

Abstract
Cancer cell invasion through physical barriers in the extracellular matrix (ECM) requires a complex synergy of traction force against the ECM, mechanosensitive feedback, and subsequent cytoskeletal rearrangement. PDMS microchannels were used to investigate the transition from mesenchymal to amoeboid invasion in cancer cells. Migration was faster in narrow 3 μm-wide channels than in wider 10 μm channels, even in the absence of cell-binding ECM proteins. Cells permeating narrow channels exhibited blebbing and had smooth leading edge profiles, suggesting an ECM-induced transition from mesenchymal invasion to amoeboid invasion. Live cell labeling revealed a mechanosensing period in which the cell attempts mesenchymal-based migration, reorganizes its cytoskeleton, and proceeds using an amoeboid phenotype. Rho/ROCK (amoeboid) and Rac (mesenchymal) pathway inhibition revealed that amoeboid invasion through confined environments relies on both pathways in a time- and ECM-dependent manner. This demonstrates that cancer cells can dynamically modify their invasion programming to navigate physically confining matrix conditions.
Keywords: Cancer cell invasion, mechanobiology, microchannels, confined migration
Cancer cell invasion is a critical step in primary tumor metastasis. In this process, proliferating cells from a tumor break free from the tumor microenvironment, often led by a single invasive cell, and invade into the surrounding stromal tissue. These cells then travel through diverse physical extracellular matrix conditions, frequently crossing several tissue boundaries before intravasating into a circulatory or lymphatic vessel. During a period of transport and interaction with other cells, surviving invasive cells are carried to capillary beds of distant organs, followed by extravasation into the tissue. This establishes a new tissue microenvironment, allowing for proliferation, angiogenesis, and further spread of the metastatic cancer.1 Although there are key differences,2 the invasion, intravasation, and extravasation processes have much in common, including the utilization of several similar signaling pathways.3 Most importantly, the metastatic process can commence with the invasion of a single cancer cell through narrow confinements in the surrounding extracellular matrix (ECM).4 Second and third harmonic intravital microscopy has revealed that individual cancer cells can traverse tracks and spaces within healthy tissue, including myofibers, collagen networks, fat tissue, and perineural tracks. In these tracks, cells were found to have diameters as low as 2 μm and speeds of around 0.25 μm per minute.5 Invading cells follow tracks of great length (>750 μm) compared to the diameter of a spread cell (∼50 μm), suggesting that sustained linear confinement, distinct from pores, is a common physical environment for invading cancer cells.5 Recently, confocal laser endomicroscopy has been used in vivo to identify similar channels with sub-10 μm diameters and lengths exceeding 150 μm in the interstitial space of a number of tissues.6 Thus, synthetic approaches to monitoring cells moving through confined spaces have great relevance in the field of cancer invasion.
Assays aimed at understanding cancer cell invasion in vitro exist along two continuums: from population-level analysis to single-cell analysis and from random confinement dimensions to well-defined dimensions (Figure 1A). Currently, the most widely adopted standard for analyzing quasi-three-dimensional confined cancer cell invasion is the Boyden chamber assay, in which populations of cells migrate through stiff pores with defined dimensions (ranging from 3 to 8 μm in diameter and 6 to 10 μm in length) into the opposite region.7 However, this assay is best suited for understanding bulk invasion, and it is difficult to observe migration on a single-cell level. Other three-dimensional (3D) assays utilize cell-permeable 3D matrix-like collagen gels8 or Matrigel,9 but cells in these gels can be difficult to image and the physical passages cells traverse are not uniform. “Pinch-point” assays can follow single-cell behavior by utilizing narrow confinements in which the length of the passage is less than the diameter of a spread cell (∼50 μm).10,11 Boyden chamber assays, in which membrane thickness is usually between 6 and 10 μm, also fall under this category.12 As pinch-point assays allow the cell to be partially “in” and partially “out” of the channel,13 they can mimic the short confinements encountered during intravasation and extravasation. Alternatively, longer channel lengths are more faithful reproductions of long ECM tracks found in vivo.5,6 Microchannel systems address this by allowing for the analysis of cancer invasion events down to a single cell in a 3D channel with defined micron-scale dimensions and have become increasingly utilized.14−18
Figure 1.

Microchannels as tools for understanding cancer cell confined migration. (A) Microchannels are both well-defined with respect to passage dimension and conducive to single-cell analysis. Parallel plate assays are capable of single-cell analysis, but cells are unconfined in two dimensions. Network invasion assays and Boyden chamber assays follow populations of cells as they invade a hydrogel (undefined pore sizes) or porous membrane (defined pore sizes), but are inconvenient for single-cell analysis. (B) The PDMS microchannel chip has 150 μm passage lengths, 11 μm heights, and are either 10 μm (wide) or 3 μm (narrow) in diameter. (C) A panel of four breast cancer cell lines and four colon cancer cell lines were analyzed in microchannels. Proportions of cells observed penetrating (blue), invading (orange), or permeating (green) are shown. In cases in which no cells interacted with the channels, gray bars are displayed. N = 408, 87, 102, 60, 64, 28, 32, 0, 10, 0, 200, 18, 71, 46, 7, and 17 cells. (D) Average cell speed during channel permeation for cell lines capable of 10 and 3 μm permeation. N = 32, 12, 11, 9, 15, 10, 8, and 19 cells from left to right, with individual cells represented as data points. Error bars represent 95% confidence intervals. (* p < 0.05, ** p < 0.01, *** p < 0.001, t test: MDA-MB-231 t = 2.126, DF = 23, HS578T t = 3.23, DF = 18, BT549 t = 2.126, DF = 23).
Few studies have explored cancer cell invasion through long, confining microchannels in the absence of a chemoattractant or pressure gradient. The migration of Panc-1 pancreatic cancer cells through fibronectin-coated microchannels was found to be heavily dependent on keratin phosphorylation state, with the reorganization of keratin around the nucleus leading to an enhancement of cell deformability and an increase in cell permeation and invasion speed.15 This was supported by similar findings on the role of intermediate filament organization on contact guidance in Panc-1 cells.19 MDA-MB-231 invasion through confined microchannels was shown to induce a change in migratory phenotype,16 but questions remain about the cytoskeletal alterations that drive this change.
