

Abstract
The small GTPase Rab7 is a key organizer of receptor sorting and lysosomal degradation by recruiting of a variety of effectors depending on its GDP/GTP‐bound state. However, molecular mechanisms that trigger Rab7 inactivation remain elusive. Here we find that, among the endosomal pools, Rab7‐positive compartments possess the highest level of PI4P, which is primarily produced by PI4K2A kinase. Acute conversion of this endosomal PI4P to PI(4,5)P2 causes Rab7 dissociation from late endosomes and releases a regulator of autophagosome–lysosome fusion, PLEKHM1, from the membrane. Rab7 effectors Vps35 and RILP are not affected by acute PI(4,5)P2 production. Deletion of PI4K2A greatly reduces PIP5Kγ‐mediated PI(4,5)P2 production in Rab7‐positive endosomes leading to impaired Rab7 inactivation and increased number of LC3‐positive structures with defective autophagosome–lysosome fusion. These results reveal a late endosomal PI4P‐PI(4,5)P2‐dependent regulatory loop that impacts autophagosome flux by affecting Rab7 cycling and PLEKHM1 association.
Keywords: autophagy, lysosome, phosphatidylinositol 4‐kinase, phosphoinositide, Rab7
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport
Introduction
Maintenance of cellular integrity demands the proper sorting of newly synthesized membrane components and the simultaneous degradation of cellular proteins or organelles through endocytic sorting pathways and autophagy. These events are controlled by the concerted actions of many protein signaling complexes often organized in specific membrane compartments by the Rab family of GTPases (Lamb et al, 2016; Novick, 2016) together with the regulatory lipids, phosphoinositides (Di Paolo & De Camilli, 2006; Balla, 2013). The importance of phosphatidylinositol 3‐phosphate (PI3P) generated by Vps34 in yeast and its homologues in metazoan cells working primarily in Rab5‐positive early endosomal compartments are well recognized in endosomal sorting (Christoforidis et al, 1999). In addition, the critical role of Vps34 in autophagosome formation and processing has been firmly established. Another small GTPase, Rab7, is distributed to the late endosome/lysosome compartment working as a key regulator of protein sorting, lysosome positioning, and autophagosome–lysosome fusion via recruitment of a variety of effectors, including RILP (Jordens et al, 2001), the retromer complex (Seaman et al, 2009), and autophagosome–lysosome fusion protein PLEKHM1 (McEwan & Dikic, 2015). Little is known about the importance of phosphoinositides in the Rab7‐mediated events.
PI4P is made by four different phosphatidylinositol 4‐kinase enzymes in mammalian cells (Minogue & Waugh, 2012; Boura & Nencka, 2015), but their contribution to the endosomal PI4P pool is not all that clear. PI4KA is mainly responsible for the plasma membrane (PM) pool of PI4P (Nakatsu et al, 2012; Bojjireddy et al, 2014), and PI4KB is primarily a Golgi‐localized enzyme (D'Angelo et al, 2012). Based on the endosomal localization of PI4K2A and PI4K2B, these two enzymes are considered the major sources of PI4P on endosomes and the TGN (Balla et al, 2002; Wang et al, 2003; Minogue et al, 2006; Jovic et al, 2014; Minogue, 2017). Studies on the role of endosomal PI4P primarily focused on its role in recruitment of adapter proteins, such as the clathrin adaptors AP‐1 (Wang et al, 2003), AP‐3 (Craige et al, 2008), and GGAs (Wang et al, 2007), or on the trafficking and regulation of SNAREs (Burgess et al, 2012; Jovic et al, 2014). PI4P and PI4K2A were also found to organize important multi‐component signaling complexes on endosomes such as the lysosome‐related organelles complex 1 (BLOC‐1) and the Wiskott‐Aldrich Syndrome Protein and SCAR Homolog (WASH) complexes (Newell‐Litwa et al, 2009; Ryder et al, 2013; Dong et al, 2016). Lastly, PI4P levels on endosomes are also regulated by lipid‐transfer proteins of the OSBP family, suggesting that endosomal PI4P is utilized to drive the transport of lipids between the ER and endosomes (Dong et al, 2016; Mesmin et al, 2017).
Functionally speaking, PI4K2A was found to be critical for EGF‐receptor degradation (Minogue et al, 2006), transferrin receptor recycling (Ketel et al, 2016), and mannose 6‐phosphate retrieval from endosomes to the Golgi by the retromer complex (Wang et al, 2003; Niu et al, 2013; Marquer et al, 2016). Also, PI4K2A was shown to interact with the family of GABARAP proteins (Jovic et al, 2014; Wang et al, 2015) and play a role in autophagosome–lysosome fusion (Wang et al, 2015). Not clear is the question as to what extent PI4P acts on its own right or can be converted to PI(4,5)P2 in endocytic compartments to regulate these events. Recent studies showed specific forms of PIP5Kγ in the control of ATG14 and autophagy flux (Tan et al, 2016) and also in regulating lysosomal degradation of e‐cadherin (Schill et al, 2014) or the EGF receptor (Sun et al, 2013). Yet, some other studies suggested that PI4P regulates autophagy unrelated to PI(4,5)P2 production (Wang et al, 2015).
In the present study, we find that conversion of PI4P synthesized by the PI4K2A enzyme to PI(4,5)P2 on Rab7‐positive endosomes facilitates Rab7 conversion to its GDP‐bound form and leads to the release of the Rab7 effector PLEKHM1, an important regulator of autophagosome–lysosome fusion. Some Rab7 effectors show less sensitivity to PI(4,5)P2 generation suggesting selective phosphoinositide regulation of different Rab7‐controlled pathways. Our studies unravel the mechanistic details of the phosphoinositide cascade that controls late endosomal events and autophagosome flux via the regulation of Rab7 cycling and PLEKHM1 recruitment and release.
Results
PI4P shows highest enrichment in Rab7‐positive compartments
PI4P has been shown to be present in various endosomal compartments showing highest co‐localization with Rab7‐positive endosomes (Hammond et al, 2014). To obtain an accurate quantitative comparison between PI4P levels of specific endosomal pools, a bioluminescence resonance energy transfer (BRET) method was introduced, which utilizes energy transfer from luciferase (donor) to Venus (acceptor). The energy transfer increases when the luciferase‐tagged lipid‐binding domain is in close proximity to the Venus protein targeted to the membrane of interest (Toth et al, 2016; Varnai et al, 2017). The intensities of luciferase and Venus can be measured by microplate readers from thousands of cells giving an unbiased measure in a population of cells. To specifically monitor PI4P in various endosomal pools, the tandem version of P4M (Hammond et al, 2014) was fused to Renilla luciferase (termed Super luciferase, or Sluc) and the Venus partner was fused to various Rab proteins (Rab7, Rab5, Rab4, and Rab11; Fig 1A). The two fusion proteins were expressed in HEK293 cells stably expressing the AT1a rat AngII receptor (HEK293‐AT1; Balla et al, 2007) from a single plasmid separated by the T2A viral sequence (Toth et al, 2016). Since the majority of the P4M‐2x reporter is bound to the PM in resting cells and only a relatively small fraction binds to endosomes (Hammond et al, 2014), we used GSK‐A1, a selective inhibitor of PI4KA, which inhibits the PI4KA enzyme responsible for the generation of the PI4P pool of the PM without affecting PI4K2A (Balla et al, 2005; Nakatsu et al, 2012; Bojjireddy et al, 2014). This treatment releases P4M2x from the PM and profoundly increases the association of the probe with the endosomal PI4P pools including the various endosomes and the Golgi as well (Fig 1B). As shown in Figs 1C and EV1C, the BRET signal showed a large increase in the Rab7 compartments and only small or no change was observed in the other Rab compartments (Rab4, Rab5, Rab11), indicating that the majority of the PI4P was found associated with the Rab7 compartment.
Figure 1. Assessment of endosome‐associated PI4P pools.

- Cartoon depicting the principle of the endosomal PI4P BRET measurement. The Venus fluorescent protein is targeted to the respective endosomes using Rab proteins, while Renilla luciferase [termed Super luciferase (Sluc)] is fused to the P4M2x PI4P reporter. This reporter binds to PI4P the majority of which is in the PM. The PI4KA inhibitor, GSK‐A1 (A1), reduces PI4P in the PM allowing more of the reporter to find the endosomal PI4P that is produced by PI4Ks that are insensitive to GSK‐A1. Relocation of the P4M2x reporter tagged with Sluc to the PI4P‐rich endosomes will increase the energy transfer between the enzyme and Venus in the presence of the coelenterazine H substrate.
- Representative confocal images show the distribution of the P4M2x reporter in HEK293‐AT1 cells before and after the addition of GSK‐A1 (30 nM). Note the signal disappearing from the PM and strongly highlighting the endosomes and Golgi. Scale bars are 20 μm.
- Representative result from a BRET experiment detecting PI4P in various endosomes (Rab7, Rab5, and Rab11) following addition of GSK‐A1. The graph shows normalized BRET values where the averages of triplicate measurements of A1‐treated cells were divided by the averages of triplicates obtained in DMSO‐treated controls. Note that these signals originate from thousands of cells.
- Similar BRET experiment showing the Rab7‐pool of PI4P both in control HEK293‐AT1 cells and in two clones of cells with PI4K2A deletion (#20 and #26). Means ± SEM are shown from three experiments performed in triplicates.
Figure EV1. Generation of PI4K2A K/O cells and contribution of PI4K2A, PI4K2B, and PI4KB to late endosomal PI4P.
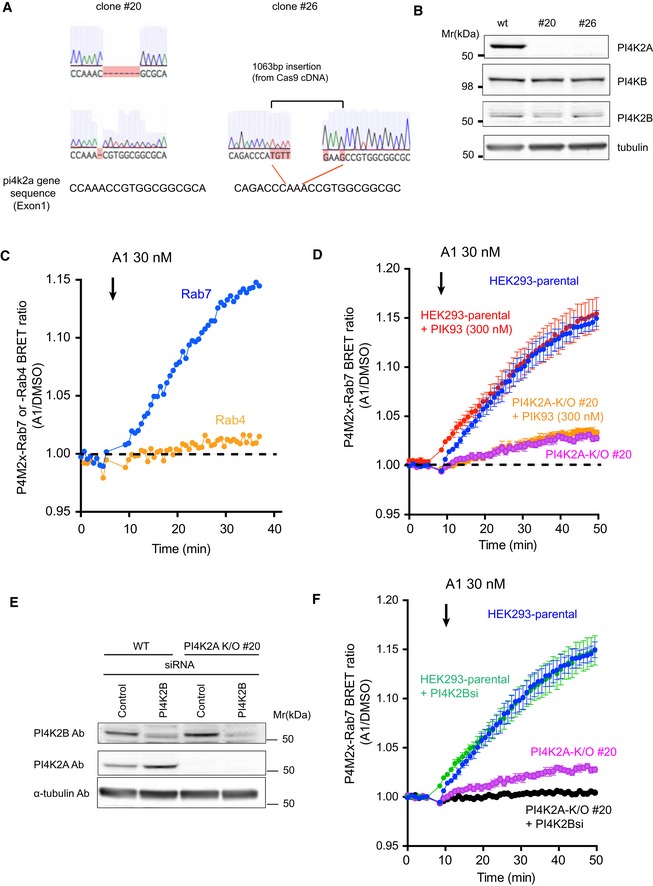
- Sequence analysis of the targeted region in PI4K2A K/O #20 or #26 cells created with CRISPR/CAS9 genome editing system. The cartoons showing the altered sequences were captured from Benchling (https://benchling.com/).
- The effect of PI4K2A K/O on the expression level of PI4KB and PI4K2B. Whole‐cell lysates prepared from wild‐type or the PI4K2A clones were subjected to Western blot analysis using anti‐PI4K2A, anti‐PI4KB, anti‐PI4K2B, and anti‐alpha‐tubulin antibodies.
