

Abstract
The generation of terminally differentiated cell lineages during organogenesis requires multiple, coordinated cell fate choice steps. However, this process has not been clearly delineated, especially in complex solid organs such as the pancreas. Here, we performed single‐cell RNA‐sequencing in pancreatic cells sorted from multiple genetically modified reporter mouse strains at embryonic stages E9.5–E17.5. We deciphered the developmental trajectories and regulatory strategies of the exocrine and endocrine pancreatic lineages as well as intermediate progenitor populations along the developmental pathways. Notably, we discovered previously undefined programs representing the earliest events in islet α‐ and β‐cell lineage allocation as well as the developmental pathway of the “first wave” of α‐cell generation. Furthermore, we demonstrated that repressing ERK pathway activity is essential for inducing both α‐ and β‐lineage differentiation. This study provides key insights into the regulatory mechanisms underlying cell fate choice and stepwise cell fate commitment and can be used as a resource to guide the induction of functional islet lineage cells from stem cells in vitro.
Keywords: cell fate choice, fate map, MAPK/ERK, pancreatic lineage, single‐cell RNA‐seq
Subject Categories: Development & Differentiation, Methods & Resources, Systems & Computational Biology
Introduction
In complex organs, the generation of a single lineage usually involves multiple steps of cell fate choice. Comprehensively, understanding the pathways of cell lineage differentiation during in vivo development, especially regulatory strategies at points of cell lineage segregation, is critical for directing stem cell differentiation into the desired cell types for regenerative medicine. However, deciphering the precise pathways of multiple‐step cell fate choices and the regulatory logic underlying the generation of complex organs requires further investigation.
The pancreas is a digestive organ with both exocrine and endocrine functions. The exocrine compartment consists of acinar and ductal cells, and the endocrine portion includes β, α, δ, ε, and PP cells clustered in the pancreatic islets. During embryogenesis, all pancreatic lineages arise from multipotent progenitor (MP) cells. The developmental potential of these progenitors is restricted in a stepwise manner, ultimately resulting in the generation of the exocrine and islet endocrine lineages (Pan & Wright, 2011; Shih et al, 2013; Bastidas‐Ponce et al, 2017; Larsen & Grapin‐Botton, 2017). In mice, the pancreatic anlagen is detected in a dorsal definitive endoderm domain as early as embryonic day 8.5 (E8.5) and in a ventral endoderm domain approximately half a day later (Gittes, 2009). The early MP cells express the key transcription factors (TFs), PDX1 and PTF1A (Burlison et al, 2008), which are regulated by other TFs such as SOX9, HNF1β, and FOXA1/2 (Haumaitre et al, 2005; Lynn et al, 2007; Gao et al, 2008). The development and maintenance of MP cells are regulated by several cell signaling pathways, including the Notch, FGF10, and BMP pathways (Bhushan et al, 2001; Tiso et al, 2002; Hald et al, 2003; Murtaugh et al, 2003; Norgaard et al, 2003).
After the pancreatic anlagen is established, the rapid growth of MP cells and cell shape changes drive the formation of nascent pancreatic buds at E9.5–E10.5. Thereafter, the pancreatic buds undergo dramatic morphological changes, resulting in multiple branched protrusions. The cells in the “tip” domain (“tip” cells) express the marker genes Ptf1a, c‐Myc, and Cpa1 (Zhou et al, 2007; Pan et al, 2013). The inner cells constituting the “trunk” domain (“trunk” cells) are marked by Sox9, Nkx6.1, Hes1, Hnf1β, Hnf6, and Glis3 expression (Jacquemin et al, 2000; Solar et al, 2009; Schaffer et al, 2010; Kopinke et al, 2011; Kopp et al, 2011; Kang et al, 2016). Genetic tracing studies have revealed that tip cells are quickly restricted into an acinar lineage, whereas trunk cells are bipotent progenitors of ductal and endocrine progenitor cells (Zhou et al, 2007; Solar et al, 2009; Kopinke et al, 2011; Kopp et al, 2011; Pan et al, 2013). Two reciprocally repressive TFs, PTF1A and NKX6.1, are master regulators of tip–trunk segregation. PTF1A promotes tip fate by repressing trunk formation, whereas NKX6.1 induces trunk fate by inhibiting the generation of tip domains (Schaffer et al, 2010). The Notch signaling pathway is critical in promoting the trunk program (Horn et al, 2012).
Starting from approximately E12.0, endocrine progenitors are specified from the bipotent trunk epithelium. Neurogenin 3 (Ngn3) is a master regulator of endocrine lineage specification, and all endocrine lineages are generated from Ngn3‐expressing progenitors (Gu et al, 2002; Schonhoff et al, 2004). A first wave of Ngn3‐expressing cells can be detected in the dorsal endoderm from E9.0 to E11.0, and the first differentiated endocrine cells at this stage are mainly glucagon‐producing α cells (first wave of α cell, also named α‐1st; Larsson, 1998). A second wave of Ngn3+ cell specification occurs in both the dorsal and ventral pancreas beginning at approximately E12.0, peaking at E15.5, and decreasing abruptly thereafter (Gradwohl et al, 2000; Schwitzgebel et al, 2000). A high level of Notch signaling represses Ngn3 expression but enhances ductal cell differentiation. Wnt, sphingosine‐1‐phosphate (S1p), and epidermal growth factor receptor (EGFR) positively regulate endocrine specification (Baumgartner et al, 2014; Lof‐Ohlin et al, 2017; Serafimidis et al, 2017).
The early‐specified endocrine progenitors express low level of Ngn3 (Ngn3low cell). The expression of Ngn3 subsequently increases in these cells (Ngn3high cell), resulting in cell cycle exit (Gu et al, 2002; Desgraz & Herrera, 2009; Miyatsuka et al, 2011; Azzarelli et al, 2017; Krentz et al, 2017), delamination from the epithelium, and activation of key TFs for endocrine development, such as NeuroD1, Pax4, Arx, Isl1, Rfx6, Insm1, and Myt1 (Petri et al, 2006; Pan & Wright, 2011; Mastracci & Sussel, 2012). The cells expressing these factors progressively differentiate into a specific islet cell lineage. Two TFs, PAX4 and ARX, play reciprocally repressive roles in regulating β‐ vs α‐lineage diversification, respectively (Collombat et al, 2003).
Many factors and cell signaling pathways that regulate various stages of pancreatic development have been revealed using classic genetic approaches. However, due to the technical difficulty of purifying the intermediate progenitor cells and their direct progeny at each juncture of the cascade of lineage choices from developing pancreatic tissue, the underlying regulatory logic has remained elusive. Here, using single‐cell transcriptomic analysis and various genetically labeled mouse strains, we comprehensively studied the precise developmental pathways of the early pancreatic lineages in vivo.