Several recent investigations have made the connection between confined cancer cell migration and the mesenchymal-to-amoeboid transition (MAT).20 Distinct from the well-known epithelial-to-mesenchymal transition (EMT), MAT is the spontaneous switch from adhesive, focal adhesion-dependent mesenchymal cell migration to poorly adhesive, contractility-dependent amoeboid cell migration.21 This transition has been induced in vitro via a number of mechanisms, including Rho activation,22 inhibition of matrix metalloproteinases,23 and altering integrin turnover.24 Mechanical induction of MAT has also been demonstrated with the use of parallel plates, both with rigid glass25 and softer hydrogels.26 Both of these studies allowed for direct observation of the MAT, gleaning important information about the role of integrins, focal adhesion proteins, and force generation. In both cases, confinement was imposed quickly onto cells, as opposed to allowing cells to impose confinement autonomously upon themselves as invading cancer cells would in vivo. This rapid mechanical deformation may induce cellular responses that are not as efficient or natural as those produced when the cell can gradually impose confinement upon itself. Both myofiber and perineural tracks have been measured with intravital imaging and found to be <5 μm in diameter on average,5 suggesting that the characteristic levels of confinement for amoeboid induction are physiologically common in the tumor microenvironment.
To model this environment, we utilize long, narrow microchannels in conjunction with a chemoattractant-free system, recapitulating the mechanical microenvironment that cancer cells face once they have left the primary tumor and are invading through ECM tracks. By comparing eight different cell lines from two different tissue origins, we demonstrate the utility of microchannels as a screening tool for invasive behavior in cancer cells. We make observations of cells transitioning from a mesenchymal phenotype to an amoeboid phenotype of their own volition, completely free of external chemotactic or mechanical stimuli. Using chemical inhibitors for several pathways theorized to play a role in this transition, we illustrate how these microchannels can be used as a key tool in understanding and addressing the MAT in cancer cells. As amoeboid phenotype cancer cells have been shown to be up to 20 times more invasive and markedly more resistant to chemotherapy than mesenchymalphenotype cancer cells,27 this platform can play an important role in future clinically relevant studies.
Channels were fabricated in a two-step photolithographic process as described previously.15 This process yields a square-shaped chip with 11 μm-high channels with lengths of 150 μm spanning 200 μm-high media reservoirs (Figure 1B). With widths of 3 or 10 μm, the channels have rectangular cross sections with cross-sectional areas of 33 or 110 μm2. As these chips are open to the incubator environment (i.e., not sealed microfluidic chips), hydrostatic pressure can be assumed to be identical on both sides, as the height of the media column is equal. The 150 μm length requires cells to completely enter the channel in order to move to the other side. No chemoattractant was supplied to any experimental conditions in order to avoid any confounding effects of chemotaxis as opposed to natural exploratory invasive cancer cell behavior.
Previous reports have suggested that the ability of cancer cells to move through confined spaces may be predictive of metastatic potential.28 Accordingly, we used eight cancer cell lines from either breast or colon tissue (Table S1, Supporting Information) to investigate their ability to migrate in confinement. Of the eight cell lines, all but one were capable of migrating through 10 μm wide channels, while only four lines were observed migrating through the narrow 3 μm channels (Figure 1C). In this panel of cell lines, the fastest migration was found in the three invasive breast cancer lines (MDA-MB-231, HS 578T, and BT 549) (Figure 1D). Of the three colon cancer cell lines, only one (HCT8) was observed moving through both 10 and 3 μm channels. Interestingly, in three of the four cell lines capable of moving through wide and narrow confinements, migration speed through the narrow channels was faster than through the wide channels, despite the 70% reduction in cross-sectional area (Figure 1D). This increase in invasion speed is consistent with a previous report of confinement-enhanced migration speed in immune cells.29
To better characterize cancer cell–microchannel interactions, three previously developed metrics of channel interaction behavior15 were used. In this system, “penetrative” cells are those which extend projections into the channel but do not fully enter, “invasive” cells are those that fully enter the channel and either remain there or turn around and exit where they entered, and “permeative” cells are those that enter the channel and exit the opposite side. In MDA-MB-231 breast cancer cells, the percentage of cells observed permeating channels decreased as confinement increased from 10 to 3 μm, suggesting that cells have a harder time moving through sustained levels of extreme confinement (Figure 2A). However, the percentage of invasive cells also decreased in 3 μm channels, revealing that once cells fully enter a narrow channel, they are more “committed” to moving through to the other side than those traversing wide channels (Figure 2A). These observations were confirmed by plotting the distance of the leading edge of the cell against time (Figure 2B) in both 10 and 3 μm channels. These traces show that in 10 μm channels, cells exhibit a much higher amount of “back-and-forth” behavior, with a higher frequency of leading edge retraction and stalling than in 3 μm channels (Figure 2C).
Figure 2.

Channel permeation dynamics of MDA-MB-231 breast cancer cells. (A) The average number of MDA-MB-231 cells observed penetrating, invading, or permeating a single microchannel in 24 h. N = 28 for 10 μm channels and 21 for 3 μm channels. Error bars represent 95% confidence intervals. (** p < 0.01, **** p < 0.0001, ANOVA, F = 11.11, DF = 108). (B) Plot of the position of the leading edge of MDA-MB-231, BT 549, and HS578T breast cancer cells vs time during microchannel permeation. N = 28 for 10 μm channels and 21 for 3 μm channels. N = 55 for 10 μm and 30 for 3 μm. (C) The proportion of “steps” taken backward, or toward the channel entrance, during the entire permeation process. Each data point represents a single cell. Cells that only moved forward during permeation are observed on the x-axis. N = 55 for 10 μm and 30 for 3 μm. Error bars represent 95% confidence intervals. (*** p < 0.001, t test, t = 4.081, DF = 83). (D, E) Kymographs illustrating representative cell dynamics during permeation in 3 μm (D) and 10 μm (E) microchannels. Time proceeds from top to bottom and the cells enter the microchannel on the left and exit on the right. Distinct phases of exploration (blue), nuclear entrance (orange), and permeation (green) are identified. Scale bar = 1 h (lower right).