- Representative BRET measurement showing the relative amount of PI4P in Rab7‐ and Rab4‐positive endosomes. The BRET analysis was performed using the P4M2x‐Rab7 sensor as described in Fig 1C and D or by using P4M2x‐Rab4 sensor in which the Rab7 was replaced by the Rab4 sequence. A1 inhibitor was added to the cell following baseline measurement. The result shown is from an experiment performed in triplicates. The graph shows normalized BRET values where the averages of triplicate measurements of A1‐treated cells were divided by the averages of triplicates obtained in DMSO‐treated controls.
- A BRET analysis was performed with the P4M2x‐Rab7 probe as described in Fig 1D. To inhibit PI4KB, 300 nM of PIK93 was added together with 30 nM of A1 following baseline measurements.
- Wild‐type or PI4K2A K/O clone #20 cells were transfected with control siRNA or one targeting PI4K2B. Western blot analysis was performed with anti‐PI4K2B, anti‐PI4K2A, and anti‐alpha‐tubulin antibodies 3 days after the transfection.
- A BRET analysis measuring late endosomal PI4P in cells after PI4K2B was knocked down in wild‐type or PI4K2A K/O cells. The values in (D) and (F) were measured in the same 96‐well plate but plotted separately for clarity (therefore, the blue curves are the same in the two panels). The BRET ratios were expressed relative to those of DMSO‐treated control cells, and means ± SEM are shown from three experiments performed in triplicates.
Source data are available online for this figure.
Since type‐II PI4Ks have been found to localize to various endosomes, we assessed the contribution of PI4K2A to the production of PI4P in the various endosomal compartments. For this, we generated PI4K2A knockout (K/O) cell lines from the HEK293‐AT1 cells using the CRISPR/Cas9 system (Ran et al, 2013). Two of the clonal lines, called #20 and #26, were selected for further analysis. Sequence analysis showed a 7 bp and a 1 bp deletion in exon 1 of PI4K2A in clone #20 and a large insertion originating from the Cas9 cDNA inserted into PIK2A exon1 in clone #26 (Fig EV1A). The PI4K2A protein was undetectable in either of the two PI4K2A K/O clones, and no changes were observed in the amounts of the other PI4Ks, PI4K2B, and PI4KB (Fig EV1B). Importantly, in both PI4K2A K/O cells, the BRET signal was greatly reduced in the Rab7 compartment (Fig 1D). To evaluate the contribution of the other PI4K enzymes, PI4KB and PI4K2B, these enzymes were inactivated either pharmacologically (for PI4KB) or by RNAi‐mediated knockdown in either naïve or PI4K2A K/O cells. Inhibition of PI4KB had no detectable effect on the PI4P content of the Rab7 compartment in either the control or PI4K2A K/O cells (Fig EV1D). PI4K2B knockdown caused no detectable reduction in the PI4P signal in the Rab7 compartment in wild‐type cells but further reduced the signal in the PI4K2A K/O cells (Fig EV1E and F). These data together suggested that type‐II PI4Ks primarily control the PI4P content of Rab7 endosomes. Importantly, the reduced PI4P pool in the Rab7 compartment was rescued by expression of HA‐tagged PI4K2A (expressed at low, close to endogenous levels, Fig 2B) but not by its kinase inactive version (Fig 2A).
Figure 2. Restoration of endosomal PI4P by re‐expression of PI4K2A and distribution of Rab7 in PI4K2A K/O cells.
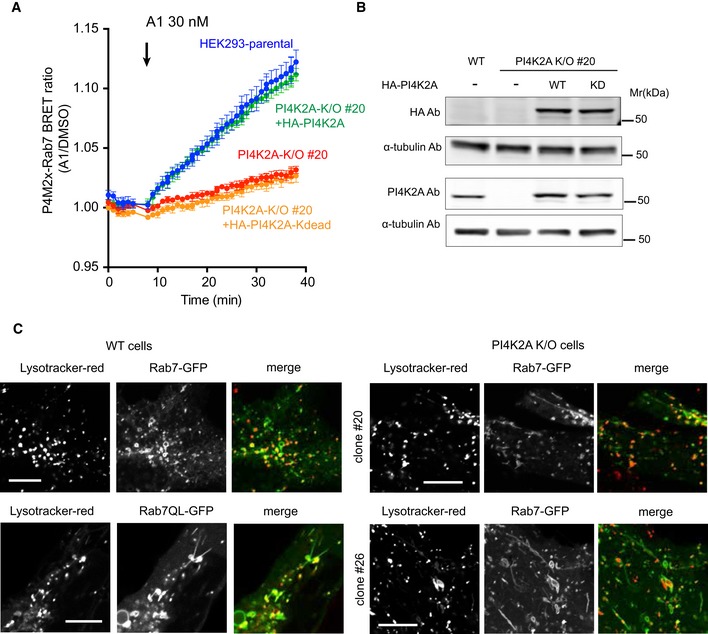
- BRET measurement of PI4P in Rab7 compartment in parental and PI4K2A K/O cells after expression of HA‐tagged PI4K2A or its kinase‐dead mutant. Means ± SEM are shown from three experiments performed in triplicates.
- Western blot analysis shows that the level of PI4K2A expression was closely matched with that of wild‐type cells.
- Distribution of GFP‐Rab7 in parental (left panels) and two clones of PI4K2A K/O cells. Representative pictures show tubulation in the K/O cells reminiscent of those seen in wild‐type cells expressing the GTP‐locked Rab7Q67L mutant. Scale bars are 10 μm.
Source data are available online for this figure.
PI4K2A K/O cells show signs of Rab7 activation
Given the presence of PI4P in the Rab7 compartment, we wanted to evaluate if this compartment showed any changes in cells lacking PI4K2A. To this end, we turned to confocal microscopy to investigate the morphology of the Rab7 compartment in cells expressing GFP‐Rab7 both in normal cells and in the two PI4K2A K/O cell lines. This analysis revealed that PI4K2A K/O cells showed prominent Rab7‐positive tubulation (Fig 2C, right panels), which was reminiscent of those observed in parental HEK293‐AT1 cells expressing low level of GFP‐Rab7 Q67L (Fig 2C, lower left panels). Importantly, there was no notable difference between the parental and PI4K2A K/O cells expressing the GFP‐Rab7Q67L mutant (not shown). These results raised the possibility that Rab7 was activated in PI4K2A K/O cells causing the tubulation. It is important to note that PI4K2A K/O cells showed no sign of lysosome redistribution, which is often associated with Rab7 cycling defects (Cantalupo et al, 2001; Jordens et al, 2001; Fig 2C).
These results have implied that PI4K2A K/O cells would show reduced Rab7 inactivation. To determine the effect of PI4K2A K/O on the Rab7 status of the cells, we quantitated the amount of Rab7 in the GTP‐bound form using a pull‐down assay with GST‐tagged Rab7‐binding domain of RILP, which is used as a trap for Rab7‐GTP (Romero Rosales et al, 2009). However, this assay was not sensitive enough to detect the difference in the amount of active Rab7 between wild‐type and PI4K2A K/O cells even after PI4K2B knockdown in PI4K2A K/O cells (Fig EV2A). When this assay was used in cells expressing GFP‐Rab7 or GFP‐Rab7‐Q67L, a small difference was found between the two Rab7 constructs, whereas no signal was detected in the pull‐down from cells expressing GFP‐Rab7‐N125I (Fig EV2B).
Figure EV2. Pull‐down assays measuring the active state of Rab7 do not show apparent changes in PI4K2A K/O cells.
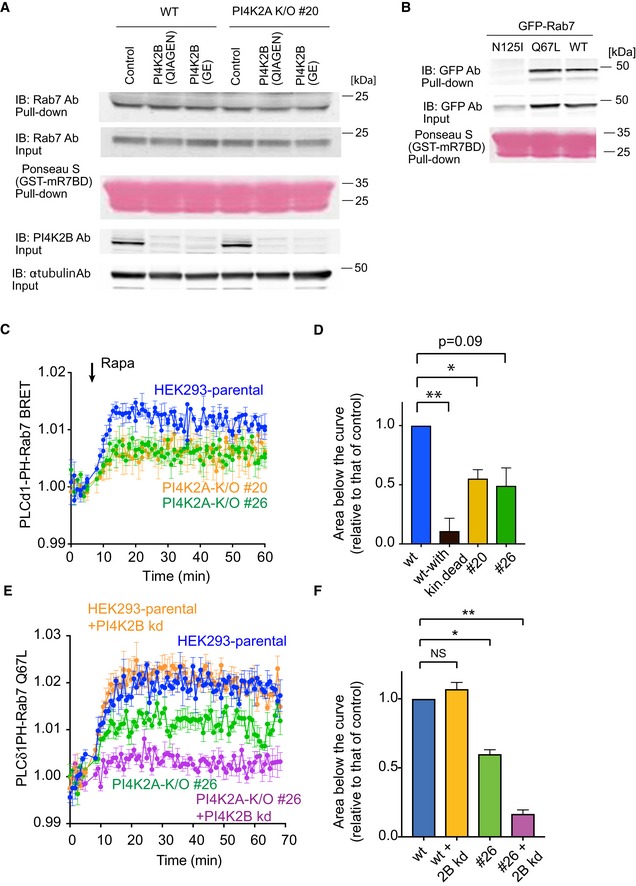
- Pull‐down assays were performed as described in Materials and Methods using lysates prepared from wild‐type or PI4K2A K/O cells #20 in which PI4K2B was knocked down by two different siRNAs (obtained from QIAGEN or GE). Whole‐cell lysates were collected 3 days after the transfection and active Rab7 captured by the Rab7‐GTP interacting motif of RILP. Western blot analysis of the captured material was performed with anti‐Rab7, anti‐PI4K2B, and anti‐alpha‐tubulin antibodies.
- Pull‐down assays showing the amount of overexpressed Rab7 wild‐type, N125I, Q67L mutant captured by the Rab7‐GTP binding domain.
- Increased production of PI(4,5)P2 in the Rab7 compartment by a recruited PIP5Kγ enzyme measured by BRET analysis in control and PI4K2A K/O cells. This panel originates from the same experiments shown in Fig 3C plotted separately for clarity (therefore, blue control curves are the same). For experimental details, see legend to Fig 3C. Means ± SEM from three separate experiments each performed in triplicates.
- This bar diagram shows the areas below the curves calculated from the time of rapamycin addition for each of four separate experiments similar to those shown in panel C (means ± SEM, n = 4). One‐way ANOVA with Dunnett's multiple comparisons was used for statistical analysis (*P = 0.0204; **P = 0.0081).
- Knockdown (kd) of PI4K2B further reduces the amount of PI(4,5)P2 generated by a recruited PIP5Kγ in the Rab7 compartment. This experiment was performed similarly to that shown in Fig 3E using the Rab7‐QL mutant in the BRET construct. Means ± SEM from three separate experiments each performed in triplicates.
- Quantitation and statistical analysis of data shown in panel (E) performed as described for panel (D) (*P = 0.0118; **P = 0.0023).
Source data are available online for this figure.