Results
Isolation of cell lineages from fetal pancreas for sc‐RNA‐seq
To comprehensively define a fate map of pancreatic lineage differentiation, we isolated pancreatic cells from different transgenic or knock‐in mouse strains from E9.5 to E17.5 via fluorescence‐activated cell sorting (FACS) and performed single‐cell RNA‐sequencing (sc‐RNA‐seq) analysis using Smart‐seq2 technology (Picelli et al, 2014). We employed the Pdx1‐GFP transgenic mouse line (Gu et al, 2004) to purify pancreatic progenitor cells from E9.5 to E11.5 (Figs 1A and EV1A and B, and Dataset EV1). In our previous study, we sequenced 126 single Pdx1‐GFP+ cells from E9.5 or E10.5 dorsal pancreas using Smart‐seq2 (Li et al, 2018), and the data were remined here (Figs 1A and EV1A, and Dataset EV1). We used a Pdx1‐Cre; Rosa‐RFP line (Hingorani et al, 2003) to isolate all pancreatic lineages on each day from E10.5 to E15.5 (Figs 1A and EV1A and B, and Dataset EV1). To avoid the omission of cell types caused by genetic labeling, our analysis included recently published datasets of unbiased enriched pancreatic lineage cells with negative selection using a blood cell marker Tie2 and an endothelial marker CD45 (Tie2−CD45−) at E13.25 or E15.25 (Sznurkowska et al, 2018; Figs 1A and EV1A, and Dataset EV1). Because the percentage of endocrine cells in the pancreas is relatively low, to enrich for endocrine lineages, we used an Ngn3‐GFP knock‐in mouse strain (Lee et al, 2002) to sort cells expressing GFP at lower (Ngn3‐GFPlow) and higher (Ngn3‐GFPhigh) levels from E13.5 to E15.5 (Figs 1A and EV1A and B, and Dataset EV1). The single‐cell datasets generated in our previous study (108 single Ngn3‐GFPlow and Ngn3‐GFPhigh cells at E13.5; Yu et al, 2018) were also included in this study (Figs 1A and EV1A, and Dataset EV1). At an earlier developmental stage (E12.5) and later stages (E16.5 and E17.5), because Ngn3‐GFPlow and Ngn3‐GFPhigh cells could not be effectively separated by FACS (Fig EV1B), we generally sorted Ngn3+ cells for single‐cell analyses (Figs 1A and EV1A and B, and Dataset EV1).
Figure 1. Identification of fetal pancreatic cell types.
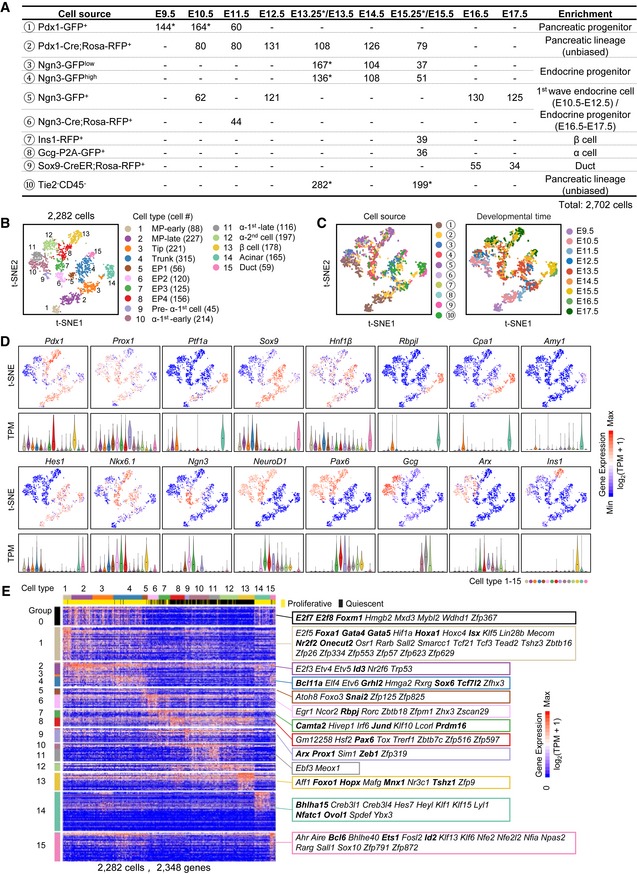
- Overview of the 2,702 fetal pancreatic cells analyzed in this study. The numbers show the cell counts from the indicated mouse strains at different developmental time points. The mouse strains (cell sources) are numbered with the circled numbers. * represents the indicated population including the cells from published resources (see Fig EV1A for details).
- The t‐SNE plot shows 15 distinct cell types. Each dot represents a single cell. Cell counts are labeled in brackets.
- The t‐SNE plots show the enriched cell source (left), the circled number indicating the cell source labeled in (A), and developmental time (right).
- Expression levels of marker genes are projected onto t‐SNE plots. The colors ranging from blue to red indicate low to high relative gene expression levels. The violin plot under the t‐SNE plot shows the expression level (TPM) of the indicated gene in each cell type. The dot within each violin plot indicates the median of expression levels.
- Heat map of cell type‐enriched genes. Each column represents a single cell, and each row represents one gene. Cell cycle‐related genes were extracted as group‐0. TFs of each gene group are labeled. The bolded TFs are known to be important for pancreas development. The colors ranging from blue to red indicate low to high relative gene expression levels.
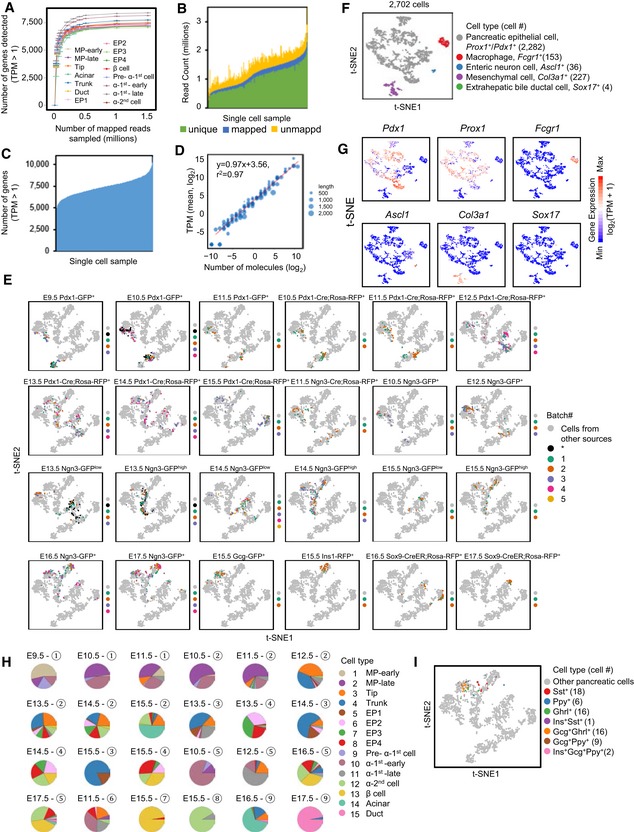
Figure EV1. The strategy for enriching pancreatic lineages from mouse embryos.

- Overview of the 2,702 fetal pancreatic cells analyzed in this study. The numbers separated by slashes represent the number of cells produced in each experiment. § represents the cells from Pdx1‐GFP highly expressing cells, *represents the single‐cell datasets from published resources (GEO: GSE86225; Li et al, 2018), **represents the single‐cell datasets from published resources (GEO: GSE84324; Yu et al, 2018), and ***represents the single‐cell datasets from published resources (GEO: GSE89798; Sznurkowska et al, 2018).
- FACS gating strategies for purifying pancreatic cells at multiple developmental stages from various mouse strains. The pancreatic tissues from WT embryos were used as negative controls (upper panel). The dashed line in the “E10.5 Pdx1‐GFP” gating plot separates the cells with higher GFP expression from cells with lower expression.
- Generation of the Gcg‐P2A‐GFP strain by inserting P2A and GFP DNA sequences before the stop codon.