This fast, smooth, and committed migratory behavior in narrow channels is also evident in kymographs, which reveal three distinct phases of cell permeation through 3 μm channels (Figure 2D). The first phase, here called “exploration”, is the initial projection of cell body, in some cases more than 100 μm long, into the channel. During the second phase, the nucleus and most of the cell body enters the channel, which is observable in phase contrast images as a marked increase in light intensity. Following this, the reorganization phase sees the cell sitting at the channel entrance almost entirely stationary. Finally, the permeation phase happens abruptly and quickly, with the entire cell moving through the 150 μm channel in <3 h. This demarcated process can be contrasted with kymographs of cell permeation through 10 μm channels, in which cell movement is slower, exhibits more back-and-forth movement, and cannot be broken down into distinct phases (Figure 2E).
As previous investigations into different forms of confined migration have identified a transition from mesenchymal style invasion to amoeboid style invasion, we looked at the cells exiting channels for signs of amoeboid invasion. While this MAT is still little understood, one classic morphological marker of amoeboid invasion is blebbing at the leading edge of the cell. Indeed, in all of the cell lines permeating 3 μm channels, bleb-like structures were observed upon exit (Figure 3A) (Videos S1–S4). Accordingly, we searched for additional characteristics of mesenchymal or amoeboid invasion in wide 10 μm and narrow 3 μm channels. Focal adhesions, as assessed by paxillin immunocytochemistry, were found to be more pronounced and localized in cells permeating 10 μm channels, while cells in narrow 3 μm channels showed diffuse paxillin expression throughout the cell body (Figure 3B). Using laser scanning confocal microscopy, the actin cytoskeleton was imaged in both channel dimensions. In 10 μm channels, cells displayed robust stress fibers both inside and outside of the channels (Figure 3C, Figure S1). Cells interacting with 3 μm channels exhibited stress fibers outside the channels, but inside the channels the cell body showed a loss of F-actin organization (Figure 3D, Figure S2). Strong blebbing was observed inside narrow channels, with blebs found at both the leading and trailing edge of the cell, although the number and size of blebs at the leading edge were higher (Figure 3E).
Figure 3.

Cytoskeletal characterization during channel invasion reveals hallmarks of mesenchymal and amoeboid invasion. (A) Bleb structures are observed in phase contrast in all four permeative cell lines upon channel exit. Scale bars = 10 μm. (B) Immunofluorescence staining in fixed cells reveals punctate focal adhesions in cells transiting 10 μm channels (left), but a loss of paxillin expression and organization in cells transiting 3 μm channels (right). Scale bars = 10 μm. (C, D) Maximum intensity projections of laser scanning confocal image stacks of fixed cells in 10 and 3 μm channels. Strong F-actin is evident in 10 μm channels, while 3 μm channels display bleb structures at the forward and rear ends of the cell and little perinuclear actin expression. Scale bars = 3 μm. (E) Individual slices through the height of the channel show diverse blebs at the leading edge. Scale bars = 3 μm.
The live-cell actin dye SiR-Actin was used in conjunction with laser scanning confocal microscopy to better understand actin dynamics in both types of channels. In 10 μm channels, cells displayed F-actin stress fibers while moving wholly in the channel (Figure 4A, Video S5). In 3 μm channels, cell body projections were initially observed up to 100 μm into the channel, mirroring observations made in Figure 2D. After this exploration phase was complete, and while the bulk of the cell body was still outside of the channel, the cytoskeleton underwent a drastic change and lost its stress fibers in a time period of around 2 h (Figure 4B,C, Video S6).
Figure 4.

Actin and integrin organization during microchannel permeation. (A–C) SiR-Actin live cell imaging of MDA-MB-231 breast cancer cells in 10 and 3 μm-wide microchannels. Strong F-actin organization is observed in 10 μm channels and at the periphery of 3 μm channels before permeation occurs (A and B, insets). After protruding over 100 μm into the narrow microchannel, the cytoskeleton at the periphery of the 3 μm channel is reorganized (C, inset). Scale bars = 10 μm, 1 μm in insets. (D–G) Maximum intensity projections of laser scanning confocal image stacks of (D–G) actin and (D′–F′) integrin β1 localization in fixed cells during various states of permeation. Unconfined (G′) and 10 μm channel (D′) cells exhibit strong integrin β1 localization at the cell–matrix interface. Cells exploring 3 μm channels (E′) exhibit similar patterns, but once cells are fully confined in the 3 μm channels (F′), integrin localization at the cell–matrix interface is greatly reduced. Scale bars = 10 μm. (H) The average integrin β1 intensity measured in saponin-permeabilized cells from all four conditions. N = 18, 17, 7, and 30. Error bars represent 95% confidence intervals. (* p < 0.05, *** p < 0.001, **** p < 0.0001. ANOVA, F = 18.08, DF = 71).
Amoeboid movement in human cells has been shown to be an integrin-independent process, allowing cells to move on surfaces that do not engage integrins.30 Using saponin-permeabilized cells, we analyzed the localization of β1 integrin in MDA-MB-231 cells and found it comparable in 10 μm channels to that in unconfined cells (Figure 4D,E,H). For cells in 3 μm channels, integrin localization remains similar to that in 10 μm channels during the exploration phase (Figure 4F,H). Once the cell has completed the reorganization phase and is fully confined in the 3 μm channel, integrin localization is lost (Figure 4G,H).
To test whether ECM binding was a requirement for channel permeation, we analyzed cell permeation through 10 and 3 μm channels with and without collagen functionalization. As plasma-activated PDMS is capable of adsorbing proteins found in serum (Figure 5A), we performed uncoated channel experiments with and without serum. The rate and speed of MDA-MB-231 permeation through 10 μm channels were reduced when uncoated channels were used with full-serum media, but interestingly, no 10 μm channel permeation was observed through uncoated channels in serum-free media (Figure 5B,C), mirroring the low levels of migration seen on the flat glass surfaces outside the channels (Video S7). Indeed, cells on the flat glass surface at the edge of the channels remained rounded and were unable to spread in the absence of serum proteins. However, a small number of cells were able to permeate narrow 3 μm channels in the absence of both collagen and serum proteins, proving that matrix adhesion is dispensable for confined migration (Figure 5D, Video S8). The finding that 10 μm permeation occurs less than 3 μm permeation lends evidence to the theory that adhesion-independent confined migration is a friction-dependent “chimneying” process, one which requires confinement to be severe enough that outward forces generate sufficient friction to provide traction for forward movement.20,26,30
Figure 5.