Endosomal PI(4,5)P2 production leads to the inactivation of Rab7 and requires type‐II PI4Ks
To assess whether PI4P affects Rab7 cycling directly or as a source of PI(4,5)P2, we used the rapamycin‐inducible heterodimerization approach to recruit a PIP5Kγ enzyme to Rab7‐positive membranes using FKBP‐fused PIP5Kγ (Suh et al, 2006) and FRB fused to Rab7 (Hammond et al, 2014; Fig 3A). Confocal images showed that PIP5Kγ acutely recruited to Rab7 membranes caused the cytosolic PLCδ1PH domain to localize to Rab7 compartment as described previously (Hammond et al, 2014; Fig 3B, middle). Importantly, the Rab7 recruiter slowly dissociated from the membranes during this manipulation (Fig 3B, top). In order to obtain a quantitative measure of these changes, we monitored PI(4,5)P2 levels in the Rab7 compartment using the BRET system. For this, we used PLCδ1PH as a PI(4,5)P2 reporter fused to Sluc and targeted Venus by fusing it to Rab7. To avoid possible interference with the BRET signal, we used iRFP in the Rab7‐targeted FRB and a mutated CFP (W66A) in the FKBP‐fused PIP5Kγ to eliminate its fluorescence. To increase the amount of PLCδ1‐PH available in the cytosol for binding to the endosome‐generated PI(4,5)P2, we also expressed a constitutively active Gq (Gq*) at low level to decrease PI(4,5)P2 in the PM. (Expression of Gq* has proven to be unnecessary and was omitted in subsequent experiments.) As shown in Fig 3C, BRET analysis detected the increased PI(4,5)P2 formation at the Rab7 compartment (Fig 3C, blue trace). Recruitment of the kinase‐dead version of the PIP5Kγ showed little if any effect (Fig 3C, black trace). These traces also showed that the BRET signal slowly returned to baseline (defined as the trace obtained with the inactive Rab7‐N125I) consistent with the Rab7 recruiter dissociating from the membrane caused by PI(4,5)P2 generation (Fig 3D blue trace). When the Rab7 component of the BRET probe (fused to Venus) was mutated either to its GTP or GDP‐locked form (Q67L and N125I, respectively), the PI(4,5)P2 increase was larger and more sustained with the GTP‐locked form, whereas no signal increase was detected with the GDP‐locked Rab7 (Fig 3D red and green traces, respectively).
Figure 3. Increased production of PI(4,5)P2 in Rab7‐positive endosomes causes Rab7 dissociation.

- Cartoon depicting the experimental design. A PIP5Kγ construct fused to FKBP12 is expressed together with an FRB module fused to Rab7 and the GFP‐tagged PLCδ1PH reporter for PI(4,5)P2. Addition of rapamycin causes recruitment of PIP5Kγ to the endosomes where it makes more PI(4,5)P2 and attracts the PI(4,5)P2 reporter.
- Representative confocal pictures showing these changes in live cells. Cells were transfected with the indicated constructs and imaged before and after addition of rapamycin (100 nM) for 10 min. Note that the Rab7 recruiter itself is dissociating from the membranes after PI(4,5)P2 generation. Left panels show some cells enlarged. Scale bars: 20 μm.
- Quantification of the changes in BRET experiments. Here the BRET sensor contained the PLCδ1PH‐fused to Sluc and the Venus targeted with Rab7 expressed from a single vector and it was co‐transfected with the Rab7‐targeted FRB (tagged with iRFP) and the FKBP12‐fused PIP5Kγ in which the CFP was mutated to eliminate its fluorescence (CFP*). Recruitment of the PIP5Kγ but not its kinase‐dead version caused an increase in the BRET signal indicating the increased PI(4,5)P2 in this compartment (means ± SEM, from three separate experiments performed in triplicates).
- PI(4,5)P2 increases were larger when the Venus part of the BRET sensor was targeted with the GTP‐locked form of Rab7 (Q67L) and was negligible with the GDP‐locked form of Rab7 (N125I) in the BRET construct. Also note that the Rab7 wild‐type‐based BRET signal slowly returned toward the green trace consistent with Rab7 falling off from the membrane both in the BRET and the recruiting constructs (means ± SEM, from three separate experiments performed in triplicates).
- The response of PI4K2A K/O cells was significantly reduced in these latter experiments. Means ± SEM are shown from three experiments performed in triplicates.
- Areas below the curves calculated from the time of rapamycin addition for each of the three separate experiments shown in panel E (means ± SEM, n = 3). One‐way ANOVA with Dunnett's multiple comparisons was used for statistical analysis (*P = 0.0478 and 0.0287 for #20 and #26 clones, respectively).
Next, we performed similar analysis in the PI4K2A K/O cells, to determine whether the reduced PI4P level in the Rab7 endosomes limits the PI(4,5)P2 that can be produced. These experiments showed that recruitment of the PIP5Kγ resulted in a ~ 50% reduction in PI(4,5)P2 production in the Rab7 compartment compared to changes observed with the parental cells (Fig 3E and F). Analysis of the areas below the curves showed significantly smaller PI(4,5)P2 generation in the knockout cells (Fig 3F). Similar changes were observed when the Rab7 wild‐type construct was used as a recruiter. In that case, the reduction in PI(4,5)P2 level was also observed in the PI4K2A K/O cells but reached significance only with clone #20 (Fig EV2C and D). To evaluate whether the remaining PI(4,5)P2 was produced from PI4P made by the PI4K2B enzyme, we used one of the PI4K2A K/O cells and knocked down PI4K2B. In such cells little if any PI(4,5)P2 was produced by the recruited PIP5K (Fig EV2E and F). These results together suggested that Rab7 could be inactivated by the generation of PI(4,5)P2 at the Rab7 compartment and that PI4K2A was an important contributor to provide PI4P as the precursor for PI(4,5)P2.
Endosomal PI4P is converted to PI(4,5)P2 by endogenous PIP5Kγ
These experiments showed that overexpressed and acutely recruited PIP5K can impact the GDP/GTP status of Rab7, but did not tell whether PI(4,5)P2 is made endogenously on Rab7 endosomes. It has been well documented that the PI(4,5)P2 reporter, PLCδ1PH‐GFP does not detect intracellular pools of this lipid presumably because of the limited size of these PI(4,5)P2 pools, yet it is able to detect them once they are expanded (Wills et al, 2018). To address this question, we turned to OSW1, one of the recently introduced inhibitors of OSBP function (also called ORPphilins; Burgett et al, 2011). It has been reported that inactivation of the ER‐localized adaptor proteins VAP‐A and VAP‐B leads to the accumulation of PI4P in endosomes due to the inability of the OSBP protein to transport PI4P from endosomes to the ER (Dong et al, 2016). Indeed, inhibition of OSBP by OSW1 was reported to induce accumulation of Golgi and endosomal PI4P (Mesmin et al, 2017). Therefore, we set out experiments to investigate the effect of OSW1 on endosomal PI4P. Confocal microscopy showed that OSW1 treatment increased PI4P both in the Golgi and in the endosomal compartments and that the endosomal PI4P increase was barely detectable in PI4K2A K/O cells (Fig 4A). This was confirmed by BRET analysis, which showed a steady accumulation of PI4P in the Rab7 compartment following OSW1 treatment (20 nM). This increase was substantially smaller in PI4K2A K/O cells (Fig 4B). This finding was in good agreement with the recent Dong study, which showed that this endosomal PI4P was largely produced by the PI4K2A enzyme (Dong et al, 2016). To investigate whether this PI4P increase was associated with an increased PI(4,5)P2 in the same Rab7 compartment, we used confocal analysis using the PLCδ1‐PH domain as the lipid sensor. As shown in Fig 4C, 60‐min OSW1 (20 nM) treatment did not reveal any PI(4,5)P2 in the endosomal compartments. Since most of the PLCδ1‐PH domain is associated with the abundant PI(4,5)P2 in the PM, we reasoned that there might not have been enough PLCδ1‐PH reporter in the cytoplasm to detect the small PI(4,5)P2 pool even after OSW1 treatment. Therefore, we used stimulation with angiotensin II (AngII) to induce hydrolysis of the PM PI(4,5)P2 and, hence, to increase the amount of lipid probe in the cytosol. This manipulation revealed the association of the lipid probe with the intracellular compartments, and this was greatly reduced in PI4K2A K/O cells (Fig 4C, right). This experiment was then performed in the BRET format where the PLCδ1‐PH domain was used as a lipid sensor fused to Sluc and the Rab7‐Q67L‐targeted Venus as an acceptor. Consistent with the confocal data, the PI(4,5)P2 BRET signal in the Rab7‐positive endosomes was below detection limits even after OSW1 treatment even in naïve HEK293‐AT1 cells without Ang II stimulation (Fig 4D, green trace). However, the BRET signal showed an easily detectable increase after AngII stimulation (which releases the PI(4,5)P2 probe from the PM), but only in cells pretreated with OSW1 (Fig 4D, red trace vs. blue trace). Again, this PI(4,5)P2 accumulation after OSW1 treatment was hardly detectable in the PI4K2A K/O cells (Fig 4E, orange trace). These experiments showed that endosomal PI4P is converted to PI(4,5)P2 in the Rab7‐positive compartment which becomes detectable after PI4P accumulation is caused by OSW1.
Figure 4. Increased level of PI4P in Rab7 endosomes is associated with PI(4,5)P2 production.
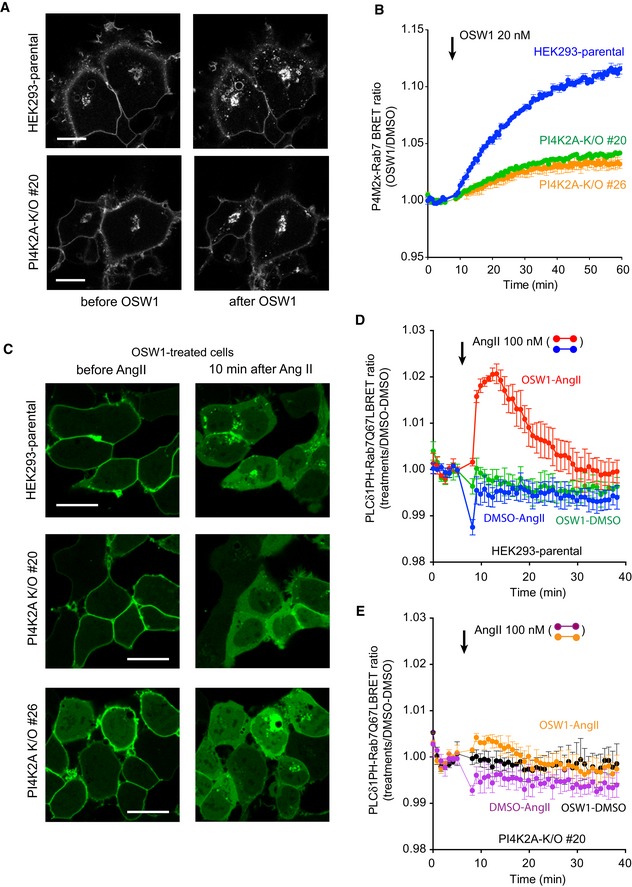
- Representative confocal images of HEK293‐AT1 cells, which show that treatment with OSW1 (20 nM), a drug that inhibits the cholesterol‐PI4P transport protein, OSBP, causes accumulation of PI4P in the Golgi and in the endosomes (upper panels). Note that the accumulation is much reduced in the endosome but less so in the Golgi in the PI4K2A K/O cells. Scale bars: 20 μm.
- Quantification of these changes by BRET analysis. The PI4P‐Rab7 BRET sensor construct was transfected into the respective cell lines and the cells treated with OSW1 (20 nM) for the indicated times. The BRET ratios were expressed relative to those of DMSO‐treated cells.
- Detection of endosomal PI(4,5)P2 in cells pretreated with OSW1 for 1 h in serum‐free medium using confocal microscopy and the PLCδ1PH‐GFP PI(4,5)P2 reporter. Angiotensin II (AngII, 100 nM; a Gq and PLC‐activating agonist) was used to liberate the PLCδ1PH‐GFP PI(4,5)P2 reporter from the PM, which was necessary to detect the endosomal PI(4,5)P2. The signal is transient as the AngII receptors show desensitization. Note the reduced signal in the two PI4K2A K/O clones. Scale bars: 20 μm.
- Quantification of the same changes by BRET analysis in wild‐type cells using the PI(4,5)P2 sensor with Rab7‐Q67L‐targeting. Note the transient signal increase after AngII stimulation only in cells treated with OSW1.
