

Abstract
Various molecular triggers define heterogeneous subsets of gastrointestinal stromal tumors (GISTs), differing in clinical behavior and drug sensitivity. KIT/PDGFRA-wild-type GISTs, including those succinate dehydrogenase (SDH)-deficient, are overall unresponsive to the tyrosine kinase inhibitors commonly used, fostering the development of specific alternative therapeutic strategies. Epigenetic inactivation of O6-methylguanine-DNA methyltransferase (MGMT) through promoter methylation leads to effectiveness of alkylating agents in several human cancers. SDH-deficient GISTs typically feature widespread DNA methylation. However, the actual occurrence of MGMT methylation in these tumors, potentially predisposing them to respond to alkylating drugs, has not been investigated so far. Here we discuss the recent findings concerning the occurrence of MGMT methylation in different GIST subgroups, including SDH-deficient ones, as a premise for a possible reappraisal of alkylating agents specifically targeting these small, otherwise overall chemorefractory, GIST subgroups.
Keywords: DNA methylation, O6-methylguanine-DNA methyltransferase, alkylating agents, gastrointestinal stromal tumor, wild-type gastrointestinal stromal tumor, molecular diagnosis
Comment on: Ricci R, Martini M, Ravegnini G, et al. Preferential MGMT methylation could predispose a subset of KIT/PDGFRA-WT GISTs, including SDH-deficient ones, to respond to alkylating agents. Clin Epigenetics. 2019;11(1):2. doi:10.1186/s13148-018-0594-9. PubMed PMID:30616628; PubMed Central PMCID:PMC6322231. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6322231/.
Introduction
Following decades during which gastrointestinal stromal tumors (GISTs) were not identified as an independent entity due to the lack of proper diagnostic tools, the groundbreaking discovery of the pivotal role of receptor tyrosine kinase KIT, both as molecular trigger and diagnostic marker in these tumors, allowed GISTs to be separated from smooth muscle tumors and other mesenchymal neoplasms.1 Shortly after, the tyrosine kinase inhibitor (TKI) imatinib proved to be dramatically active in a patient affected by unresectable metastatic GIST,2 paving the way for the approval of this drug for treating GIST in record time. However, it was clear, within a short time, that not all GISTs respond to imatinib and that imatinib-responsive tumors eventually develop secondary resistance. In addition, over the years, GIST triggers alternative to KIT have been progressively discovered, and, increasingly, rare molecular defects are still being detected.3 For these reasons, several drugs alternative to imatinib have been tested with the aim of overcoming imatinib resistance, either primary or secondary, in GIST (for reference, a list of drugs considered for the treatment of GIST, including their mechanisms of action, is presented in Table 1). However, coherent with the epoch of the studies, past GIST clinical trials often suffered from the lack of a proper selection of cases on the one hand, and uncommon GIST subsets, possibly ignored at the time of the referred trials, were barely represented among the tested patients on the other. This fact possibly disguised the activity of experimental drugs, which were ineffective on most of the GISTs, on rare GIST types.
Table 1.
Main classes of drugs considered in GIST treatment.