ECM adhesion is required in wide channels but not in narrow channels. (A) Collagen immunofluorescence staining in channels that have been functionalized with collagen (left), exposed to 10% FBS in cell culture media for 24 h (center), and exposed to serum free cell culture media for 24 h (right). Scale bars = 10 μm. (B) The number of cells observed permeating channels functionalized with collagen, FBS, or no protein. Permeation was not observed in protein-free 10 μm channels. Error bars represent 95% confidence intervals. N = 28, 21, 10, 16, 9, and 8 from left to right. (* p < 0.05, ANOVA, F = 13.71, DF = 141). (C) Average cell speed during channel permeation. N = 32, 12, 7, 22, 0, and 1 from left to right. Error bars represent 95% confidence intervals. (*** p < 0.001, ANOVA, F = 6.602, DF = 69). (D) Phase contrast, background subtracted images of a cell permeating a serum-free channel (cell moves from left to right).
Interplay between the RhoGTPases Rac and Rho are crucial for generalized cell migration,31 and antagonistic signaling between Rac and Rho has been shown to play a role in the MAT.32 Chemical inhibitors for Rac1 (NSC23766) and p160ROCK (Y27632) were applied to MDA-MB-231 cells to better understand their role in self-induced confined migration. We found that Rac1 inhibition completely eliminated 10 μm permeation, as expected due to its role in the primarily mesenchymal invasion observed in wide channels. However, Rac inhibition also abrogated 3 μm permeation, which exhibits more amoeboid characteristics (Figure 6A). This is likely due to the mesenchymal nature of the exploration phase of 3 μm permeation, in which the cell extends projections into the channel. Furthermore, Rac1 inhibition completely eliminated invasion into the 3 μm channels, as no cellular projections were observed inside the narrow channels. Alternatively, p160ROCK inhibition did not prevent cell permeation in 3 or 10 μm channels, but the percentage of cells only invading the channels, but not fully permeating, increased. This suggests that fewer cells were able to make the transition from Rac1-driven invasion to Rho-dependent permeation (Figure 6B). Cell speed was negatively affected in both conditions, underlining the role of ROCK in generation of actomyosin force for both mesenchymal and amoeboid movement. The sustained ability of ROCK-inhibited cells to traverse 3 μm channels, which we believe to be a contractility-dependent process, is likely due to the compensatory role of other contractile cytoskeletal components, including microtubules and intermediate filaments, both of which have been shown to play a role in self-induced confined migration.15,16
Figure 6.

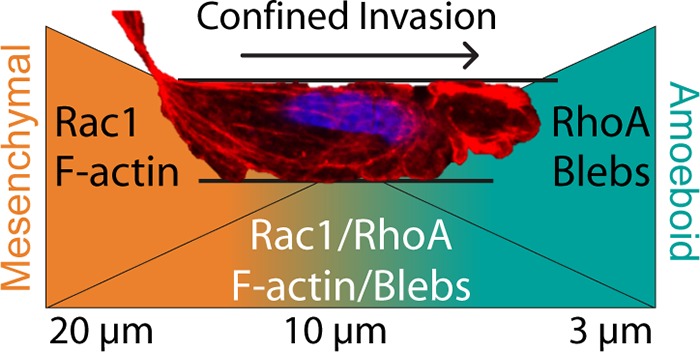
The MAT is a dynamic interplay between Rac and Rho signaling. (A) Proportional representation of cells penetrating, invading, or permeating 10 or 3 μm microchannels. (B) Average cell speed during channel permeation. N = 32, 12, 8, and 4 for WT-10, WT-3, Y27632-10, and Y27632-3, respectively. Error bars represent 95% confidence intervals. (*** p < 0.001, ANOVA, F = 13.97, DF = 52). (C) A model for gradual transition from mesenchymal-to-amoeboid migration as confinement level increases. At nonconfining channel widths >20 μm, cells exclusively utilize mesenchymal migration. As channel diameter decreases (∼10 μm), cells remain able to utilize mesenchymal machinery (F-Actin, focal adhesions), but can exhibit blebbing in regions where the cell completely fills the channel. At extreme levels of confinement (∼3 μm), cells fully transition to amoeboid migration. The image shown is a cell moving through a 10 μm channel displaying both F-actin bundles and blebbing at the leading edge.
Characteristics of Amoeboid Invasion Are Apparent in Narrow Microchannels
Invasive cancer cells are capable of an adhesion-dependent migration pattern known as mesenchymal motility. In this invasion modality, actomyosin force generated against the ECM is transmitted and transduced at large, integrin-rich focal adhesions, with leading edge focal contacts serving as positive feedback with regards to the invasion direction.33 It is thought that MMP activity is a prerequisite for mesenchymal motility in vivo, and thus many efforts to combat invasion have focused on decreasing MMP levels. However, some particularly insidious types of cancer cells are capable of modifying their invasion modality to overcome this loss of MMP activity. Single cancer cells can change their invasive programming and shift from adhesion-dependent mesenchymal motility to adhesion-independent amoeboid motility. To achieve this, cortical actin contraction leads to hydrostatic pressure gradients, causing changes in cell shape and an enhancement in the ability of the cell to squeeze through physical gaps in the ECM.34 During this process, focal adhesions remain small and diffuse. This plasticity of migration presents a challenge for future cancer metastasis treatments, and thus direct observation of the MAT is invaluable to the development of clinical strategies. Here, we present evidence that MATs happen in confined microchannels and can be recognized via increases in cell velocity, the appearance of blebs, the loss of focal adhesion and integrin localization, and the dramatic reorganization of the cytoskeleton.