- Immunofluorescence staining of the GCG in 8‐week‐old mouse islets verified the high quality of the Gcg‐P2A‐GFP strain. Scale bars: 50 μm.
To obtain differentiated α cells, we generated a Gcg‐P2A‐GFP mouse strain (Fig EV1C) in which α‐cell identity was validated by immunostaining of glucagon in islets (Fig EV1D) and by sc‐RNA‐seq of E17.5 GFP+ cells from this strain. Principal component analysis (PCA) showed that these cells clustered with the E17.5 α cells sequenced in our previous study (Qiu et al, 2017a; Fig EV1E). Therefore, this Gcg‐P2A‐GFP mouse strain can be used to label α cells.
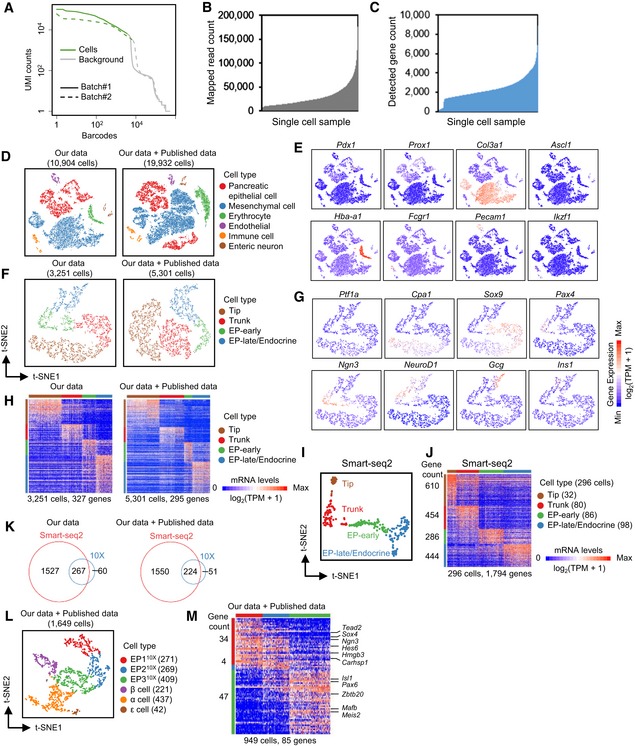
From Gcg‐P2A‐GFP embryos, we sorted the early‐stage α‐2nd cells at E15.5. We employed an Ins1‐RFP line (Piccand et al, 2014) to sort the early‐stage differentiated β cells at E15.5 (Figs 1A and EV1A and B, and Dataset EV1). We enriched pancreatic ductal cells using Sox9‐CreER; Rosa‐RFP mice (Kopp et al, 2011) and collected RFP+ cells at E16.5 and E17.5 (Figs 1A and EV1A and B, and Dataset EV1). To investigate the characteristics of the earlier‐differentiated endocrine cells, also known as “first wave” of endocrine cells, we purified Ngn3‐GFP+ cells at E10.5 and the descendants of Ngn3‐expressing cells by sorting Ngn3‐Cre; Rosa‐RFP+ cells at E11.5 (Schonhoff et al, 2004), when Ngn3 expression was turned off (Figs 1A and EV1A and B, and Dataset EV1). To estimate the technical noise in the sc‐RNA‐seq experiments, we applied an ERCC spike‐in control (Brennecke et al, 2013). A total of 2,702 transcriptomes of single cells passed the quality control criteria. On average, 6,000–9,000 genes were detected in each cell, with > 0.2 million mapped reads (Fig EV2A–D and Dataset EV1). The cells at each time point were pooled from multiple embryos, and at least two biological replicates were performed to generate all single‐cell transcriptomic data (Fig EV1A and Dataset EV1). The batch effects between these replicates were not obvious (Fig EV2E).
Figure EV2. Quality control analysis of sc‐RNA‐seq data generated by Smart‐seq2.
-
ASaturation analysis indicated that 0.2 million mapped reads were sufficient to cover most of the genes detected in a single cell. Each point represents the mean of four independent subsamplings. Error bars: SEM.
-
B, CStatistical analysis of the alignment (B) and detected genes (C) for available single‐cell samples. Each bar represents one sample.
-
DCorrelations between the number of mRNA molecules and TPM values. Each point represents an ERCC spike‐in. Point size reflects transcript length.
- E
-
Ft‐SNE analysis identified nonpancreatic and pancreatic epithelial lineage cells. The color code, marker gene, and cell count for each cell type are provided next to the t‐SNE plot.
-
GExpression patterns of marker genes in (F).
- H
-
ICells expressing Sst, Ghrl and Ppy (TPM > 10,000) are projected onto the t‐SNE plot of Fig 1B.
Identification of cell types in fetal pancreatic development
To identify cell types among the sequenced single cells, we performed a t‐distributed stochastic neighbor‐embedding (t‐SNE) analysis (Satija et al, 2015). After excluding cells expressing the macrophage marker gene Fcgr1 (Gautier et al, 2012), the enteric neuron marker gene Ascl1 (Memic et al, 2016), the mesenchymal cell marker gene Col3a1 (Byrnes et al, 2018), and the extrahepatic bile ductal cell marker gene Sox17 (Spence et al, 2009; Fig EV2F and G, and Dataset EV1), we identified fifteen distinct cell types (clusters 1–15) among the remaining 2,282 cells (Figs 1B and C, and EV2H, and Dataset EV1), which generally expressed the pancreatic marker genes Pdx1 and/or Prox1 (Figs 1D and EV2G).