- This increase is barely detectable in the PI4K2A K/O cells.
To investigate which PIP5K was responsible for this effect, we repeated the above‐described experiments in cells where PIPγ or PIP5Kβ was knocked down. These two enzymes were chosen as they have been shown to operate in the late‐endosome lysosome compartments (Rong et al, 2012; Tan et al, 2016). Knockdown of PIP5Kγ (all isoforms, Fig 5B) significantly decreased the PI(4,5)P2 signal detected in OSW1‐treated parental HEK293‐AT1 cells, while PIP5Kβ knockdown showed a smaller effect (Fig 5A and B). It has to be noted, though, that Western blot analysis showed no detectable PIP5Kβ in these cells using two different antibodies both of which detected the expressed protein (Fig EV3A and C). The effectiveness of the PIP5Kβ siRNA was tested using a GFP‐fused human PIP5Kβ (Fig EV3B). The small effect of PIP5Kβ knockdown could be attributed to the small decrease it caused in PIP5Kγ levels (Fig 5C). Knockdown of either PIP5Kβ or PIP5Kγ did not change the expression of PIP5Kα (Fig EV3D). Since our BRET analysis of the PI(4,5)P2 signal relied upon the release of the PLCδ1PH from the PM, we ran parallel experiments to monitor PM PI(4,5)P2 changes in the cells in which the PIP5Ks were knocked down (Fig EV3E). These experiments showed that neither PIP5Kγ nor PIP5Kβ knockdown had detectable effects on PI(4,5)P2 changes in the PM during AngII stimulation. This also suggested that these HEK293 cells can maintain their PI(4,5)P2 levels by using the remaining (non‐targeted) PIP5K enzymes, consistent with conclusions drawn by studies on knockout animals (Volpicelli‐Daley et al, 2010). Together, these experiments suggested that PIP5Kγ but not PIP5Kβ was responsible for the majority of the PI(4,5)P2 that was generated in the Rab7‐positive compartment.
Figure 5. PIP5Kγ is the primary source of PI(4,5)P2 in late endosomes and LC3 accumulation detected in PI4K2A K/O cells.
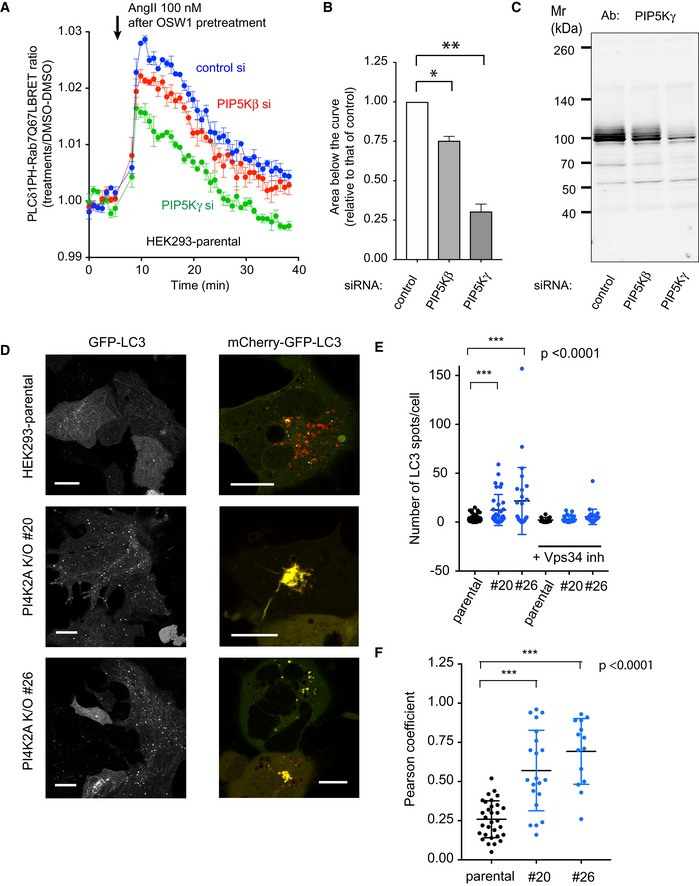
- BRET experiment in parental HEK293‐AT1 cells in OSW1‐pretreated cells after RNAi‐mediated knockdown of PIP5Kβ or PIP5Kγ. The BRET experiment was performed the same way as described in the legend to Fig 4D (means ± SEM from three experiments, each performed in triplicates). Note the reduced response in the PIP5Kγ knockdown cells and the slight reduction in the case of PIP5Kβ knockdown.
- Areas below the curves calculated from the time of rapamycin addition for each of the three separate experiments shown in panel A (means ± SEM, n = 3). One‐way ANOVA with Dunnett's multiple comparisons was used for statistical analysis (*P = 0.0241; **P = 0.0088).
- These reductions are proportional to the reduction in PIP5Kγ levels as assessed by Western blot analysis of cell lysates obtained from cells treated with siRNAs at the same time.
- Increased appearance of GFP‐LC3‐positive vesicles in PI4K2A K/O cells and reduced acidification of the LC3‐positive vesicles using the GFP‐mCherry‐LC3 reporter. Cells were transfected with the indicated constructs for 1 day and observed by confocal microscopy. Scale bars: 20 μm.
- Quantification of the LC3‐positive vesicles (n = 67, 34, 26, 13, 30, 27 for the various groups, respectively, from left to right). *** designates P < 0.0001 using unpaired t‐test. The same data set was also analyzed with the non‐parametric Mann–Whitney U‐test showing a P = 0.0004 and 0.0012 for #20 and #26 clones relative to control, respectively.
- Co‐localization analysis using the Pearson coefficient (n = 31, 21, and 14 cells analyzed for the different groups from left to right). *** designates P < 0.0001 using unpaired t‐test. The same data set was also analyzed with the non‐parametric Mann–Whitney U‐test showing a P value < 0.0001 for both groups.
Source data are available online for this figure.
Figure EV3. Knockdown of PIP5Kβ and PIP5Kγ did not affect the level of PIP5Kα or plasma membrane PI(4,5)P2 .

-
A, CWestern blot analysis with two different PIP5Kβ antibodies (AB1‐ or AB2‐PIP5Kβ) performed with whole‐cell lysates prepared from wild‐type cells or from cells expressing myc‐ or mRFP‐tagged PIP5Kβ. Note that both antibodies detect the expressed proteins but failed to detect the endogenous PIP5Kβ.
-
BsiRNA against PIP5Kβ is effective as assessed by Western blotting using a GFP‐humanPIP5Kβ enzyme. The amounts of transfected PIP5Kβ plasmid DNA are indicated.
-
DWestern blot analysis showing the lack of effect of PIP5Kβ or PIPK5γ siRNA on the amount of PIP5Kα.
-
EBRET analysis showing the level of changes in plasma membrane PI(4,5)P2 during stimulation by AngII in cells depleted in PIP5Kβ or PIPK5γ. AngII was added to the cells for monitoring the rate of PI(4,5)P2 breakdown and subsequent re‐synthesis. The BRET ratios were expressed relative to those of DMSO‐treated cells, and means ± SEM are shown from three experiments performed in triplicates.
Source data are available online for this figure.
Impaired production of PI(4,5)P2 in Rab7 compartment causes autophagosome–lysosome fusion defects
We have found previously that PI4K2A interacts with GABARAP (Jovic et al, 2014). Moreover, Wang et al (2015) has recently reported that PI4K2A not only binds to the LC3 homologue, GABARAPs, but also regulates autophagosome–lysosome fusion. We also observed increased number of GFP‐LC3‐positive puncta PI4K2A K/O cells which were eliminated after inhibiting hVps34 (Fig 5D and E). Also, we found that a significantly higher number of mCherry‐GFP‐LC3‐positive vesicles showed acidification problems and in some cells prominent tubulation (Fig 5D and F). This reporter decreases its GFP signal once in the acidic compartment, i.e., following fusion of autophagosomes with lysosomes, while still retaining its red fluorescence due to the resistance of mCherry fluorescence to acidification. These findings together suggested that PI4K2A K/O cells did show a defect in acidification of autophagosomes as described by Wang et al (2015) in PI4K2A knockdown cells. However, this block was not absolute since cells still cleared their LC3‐positive vesicles after Vps34 inhibition.
Next, we investigated whether PIP5Kγ knockdown in wild‐type cells caused any defect in autophagosome processing. As shown in Fig 6A–C, PIP5Kγ knockdown caused similar acidification defects in LC3 vesicles as did PI4K2A K/O. Moreover, such cells also displayed prominent tubulation of Rab7 endosomes, which was similar to those observed in PI4K2A K/O cells (Fig 6D). Several cells also showed tubulation of the LC3‐positive structures (Fig 6B lower) similar to those found in PI4K2A K/O cells. These experiments suggested that PIP5Kγ deficiency caused defects in Rab7 cycling and autophagosome processing similar to those found in PI4K2A K/O cells.
Figure 6. PIP5Kγ knockdown causes defects in autophagosome–lysosome fusion and tubulation of Rab7 compartments.

-
A, BDistribution of mCherry‐GFP‐LC3 in HEK293‐AT1 cells (without starvation) after treatment with control siRNA (A) or siRNA for PIP5Kγ (PIP5K1C). Scale bars: 20 μm. Note the massive tubulation of the LC3 compartment in some of the knock‐down cells (B, lower images, scale bar: 10 μm).
-
CComparison of Pearson coefficients from cells treated with control or PIP5Kγ RNAi. For statistical analysis, the unpaired t‐test was used (n = 50 and 45 cells for control a PIP5Kγ knockdown groups, respectively; ***P < 0.0001).
-
DRepresentative confocal images of live HEK293‐AT1 cells expressing GFP‐Rab7 treated with control or PIP5Kγ RNAi. Note the tubulation in the PIP5Kγ RNAi‐treated cells. Scale bars: 10 μm.
The production of PI(4,5)P2 dissociated PLEKHM1 from late endosomes/lysosomes
It has been reported that PLEKHM1, a Rab7 effector protein, tethers autophagosomes with lysosomes (McEwan & Dikic, 2015). PLEKHM1 is a multivalent adaptor protein that possesses two PH domains and interacts with both LC3/GABARAP proteins and members of the HOPS (homotypic fusion and protein sorting) complex (McEwan & Dikic, 2015). Having seen that PI(4,5)P2 production upon PIP5Kγ recruitment promotes Rab7 inactivation, together with the role of PI4K2A in this process, we wanted to examine whether PLEKHM1 association with Rab7 endosomes is altered by manipulations of PI(4,5)P2 levels and whether it is affected in PI4K2A K/O cells. For this, we recruited FKBP‐fused PIP5Kγ (Suh et al, 2006) to Rab7 compartments using the rapamycin system and monitored PLEKHM1 association with the Rab7 compartment using confocal imaging and BRET analysis. First, wild‐type or PI4K2A K/O cells were transfected with plasmids encoding CFP‐FKBP‐PIP5Kγ, iRFP‐FRB‐Rab7, and PLEKHM1‐GFP. Cells were examined by confocal microscopy and were subjected to rapamycin treatment to generate PI(4,5)P2 in the Rab7 compartment. This treatment induced PLEKHM1‐GFP dissociation from the Rab7 compartment (Fig 7A). PI4K2A K/O cells showed much smaller release of PLEKHM1‐GFP after rapamycin treatment (Fig 7B). To have a quantitative assessment of these changes, we used BRET analysis. We constructed a BRET construct encoding PLEKHM1‐fused to Sluc and Venus fused to Rab7 to measure PLEKHM1 level on Rab7‐positive membranes (Fig 7C). Consistent with the confocal data, PI(4,5)P2 generation released PLEKHM1 from the endosomes as indicated by the drop in the BRET signal (Fig 7C, blue trace). No change was observed when the kinase‐dead PIP5Kγ was recruited to the membrane (Fig 7C, red trace). In both PI4K2A K/O lines, the dissociation of PLEKHM1 was greatly reduced (Fig 7C, green and yellow traces). Similar results were obtained when the Rab7 interacting Rubicon homology domain of PLEKHM1 (Tabata et al, 2010) was used instead of the full‐length protein in the BRET measurements (Fig EV4A). Importantly, PI(4,5)P2 generation failed to release PLEKHM1 from endosomes of cells expressing the Rab7‐Q67L mutant as the BRET acceptor (Fig EV4B). These results raised the possibility that autophagosome–lysosome fusion requires the cycling of PLEKHM1 in the Rab7 compartments and that PI(4,5)P2 exerts a control over this process.