We observed marked increases in cell velocity in narrow 3 μm channels compared to wider 10 μm channels (Figure 1D). Increases in cell velocity as a function of confinement have been seen in several studies, both in microchannels35 and in other confinement assays.25 Increases in cell migration velocities in tightly confined spaces have been related to constant volume flow: As the cross-sectional area decreases, velocity must increase commensurately.10 Our observations of cells in narrow 3 μm channels support this assertion, as position traces indicate smooth, consistent migration profiles for tightly confined cells (Figure 2B). Other studies have found that cell volume can change within channels, a phenomena tied to aquaporin-regulated osmosis changes.17
Another key sign of amoeboid invasion is a loss of punctate focal adhesion localization, which is likely related to lower importance of integrin-mediated matrix attachment in amoeboid cells. Here, we observe far fewer mature focal adhesions in narrow channels compared to wide channels, both in exploring cells and permeating cells. Focal adhesion protein expression was still observed, but in a diffuse cytoplasmic pattern, suggesting that focal adhesion components are not downregulated on an expression level, or simply that invasion occurs so quickly that gene expression dynamics are not reflected. Additionally, we saw a marked reduction in integrin localization at the cell–matrix interface when cells were completely within narrow microchannels. However, when the cell body remained mostly outside the narrow channels during the exploration phase, integrin localization was observed at similar levels as in wide channels. Together, this suggests that cells exploring a narrow channel will engage their integrins, but do not actively assemble focal adhesions. Then, once the cell fully enters the channel, integrins become dispensable and are not observed at the cell-ECM interface.
This dispensability was confirmed by observing cells traversing narrow microchannels that had not been coated with ECM protein. As serum within culture media can adsorb to PDMS and provide ECM proteins to anchor to, we performed these experiments in serum-free media. While cells outside the channels displayed a rounded morphology and did not appear to bind to the surface (Video S7), those that entered the narrow microchannels were able to quickly move from one side to the other with a similar velocity as those moving through ECM-coated channels. Interestingly, this phenomena was only observed in narrow 3 μm channels, not in wider 10 μm channels, giving credence to the concept of “chimneying”, in which a cell exerts outward force, providing friction against which it can propel forward.36 The characteristic width for chimneying behavior may be cell line- or even cell size-dependent, as amoeboid-style migration was observed in some cell lines in 10 μm channels (Video S9).
The transition from mesenchymal to amoeboid invasion is accompanied by a loss of F-actin stress fibers.16 We were able to directly observe this cytoskeletal reorganization, which took around 2 h and was complete once the cell had extended projections over 100 μm into the channel. Another cytoskeletal sign of amoeboid invasion was the appearance of blebs, regions where the cell membrane detaches from the cortex as a result of hydrostatic pressure. Blebs were markedly increased in 3 μm channels and observed in all invasive cell types. These blebs are a hallmark of amoeboid cell invasion,21 which are also observed in response to confinement in Dictyostelium cells.37 Taken together, these cell behavior changes and morphological patterns strongly suggest that cells undergo a MAT without any external chemical or mechanical stimuli.
While there is no protein “marker” for amoeboid invasion, the biochemical pathways implicated in this switch were also analyzed. Rac1 plays a large role in lamellipodia protrusion and mesenchymal migration.38,39 ROCK is a downstream effector of RhoA, which promotes stress fiber formation and actomyosin contractile force necessary for blebbing and amoeboid migration.39,40 Y27632-mediated ROCK inhibition has been shown to prevent the switch from mesenchymal to amoeboid invasion,41 while inactivation of Rac1 is sufficient to drive amoeboid movement.39 We found that both Rac1 and p160ROCK play a role in self-induced MAT, with Rac1 responsible for generating initial protrusions into the confined space and the Rho/ROCK pathway providing contractile force necessary for the high cell speeds observed in narrow channels.
Reorganization Is a Dynamic Process Requiring Hours, Not Minutes
While the connection between confinement and the MAT has been examined, questions about the dynamic nature of this process remain. Previous studies have also observed a “stalling” effect at the entrance of pinch-point channels with a cross-sectional area below 30 μm2, concluding that nuclear steric hindrance serves as an initial barrier to invasion.10 Kymographs of cells entering narrow 3 μm channels reveal a similar stalling effect, which we have called the “exploration period” (Figure 2D). Indeed, previous studies have observed similar distinct exploration phases in dendritic cells undergoing confined migration.42 While our observations of increased migration speed within more tightly confined environments are in agreement with this observation, we have also observed cells in which extreme nuclear deformation into the channel occurs, while the nucleus is still in the middle region of the cell, as opposed to the trailing edge where it can be observed in pinch-point studies10 (Figure 4F). We speculate that this is a function of channel design: As pinch-point channels are shorter than the length of a spread cell, they allow the cell to protrude through the channel, build focal adhesions, and generate force on the opposite side, all while the nucleus remains “left behind”. This actomyosin contractility-dependent process has been observed in channels as long as 50 μm.13 As this process can occur relatively quickly, nuclear segregation is the likely result. Spring-like nuclear behavior in which the leading edge moves with a constant velocity but the trailing edge stalls then “bounces back”10 also suggests that cells actively pull their nucleus through “pinch-point” obstructions, as opposed to cells in long channels that must reorganize their cytoskeleton to successfully traverse. Indeed, recent work has shown that DNA damage and nuclear envelope rupture occurs as cancer cells pull their nuclei through pinch-points, with mobile repair factor segregation and envelope repair complex localization observed.11,12 Thus, our findings are most relevant for understanding cancer cell migration through the interstitial matrix surrounding tumors, not in the short barrier penetration that occurs during intravasation and extravasation, which may be more closely modeled as pinch-point events.
Here, we show that the exploration and reorganization phases of cancer cell invasion into microchannels can take as long as 10 h, after which cells move quickly and efficiently to the opposite side of the channel. This is in contrast with parallel plate experiments that impose confinement onto cells in a matter of seconds. While these assays do yield clear confinement-dependent MATs, they also result in a number of “stalled” cells that did not polarize or move in an amoeboid fashion.25 It is possible that in cells that have already formed strong focal adhesions, the disassembly process under confinement is suboptimal, preventing the cytoskeletal rearrangement that happens during the reorganization phase of microchannel permeation.
Mesenchymal-to-Amoeboid Transition Is Likely a Continuous Gradient, Not a Binary Switch
While mesenchymal invasion is most common in 10 μm channels and amoeboid invasion is most common in 3 μm channels, some cells in 10 μm channels display signs of both mesenchymal and amoeboid invasion simultaneously (Figure 6C). Previous studies using very long microchannels (500 μm) also observed similar phenomena, with two cells within the same channel exhibiting opposite invasion phenotypes.43 Within parallel confinements, Dictyostelium cells have been observed migrating with a combination of blebs and pseudopods.37 Fibroblasts in similar confinements were found to have mesenchymal morphology in all conditions, but the percentage of mesenchymal cells decreased as confinement increased.25 Furthermore, for nearly all cell lines and channel width conditions, the distribution of average cell speed appears slightly bimodal, suggesting that subsets of the cells may display more mesenchymal phenotypes, while others more efficiently switch to amoeboid migration (Figure S3). Thus, we propose a gradient model of MAT (Figure 6C) in which loose confinement results in nearly complete mesenchymal invasion and extreme confinement results in nearly complete amoeboid invasion. In between these extremes, the likelihood of observing one or the other depends on how close the confinement is to either extreme. While it is logical to speculate that these extremes are dictated by intrinsic cellular properties, both biological and mechanical, further research is necessary to unravel the contribution of each.