| Class of drug | Drug | Mechanism of action | Reference |
|---|---|---|---|
| Tyrosine kinase inhibitors | Imatinib First line in GISTs |
TKI: It occupies the ATP-binding pocket of KIT/PDGFRA receptor and prevents substrate phosphorylation, thus inhibiting downstream signaling, cellular proliferation, and cell survival. | Quek and George4 |
| Sunitinib Second line in GISTs after imatinib failure |
Multitarget TKI: Similarly to imatinib, it occupies the ATP-binding pocket of KIT/PDGFRA receptor but inhibits also diverse tyrosine kinases including VEGFRs, FLT3, or RET. | Aparicio-Gallego et al5 | |
| Regorafenib Third line in GISTs after imatinib and sunitinib failure |
Multitarget TKI: similarly to sunitinib, it inhibits diverse tyrosine kinases (along with KIT/PDGFRA receptors), including FGFR1, FGFR2, TIE2, DDR2, TrkA, Eph2A, RAF-1, SAPK2, PTK5, and Abl. | Ettrich and Seufferlein6 | |
| Nilotinib | Second-generation KIT and PDGFRA inhibitor derived from imatinib, showing greater in vitro activity against BRC-ABL. Results from one phase I study, two phase II studies, and a compassionate use program showed preliminary evidence of clinical benefit in TKI-refractory GISTs. However, 2 randomized phase III studies have failed to demonstrate significant activity in either the first- or third-line setting. | Serrano and George7 | |
| Sorafenib | Multikinase inhibitor closely related to regorafenib, with activity against KIT and PDGFRA among several other kinases. Tested in GISTs after imatinib and sunitinib failure or even in fourth line. | Serrano and George7 | |
| Linsitinib | IR/IGF1R inhibitor, in trial for advanced WT GIST (trial NCT01560260). | NCI8 | |
| PI3K/AKT/mTOR inhibitors | They inhibit the PI3K/Akt/mTOR pathway, which is highly active in imatinib-resistant GISTs, as a result of secondary mutations in the KIT/PDGFRA kinase domains. Studies showed that mTOR inhibitors have limited success, which may be due to the activation of Akt that occurs following mTORC1 inhibition. Therefore, targeting PI3K or Akt, upstream of mTORC1, may result in a more efficient pathway inhibition. | Patel9 | |
| Heat-shock protein (HSP) inhibitors | HSPs are chaperone proteins that act on numerous so-called “client proteins,” including KIT and PDGFRA, stabilizing protein folding and assembly. | Songdej and von Mehren10 | |
| Demethylating agents | Agents capable of altering epigenetic states, including DNA methylation patterns and histone modification states. Hypermethylation is an attractive target with the aim of influencing tumor biology and overcoming therapy resistance. | Linnekamp et al11 |
ATP, adenosine triphosphate; GIST, gastrointestinal stromal tumor; TKI, tyrosine kinase inhibitor; VEGFR, vascular endothelial growth factor receptor.
Succinate-Dehydrogenase-Deficient GISTs: Let Sleeping Dogs Lie
Succinate dehydrogenase (SDH)-deficient GISTs are rare, accounting for about 5% of GISTs as a whole. Their tumorigenesis hinges on either genomic or epigenetic defects of the heterotetrameric SDH complex, which participates in the Krebs cycle. These defects lead to succinate accumulation, which inhibits prolyl-hydroxylase and TET DNA-hydroxylases, causing hypoxia-inducible factor 1-α (HIF1-α) accumulation and impairment of the conversion of 5-methylcytosine to 5-hydroxymethylcytosine, respectively. These 2 events are oncogenic, on the one hand, through nuclear translocation and induced transcription of through tumorigenic genes by HIF1-α, and, on the other hindrance of DNA demethylation, ultimately leading to widespread DNA methylation.12,13 Clinically, SDH-deficient GISTs are often indolent, even though relatively often displaying metastases, likely because of their metabolic handicap secondary to SDH deficit. Metastases may present as late as 42 years after primary tumors. However, some cases are fatal in a few years, and accumulating data from increasingly extended follow-ups seem to indicate an overall mortality rate as high as 15%.14 In case of clinically aggressive SDH-deficient GISTs, the room for maneuver currently offered to the oncologist is rather limited, as these tumors are usually unresponsive to the TKI therapies commonly used.15,16
State-of-the-Art of Therapy of SDH-Deficient and Other KIT/PDGFRA-Wild-Type GISTs: Bogs and Quicksand
Satisfactory clinical research on pure populations of SDH-deficient GISTs is rare because these tumors are uncommon and their identification is relatively recent.1,17 These events, in conjunction with the commonly extremely slow growth of these tumors, make it very difficult to collect significant data concerning not only the deceptive biology of SDH-deficient GISTs, but also their response to drugs, as time lapses of apparent disease stability could be independent of the latter’s effect.18 Data on SDH-deficient GISTs have been therefore often inferred from cohorts of KIT/PDGFRA-wild-type (WT) GISTs, which are enriched in the former tumors. KIT/PDGFRA-WT GISTs as a whole constitute a pathogenetically heterogeneous subset accounting for about 10% to 15% of GISTs, which, similarly to SDH-deficient GISTs when taken separately, combine an overall indolent behavior with unresponsiveness to the commonly employed TKI in malignant cases.15,16 Based on the biology of these tumors, drugs of potential interest for their treatment include linsitinib, PI3K/AKT/mTOR inhibitors, nilotinib, sorafenib, heat-shock protein inhibitors, chemicals targeting HIF1-α, and demethylating agents.19 Furthermore, different studies have recently shown that epigenetic modifications may represent alternative mechanisms to evade the pharmacologic response.20–22 The growing importance of epigenetics in cancer development has been recently recognized by the European Society of Medical Oncology (ESMO), which included it among the new horizons of soft tissue sarcoma and GIST research (ESMO Sarcoma and GIST Symposium 2018).23 In this context, different trials are evaluating epigenetic therapies as modulators of drug resistance in solid tumors.22 Regarding GIST, in recent years, epigenetic treatments have been proposed as alternative treatments to develop in the future, to bypass TKIs’ resistance.24,25 However, results are still at an early stage and further research will be pivotal in this novel field. In the current landscape, in which a clearly effective therapy for SDH-deficient GISTs is lacking, regorafenib and sunitinib might constitute possible exceptions.18,26
Alkylating Agents’ Ineffectiveness in GISTs: A “Bona Fide” Misdirection?