Figure 7. PI(4,5)P2 production releases PLEKHM1 from the Rab7 compartment.

- HEK293‐AT1 cells were transfected with PLEKHM1‐GFP‐Rab7 along with CFP‐FKBP‐PIP5Kγ and iRFP‐FRB‐Rab7 constructs for 1 day and examined live by confocal microscopy. Representative cells show reduced PLEKHM1 localization when PIP5Kγ was recruited to the Rab7 endosomes (lower row images).
- This response is reduced in PI4K2A K/O cells.
- Quantification of these changes by BRET analysis where the full‐length PLEKHM1 was fused to Sluc and Venus was fused to Rab7 in the BRET construct. For PIP5Kγ recruitment, a mutant dark CFP(W66A)‐FKBP‐PIP5Kγ was used with an iRFP‐FRB‐Rab7. BRET values were normalized to those of DMSO‐treated cells. Comparisons were made for parental (blue) and PI4K2A K/O (green and orange) cells.
- Similar BRET analysis where the PLEKHM1 was replaced by either RILP or Vps35 in the BRET construct.
Figure EV4. PI(4,5)P2 production releases the PLEKHM1 RH domain from Rab7‐positive compartments.

-
ABRET analysis was performed as described for Fig 7C except that the RH domain of PLEKHM1 instead of the full‐length PLEKHM1 protein was used in the BRET construct. Recruitment of PIP5Kγ to the Rab7 endosomes caused dissociation of the RH domain from late endosomes. This response was larger in wild‐type cells than in PI4K2A K/O #20 cells. Means ± SEM from three separate experiments each performed in triplicates.
-
BBRET analysis showing that when Rab7Q67L mutant was used in the BRET construct it failed to release either the full‐length PLEKHM1 or its RH domain from late endosomes when PI(4,5)P2 was acutely produced. CFP(W66A)‐FKBP‐PIP5Kγ and iRFP‐FRB‐Rab7 was used as the recruitment constructs and Sluc‐fused full‐length PLEKHM1 or its RH domain together with Venus‐tagged Rab7wt or Q67L mutant as the BRET sensor. Means ± SEM from three separate experiments each performed in triplicates.
-
C, DLive‐cell confocal imaging showing the distribution of GFP‐RILP or GFP‐Vps35 upon acute production of PI(4,5)P2 in the Rab7 compartment. HEK293‐AT1 cells were transfected with CFP‐FKBP‐PIP5Kγ, iRFP‐FRB‐Rab7, and GFP‐RILP(C) or GFP‐Vps35(D). Next day, the images were captured by confocal microscopy before and after 100 nM rapamycin treatment. Scale bars: 20 μm.
The production of PI(4,5)P2 fails to dissociate RILP or Vps35 from late endosomes/lysosomes
Next we examined whether generation of PI(4,5)P2 at the Rab7 compartment also dissociates other Rab7 effectors from late endosomes. For this, we used RILP, a strong Rab7 interactor that regulates the transport of late endosomes/lysosomes along the microtubules (Jordens et al, 2001) or Vps35 a member of the trimeric retromer complex (Seaman et al, 2009). This was tested both by confocal microscopy and BRET analysis. As shown in Fig EV4C, expression of RILP‐GFP caused a collection of Rab7‐positive endosomes at the juxtanuclear area and recruitment of PIP5Kγ hardly affected its dissociation. In fact, RILP association with Rab7 endosomes increased when PI(4,5)P2 was produced in the endosomes (Fig 7D). This finding suggested that strong association of RILP with Rab7 endosomes locks and sequesters a significant fraction of Rab7 in its GTP form preventing access to the respective Rab7‐GAP proteins and when Rab7 is released from its remaining less avid effectors by PI(4,5)P2 production, it is rapidly captured by RILP.
Expression of Vps35 showed only partial co‐localization with the Rab7‐positive endosomes consistent with its primary role in retrograde trafficking and recruitment of the PIP5Kγ had little if any effect on its localization. The more quantitative BRET analysis showed that in contrast to PLEKHM1, Vps35 failed to show any change after PIP5Kγ recruitment. This was not unexpected given the localization of Vps35 only to a small fraction of Rab7 endosome. These results suggested that strong binding of some Rab7 effectors can either prevent PI(4,5)P2 to cause Rab7 GTP hydrolysis or the PI(4,5)P2‐regulated process is selective to influence some but not all Rab7‐controlled pathways.
Multiple Rab7 GAP proteins affect autophagosome maturation
These results suggested that PI(4,5)P2 affects the GDP‐GTP cycling of Rab7 in late endosomes yielding more active Rab7 when PI(4,5)P2 levels are reduced. Conversely, increased PI(4,5)P2 production caused Rab7 inactivation, which indicates either activation of a Rab7 GAP protein, or inactivation of the Rab7 GEF protein. It has been well documented that many GAP proteins mostly acting on Arf proteins are activated by PI(4,5)P2 (Roy et al, 2016). On the other hand, to our knowledge no study has reported inositol lipids inhibiting a GEF protein. For this reason, we explored whether any of the known Rab7 GAP proteins would be regulated by PI(4,5)P2. There are a large number of candidate proteins that belong to the TBC1D family that possess GAP activity against Rab proteins (Frasa et al, 2012). Some of these proteins, such as Armus/TBC1D2A (Frasa et al, 2010; Jaber et al, 2016) or TBC1D15 (Yamano et al, 2014), have been shown to act on Rab7 and play important roles in autophagy and also in the fission of healthy mitochondria (Wong et al, 2018). However, in many cases, it is not clear which TBC1D protein(s) act on Rab7 and whether they affect functionally different Rab7 pools.
We selected a panel of TBC1D proteins (TBC1D9; TBC1D9B; GRTP1; TBC1D7; TBC1D15; TBC1D25; TBC1D2 (Armus); TBC1D5; TBC1D24) and performed knockdown experiments to test if they could prevent the PI(4,5)P2‐induced release of PLEKHM1. None of these manipulations has identified any single TBC1D protein that would be responsible for mediating this effect (not shown). We also tested the localizations of the GFP‐tagged versions of these proteins and their impact on Rab7 distribution and found that only TBC1D9A and TBC1D5 showed localization to endosomes and several of them (TBC1D2, TBC1D5, TBC1D9B and TBC1D15) caused Rab7 endosomes to localize at the cell periphery (Fig EV5). The Drosophila orthologue of TBC1D24 (Skywalker) has been shown to bind PI(4,5)P2 (Fischer et al, 2016), and indeed, it showed prominent plasma membrane and some endosomal localization (Fig EV5). Moreover, generation of PI(4,5)P2 on Rab7 vesicles by the recruitable PIP5K caused localization of TBC1D24 to Rab7 endosomes (Appendix Fig S1A). A similar but more subtle appearance of TBC1D7 in the Rab7 compartment was detectable upon PI(4,5)P2 generation (Appendix Fig S1B).
Figure EV5. Distribution of selected GFP‐fused TBC1D proteins and mCherry Rab7 co‐expressed in HEK293‐AT1 cells.
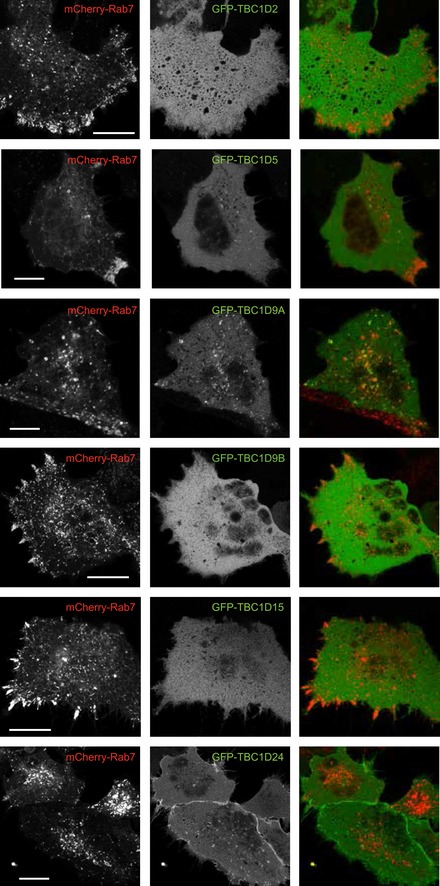
Cells were transfected with the indicated DNAs for 24 h and imaged live by confocal microscopy. Cells expressing low levels of the constructs were chosen to evaluate TBC1D endosomal localization. Note the peripheral distribution of Rab7 after expression of some of the TBC1D proteins. Scale bar: 20 μm.
Finally, we investigated whether knockdown of any of these TBC1D proteins had an impact on autophagosome–lysosome fusion. These experiments showed that knockdown of several TBC1D proteins (namely, TBC1D2, TBC1D7, TBC1D9A, TBC1D9B, and GRTP1) caused fusion defects and TBC1D9B and TBC1D7 also showed strong tubulation in a small fraction of cells (Appendix Fig S2).
Taken all these data together, it is unlikely that a single Rab7‐GAP protein would serve as the mediator of the robust PI(4,5)P2 effect. It is more likely that multiple TBC1D proteins control autophagy flux, a conclusion also reached by previous studies that analyzed their LIR domain‐mediated interactions with Atg8/GABARAP proteins (Popovic et al, 2012). Clearly more research will be required to clarify these questions.
Discussion
This study was designed to explore the role(s) of endosomal PI4P‐PI(4,5)P2 generation in the control of trafficking pathways with special emphasis on late endosome–lysosome function. The key findings presented here identified a chain of events whereby PI4P synthesized by PI4K2A in late endosomes is converted to PI(4,5)P2 which, in turn, causes Rab7 inactivation and the release of PLEKHM1, an adaptor protein critical for autophagosome–lysosome fusion (McEwan et al, 2015). This newly recognized connection between PI4K2A, PI(4,5)P2, Rab7, and PLEKHM1 uncovers a molecular sequence by which PI4K2A can be linked to regulation of autophagic flux and may help us understand some earlier findings that showed the functional importance of type‐II PI4Ks in early and late endosomes (Minogue, 2017).
A recent study showed that PI4K2A was recruited to autophagosome membranes by direct interaction with GABARAP and it was necessary for autophagosome–lysosome fusion (Wang et al, 2015). Moreover, in subsequent studies, co‐trafficking of GABARAP and PI4K2A was also reported (Chen et al, 2018). Although these studies suggested a direct role of PI4P or the PI4K2A protein itself in the control of these trafficking steps, several studies have shown that PI(4,5)P2 also controls lysosomal degradation of integral membrane proteins (Sun et al, 2013; Schill et al, 2014; Tan et al, 2015). PI(4,5)P2 was found to regulate autophagy initiation (Tan et al, 2016) and lysosome reformation from autophagosomes (Rong et al, 2012). The Tan study has established a physical connection between the PIP5Kγi5 isoform and the ATG14 protein and showed that PI(4,5)P2 binding to the C‐terminal Barkor/ATG14(L) autophagosome‐targeting sequence of ATG14 promotes its assembly with Vps34 and Beclin1 and hence is critical for autophagosome initiation (Tan et al, 2016). Our studies revealed an additional role of PI4K2A, PIP5Kγ, and PI(4,5)P2 at the level of the Rab7 GTPase, namely the control of a late step in autophagosome maturation and fusion with lysosomes. Our findings were in agreement with those of Wang et al (2015) regarding the defect in autophagosome–lysosome fusion in PI4K2A‐depleted or PI4K2A‐deleted cells, but also suggested that PI(4,5)P2 is the ultimate regulatory lipid in the process. Our studies concluded that down‐regulation of PIP5Kβ is not responsible for endosomal PI(4,5)P2 while depletion of PIP5Kγ had a strong effect in our studies. This was in agreement with previous reports regarding the lack of PIP5Kβ involvement (Wang et al, 2015), and that a specific isoform of PIP5Kγ, PIP5Kγi5, identified by the Anderson group (Sun et al, 2013; Schill et al, 2014) is the PIP5K that converts PI4P to PI(4,5)P2 in the late endosome.