Motivating Migration: Chemotaxis, Haptotaxis, and Contact Guidance
One persistent question proceeding from microchannel invasion studies is what exactly drives cancer cells to enter a confined space and traverse it. With several notable exceptions,15,16 most microchannel invasion studies have utilized chemoattractants to stimulate cell migration.11,13,17,37,44,45 Chemotaxis requires the activation of a number of chemokine receptors and the subsequent initiation of chemotactic pathways. Thus, exploratory cell migration, in the absence of any chemoattractant, is likely a distinct migratory phenomenon from chemotactic migration. Indeed, the magnitude of either soluble or bound chemokines in vivo is difficult to determine,46 and in the ECM niche our system replicates (stiff tracks in healthy ECM extending hundreds of microns),5,6 chemokine gradients may be too low for invading cancer cells to detect. Interstitial measurements of oxygen and pH gradients in both tumors and healthy tissue reveal extremely heterogeneous patterns within tumors, including some regions with very low gradient magnitudes, as well as a general pattern that gradient magnitude is higher in tumor perivascular space than in healthy perivascular space.47 The ability of cells to form self-directed chemotactic gradients by degrading basal media components has been explored recently,48 with evidence that self-generated gradients result in migratory waves and long-range directed migration.49 This phenomenon is worth considering in the context of microchannels, as the great reduction in cross-sectional area encountered when moving into the channel causes a concordant decrease in media volume surrounding the cell. Thus, it is conceivable that invading cells quickly degrade nutrients in the microchannel, causing a positive chemotactic stimulus through the channel. It must be noted, however, that in this study (Video S10) and others,35 cells fully within narrow microchannels have been observed changing direction, which is not compatible with a self-directed chemotaxis model.
Self-directed chemotaxis has also been shown to be augmented by a polarized sequestration of receptor ligands.50 This is in agreement with the proposal of the “Osmotic Engine Model”, in which polarized Na+/H+ pumps and aquaporin channels result in a net flux of water through the cell, yielding cell movement in the absence of actin polymerization and actomyosin contractility.17 These observations were made in a chemotaxis-based microchannel model. Indeed, experiments performed without a chemotactic gradient found that cell migration in narrow microchannels was over 60% slower, with the researchers speculating that the chemoattractant gradient may help cells effectively polarize ion pump distribution.17 Furthermore, aquaporin expression has been shown to increase cancer cell migration in unconfined environments, with aquaporin localization polarized in lamellipodia at the leading edge of collectively migrating cells, effectively responding to a chemotactic gradient.51 As the experimental setup in this study does not utilize any chemoattractant gradient, the osmotic engine likely plays a smaller role.
Haptotaxis, or directed migration toward areas with higher concentrations of substrate-bound ligands, is another potential driver of self-induced confinement that has been found to be relevant for cancer cells in vivo.52 As cells are capable of engaging all four walls of the channel (Figure S2), the local concentration of bound ligands would be higher inside a narrow 3D channel than on the 2D surfaces outside the channels. This local increase in integrin-binding sites provides a stimulus for cell polarization, which has been shown to be dependent on local ligand concentration gradients.53 Furthermore, 1D topographical cues have been shown to drive polarization in a process that mirrors the cues presented by 3D ECM fiber bundles. Thus, when a cell makes a random protrusion into the channel, the channel forces the protrusion to continue in a unidirectional fashion, which results in a change in polarity and drives further protrusions.54 However, the eventual escape from channels would provide a reversion back to 2D surface ligands, meaning that haptotaxis alone also cannot explain the entire process of microchannel permeation.
Contact guidance refers to the ability of topographical features of the matrix to direct cell migration.55 It has been observed in cancer cells19 and been found to influence amoeboid migration in T-lymphocytes.56 In this case, the planar intersections at the interface between glass and PDMS provide a degree of topographical stimulus for cells emerging from the channels. This would explain how, in many cases, cells emerging from the channels will follow the PDMS–glass interface perpendicular to the channel (Video S11). Ultimately, it is likely that a complex combination of chemotaxis, haptotaxis, and contact guidance all contribute to self-induced cancer cell permeation, and future work focused on preventing confined migration will need to identify and isolate key pathways required for cancer cell invasion.
Acknowledgments
We thank Tina Wiegand and Dr. Rebecca Medda for microscopy assistance. This work was supported by Max Planck Institute Postdoctoral Fellowships (to A.W.H.), the 2016 AACR Basic Cancer Research Fellowship, Grant Number 16-40-01-HOLL (to A.W.H.), European Research Council Grant 294852 (to J.P.S.), the Federal Ministry of Education and Research of Germany and the Max Planck Society (to J.P.S.), the excellence cluster CellNetworks at the University of Heidelberg (to J.P.S.), and the MWK Baden-Württemberg Innovative Projekte (MigChip) (to J.P.S. and R.K.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.8b04720.