Over the years, SDH-deficient GISTs have been extensively characterized from a genetic and an epigenetic point of view,14 and a wealth of evidence has markedly corroborated that SDH-deficient GISTs are deeply different from KIT/PDGFRA mutant GISTs. Among the distinguishing features of SDH-deficient GISTs, widespread DNA methylation is the most evident.12,13 SDH-deficient GISTs show a general hypermethylation, which, in turn, promotes the loss of expression of different proteins. The impaired activity of O6-methylguanine-DNA methyltransferase (MGMT) due to promoter methylation allows the effective employment of alkylating agents in human glioma, colorectal cancer, and diffuse large B cell lymphoma.19,27 Given these premises, the hypothesis that DNA methylation could affect MGMT in SDH-deficient GISTs, thereby predisposing them to respond to alkylating agents, appears reasonable. However, does existing evidence support it? Following reports on MGMT in GISTs, which did not consider SDH status,28–31 MGMT methylation and MGMT expression were recently studied for the first time in SDH-deficient GISTs, comparing them with other pathogenetic GIST subsets. Consistent with the above-mentioned expectations, SDH-deficient GISTs were significantly enriched in MGMT-methylated cases, which, in turn, displayed loss of MGMT protein expression, as detected by immunohistochemistry. KIT/PDGFRA-WT GISTs were significantly MGMT-methylated as well; but, unlike SDH-deficient GISTs, this result was not indicated by any particular theoretic premise, except the consideration that SDH-deficient GISTs are the largest, pathogenetically characterized subset of KIT/PDGFRA-WT GISTs, thereby significantly influencing the features of the latter tumors when globally considered. Conversely, MGMT methylation turned out to be a relatively rare event among KIT/PDGFRA mutant GISTs (i.e., the vast majority of GISTs19; Figure 1). Therefore, although confirmatory studies on larger GIST series are warranted, it is most likely that not only SDH-deficient GISTs, but also KIT/PDGFRA-WT ones as a whole (i.e., GISTs commonly resistant to canonical TKIs) often bear an inactivated MGMT, a feature potentially permissive for the efficacy of alkylating agents. What has been the clinical outcome of alkylating agents in GISTs? To the best of our knowledge, the latter approach has been tested in a total of 36 GISTs, enrolled in trials studying temozolomide and carmustine in heterogeneous sarcomas, with negative results.30,32,33 However, these GISTs were neither selected by genotype nor, coherent with the time of the studies, selected and/or investigated for SDH. In addition, the only 2 cases in which MGMT was studied revealed baseline activity, without verifying its residual function after administering its inactivating substrate, O6-benzylguanine.33 On top of that, none of these trials employed Choi criteria, the most sensitive method for assessing GIST response to therapy.34 Therefore, the possible efficacy of alkylating agents in a predisposed fraction of GISTs could have escaped detection due to the lack of a proper case selection added to the rarity of these potentially predisposed tumors and to suboptimal criteria for evaluating tumor response.
Figure 1.