Notably, we found no effect of acute PI(4,5)P2 generation in late endosomes on RILP‐ or Vps35‐Rab7 association contrasting the strong effect on PLEKHM1. This may reflect the fact that not all Rab7 effectors are equally regulated by PI(4,5)P2 in late endosomes. Curiously, we found no obvious difference in lysosome distribution in our PI4K2A K/O cells even though Rab7 and PLEKHM1 were found important in lysosomal positioning (Mrakovic et al, 2012; Fujiwara et al, 2016; Willett et al, 2017). Several other trafficking steps that involve Rab7 GTPases were shown previously to be controlled by PI4P. For example, the retrograde transport of CI‐M6PR was impaired in PI4K2A knockdown cells (Wang et al, 2003) and it required PI4P‐mediated recruitment of the sorting nexin, SNX6 (Marquer et al, 2016). It was also reported that PI4P allowed release of retromer cargoes from their dynein motors at the TGN (Niu et al, 2013). In the TGN, the retromer‐specific Rab7 GAP protein, TBC1D5, was found to control Rab7 distribution and function (Jimenez‐Orgaz et al, 2018). Interestingly, we found no obvious difference in the distribution of CI‐M6P receptor in the PI4K2A K/O cells (Appendix Fig S3A). Similarly, siRNA‐mediated knockdown of PI4K2A was shown to cause a defect in EGF‐receptor degradation (Minogue et al, 2006), yet we found no problem with EGF‐receptor degradation in the PI4K2A K/O cells (Appendix Fig S3B). These apparent discrepancies reflect a significant level of plasticity and adaptation of the cells that have to live without PI4K2A and that siRNA‐mediated knockdown and total gene inactivation may result in different phenotypic outcomes. This adaptation may also explain why the PI4K2A gene‐trap mice develop spinocerebellar degeneration only at a later stage in life (Simons et al, 2009). It is also notable that PLEKHM1 K/O mice have a restricted phenotype mostly affecting bone density and osteoclast function (Fujihara et al, 1993) and human studies also pointed to osteoclast dysfunction in individuals with PLEKHM1 mutation without a more generalized phenotype (Bo et al, 2016).
A puzzling question that is raised by these results is the apparent need of PLEKHM1 to be released from the bulk Rab7 compartment by the PI(4,5)P2‐dependent process. This finding is counterintuitive given the proposed role of PLEKHM1 to tether autophagosomes to lysosomes. Our results would argue that PLEKHM1 cycling is important for its function, but we cannot rule out that PI(4,5)P2 and Rab7‐regulated effectors other than PLEKHM1 are equally or even more important in the control of autophagosome–lysosome fusion. The complexity of these processes and their PI(4,5)P2 regulation is highlighted by recent studies showing that PI(4,5)P2 produced from PI5P by type‐II PIP‐kinases (also called PI5P 4‐kinases) is also important for clearance of autophagosomes during starvation (Lundquist et al, 2018). In another study, PI5P was found to control autophagosome maturation under conditions when the Vps34/PI3P canonical pathway is inactivated (Vicinanza et al, 2015).
It is important to note that few effector proteins or mechanism have been identified that would mediate the effects of inositol lipids, PI(4,5)P2 in particular, in autophagosome–lysosome fusion. Our studies show for the first time a PI(4,5)P2 regulation of Rab7 cycling in an intact cell and point to the possible need of PI(4,5)P2 to activate a Rab7 GAP protein. While our efforts to identify the GAP protein(s) mediating this effect were unsuccessful, the strong effect of PI(4,5)P2 on Rab7 cycling can facilitate further research in that direction.
If PI(4,5)P2 is an important regulator of Rab7 cycling, it is expected that a PI(4,5)P2 phosphatase is also part of this regulatory loop. OCRL, one of the PI(4,5)P2 5‐phosphatases, is activated in the lysosome during autophagosome–lysosome fusion and its depletion results in PI(4,5)P2 accumulation and inhibition of the Mucolipin 1 channel and hence defective autophagosome–lysosome fusion (De Leo et al, 2016). Interestingly, while studies in OCRL‐depleted cells reported increased LC3‐positive puncta, knockdown of either PIP5Kα or PIP5Kβ was able to reverse this phenotype, suggesting that the OCRL‐controlled PI(4,5)P2 pool might originate from different PIP5Ks (De Leo et al, 2016). Therefore, the OCRL function may also be related to the clathrin and PIP5Kβ‐dependent reformation of lysosomes (Rong et al, 2012) for which a different PI4K, PI4KB appears to provide the PI4P lipid precursor (Sridhar et al, 2013). These data together suggest that PI4P and PI(4,5)P2 may control more than one step in the autophagy pathway using different lipid kinases and phosphatases (also see discussion above). In this context, it is important to state that we found no difference in the level of OCRL between the PI4K2A K/O and parental cells (T. Baba and T. Balla unpublished observation). Whether other 5‐phosphatase enzymes such as the PI(4,5)P2 and PI(3,4,5)P3 5‐phosphatase, SKIP/INPP5K found in the ER (Gurung et al, 2003) serves as a regulator of PI(4,5)P2 in endosomes remain to be investigated.
In summary, the present studies have identified a role of PI4K2A and PIP5Kγ in the control Rab7 cycling that affects PLEKHM1 association with late endosomes and autophagosome–lysosome fusion. Importantly, this control is exerted with localized production of a small pool of PI(4,5)P2, presumably acting on Rab7 GAP proteins and displays selectivity among different Rab7 effectors (see summary model in Appendix Fig S4). The discrepancy between reported defects in RNAi‐mediated knockdown of PI4K2A in cultured cells that are not featured in our PI4K2A K/O cells reminds us that cells show resilience and adaptation during long‐term gene inactivation. The remarkable assortment of checkpoints regulated by diverse phosphoinositides along the autophagy pathway underscores the unique importance of these lipids in eukaryotic cells.
Materials and Methods
Antibodies and chemicals
Rabbit polyclonal antibody to PI4K2A was a kind gift from Dr. Pietro De Camilli (Yale School of Medicine, New Haven, CT). Rabbit polyclonal antibodies against PI4K2B (cat. no. HPA004099‐100UL) and GFP (cat. no. A11122) were purchased from Sigma‐Aldrich (St. Louis, MO) and Thermo Fisher Scientific (Waltham, MA), respectively. A mouse monoclonal antibody against α‐tubulin (cat. no. 2144S), rabbit monoclonal antibodies against Rab7 and LC3 (Cat. no. 9367S and 4267S, respectively), and a rabbit polyclonal antibody against PIP5K1C (cat. no. 329S) were obtained from Cell Signaling Technology (Danvers, MA). Mouse monoclonal antibody against PI4KB (cat. no. 611816) was from BD Transduction Laboratory (San Jose, CA). Mouse monoclonal antibody against the CI‐M6PR (cat. no. ab2733) was from Abcam (Cambridge, MA). Goat anti‐mouse IgG conjugated with Alexa 488 and Transferrin from human serum conjugated with Alexa Fluor 594 were obtained from Thermo Fisher Scientific. Rabbit polyclonal antibodies to PIP5K1A (cat. no. 15713‐1‐AP) and PIP5K1B (termed AB1; cat. no. 12541‐1‐AP) were from Proteintech (Rosemont, IL). Another PIP5K1B antibody (termed AB2) was a kind gift from Dr. Richard A. Anderson (University of Wisconsin‐Madison, Madison, WI). Mouse HA antibody (cat. no. MMS‐101P) was from COVANCE (Denver, PA). Rapamycin and wortmannin were purchased from Calbiochem (Burlington, MA). GSK‐A1 inhibitor was described previously (Bojjireddy et al, 2014). VPS34‐IN1 and PIKIII were obtained from Selleckchem (Houston, TX). PIK93 was from Sigma‐Aldrich. LysoTracker Red DND‐99 was purchased from Thermo Fisher Scientific.
DNA constructs and siRNAs
The source of DNA constructs used is listed in Appendix Table S1. GFP‐VPS35, GFP‐RILP, humanPIP5Kβ, and TBC1D proteins were created by inserting the coding region of the respective cDNAs amplified by PCR into the SmaI site in the pEGFP‐C1 vector. The Sluc‐P4M2X‐T2A‐Venus‐Rab7 plasmid was created in multiple steps. First, a Venus‐Rab7 construct was made using the iRFP‐FRB‐Rab7 (described in Hammond et al, 2014) by replacing the iRFP‐FRB part of this construct with mVenus (from pmVenus‐C1) using the NheI‐HindIII fragments of the respective plasmids. In the second step, the Sluc‐P4M2x‐T2A sequence was created by PCR using a forward primer containing a NheI site and a reverse primer containing the T2A sequence and an AgeI site. The L10‐Venus‐T2A‐Sluc‐P4M2x plasmid (described in Toth et al, 2016) was used as template after removing an internal NheI site form this original construct by mutagenesis. The PCR fragment was cloned into TOPO vector and the NheI‐AgeI digested insert was placed in the mVenus‐Rab7 plasmid digested with NheI‐AgeI to get the final construct. The other P4M2x‐Sluc‐T2A‐Rab constructs were generated by replacing the Rab7 with the other Rab versions using HindIII/XbaI (for Rab5) and HindIII/BamHI (for Rab11) restriction digests. To obtain Sluc‐P4M2x‐T2A‐Venus‐Rab4, Rab7 cDNA was replaced with Rab4 in Sluc‐P4M2x‐T2A‐Venus‐Rab7 probe using PvuI and SalI site. BRET sensors containing FYVE domain, PLCδ1‐PH, PLEKHM‐full length, PLEKHM1‐RH domain, Vps34, and RILP were constructed by replacing the P4M2x in Sluc‐P4M2x‐T2A‐Venus‐Rab7 or Sluc‐P4M2x‐T2A‐Venus‐Rab5 (in the case of FYVE domain) with the respective fragments using PCR products and T4 polynucleotide kinase for blunt ligation. Mutagenesis was performed using primers listed in Appendix Table S3.
siRNA‐SMARTpools purchased from GE Dharmacon are listed in Appendix Table S2. PI4K2B siRNA (offset 892 of AY_065990) and control siRNA (Catalog number 1027281) were obtained from QIAGEN.
Cell culture and transfection
HEK293 cells stably expressing the AT1a rat AngII receptor (Balla et al, 2007) were maintained in Dulbecco's modified Eagle's medium (DMEM—high glucose) containing 10% FBS and 1% penicillin‐streptomycin. The cell line has been regularly tested for Mycoplasma contamination using InvivoGen mycoplasma detection kit each time after thawing and treated with Plasmocin prophylactic (InvivoGen) at 500 μg/ml for 1 week. The subsequent passages were maintained at 5 μg/ml of Plasmocin. Lipofectamine 2000 and Lipofectamine RNAiMAX were used for plasmid and siRNA transfection, respectively, according to manufacturer's protocols.