Video S1: Phase contrast time lapse imaging of an MDA-MB-231 cell permeating a 3 μm channel (AVI)
Video S2: Phase contrast time lapse imaging of an HS578T cell permeating a 3 μm channel (AVI)
Video S3: Phase contrast time lapse imaging of a BT549 cell permeating a 3 μm channel (AVI)
Video S4: Phase contrast time lapse imaging of an HCT-8 cell permeating a 3 μm channel (AVI)
Video S5: Maximum intensity projection of a confocal time lapse image of an SiR-Actin labeled MDA-MB-231 cell moving through a 10 μm channel (AVI)
Video S6: Maximum intensity projection of a confocal time lapse image of an SiR-Actin labeled MDA-MB-231 cell moving through a 3 μm channel (AVI)
Video S7: Phase contrast time lapse imaging of MDA-MB-231 cells in a protein-free PDMS chip with 10 μm widths in 0% FBS media (AVI)
Video S8: Phase contrast time lapse imaging of MDA-MB-231 cells in a protein-free PDMS chip with 3 μm widths in 0% FBS media (AVI)
Video S9: Phase contrast time lapse imaging of an MCF-7 cell in a 10 μm channel (AVI)
Video S10: Phase contrast time lapse imaging of MDA-MB-231 cells in a 10 μm channel (AVI)
Video S11: Phase contrast time lapse imaging of MDA-MB-231 cells in a 3 μm channel displaying contact guidance along the edges of the chip after exiting (AVI)
Materials and methods and supplementary figures (PDF)
Author Contributions
⊥ These authors contributed equally to this work. A.H., R.K., and J.S. conceived the project and designed the experiments. A.H., N.G.K.D., and K.C. conducted experiments and analyzed data. A.H., A.F., and T.S. performed SiR-Actin optimization, imaging, and processing. A.H., R.K., and J.S. wrote the manuscript. A.H. made the figures. A.H., N.G.K.D, K.C., A.F., T.S., R.K., and J.S. commented on and/or edited the manuscript and figures.
The authors declare no competing financial interest.
Supplementary Material
References
- Fidler I. J. Nat. Rev. Cancer 2003, 3 (6), 453–458. 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- Duffy M. J.; McGowan P. M.; Gallagher W. M. Journal of Pathology 2008, 214 (3), 283–293. 10.1002/path.2282. [DOI] [PubMed] [Google Scholar]
- Spano D.; Heck C.; De Antonellis P.; Christofori G.; Zollo M. Semin. Cancer Biol. 2012, 22 (3), 234–249. 10.1016/j.semcancer.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Quintana E.; Shackleton M.; Sabel M. S.; Fullen D. R.; Johnson T. M.; Morrison S. J. Nature 2008, 456 (7222), 593–598. 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelin B.; Bakker G.-J.; Friedl P. IntraVital 2012, 1 (1), 32–43. 10.4161/intv.21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benias P. C.; Wells R. G.; Sackey-Aboagye B.; Klavan H.; Reidy J.; Buonocore D.; Miranda M.; Kornacki S.; Wayne M.; Carr-Locke D. L.; Theise N. D. Sci. Rep. 2018, 8 (1), 4947. 10.1038/s41598-018-23062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngalim S. H.; Magenau A.; Le Saux G.; Gooding J. J.; Gaus K. J. Oncol. 2010, 2010 (170), 1–7. 10.1155/2010/363106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P.; Sahai E.; Weiss S.; Yamada K. M. Nat. Rev. Mol. Cell Biol. 2012, 13 (11), 743–747. 10.1038/nrm3459. [DOI] [PubMed] [Google Scholar]
- Benton G.; Arnaoutova I.; George J.; Kleinman H. K.; Koblinski J. Adv. Drug Delivery Rev. 2014, 79–80, 3–18. 10.1016/j.addr.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Lautscham L. A.; Kämmerer C.; Lange J. R.; Kolb T.; Mark C.; Schilling A.; Strissel P. L.; Strick R.; Gluth C.; Rowat A. C.; Metzner C.; Fabry B. Biophys. J. 2015, 109 (5), 900–913. 10.1016/j.bpj.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denais C. M.; Gilbert R. M.; Isermann P.; McGregor A. L.; Lindert M.; Weigelin B.; Davidson P. M.; Friedl P.; Wolf K.; Lammerding J. Science 2016, 352 (6283), 353–358. 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irianto J.; Xia Y.; Pfeifer C. R.; Athirasala A.; Ji J.; Alvey C.; Tewari M.; Bennett R. R.; Harding S. M.; Liu A. J.; Greenberg R. A.; Discher D. E. Curr. Biol. 2017, 27 (2), 210–223. 10.1016/j.cub.2016.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge M. T.; Egelhoff T. T.; Baskaran H. Biomed. Microdevices 2010, 12 (3), 543–553. 10.1007/s10544-010-9411-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huda S.; Pilans D.; Makurath M.; Hermans T. M.; Kandere Grzybowska K.; Grzybowski B. A. Adv. Mater. Interfaces 2014, 1 (7), e1400158 10.1002/admi.201470044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolli C. G.; Seufferlein T.; Kemkemer R.; Spatz J. P. PLoS One 2010, 5 (1), e8726 10.1371/journal.pone.0008726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzer E. M.; Tong Z.; Paul C. D.; Hung W.-C.; Stroka K. M.; Boggs A. E.; Martin S. S.; Konstantopoulos K. FASEB J. 2012, 26 (10), 4045–4056. 10.1096/fj.12-211441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroka K. M.; Jiang H.; Chen S.-H.; Tong Z.; Wirtz D.; Sun S. X.; Konstantopoulos K. Cell 2014, 157 (3), 611–623. 10.1016/j.cell.2014.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergert M.; Erzberger A.; Desai R. A.; Aspalter I. M.; Oates A. C.; Charras G.; Salbreux G.; Paluch E. K. Nat. Cell Biol. 2015, 17 (4), 524–529. 10.1038/ncb3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holle A. W.; Kalafat M.; Ramos A. S.; Seufferlein T.; Kemkemer R.; Spatz J. P. Sci. Rep. 2017, 7, 45152. 10.1038/srep45152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluch E. K.; Aspalter I. M.; Sixt M. Annu. Rev. Cell Dev. Biol. 2016, 32 (1), 469–490. 10.1146/annurev-cellbio-111315-125341. [DOI] [PubMed] [Google Scholar]