The cells in cluster‐1 mainly consisted of cells from E9.5 Pdx1‐GFP embryos and expressed pancreatic progenitor feature genes such as Pdx1, Sox9, and Hnf1β, but not the endocrine cell marker gene NeuroD1 (Fig 1B–D). Therefore, the cells in cluster‐1 were considered MP‐early cells. The cluster‐2 cells primarily included E10.5 and E11.5 pancreatic cells from Pdx1‐GFP or Pdx1‐Cre; Rosa‐RFP embryos and expressed Ptf1a, Sox9, and Hnf1β at high levels (Fig 1B–D). Therefore, the cells in cluster‐2 were MP‐late cells. The cluster‐3 cells were primarily obtained at E11.5–E14.5 and expressed Ptf1a, Rbpjl, and Cpa1 at high levels but not the acinar marker gene Amy1, which indicated that the cluster‐3 cells were tip cells (Fig 1B–D). The cluster‐4 cells were RFP+ cells from Pdx1‐Cre; Rosa‐RFP embryos and Ngn3‐GFPlow cells at E12.5‐E16.5. These cells showed high expression levels of Sox9, Nkx6.1, and Hes1 but not Ptf1a expression. Based on this information, we concluded that the cluster‐4 cells were trunk cells (Fig 1B–D). The cluster‐5 cells mainly contained Ngn3‐GFPlow and a fraction of Ngn3‐GFPhigh cells, and the cluster‐6 and cluster‐7 cells mainly consisted of Ngn3‐GFPhigh cells at E13.5–E15.5 (Fig 1B and C). Ngn3 expression was upregulated in the cluster‐5 cells and peaked in the cluster‐6 and cluster‐7 cells (Fig 1D). We concluded that the cells in cluster‐5, cluster‐6, and cluster‐7 represented continuous developmental stages of endocrine precursor (EP) cells. We therefore named these populations EP1, EP2, and EP3, respectively. In cluster‐8, Ngn3 expression was decreased, whereas the expression levels of the Ngn3 downstream genes NeuroD1 and Pax6 were maintained at high levels. In addition, islet hormone genes were not expressed in these cells (Fig 1B–D). Taken together, the results supported the identification of the cluster‐8 cells as late‐stage EP cells (EP4). The cluster‐9 cells expressed the endocrine and α‐cell feature TFs NeuroD1 and Arx but not Gcg and included cells at E9.5–E10.5 (Fig 1B–D), indicating that these cells were early‐specified “first wave” of α cells (pre‐α‐1st cells). The cluster‐10 and cluster‐11 cells expressed the α‐cell markers Gcg and Arx and included many E10.5–E13.5 cells (Fig 1B–D), indicating that these cells were differentiated α‐1st cells but at different developmental stages. We therefore named the cluster‐10 and cluster‐11 cells α‐1st‐early and α‐1st‐late, respectively. Notably, a fraction of α‐1st cells also expressed Ins1 but at a much lower level than in later differentiated β cells (Fig 1D). This result is consistent with a previous finding that the earliest endocrine cells express polyhormones (Larsson, 1998). The other Gcg + Arx + cells were located in cluster‐12, and because E15.5 Gcg‐GFP+ cells were found in this cluster (Fig 1B–D), we referred to these cells as “second wave” of α cells (α‐2nd cells). The cluster‐13 cells were clearly β cells because they included E15.5 Ins1‐RFP+ cells and expressed a high level of the Ins1 gene (Fig 1B–D). The cluster‐14 cells were acinar cells because they exclusively expressed the differentiated acinar markers Amy1 and showed a high expression level of Rbpjl and Ptf1a (Fig 1B–D). The cluster‐15 cells were primarily isolated from E17.5 Sox9‐CreER; Rosa‐RFP embryos, in which Cre recombinase expression was induced by tamoxifen injection 1.5 days before embryo harvest. After E15.5, Sox9 expression is gradually restricted to differentiated ductal cells in the pancreas (Fig 1B–D). Therefore, the cluster‐15 cells were identified as ductal cells. In the EP4, α‐2nd, and β‐cell populations, we also detected some cells expressing Sst (18 cells), Ghrl (16 cells), Ppy (6 cells), or polyhormonal genes (28 cells; Fig EV2I). However, due to the insufficient cell number, these cells were not identified as specific cell clusters. To characterize the transcriptomic features of each cluster, we identified the genes showing enriched expression in each cell cluster and extracted cell cycle‐related genes (group‐0 in Fig 1E) through hierarchical clustering analysis. These genes were used to distinguish proliferative cells (Fig 1E and Dataset EV2). Collectively, these comprehensive single‐cell transcriptomic analyses identified all pancreatic lineages and their major intermediate progenitors.
The cell throughput of the Smart‐seq2 method was typically lower than that of the most recent droplet‐based sc‐RNA‐seq technologies, such as 10X Genomics, which can sample thousands of cells on a microfluidic chip (Pijuan‐Sala et al, 2018). To verify whether the number of cells in our study was sufficient to identify the cell subpopulations of pancreatic lineages and the differentially expressed genes, we performed sc‐RNA‐seq using the 10X Genomics platform on E14.5 whole mouse dorsal pancreas. After filtering of low‐quality cells (Fig EV3A), 10,904 cells were retained for further analyses. On average, more than 2,000 genes were detected in each cell, with ~3 × 104 mapped reads (Fig EV3B and C, and Dataset EV1). After t‐SNE analysis, groups expressing markers of mesenchymal cells, erythrocytes, endothelial cells, immune cells, or neurons were excluded from downstream analyses (Fig EV3D and E). Finally, a cluster containing 3,251 cells that generally expressed Prox1 was identified as pancreatic epithelial lineage cells (Fig EV3D and E). Focusing on this cluster, we performed another round of t‐SNE analysis and identified four groups representing tip cells (1,280 cells), trunk cells (793 cells), the early stage of EP cells (EP‐early) (551 cells), and the late stage of EP cells (EP‐late)/endocrine cells (627 cells) based on the expression patterns of the TFs Ptf1a, Cpa1, Sox9, Ngn3, NeuroD1, and Pax4 and the hormone genes Gcg and Ins1 (Fig EV3F and G). From 3,251 single‐cell datasets produced by 10X Genomics, we identified 327 genes specifically enriched in each cell cluster (Fig EV3H and Dataset EV3). We also included the datasets of E14.5 pancreata generated using the 10X Genomics platform from a recently published work (Byrnes et al, 2018). Although the number of cells increased, the number of differentially expressed genes detected between the four cell populations did not increase (Fig EV3D, F, H, and Dataset EV3), indicating that for 10X Genomics, a cell number of 2,000–3,000 should be “saturated” for identifying differentially expressed gene sets. Similarly, t‐SNE analysis of the E14.5 sequencing datasets generated by Smart‐seq2 (296 cells) was performed and revealed four cell clusters representing tip (32 cells), trunk (80 cells), EP‐early (86 cells), and EP‐late/endocrine (98 cells) cells (Fig EV3I). However, 1,794 cluster‐enriched genes were identified in the 296 Smart‐seq2 single‐cell datasets (Fig EV3J and Dataset EV3). Notably, approximately 80% of the differentially expressed genes identified by 10X Genomics overlapped with the differentially expressed genes identified by Smart‐seq2 (Fig EV3K). Therefore, our analyses using Smart‐seq2 efficiently identified more cell population‐specific expressed genes, thereby enabling us to characterize the features and map the trajectory of pancreatic lineage development.
Figure EV3. Comparison of E14.5 pancreatic sc‐RNA‐seq data between analyses using Smart‐seq2 and the 10X Genomics platform.
-
ABarcode‐labeled cells (green) were identified using the cellranger program (10X Genomics). The y‐axis represents the count of UMI (unique molecular identifiers) associated with each individual barcode.
-
B, CStatistical analyses of mapped reads (B) and detected genes (C) for available 10X Genomics samples.
- D
-
EExpression pattern of marker genes of various lineages in our data in (D).
- F
-
GExpression patterns of marker genes in different groups in (F).
- H
-
ISmart‐seq2 data identified four pancreatic cell groups in E14.5 pancreas.
-
JHeat map showing the expression of the group‐enriched genes identified from Smart‐seq2 data. Each column represents a single cell, and each row represents one gene. Gene counts are on the left.
-
KVenn diagrams showing the overlapped gene counts of differentially expressed genes in (H) and (J).
- L
-
MHeat map showing the expression of the EP subgroup‐enriched genes from the 10X Genomics data. TFs are listed on the right. Gene counts are on the left.
