DOI] [PubMed] [Google Scholar]
- Harlan DM, Graff JM, Stumpo DJ, Eddy RL Jr, Shows TB, Boyle JM, Blackshear PJ (1991) The human myristoylated alanine‐rich C kinase substrate (MARCKS) gene (MACS). Analysis of its gene product, promoter, and chromosomal localization. J Biol Chem 266: 14399–14405 [PubMed] [Google Scholar]
- Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A (1992) Marcks is an actin filament cross‐linking protein regulated by protein‐kinase‐C and calcium calmodulin. Nature 356: 618–622 [DOI] [PubMed] [Google Scholar]
- Henneke P, Dramsi S, Mancuso G, Chraibi K, Pellegrini E, Theilacker C, Hubner J, Santos‐Sierra S, Teti G, Golenbock DT, Poyart C, Trieu‐Cuot P (2008) Lipoproteins are critical TLR2 activating toxins in group B streptococcal sepsis. J Immunol 180: 6149–6158 [DOI] [PubMed] [Google Scholar]
- Hezroni H, Ben‐Tov Perry R, Meir Z, Housman G, Lubelsky Y, Ulitsky I (2017) A subset of conserved mammalian long non‐coding RNAs are fossils of ancestral protein‐coding genes. Genome Biol 18: 162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housman G, Ulitsky I (2016) Methods for distinguishing between protein‐coding and long noncoding RNAs and the elusive biological purpose of translation of long noncoding RNAs. Biochem Biophys Acta 1859: 31–40 [DOI] [PubMed] [Google Scholar]
- Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K, Kato A, Kawaguchi Y, Sato H, Yoneda M, Kai C, Yada T, Suzuki Y, Yamada T, Ozawa T, Kaneki K, Inoue T, Kobayashi M, Kodama T, Wada Y et al (2014) Long noncoding RNA NEAT1‐dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol Cell 53: 393–406 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS (2009) Genome‐wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R (2015) Control of adaptive immunity by the innate immune system. Nat Immunol 16: 343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Engreitz JM, Konermann S, Abudayyeh OO, Verdine VK, Aguet F, Gootenberg JS, Sanjana NE, Wright JB, Fulco CP, Tseng YY, Yoon CH, Boehm JS, Lander ES, Zhang F (2017) Genome‐scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 548: 343–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S (2010) The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 11: 373–384 [DOI] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL (2009) Many human large intergenic noncoding RNAs associate with chromatin‐modifying complexes and affect gene expression. Proc Natl Acad Sci USA 106: 11667–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp F, Mendell JT (2018) Functional classification and experimental dissection of long noncoding RNAs. Cell 172: 393–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HM, Yuk JM, Shin DM, Yang CS, Kim KK, Choi DK, Liang ZL, Kim JM, Jeon BH, Kim CD, Lee JH, Jo EK (2009) Apurinic/apyrimidinic endonuclease 1 is a key modulator of keratinocyte inflammatory responses. J Immunol 183: 6839–6848 [DOI] [PubMed] [Google Scholar]
- Lee SM, Suk K, Lee WH (2015) Myristoylated alanine‐rich C kinase substrate (MARCKS) regulates the expression of proinflammatory cytokines in macrophages through activation of p38/JNK MAPK and NF‐kappaB. Cell Immunol 296: 115–121 [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T (2006) A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol 177: 583–592 [DOI] [PubMed] [Google Scholar]
- Lemaitre B (2004) The road to toll. Nat Rev Immunol 4: 521–527 [DOI] [PubMed] [Google Scholar]
- Li Z, Chao TC, Chang KY, Lin N, Patil VS, Shimizu C, Head SR, Burns JC, Rana TM (2014) The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc Natl Acad Sci USA 111: 1002–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin N, Chang K‐Y, Li Z, Gates K, Rana Zacharia A, Dang J, Zhang D, Han T, Yang C‐S, Cunningham TJ, Head SR, Duester G, Dong PDS, Rana TM (2014) An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol Cell 53: 1005–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Lu JY, Liu L, Yin Y, Chen C, Han X, Wu B, Xu R, Liu W, Yan P, Shao W, Lu Z, Li H, Na J, Tang F, Wang J, Zhang YE, Shen X (2016) Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell 18: 637–652 [DOI] [PubMed] [Google Scholar]