- Friedl P.; Alexander S. Cell 2011, 147, 992. 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Nimnual A. S.; Taylor L. J.; Bar-Sagi D. Nat. Cell Biol. 2003, 5, 236. 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- Wolf K.; Mazo I.; Leung H.; Engelke K.; von Andrian U. H.; Deryugina E. I.; Strongin A. Y.; Bröcker E.-B.; Friedl P. J. Cell Biol. 2003, 160 (2), 267–277. 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P. A. J.; Caswell P. T.; Doyle B.; Iwanicki M. P.; Tan E. H.; Karim S.; Lukashchuk N.; Gillespie D. A.; Ludwig R. L.; Gosselin P.; Cromer A.; Brugge J. S.; Sansom O. J.; Norman J. C.; Vousden K. H. Cell 2009, 139 (7), 1327–1341. 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- Liu Y.-J.; Le Berre M.; Lautenschlaeger F.; Maiuri P.; Callan-Jones A.; Heuzé M.; Takaki T.; Voituriez R.; Piel M. Cell 2015, 160 (4), 659–672. 10.1016/j.cell.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Yip A. K.; Chiam K.-H.; Matsudaira P. Integr. Biol. 2015, 7 (10), 1196–1211. 10.1039/C4IB00245H. [DOI] [PubMed] [Google Scholar]
- Vig N.; Rahman M.; Gammon L.; Peyric E.; Mackenzie I. Lancet 2017, 389, S97. 10.1016/S0140-6736(17)30493-2. [DOI] [Google Scholar]
- Hendrix M. J.; Seftor E. A.; Seftor R. E.; Fidler I. J. Cancer Lett. 1987, 38 (1–2), 137–147. 10.1016/0304-3835(87)90209-6. [DOI] [PubMed] [Google Scholar]
- Jacobelli J.; Friedman R. S.; Conti M. A.; Lennon-Dumenil A.-M.; Piel M.; Sorensen C. M.; Adelstein R. S.; Krummel M. F. Nat. Immunol. 2010, 11 (10), 953–961. 10.1038/ni.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malawista S. E.; de Boisfleury Chevance A. Proc. Natl. Acad. Sci. U. S. A. 1997, 94 (21), 11577–11582. 10.1073/pnas.94.21.11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K.; Doughman R. Nat. Cell Biol. 2006, 8 (8), 781–782. 10.1038/ncb0806-781. [DOI] [PubMed] [Google Scholar]
- Paňková K.; Rosel D.; Novotný M.; Brábek J. Cell. Mol. Life Sci. 2010, 67 (1), 63–71. 10.1007/s00018-009-0132-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E. Curr. Opin. Genet. Dev. 2005, 15 (1), 87–96. 10.1016/j.gde.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Sabeh F.; Shimizu-Hirota R.; Weiss S. J. J. Cell Biol. 2009, 185 (1), 11–19. 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia D. Methods Cell Biol. 2014, 121, 141–153. 10.1016/B978-0-12-800281-0.00010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malawista S. E.; de Boisfleury Chevance A.; Boxer L. A. Cell Motil. Cytoskeleton 2000, 46 (3), 183–189. 10.1002/1097-0169(200007)46:3<183::AID-CM3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Ibo M.; Srivastava V.; Robinson D. N.; Gagnon Z. R. PLoS One 2016, 11 (10), e0163866 10.1371/journal.pone.0163866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A. J.; Paterson H. F.; Johnston C. L.; Diekmann D.; Hall A. Cell 1992, 70 (3), 401–410. 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V.; Gadea G.; Ahn J.; Paterson H.; Marra P.; Pinner S.; Sahai E.; Marshall C. J. Cell 2008, 135 (3), 510–523. 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- Amano M.; Chihara K.; Kimura K.; Fukata Y.; Nakamura N.; Matsuura Y.; Kaibuchi K. Science 1997, 275 (5304), 1308–1311. 10.1126/science.275.5304.1308. [DOI] [PubMed] [Google Scholar]
- Sahai E.; Marshall C. J. Nat. Cell Biol. 2003, 5, 711. 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- Faure-André G.; Vargas P.; Yuseff M.-I.; Heuzé M.; Diaz J.; Lankar D.; Steri V.; Manry J.; Hugues S.; Vascotto F.; Boulanger J.; Raposo G.; Bono M.-R.; Rosemblatt M.; Piel M.; Lennon-Dumenil A.-M. Science 2008, 322 (5908), 1705–1710. 10.1126/science.1159894. [DOI] [PubMed] [Google Scholar]
- Irimia D.; Toner M. Integrative Biology 2009, 1 (8–9), 506–512. 10.1039/b908595e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Z.; Balzer E. M.; Dallas M. R.; Hung W.-C.; Stebe K. J.; Konstantopoulos K. PLoS One 2012, 7 (1), e29211 10.1371/journal.pone.0029211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaw K. C.; Manimaran M.; Tay F. E. H.; Swaminathan S. Microvasc. Res. 2006, 72 (3), 153–160. 10.1016/j.mvr.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Roussos E. T.; Condeelis J. S.; Patsialou A. Nat. Rev. Cancer 2011, 11 (8), 573–587. 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger G.; Yuan F.; Dellian M.; Jain R. K. Nat. Med. 1997, 3 (2), 177–182. 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- Tweedy L.; Knecht D. A.; Mackay G. M.; Insall R. H. PLoS Biol. 2016, 14 (3), e1002404 10.1371/journal.pbio.1002404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedy L.; Susanto O.; Insall R. H. Curr. Opin. Cell Biol. 2016, 42, 46–51. 10.1016/j.ceb.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Boldajipour B.; Mahabaleshwar H.; Kardash E.; Reichman-Fried M.; Blaser H.; Minina S.; Wilson D.; Xu Q.; Raz E. Cell 2008, 132 (3), 463–473. 10.1016/j.cell.2007.12.034. [DOI] [PubMed] [Google Scholar]
- Hu J.; Verkman A. S. FASEB J. 2006, 20 (11), 1892–1894. 10.1096/fj.06-5930fje. [DOI] [PubMed] [Google Scholar]
- Gritsenko P. G.; Ilina O.; Friedl P. Journal of Pathology 2012, 226 (2), 185–199. 10.1002/path.3031. [DOI] [PubMed] [Google Scholar]
- Smith J. T.; Elkin J. T.; Reichert W. M. Exp. Cell Res. 2006, 312 (13), 2424–2432. 10.1016/j.yexcr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Petrie R. J.; Doyle A. D.; Yamada K. M. Nat. Rev. Mol. Cell Biol. 2009, 10 (8), 538–549. 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira A. I.; Abrams G. A.; Bertics P. J.; Murphy C. J.; Nealey P. F. J. Cell Sci. 2003, 116 (10), 1881–1892. 10.1242/jcs.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K.; Müller R.; Borgmann S.; Bröcker E.-B.; Friedl P. Blood 2003, 102 (9), 3262–3269. 10.1182/blood-2002-12-3791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.