Defining the developmental pathways of pancreatic lineage differentiation
To define the developmental trajectory of these cell populations, we performed a three‐dimensional (3D) PCA of all 2,282 pancreatic cells (Fig 2A and B). Simultaneous principal curves (Fletcher et al, 2017; Street et al, 2018; preprint: Saelens et al, 2018) were used to depict the differentiation pathways of all pancreatic lineages (Fig 2A and B). In the PCA plot, the MP‐early population was considered the starting point of all pancreatic cell lineage pathways and the first branching node of MP differentiation (Fig 2A and B). From this point, MP‐early cells along one branch entered into the MP‐late stage and further developed into tip cells, and along the other branch, MP‐early cells developed into α‐1st cells. The tip cells represented the second branching node of the pancreatic lineage differentiation pathway, generating acinar and trunk cells (Fig 2A and B). At the third differentiation node, the trunk cells produced two cell branches: ductal and EP cells (Fig 2A and B). The EP cell developmental process was divided into four stages (EP1–EP4) in the t‐SNE plot and PCA plot (Figs 1B and 2A). The EP4 population was identified as the fourth major branching node during the development of the pancreas (Fig 2A and B). The segregation of islet β cells and α‐2nd cells occurred at this point (Fig 2A and B). Thus, our comprehensive single‐cell transcriptomic analyses constructed a precise developmental trajectory of exocrine and endocrine pancreatic lineages as well as their major intermediate progenitors. Next, we sought to decipher the regulatory features of each branch of cell differentiation along the pathway of pancreatic development.
Figure 2. Roadmap of pancreatic development.
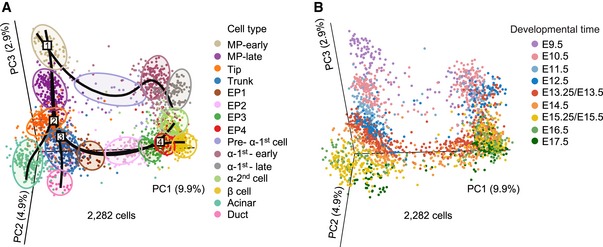
-
A, B3D PCA plots of pancreatic cells. Each dot represents a single cell. The colors denote cell types (A) and developmental time (B). The simultaneous principal curves in (A) indicate the pathways of pancreatic lineage development. The numbers on the curves in (A) indicate a series of branching nodes during pancreas development. The shadows in (A) represent each cell population.
Defining the developmental pathway of multipotent progenitor differentiation
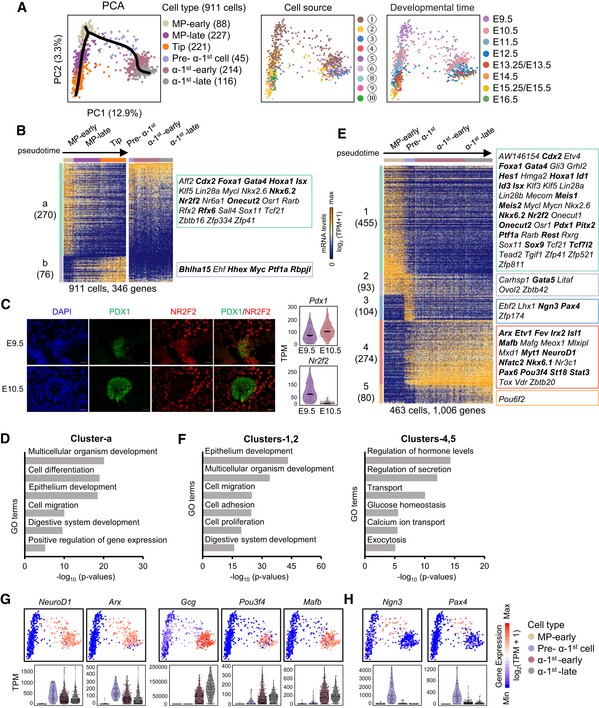
To define the pathway and regulatory logic of MP cell differentiation, we performed PCA of MP‐early cells, MP‐late cells, tip cells, and various stages of α‐1st cells. PCA and a simultaneous principal curve showed that the populations of MP‐early, MP‐late, and tip cells generally formed a linear developmental pathway along which the individual cells were arranged according to a pseudochronological order (pseudotime; Fig 3A). Hierarchical analysis identified two major gene clusters that displayed reverse expression patterns from MP‐early cell to tip cell development. Cluster‐a genes were downregulated, while cluster‐b genes were upregulated along the developmental pathway (Fig 3B and Dataset EV4). Curiously, many genes highly expressed in MP‐early or tip cells, such as Nkx6.2 and Lin28a or Myc and Hhex, were also expressed in MP‐late cells but at relatively lower levels (Figs 3B and EV4A). Moreover, MP‐late cell‐specific genes were barely identified in the heat map, which suggests that MP‐late cells are at an intermediate developmental stage between the MP‐early and tip cell stages (Fig 3B).
Figure 3. Differentiation pathway of the multipotent progenitors.
-
APCA plots of MP‐early, MP‐late, tip, and various stages of α‐1st cells. The colors denote cell types (left) and cell source (middle), the circled number indicating the cell source labeled in Fig 1A, and the developmental time (right). The simultaneous principal curves indicate the developmental pathways of MP‐early cells (left).
-
BHierarchical clustering of pseudotemporally ordered cells plotted in PCA (A). High‐loading genes in the PCA of MP‐early, MP‐late, and tip cells were employed for hierarchical clustering (PC1, P < 10−20). TFs are listed on the right. The bolded TFs are known to be important for pancreas development. Gene counts are labeled in brackets.
-
CImmunofluorescence staining of PDX1 and NR2F2 in paraffin sections of E9.5 and E10.5 pancreatic tissues. Scale bars: 20 μm. The expression levels (TPM) of Pdx1 and Nr2f2 are shown on the right. The line within each violin plot indicates the median of expression levels.
-
DSelected GO terms of gene cluster‐a in (B).
-
EHierarchical clustering of pseudotemporally ordered MP‐early and various stages of α‐1st cells plotted in PCA (A). High‐loading genes of the corresponding PCA were used for hierarchical clustering (PC1 and PC2, P < 10−12). TFs are listed on the right. The bolded TFs are known to be important for pancreas development. Gene counts are labeled in brackets.
-
FSelected GO terms of gene clusters‐1, 2 and clusters‐4, 5 in (E).
-
G, HExpression levels of marker genes are projected onto the PCA plot in (A) (upper). The violin plot shows the expression level (TPM) of the indicated gene in each cell type (down).
Figure EV4. Dynamics of cell type feature genes during multipotent pancreatic progenitor differentiation.
- A
-
BE11.5 Ptf1a‐CreER; Rosa‐RFP+ cells were intermingled with E11.5 Pdx1‐Cre; Rosa‐RFP+ or Pdx1‐GFP+ cells.
- C
-
D, EViolin plots showing the expression levels (TPM) of selected genes in clusters‐1, 2 (D) and clusters‐4, 5 (E) described in Fig 3E in MP‐early and various stages of α‐1st cells.
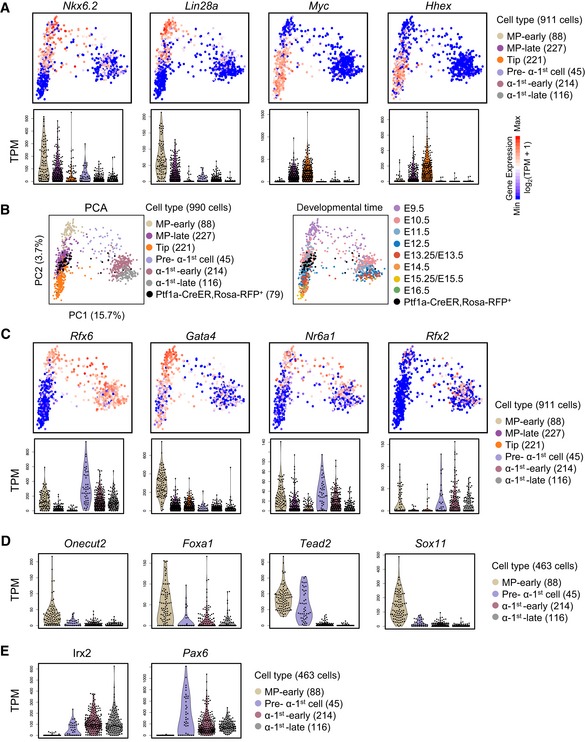
The MP populations were enriched from the Pdx1‐traced mouse models (Fig 3A). As an additional confirmation of MP cell identity, we used another MP cell marker, Ptf1a (Burlison et al, 2008; Kopinke et al, 2012), to trace and enrich MP cells. We sorted E11.5 RFP+ cells from Ptf1a‐CreER; Rosa‐RFP pancreata for sc‐RNA‐seq analysis. We found that the E11.5 cells from Ptf1a‐CreER; Rosa‐RFP, Pdx1‐Cre; Rosa‐RFP, or Pdx1‐GFP mice were intermingled in the MP‐late population. This finding independently confirmed the identity of the MP cells (Fig EV4B).