Production of PI4K2A K/O cells with CRISPR/Cas9 system
Three targeting sequences were designed with Optimized CRUSPR Design (http://crispor.tefor.net) from exon 1 sequence in PI4K2A gene. The target sequences were inserted into a modified plentiCRISPR v2 plasmid (Jones et al, 2017) with BsmBI site. This plasmid was used straight (not in the form of a virus) for transfection and puromycin selection. Gene editing efficiency of the three gRNAs was tested with T7 Endonuclease I (New England Biolab) digestion of PCR fragments containing the target sequence, and one of the gRNAs was chosen to create PI4K2A K/O cells: 5′‐GGCCTGCGCCGCCACGGTTT‐3′. 5 × 105 HEK293‐AT1 cells were seeded into a 6‐well plate and transfected with 1 μg of the target plasmid the next day. After incubation overnight, the cells were cultured with 15 μg/ml puromycin in the media for 5 days. The cells further were seeded at very low density into 10‐cm dishes and cultured for 11 days in the medium without puromycin to grow colonies. Thirty‐four colonies were isolated and scaled up until each clone was confluent in 6‐well plates. The clones were screened with Western blotting analysis using the anti‐PI4K2A antibody. Genomic DNA was prepared from selected clones and used as a template to generate PCR products, which were then cloned into the PCR2.1 TOPO plasmid for sequencing.
BRET measurements
2,500 or 5,000 cells were seeded to white 96‐well plates and cultured overnight. Cells were then transfected with 100 ng of a BRET sensor using Lipofectamine 2000 according to the manufacturer's protocol. If necessary, additional plasmids, such as those for FKBP/FRB dimerization system, were transfected together with the BRET sensor. Twenty‐four hours after transfection, the medium was replaced, and cells were preincubated with 50 μl of modified Krebs‐Ringer buffer (120 mM NaCl, 4.7 mM KCl, 0.7 mM MgSO4, 10 mM HEPES pH 7.3, 10 mM D+ glucose, 2 mM CaCl2, with a final pH adjusted to 7.4) at room temperature for 30 or 60 min. At that point, 40 μl of coelenterazine h (final concentration 5 μM) in the same buffer was added to the cells, and both Venus fluorescence and Sluc luminescence were monitored with a Tristar2 LB 942 Multimode Microplate Reader (Berthold Technologies) customized with emission filters (540/40 nm and 475/20 nm) for base line measurement for 5 min. The plates were removed for the addition of various treatments in 10 μl volume and measurements continued (one per 30–50 s) for 30 or 60 min.
Live‐cell imaging
300,000 cells were seeded into 30‐mm glass bottom culture dishes and transfected with the indicated plasmids using Lipofectamine 2000. Next day, the media was replaced with 1 ml modified Krebs‐Ringer buffer and the cells were observed at room temperature with a Zeiss confocal microscope (LSM710 or 780, Carl Zeiss MicroImaging).
GST‐R7BD pull‐down assay
BL21 Escherichia coli cells that were transformed with GST‐mR7BD were cultured in 250 ml LB with 100 μg/ml Amp at 37° for 2.5–3 h. GST‐mR7BD expression was induced with 0.5 mM IPTG for 3 h at 30°C. The cells were collected and washed with cold PBS once. Cells were then suspended and sonicated in 5 ml of lysis buffer [25 mM Tris–HCl (pH 7.4), 625 mM NaCl, 0.1% Triton X‐100, 1 mM DTT with a protease inhibitor cocktail (Sigma)]. After centrifugation at 4,000 × g for 30 min, the supernatant was added to 190 μl of a pre‐equilibrated 80% slurry of Glutathione‐Sepharose 4B (GE Healthcare) and incubated for 30 min at room temperature. The beads were washed with the cold lysis buffer (7 ml) three times, and 150 μl of lysis buffer was added to the beads. Concentration of the protein attached to the beads was measured using BCA protein assay kit (Thermo Fisher Scientific). For preparing WT and PI4K2A K/O cell lysates, those cells (from a 6 cm Culture Dish) were lysed with binding buffer (20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 1% Triton X‐100 with a protease inhibitor cocktail). 300 μg of lysates from the HEK293‐AT1 cells was incubated with 30 μg of the beads overnight at 4°C. The beads were washed with the binding buffer without inhibitors three times (1 ml), and equal volume of 2× sample buffer was added. The samples were boiled at 72°C for 10 min and subjected to Western blotting.
EGFR degradation assay
500,000 cells were seeded to 6‐well plate and cultured overnight in the culture medium including 10% FBS. The cells were serum‐starved with DMEM‐high glucose without FBS overnight. After the starvation, the cells were incubated with 100 ng/ml of EGF in the starvation medium for 0, 1, and 3 h and lysed. The lysates were subjected to Western blotting with anti‐EGFR antibody.
Immunocytochemistry
Wild‐type or PI4K2A K/O cells cultured in 30‐mm glass bottom culture dishes were washed with PBS once and fixed by 4% paraformaldehyde for 10 min at room temperature. The fixed cells were incubated in permeabilization buffer (0.5% bovine serum albumin, 0.2% saponin) for 30 min at room temperature. Primary antibody was diluted with the permeabilization buffer and reacted with the antigen in the cells overnight at 4°C. After three washes with 1 ml PBS, the cells were stained with secondary antibody (1:1,000) for 60 min at 37°C or room temperature. After three more washes with PBS, a round coverslip was mounted on top of the cells and sealed using a drop of Mounting medium (Southern Biotech).
Image analysis and statistics
Confocal images were imported to Adobe Photoshop for cropping and adjustments to use the whole dynamic range. No changes in gamma were allowed. For counting particles, images were imported into FIJI, and after thresholding, particles were counted using the Analyze Particles module. Coloc 2 in FIJI was used for Pearson's correlation coefficient. For statistical analysis, the two‐sample t‐test was used and significant difference was considered with P < 0.05 using the two‐tail method. In cases where the F‐test showed significantly different variations, the non‐parametric Mann–Whitney U‐test was also performed.
Author contributions
TBab and TBal designed the experiments and TBab conducted most of the experiments. DJT and YJK generated the PI4K2A K/O cells. NS generated some of the BRET sensors. TBal and TBab performed all microscopy analysis. TBab and TBal analyzed the data and wrote the manuscript in consultation with the other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Acknowledgements
We are grateful for DNA constructs provided by the Varnai laboratory (Semmelweis University Medical School) and by Dr. Francis Barr (University of Oxford, UK). We would like to thank Dr. Richard A. Anderson (University of Wisconsin) and Dr. Pietro De Camilli (Yale University) for the PIP5Kβ and PI4K2A antibodies, respectively. The expert advice and guidance of Dr. Choi Uimook (NIAID, NIH, Bethesda, MD) during the CRISPR/Cas9 gene‐editing experiments is highly appreciated. Confocal imaging was performed at the Microscopy & Imaging Core of the National Institute of Child Health and Human Development, NIH with the kind assistance of Dr. Vincent Schram and Lynne Holtzclaw. We would also like to thank Dr. Joshua Pemberton for critical reading of the manuscript. This work was supported in part by the intramural research program of the Eunice Kennedy Shriver NICHD, at the National Institutes of Health.
The EMBO Journal (2019) 38: e100312
References
- Balla A, Tuymetova G, Barshishat M, Geiszt M, Balla T (2002) Characterization of type II phosphatidylinositol 4‐kinase isoforms reveals association of the enzymes with endosomal vesicular compartments. J Biol Chem 277: 20041–22050 [DOI] [PubMed] [Google Scholar]
- Balla A, Tuymetova G, Tsiomenko A, Varnai P, Balla T (2005) A plasma membrane pool of phosphatidylinositol 4‐phosphate is generated by phosphatidylinositol 4‐kinase type‐III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol Biol Cell 16: 1282–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A, Kim YJ, Varnai P, Szentpetery Z, Knight Z, Shokat KM, Balla T (2007) Maintenance of hormone‐sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4‐kinase III{alpha}. Mol Biol Cell 19: 711–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93: 1019–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bo T, Yan F, Guo J, Lin X, Zhang H, Guan Q, Wang H, Fang L, Gao L, Zhao J, Xu C (2016) Characterization of a relatively malignant form of osteopetrosis caused by a novel mutation in the PLEKHM1 gene. J Bone Miner Res 31: 1979–1987 [DOI] [PubMed] [Google Scholar]
- Bojjireddy N, Botyanszki J, Hammond G, Creech D, Peterson R, Kemp DC, Snead M, Brown R, Morrison A, Wilson S, Harrison S, Moore C, Balla T (2014) Pharmacological and genetic targeting of pPI4KA reveals its important role in maintaining plasma membrane PtdIns4p and PtdIns(4,5)p2 levels. J Biol Chem 289: 6120–6132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boura E, Nencka R (2015) Phosphatidylinositol 4‐kinases: function, structure, and inhibition. Exp Cell Res 337: 136–145 [DOI] [PubMed] [Google Scholar]
- Burgess J, Del Bel LM, Ma CI, Barylko B, Polevoy G, Rollins J, Albanesi JP, Kramer H, Brill JA (2012) Type II phosphatidylinositol 4‐kinase regulates trafficking of secretory granule proteins in Drosophila . Development 139: 3040–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgett AW, Poulsen TB, Wangkanont K, Anderson DR, Kikuchi C, Shimada K, Okubo S, Fortner KC, Mimaki Y, Kuroda M, Murphy JP, Schwalb DJ, Petrella EC, Cornella‐Taracido I, Schirle M, Tallarico JA, Shair MD (2011) Natural products reveal cancer cell dependence on oxysterol‐binding proteins. Nat Chem Biol 7: 639–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C (2001) Rab‐interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J 20: 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sun HQ, Eichorst JP, Albanesi JP, Yin H, Mueller JD (2018) Comobility of GABARAP and phosphatidylinositol 4‐kinase 2A on cytoplasmic vesicles. Biochemistry 57: 3556–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforidis S, Miaczynska M, Ashman K, Wilm M, Zhao L, Yip SC, Waterfield MD, Backer JM, Zerial M (1999) Phosphatidylinositol‐3‐OH kinases are Rab5 effectors. Nat Cell Biol 1: 249–252 [DOI] [PubMed] [Google Scholar]
- Craige B, Salazar G, Faundez V (2008) Phosphatidylinositol‐4‐kinase type II alpha contains an AP‐3 sorting motif and a kinase domain that are both required for endosome traffic. Mol Biol Cell 19: 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo G, Vicinanza M, Wilson C, De Matteis MA (2012) Phosphoinositides in Golgi complex function. Subcell Biochem 59: 255–270 [DOI] [PubMed] [Google Scholar]
- De Leo MG, Staiano L, Vicinanza M, Luciani A, Carissimo A, Mutarelli M, Di Campli A, Polishchuk E, Di Tullio G, Morra V, Levtchenko E, Oltrabella F, Starborg T, Santoro M, Di Bernardo D, Devuyst O, Lowe M, Medina DL, Ballabio A, De Matteis MA (2016) Autophagosome‐lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat Cell Biol 18: 839–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443: 651–657 [DOI] [PubMed] [Google Scholar]
- Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, De Camilli P (2016) Endosome‐ER contacts control actin nucleation and retromer function through VAP‐dependent regulation of PI4P. Cell 166: 408–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B, Luthy K, Paesmans J, De Koninck C, Maes I, Swerts J, Kuenen S, Uytterhoeven V, Verstreken P, Versees W (2016) Skywalker‐TBC1D24 has a lipid‐binding pocket mutated in epilepsy and required for synaptic function. Nat Struct Mol Biol 23: 965–973 [DOI] [PubMed] [Google Scholar]