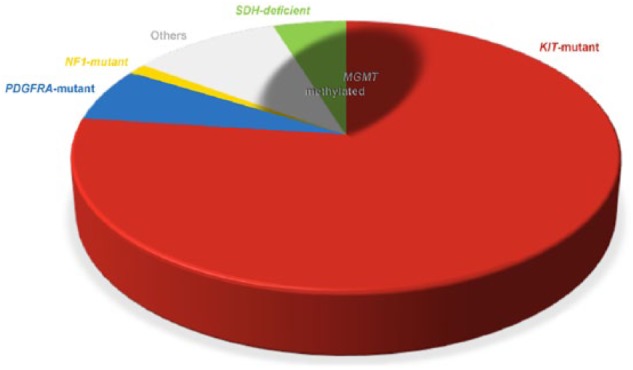
Approximate distribution of MGMT methylation in GISTs, according to pathogenetic type. The pie chart illustrates the distribution of GISTs according to the molecular trigger.1 The shaded area, representing GISTs featuring MGMT methylation, involves about two thirds of SDH-deficient GISTs, one fifth of NF1-associated GISTs, two fifths of the other KIT/PDGFRA-WT GISTs, and slightly more than one twentieth of KIT mutant GISTs; no MGMT-methylated cases have been reported so far among PDGFRA mutant GISTs.
Source: Data from Ricci et al.19
GIST, gastrointestinal stromal tumor; MGMT, O6-methylguanine-DNA methyltransferase; SDH, succinate dehydrogenase.
Future Perspectives
The presented reasoning justified the reappraisal of alkylating agents in GIST therapy to the point that a phase II trial investigating temozolomide in metastatic SDH-deficient GIST was recently launched and is currently underway (ClinicalTrials.gov/NCT03556384). In case of positive results, an important new therapeutic approach will be available to oncologists, further expanding the efficacy of GIST treatment.
Footnotes
Funding:This work was supported by grants from the Università Cattolica del Sacro Cuore (Linea D1 grants nos R4124500212 and R4124500331) to R.R.
Declaration of conflicting interests:R.R. received speaker’s honoraria from Novartis. G.R. declares no conflicts of interest.
Author Contributions: GR and RR wrote and revised the manuscript.
ORCID iDs: Gloria Ravegnini  https://orcid.org/0000-0002-7774-402X
https://orcid.org/0000-0002-7774-402X
Riccardo Ricci
https://orcid.org/0000-0002-9089-5084
References
- 1. Ricci R, Dei Tos AP, Rindi G. GISTogram: a graphic presentation of the growing GIST complexity. Virchows Arch. 2013;463:481–487. [DOI] [PubMed] [Google Scholar]
- 2. Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344:1052–1056. [DOI] [PubMed] [Google Scholar]
- 3. Nannini M, Urbini M, Astolfi A, Biasco G, Pantaleo MA. The progressive fragmentation of the KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GIST). J Transl Med. 2017;15:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Quek R, George S. Update on the treatment of gastrointestinal stromal tumors (GISTs): role of imatinib. Biologics. 2010;4:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aparicio-Gallego G, Blanco M, Figueroa A, et al. New insights into molecular mechanisms of sunitinib-associated side effects. Mol Cancer Ther. 2011;10:2215–2223. [DOI] [PubMed] [Google Scholar]
- 6. Ettrich TJ, Seufferlein T. Regorafenib. Recent Results Cancer Res. 2018;211:45–56. [DOI] [PubMed] [Google Scholar]
- 7. Serrano C, George S. Recent advances in the treatment of gastrointestinal stromal tumors. Ther Adv Med Oncol. 2014;6:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. NCI. Linsitinib in treating patients with gastrointestinal stromal tumors. ClinicalTrials.gov Website. https://clinicaltrials.gov/ct2/show/results/NCT01560260. Published 2017. Up-dated September 21, 2018. Accessed February 28, 2019.