The separation of MP‐early and MP‐late cells mainly occurred at the stage between E9.5 and E10.5 (Fig 3A). In the flow cytometry gating, Pdx1‐GFP fluorescence intensity was lower in E9.5 cells than in E10.5 and E11.5 cells (Fig EV1B). This finding was consistent with the lower expression level of Pdx1 in E9.5 cells (Fig 3C). Furthermore, we identified Nr2f2 was highly expressed in the MP‐early population. We validated this difference by performing immunofluorescence staining using the antibody against NR2F2 in the E9.5 and E10.5 pancreata. We found that the signal of NR2F2 was observed in E9.5 but were not in E10.5 dorsal pancreatic buds. However, the PDX1 signal in E10.5 pancreatic buds was higher than that in E9.5 (Fig 3C). Taken together, these results confirmed the difference in MP cells between E9.5 and E10.5.
Cluster‐a contained 78% (270 genes) of all variously expressed genes during MP‐early to tip cell differentiation and included many TFs (Fig 3B). Gene Ontology (GO) analysis revealed that the cluster‐a genes were enriched for terms related to regulation of cell fate differentiation and morphogenesis (Fig 3D and Dataset EV4); however, almost no GO terms were significantly enriched in cluster‐b genes. As development progressed, the capacity of MP cells to differentiate into α‐1st cells was lost, consistent with the downregulation of cluster‐a genes (Fig 3B). We therefore examined whether the expression of cluster‐a genes was accompanied by the differentiation of α‐1st cells. Curiously, a large proportion of cluster‐a genes were expressed in various stages of α‐1st cells, whereas the expressions of cluster‐b genes were almost absent in the α‐1st cells (Fig 3B). Moreover, cluster‐a genes included several TFs, such as Rfx6 and Nkx6.2, which are known to regulate pancreatic endocrine differentiation (Nelson et al, 2007; Smith et al, 2010; Figs 3B and EV4A and C). This finding suggests that cluster‐a genes are associated with the differentiation potential of MP‐early cells to generate α‐1st cells.
On the PCA plot, we observed the developmental pathway of the α‐1st cells from the MP‐early population and identified five clusters of genes variously expressed during the differentiation process (Fig 3A and E). Cluster‐1 and cluster‐2 genes were highly expressed in MP‐early cells and included GO terms related to cell adhesion, migration, and differentiation, likely related to their dramatic morphogenesis and cell fate transition (Figs 3E and F, and EV4D, and Dataset EV4). Cluster‐4 and cluster‐5 genes, including NeuroD1, Arx, Gcg, Pou3f4, and Mafb, were upregulated in the differentiated α‐1st cells and enriched in GO terms for endocrine cell functions (Figs 3E–G and EV4E, and Dataset EV4). Curiously, pre‐α‐1st cells expressed both MP‐early (cluster‐2) and α‐1st‐early/α‐1st‐late cells (cluster‐4) enriched genes (Fig 3E). On the PCA plot, pre‐α‐1st cells connected MP‐early cells and a later developmental stage of α‐1st cells (Fig 3A). Moreover, pre‐α‐1st cells also transiently expressed cluster‐3 genes, including key TFs for endocrine fate transition such as Ngn3 and Pax4 (Fig 3E and H). All these data suggest that the pre‐α‐1st cells were in an intermediate state in the cell fate transition from MP‐early to α‐1st cells. Therefore, our studies defined the pathways of early multipotent pancreatic progenitor differentiation and identified intermediate cell states on both branches.
Defining the developmental pathway of tip cell differentiation
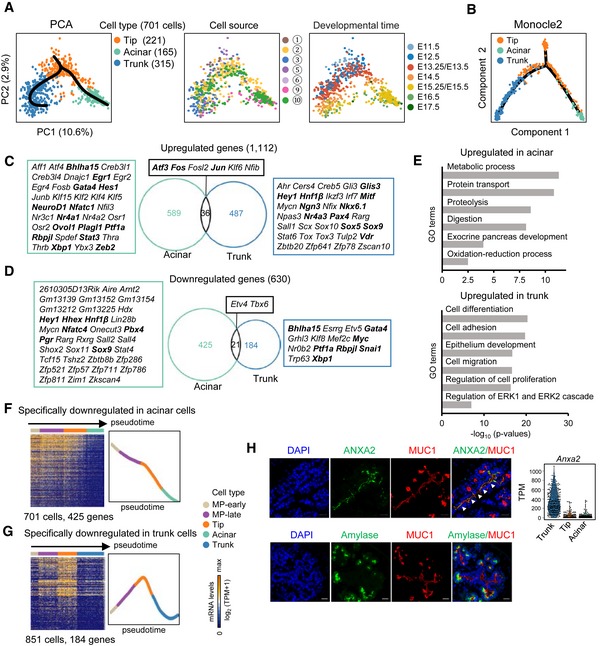
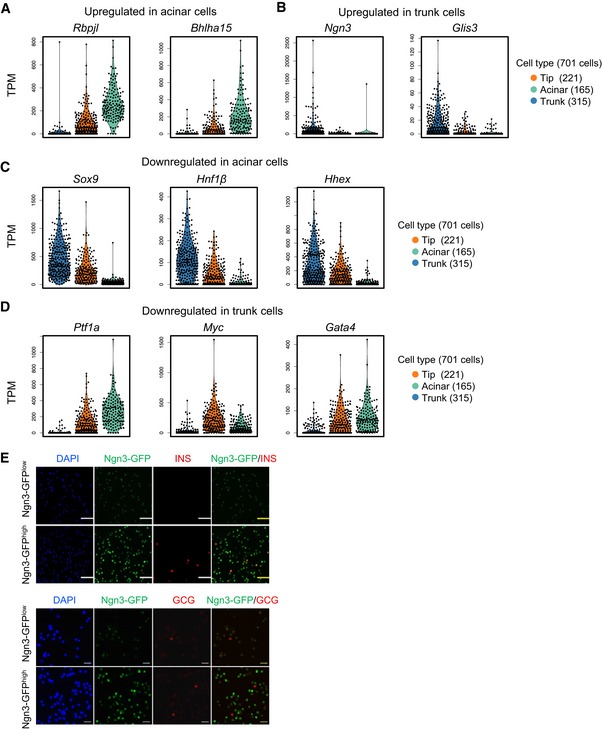
A second branch of lineage fate choice occurs when the tip cells differentiate into acinar and trunk cells (Fig 2A). Principal component analysis identified a triangular distribution on the plot in which tip, trunk, and acinar cells each occupied a single corner (Fig 4A). Consistently, using Monocle2 (Qiu et al, 2017b), another analysis algorithm for pseudotime path construction, we mapped this branching trajectory (Fig 4B). The tip cells primarily included cells at the E11.5–E14.5 developmental stages. Although a recent study detected acinar‐committed cells from E11.0 to E12.0 (Larsen et al, 2017), in our study, most trunk or acinar cell differentiation occurred after E12.5 (Fig 4A). To examine the dynamics of gene expression during the process of tip cell differentiation, we performed differential expression analyses between tip and trunk cells as well as between tip and acinar cells. We identified 1,742 differentially expressed genes (Fig 4C and D, and Dataset EV5), which suggested a dramatic change in cell characteristics during tip cell differentiation. Among the genes upregulated during tip‐to‐acinar and tip‐to‐trunk cell fate transition, overlapping genes only accounted for a small portion (Fig 4C), indicating that the differentiation of acinar or trunk cells is specifically regulated. Two TFs related to acinar cell differentiation, Rbpjl and Bhlha15 (Pin et al, 2001; Masui et al, 2010), were identified in the group of genes upregulated during the tip‐to‐acinar transition (Figs 4C and EV5A). The GO terms enriched in this group included “exocrine pancreas development” and digestive activities related to the characteristics of acinar cells (Fig 4E and Dataset EV5). During tip‐to‐trunk differentiation, the upregulated genes included GO terms related to regulation of cell proliferation, migration, and differentiation (Fig 4E and Dataset EV5). Curiously, TFs for endocrine differentiation, such as Ngn3 and Glis3 (Gu et al, 2002; Schonhoff et al, 2004; Kang et al, 2016), were upregulated during the tip‐to‐trunk fate transition (Figs 4C and EV5B).