- Frasa MA, Maximiano FC, Smolarczyk K, Francis RE, Betson ME, Lozano E, Goldenring J, Seabra MC, Rak A, Ahmadian MR, Braga VM (2010) Armus is a Rac1 effector that inactivates Rab7 and regulates E‐cadherin degradation. Curr Biol 20: 198–208 [DOI] [PubMed] [Google Scholar]
- Frasa MA, Koessmeier KT, Ahmadian MR, Braga VM (2012) Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat Rev Mol Cell Biol 13: 67–73 [DOI] [PubMed] [Google Scholar]
- Fujihara H, Fukuda S, Tanaka T, Kanazawa H, Fujiwara N, Shimoji K (1993) Arginine vasopressin increases perinuclear [Ca2+] in single cultured vascular smooth muscle cells of rat aorta. J Vasc Res 30: 231–238 [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Ye S, Castro‐Gomes T, Winchell CG, Andrews NW, Voth DE, Varughese KI, Mackintosh SG, Feng Y, Pavlos N, Nakamura T, Manolagas SC, Zhao H (2016) PLEKHM1/DEF8/RAB7 complex regulates lysosome positioning and bone homeostasis. JCI Insight 1: e86330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung R, Tan A, Ooms LM, McGrath MJ, Huysmans RD, Munday AD, Prescott M, Whisstock JC, Mitchell CA (2003) Identification of a novel domain in two mammalian inositol‐polyphosphate 5‐phosphatases that mediates membrane ruffle localization. The inositol 5‐phosphatase skip localizes to the endoplasmic reticulum and translocates to membrane ruffles following epidermal growth factor stimulation. J Biol Chem 278: 11376–11385 [DOI] [PubMed] [Google Scholar]
- Hammond GR, Machner MP, Balla T (2014) A novel probe for phosphatidylinositol 4‐phosphate reveals multiple pools beyond the Golgi. J Cell Biol 205: 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber N, Mohd‐Naim N, Wang Z, DeLeon JL, Kim S, Zhong H, Sheshadri N, Dou Z, Edinger AL, Du G, Braga VM, Zong WX (2016) Vps34 regulates Rab7 and late endocytic trafficking through recruitment of the GTPase‐activating protein Armus. J Cell Sci 129: 4424–4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez‐Orgaz A, Kvainickas A, Nagele H, Denner J, Eimer S, Dengjel J, Steinberg F (2018) Control of RAB7 activity and localization through the retromer‐TBC1D5 complex enables RAB7‐dependent mitophagy. EMBO J 37: 235–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KR, Choi U, Gao JL, Thompson RD, Rodman LE, Malech HL, Kang EM (2017) A novel method for screening adenosine receptor specific agonists for use in adenosine drug development. Sci Rep 7: 44816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordens I, Fernandez‐Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, Janssen H, Wubbolts R, Neefjes J (2001) The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein‐dynactin motors. Curr Biol 11: 1680–1685 [DOI] [PubMed] [Google Scholar]
- Jovic M, Kean MJ, Dubankova A, Boura E, Gingras AC, Brill JA, Balla T (2014) Endosomal sorting of VAMP3 is regulated by PI4K2A. J Cell Sci 127: 3745–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketel K, Krauss M, Nicot AS, Puchkov D, Wieffer M, Muller R, Subramanian D, Schultz C, Laporte J, Haucke V (2016) A phosphoinositide conversion mechanism for exit from endosomes. Nature 529: 408–412 [DOI] [PubMed] [Google Scholar]
- Lamb CA, Longatti A, Tooze SA (2016) Rabs and GAPs in starvation‐induced autophagy. Small GTPases 7: 265–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist MR, Goncalves MD, Loughran RM, Possik E, Vijayaraghavan T, Yang A, Pauli C, Ravi A, Verma A, Yang Z, Johnson JL, Wong JCY, Ma Y, Hwang KS, Weinkove D, Divecha N, Asara JM, Elemento O, Rubin MA, Kimmelman AC et al (2018) Phosphatidylinositol‐5‐phosphate 4‐kinases regulate cellular lipid metabolism by facilitating autophagy. Mol Cell 70: 531–544 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquer C, Tian H, Yi J, Bastien J, Dall'Armi C, Yang‐Klingler Y, Zhou B, Chan RB, Di Paolo G (2016) Arf6 controls retromer traffic and intracellular cholesterol distribution via a phosphoinositide‐based mechanism. Nat Commun 7: 11919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Dikic I (2015) PLEKHM1: adapting to life at the lysosome. Autophagy 11: 720–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I (2015) PLEKHM1 regulates autophagosome‐lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57: 39–54 [DOI] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Polidori J, Jamecna D, Lacas‐Gervais S, Antonny B (2017) Sterol transfer, PI4P consumption, and control of membrane lipid order by endogenous OSBP. EMBO J 36: 3156–3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minogue S, Waugh MG, De Matteis MA, Stephens DJ, Berditchevski F, Hsuan JJ (2006) Phosphatidylinositol 4‐kinase is required for endosomal trafficking and degradation of the EGF receptor. J Cell Sci 119: 571–581 [DOI] [PubMed] [Google Scholar]
- Minogue S, Waugh MG (2012) The phosphatidylinositol 4‐kinases: don't call it a comeback. Subcell Biochem 58: 1–24 [DOI] [PubMed] [Google Scholar]
- Minogue S (2017) The many roles of type II phosphatidylinositol 4‐kinases in membrane trafficking: new tricks for old dogs. BioEssays 40: 1700145 [DOI] [PubMed] [Google Scholar]
- Mrakovic A, Kay JG, Furuya W, Brumell JH, Botelho RJ (2012) Rab7 and Arl8 GTPases are necessary for lysosome tubulation in macrophages. Traffic 13: 1667–1679 [DOI] [PubMed] [Google Scholar]
- Nakatsu F, Baskin JM, Chung J, Tanner LB, Shui G, Lee SY, Pirruccello M, Haio M, Ingolia NT, Wenk MR, De Camilli P (2012) PtdIns4P synthesis by PI4KIIIa at the plasma membrane and its impact on plasma membrane identity. J Cell Biol 199: 1003–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell‐Litwa K, Salazar G, Smith Y, Faundez V (2009) Roles of BLOC‐1 and adaptor protein‐3 complexes in cargo sorting to synaptic vesicles. Mol Biol Cell 20: 1441–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y, Zhang C, Sun Z, Hong Z, Li K, Sun D, Yang Y, Tian C, Gong W, Liu JJ (2013) PtdIns(4)P regulates retromer‐motor interaction to facilitate dynein‐cargo dissociation at the trans‐Golgi network. Nat Cell Biol 15: 417–429 [DOI] [PubMed] [Google Scholar]
- Novick P (2016) Regulation of membrane traffic by Rab GEF and GAP cascades. Small GTPases 7: 252–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D, Akutsu M, Novak I, Harper JW, Behrends C, Dikic I (2012) Rab GTPase‐activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol Cell Biol 32: 1733–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero Rosales K, Peralta ER, Guenther GG, Wong SY, Edinger AL (2009) Rab7 activation by growth factor withdrawal contributes to the induction of apoptosis. Mol Biol Cell 20: 2831–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y, Liu M, Ma L, Du W, Zhang H, Tian Y, Cao Z, Li Y, Ren H, Zhang C, Li L, Chen S, Xi J, Yu L (2012) Clathrin and phosphatidylinositol‐4,5‐bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol 14: 924–934 [DOI] [PubMed] [Google Scholar]
- Roy NS, Yohe ME, Randazzo PA, Gruschus JM (2016) Allosteric properties of PH domains in Arf regulatory proteins. Cell Logist 6: e1181700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder PV, Vistein R, Gokhale A, Seaman MN, Puthenveedu MA, Faundez V (2013) The WASH complex, an endosomal Arp2/3 activator, interacts with the Hermansky‐Pudlak syndrome complex BLOC‐1 and its cargo phosphatidylinositol‐4‐kinase type IIalpha. Mol Biol Cell 24: 2269–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schill NJ, Hedman AC, Choi S, Anderson RA (2014) Isoform 5 of PIPKIgamma regulates the endosomal trafficking and degradation of E‐cadherin. J Cell Sci 127: 2189–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN, Harbour ME, Tattersall D, Read E, Bright N (2009) Membrane recruitment of the cargo‐selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab‐GAP TBC1D5. J Cell Sci 122: 2371–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons JP, Al‐Shawi R, Minogue S, Waugh MG, Wiedemann C, Evangelou S, Loesch A, Sihra TS, King R, Warner TT, Hsuan JJ (2009) Loss of phosphatidylinositol 4‐kinase 2alpha activity causes late onset degeneration of spinal cord axons. Proc Natl Acad Sci USA 106: 11535–11539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar S, Patel B, Aphkhazava D, Macian F, Santambrogio L, Shields D, Cuervo AM (2013) The lipid kinase PI4KIIIbeta preserves lysosomal identity. EMBO J 32: 324–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Inoue T, Meyer T, Hille B (2006) Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314: 1454–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Hedman AC, Tan X, Schill NJ, Anderson RA (2013) Endosomal type Igamma PIP 5‐kinase controls EGF receptor lysosomal sorting. Dev Cell 25: 144–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata K, Matsunaga K, Sakane A, Sasaki T, Noda T, Yoshimori T (2010) Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway via a novel Rab7‐binding domain. Mol Biol Cell 21: 4162–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Sun Y, Thapa N, Liao Y, Hedman AC, Anderson RA (2015) LAPTM4B is a PtdIns(4,5)P2 effector that regulates EGFR signaling, lysosomal sorting, and degradation. EMBO J 34: 475–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Thapa N, Liao Y, Choi S, Anderson RA (2016) PtdIns(4,5)P2 signaling regulates ATG14 and autophagy. Proc Natl Acad Sci USA 113: 10896–10901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth JT, Gulyas G, Toth DJ, Balla A, Hammond GR, Hunyady L, Balla T, Varnai P (2016) BRET‐monitoring of the dynamic changes of inositol lipid pools in living cells reveals a PKC‐dependent PtdIns4P increase upon EGF and M3 receptor activation. Biochim Biophys Acta 1861: 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Gulyas G, Toth DJ, Sohn M, Sengupta N, Balla T (2017) Quantifying lipid changes in various membrane compartments using lipid binding protein domains. Cell Calcium 64: 72–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicinanza M, Korolchuk VI, Ashkenazi A, Puri C, Menzies FM, Clarke JH, Rubinsztein DC (2015) PI(5)P regulates autophagosome biogenesis. Mol Cell 57: 219–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli‐Daley LA, Lucast L, Gong LW, Liu L, Sasaki J, Sasaki T, Abrams CS, Kanaho Y, De Camilli P (2010) Phosphatidylinositol‐4‐phosphate 5‐kinases and phosphatidylinositol 4,5‐bisphosphate synthesis in the brain. J Biol Chem 285: 28708–28714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Wang J, Sun HQ, Martinez M, Sun YX, Macia E, Kirschhausen T, Albanesi JP, Roth MG, Yin HL (2003) Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP‐1 complexes to the Golgi. Cell 114: 299–310 [DOI] [PubMed] [Google Scholar]
- Wang J, Sun HQ, Macia E, Kirchhausen T, Watson H, Bonifacino JS, Yin HL (2007) PI4P promotes the recruitment of the GGA adaptor proteins to the trans‐golgi network and regulates their recognition of the ubiquitin sorting signal. Mol Biol Cell 18: 2646–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun HQ, Zhu X, Zhang L, Albanesi J, Levine B, Yin H (2015) GABARAPs regulate PI4P‐dependent autophagosome:lysosome fusion. Proc Natl Acad Sci USA 112: 7015–7020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Martina JA, Zewe JP, Wills R, Hammond GRV, Puertollano R (2017) TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat Commun 8: 1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills RC, Goulden BD, Hammond GRV (2018) Genetically encoded lipid biosensors. Mol Biol Cell 29: 1526–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Ysselstein D, Krainc D (2018) Mitochondria‐lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554: 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ (2014) Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife 3: e01612 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5