- Mancek‐Keber M, Bencina M, Japelj B, Panter G, Andra J, Brandenburg K, Triantafilou M, Triantafilou K, Jerala R (2012) MARCKS as a negative regulator of lipopolysaccharide signaling. J Immunol 188: 3893–3902 [DOI] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling‐Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D et al (2012) Systematic localization of common disease‐associated variation in regulatory DNA. Science 337: 1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22: 240–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal T, Rasmussen M, Pandey GK, Isaksson A, Kanduri C (2010) Characterization of the RNA content of chromatin. Genome Res 20: 899–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R (2010) Long noncoding RNAs with enhancer‐like function in human cells. Cell 143: 46–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacis A, Tailleux L, Morin AM, Lambourne J, MacIsaac JL, Yotova V, Dumaine A, Danckaert A, Luca F, Grenier JC, Hansen KD, Gicquel B, Yu M, Pai A, He C, Tung J, Pastinen T, Kobor MS, Pique‐Regi R, Gilad Y et al (2015) Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res 25: 1801–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JJ, Chang HY (2016) Unique features of long non‐coding RNA biogenesis and function. Nat Rev Genet 17: 47–62 [DOI] [PubMed] [Google Scholar]
- Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY (2013) A mammalian pseudogene lncRNA at the interface of inflammation and anti‐inflammatory therapeutics. Elife 2: e00762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Pena MJ, Castillo Bennett JV, Soler OM, Mayorga LS, Michaut MA (2013) MARCKS protein is phosphorylated and regulates calcium mobilization during human acrosomal exocytosis. PLoS ONE 8: e64551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seykora JT, Ravetch JV, Aderem A (1991) Cloning and molecular characterization of the murine macrophage “68‐kDa” protein kinase C substrate and its regulation by bacterial lipopolysaccharide. Proc Natl Acad Sci USA 88: 2505–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigova AA, Mullen AC, Molinie B, Gupta S, Orlando DA, Guenther MG, Almada AE, Lin C, Sharp PA, Giallourakis CC, Young RA (2013) Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci USA 110: 2876–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song DH, Lee JO (2012) Sensing of microbial molecular patterns by Toll‐like receptors. Immunol Rev 250: 216–229 [DOI] [PubMed] [Google Scholar]
- Thelen M, Rosen A, Nairn AC, Aderem A (1990) Tumor necrosis factor alpha modifies agonist‐dependent responses in human neutrophils by inducing the synthesis and myristoylation of a specific protein kinase C substrate. Proc Natl Acad Sci USA 87: 5603–5607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J (2009) Bayes factors for genome‐wide association studies: comparison with P‐values. Genet Epidemiol 33: 79–86 [DOI] [PubMed] [Google Scholar]
- Wang L, Liu X, Lenox RH (2002) Transcriptional regulation of mouse MARCKS promoter in immortalized hippocampal cells. Biochem Biophys Res Comm 292: 969–979 [DOI] [PubMed] [Google Scholar]
- Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, Jiang Z, Xu J, Liu Q, Cao X (2014) The STAT3‐binding long noncoding RNA lnc‐DC controls human dendritic cell differentiation. Science 344: 310–313 [DOI] [PubMed] [Google Scholar]
- Wang P, Xu J, Wang Y, Cao X (2017) An interferon‐independent lncRNA promotes viral replication by modulating cellular metabolism. Science 358: 1051–1055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
Data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE109084 (related to Fig 1) and GSE108680 (related to Fig 6).