Figure 4. Differentiation pathway of the tip cells.
-
APCA plots of tip, acinar, and trunk cells. The colors denote the cell types (left) and cell source (middle), the circled number indicating the cell source labeled in Fig 1A, and the developmental time (right). The simultaneous principal curves indicate the developmental pathways of tip cell differentiation (left).
-
BDevelopmental trajectory of tip, acinar, and trunk cells produced by Monocle2 analysis. The colors denote cell types.
-
C, DUpregulated (C) and downregulated (D) genes in acinar and trunk cells compared with tip cells. The numbers indicate gene counts. TFs are listed next to the Venn diagrams. The bolded TFs are known to be important for pancreas development.
-
ESelected GO terms of upregulated genes in acinar or trunk cells.
-
F, GHeat maps showing the expression of acinar‐ (F) or trunk‐ (G) specifically downregulated genes in pseudotemporally ordered cells. The mean relative expression levels of the genes are shown on the right.
-
HImmunofluorescence staining of MUC1 and ANXA2 or amylase in paraffin sections of E14.5 pancreatic tissues. Scale bars: 20 μm. The gene expression level (TPM) of Anxa2 is shown on the right. The arrowheads indicate the trunk domain.
Figure EV5. Dynamics of cell type feature genes during tip cell differentiation.
-
A, BViolin plots showing the expression levels (TPM) of selected upregulated genes in acinar (A) and trunk (B) cells.
-
C, DViolin plots showing the expression levels (TPM) of selected downregulated genes in acinar (C) and trunk (D) cells.
-
EImmunofluorescence staining of insulin and glucagon in sorted Ngn3‐GFPlow and Ngn3‐GFPhigh cells at E14.5. Scale bars: 50 μm (upper), 20 μm (down).
We next focused on the genes downregulated during tip cell differentiation. Notably, during tip‐to‐acinar transition, 425 genes were specifically downregulated, including many TFs expressed in the MP stage, such as Sox9, Hnf1β, and Hhex (Figs 4D and EV5C, and Dataset EV5). We therefore examined the expression patterns of these genes along the developmental process from MP‐early to acinar cells. Strikingly, we found that this group of genes was expressed in cells from the MP‐early to tip stage but was downregulated in the terminally differentiated acinar cells (Fig 4F), suggesting that the expression of these genes accompanied multipotent potential. By contrast, only 184 genes were specifically downregulated during the tip‐to‐trunk transition (Fig 4D and Dataset EV5). Curiously, these genes underwent transient upregulation from the MP‐early to tip differentiation stage before they were turned off in the trunk cells (Fig 4G). Key TFs for tip and/or acinar differentiation, such as Ptf1a and Rbpjl, were included in this group (Figs 4D and EV5A and D), suggesting that this group of genes is functionally related to the generation of tip cells. Taken together, our single‐cell analyses defined a binary pathway of cell fate segregation and regulatory characteristics during tip cell differentiation.
Based on the differential gene expression analysis, we attempted to identify novel markers for distinguishing tip and trunk cells in pancreatic tissue. We found that Anxa2 (encoding a member of the annexin family) was highly expressed in trunk cells (Fig 4H). We performed immunofluorescence staining to detect the distribution of ANXA2 in MUC1+ epithelial cells of the E14.5 pancreas, in which the tip cells were labeled with the amylase. We found that ANXA2 specifically labeled the trunk luminal regions (Fig 4H). This result indicates that ANXA2 can be used to label trunk cells in the developing pancreas.
Defining the subpopulations and developmental pathway of trunk cell differentiation
A third branch of lineage fate choice occurred when the bipotent trunk‐progenitor cells differentiate into ductal cells and EP cells (Fig 2A). PCA and Monocle2 analysis showed that the trunk cells formed a branch pointing to ductal (trunk‐duct) or EP1 (trunk‐EP) cells in the developmental path (Fig 5A and B). On the Monocle2 plot, the broad distribution of trunk cells on the node and two branches indicated heterogeneity of trunk cells (Fig 5B). To confirm this heterogeneity, we performed differential gene expression analysis between the trunk‐duct and trunk‐EP groups. We identified the genes in cluster‐1 (67 genes) and cluster‐2 (18 genes) that were highly expressed in trunk‐duct or trunk‐EP cells, respectively (Fig 5C and Dataset EV6). The Ngn3 expression level was significantly higher in trunk‐EP and EP1 cells than in the cells of other groups (Fig 5D). Therefore, trunk‐EP cells were considered precursors of EP cells expressing Rfx6 and Dll1 (Appendix Fig S1A), and trunk‐duct cells were considered duct precursors due to the continuation of trunk‐duct cells and ductal cells on the developmental trajectory. A third group of trunk cells was distributed around the branching node on the Monocle2 plot (Fig 5B) and mainly included cells at earlier developmental stages, suggesting that these cells were the earlier progenitors of trunk‐duct and trunk‐EP cells. We named these cells trunk‐early cells. We also identified cell subgroup‐specific genes by comparative analyses of trunk‐duct and trunk‐early cells and of trunk‐EP and trunk‐early cell populations (Appendix Fig S1B and Dataset EV6). Therefore, our study revealed the intermediate trunk progenitors and pathways of trunk cell differentiation.
Figure 5. Differentiation pathway of the trunk cells.