- 9. Patel S. Exploring novel therapeutic targets in GIST: focus on the PI3K/Akt/mTOR pathway. Curr Oncol Rep. 2013;15:386–395. [DOI] [PubMed] [Google Scholar]
- 10. Songdej N, von Mehren M. GIST treatment options after tyrosine kinase inhibitors. Curr Treat Options Oncol. 2014;15:493–506. [DOI] [PubMed] [Google Scholar]
- 11. Linnekamp JF, Butter R, Spijker R, Medema JP, van Laarhoven HWM. Clinical and biological effects of demethylating agents on solid tumours—a systematic review. Cancer Treat Rev. 2017;54:10–23. [DOI] [PubMed] [Google Scholar]
- 12. Ricci R. Syndromic gastrointestinal stromal tumors. Hered Cancer Clin Pract. 2016;14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ricci R, Saragoni L. Everything you always wanted to know about GIST (but were afraid to ask)—an update on GIST pathology. Pathologica. 2016;108:90–103. [PubMed] [Google Scholar]
- 14. Miettinen M, Lasota J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs)—a review. Int J Biochem Cell Biol. 2014;53:514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weldon CB, Madenci AL, Boikos SA, et al. Surgical management of wild-type gastrointestinal stromal tumors: a report from the National Institutes of Health Pediatric and Wildtype GIST Clinic. J Clin Oncol. 2017;35:523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boikos SA, Pappo AS, Killian JK, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors: a report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016;2:922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McWhinney SR, Pasini B, Stratakis CA. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357:1054–1056. [DOI] [PubMed] [Google Scholar]
- 18. Pantaleo MA, Lolli C, Nannini M, et al. Good survival outcome of metastatic SDH-deficient gastrointestinal stromal tumors harboring SDHA mutations. Genet Med. 2015;17:391–395. [DOI] [PubMed] [Google Scholar]
- 19. Ricci R, Martini M, Ravegnini G, et al. Preferential MGMT methylation could predispose a subset of KIT/PDGFRA-WT GISTs, including SDH-deficient ones, to respond to alkylating agents. Clin Epigenetics. 2019;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer. 2014;14:747–753. [DOI] [PubMed] [Google Scholar]
- 21. Knoechel B, Roderick JE, Williamson KE, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salgia R, Kulkarni P. The genetic/non-genetic duality of drug “resistance” in cancer. Trends Cancer. 2018;4:110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frezza AM, Lee ATJ, Nizri E, et al. 2018 ESMO Sarcoma and GIST Symposium: “take-home messages” in soft tissue sarcoma. ESMO Open. 2018;3:e000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nervi C, De Marinis E, Codacci-Pisanelli G. Epigenetic treatment of solid tumours: a review of clinical trials. Clin Epigenetics. 2015;7:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bauer S, Hilger RA, Muhlenberg T, et al. Phase I study of panobinostat and imatinib in patients with treatment-refractory metastatic gastrointestinal stromal tumors. Br J Cancer. 2014;110:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ben-Ami E, Barysauskas CM, von Mehren M, et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann Oncol. 2016;27:1794–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–1354. [DOI] [PubMed] [Google Scholar]
- 28. House MG, Guo M, Efron DT, et al. Tumor suppressor gene hypermethylation as a predictor of gastric stromal tumor behavior. J Gastrointest Surg. 2003;7:1004–1014. [DOI] [PubMed] [Google Scholar]
- 29. Saito K, Sakurai S, Sano T, et al. Aberrant methylation status of known methylation-sensitive CpG islands in gastrointestinal stromal tumors without any correlation to the state of c-kit and PDGFRA gene mutations and their malignancy. Cancer Sci. 2008;99:253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trent JC, Beach J, Burgess MA, et al. A two-arm phase II study of temozolomide in patients with advanced gastrointestinal stromal tumors and other soft tissue sarcomas. Cancer. 2003;98:2693–2699. [DOI] [PubMed] [Google Scholar]
- 31. Okamoto Y, Sawaki A, Ito S, et al. Aberrant DNA methylation associated with aggressiveness of gastrointestinal stromal tumour. Gut. 2012;61:392–401. [DOI] [PubMed] [Google Scholar]
- 32. Garcia del Muro X, Lopez-Pousa A, Martin J, et al. A phase II trial of temozolomide as a 6-week, continuous, oral schedule in patients with advanced soft tissue sarcoma: a study by the Spanish Group for Research on Sarcomas. Cancer. 2005;104:1706–1712. [DOI] [PubMed] [Google Scholar]
- 33. Ryan CW, Dolan ME, Brockstein BB, et al. A phase II trial of O6-benzylguanine and carmustine in patients with advanced soft tissue sarcoma. Cancer Chemother Pharmacol. 2006;58:634–639. [DOI] [PubMed] [Google Scholar]
- 34. Benjamin RS, Choi H, Macapinlac HA, et al. We should desist using RECIST, at least in GIST. J Clin Oncol. 2007;25:1760–1764. [DOI] [PubMed] [Google Scholar]