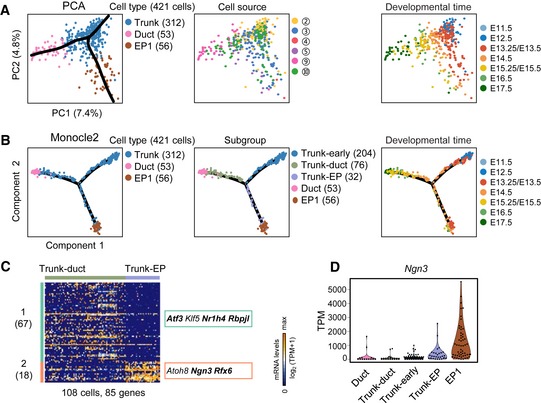
- PCA plots of trunk, ductal, and EP1 cells. The colors denote cell types (left) and the cell source (middle), the circled number indicating the cell source labeled in Fig 1A, and the developmental time (right). The simultaneous principal curves indicate the developmental pathways of trunk cell differentiation (left). Cell counts are labeled in brackets.
- Developmental trajectory of trunk, ductal, and EP1 cells produced by Monocle2 analysis. The colors denote cell types (left), subgroups (middle), and developmental time (right).
- Differential expression analysis between trunk‐duct and trunk‐EP cells. TFs are listed on the right. The bolded TFs are known to be important for pancreas development. Gene counts are shown in brackets.
- The violin plot shows the Ngn3 expression level (TPM) in each of the subgroups in (B, middle).
Dynamics and regulation during the transition from trunk to EP cells
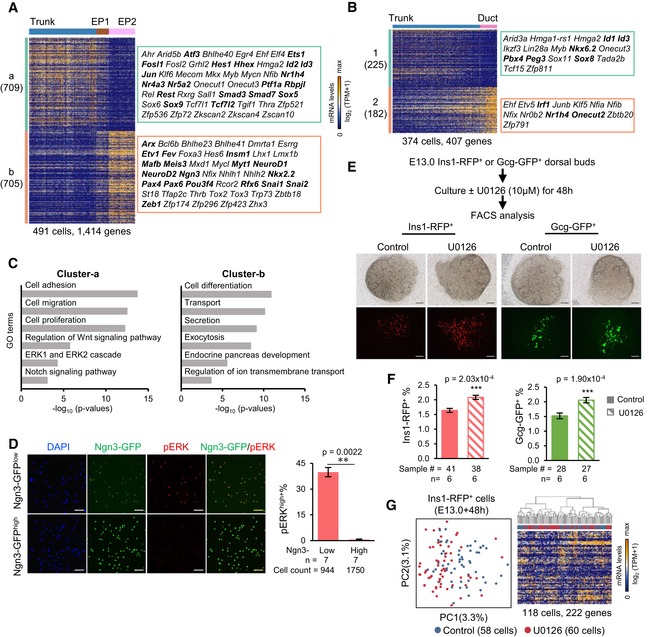
The clear identification of distinct cell populations enabled us to decipher the transcriptomic differences between trunk‐progenitor cells and their descendant cells. However, due to the low gene expression variability among the subgroups of trunk cells (Fig 5C and Appendix Fig S1B), these subgroups were analyzed as a whole in the following differential expression analyses. We compared the gene expression patterns between trunk and EP early‐stage cells (EP1 and EP2) and between trunk and ductal cells, and discovered cell type‐specific genes (Fig 6A and B, and Dataset EV7). The number of differentially expressed genes between trunk and EP early cells (1,414 genes) was three times the number of those between trunk and ductal cells (407 genes), and a large number of TFs were involved in the process of trunk‐to‐EP cell differentiation (Fig 6A and B, and Dataset EV7). These findings indicated that dramatic changes in gene expression occurred between trunk and EP fate transition. No significant GO terms were identified among the genes differentially expressed between trunk and ductal cells. However, among the genes differentially expressed between trunk and EP early cells, GO analysis revealed that genes highly expressed in trunk cells showed enrichment for terms such as “cell adhesion” and “cell migration” (Fig 6C and Dataset EV7), which are associated with the properties of trunk epithelial cells. Genes highly expressed in EP early cells were enriched in GO terms associated with endocrine pancreatic development (Fig 6C and Dataset EV7). Curiously, several signaling pathways, such as the Notch, Wnt, and extracellular signal‐regulated kinase (ERK) pathways, were expressed in trunk cells but downregulated in EP early cells (Fig 6C and Dataset EV7). The role of Notch signaling in negatively regulating endocrine progenitor differentiation has been well‐studied (Apelqvist et al, 1999; Li et al, 2015), and regulation of pancreatic progenitor expansion by the canonical Wnt pathway has also been demonstrated (Baumgartner et al, 2014). However, whether repression of the ERK pathway is necessary for endocrine differentiation is unclear.
Figure 6. The regulation of the cell fate transition from trunk to EP cells.
-
A, BDifferential expression analysis between trunk and EP (1 and 2) cells (A) or between trunk and ductal cells (B). TFs are listed on the right. The bolded TFs are known to be important for pancreas development. Gene counts are labeled in brackets.
-
CSelected GO terms of gene cluster‐a and cluster‐b in (A).
-
DImmunofluorescence staining of pERK and DAPI in sorted Ngn3‐GFPlow and Ngn3‐GFPhigh cells at E14.5. Scale bars: 50 μm. Statistical analysis of the percentage of pERK+ cells within Ngn3‐GFPlow and Ngn3‐GFPhigh cells (right). The data are presented as the mean ± SEM. n: number of biological replicates. **P‐value < 0.01, two‐sided t‐test.
-
ESchematic workflow of E13.0 dorsal pancreatic explant cultures (upper). Pancreata from Ins1‐RFP or Gcg‐GFP mice were or were not treated with U0126 for 48 h. Images of explants after treatment (down). Scale bars: 200 μm.
-
FStatistical analysis of the percentage of Ins1‐RFP+ or Gcg‐GFP+ cells after treatment. The data are presented as the mean ± SEM. n: number of independent biological replicates. Sample #: total number of samples (pancreatic dorsal buds) in all biological replicates. ***P‐value < 0.001, two‐sided t‐test.
-
GPCA (left) of E13.0 Ins1‐RFP+ cells after 48 h of treatment and the corresponding hierarchical clustering (right). High‐loading genes in PCA were used for hierarchical clustering (PC1 and PC2, P < 5 × 10−2).
To confirm that the ERK pathway is repressed during pancreatic endocrine differentiation, we performed immunofluorescence analysis of active phosphorylated ERK1/2 (pERK) in sorted E14.5 Ngn3‐GFPlow and Ngn3‐GFPhigh cells. We found that the percentage of cells highly expressed pERK (pERKhigh) decreased significantly in Ngn3high cells, in which the pERKhigh cells were almost undetectable (Fig 6D). Immunofluorescence staining experiments showed that Ngn3‐GFPlow cells generally lacked insulin and glucagon expression, whereas a few Ngn3‐GFPhigh cells expressed hormones (Fig EV5E), consistent with our finding that the late stage of Ngn3‐GFPhigh cells had begun to differentiate into islet lineages. These results confirmed that Ngn3‐GFPlow cells are earlier than Ngn3‐GFPhigh cells in developmental time. To examine the function of the ERK pathway in endocrine fate transition, we treated dorsal pancreatic explants at E13.0, when the second wave of pancreatic endocrine specification begins, with the MAPK/ERK kinase 1/2 (MEK1/2) inhibitor U0126 (Favata et al, 1998; Fig 6E). The pancreatic tissues were isolated from Ins1‐RFP and Gcg‐GFP reporter mice and cultured for 48 h. After culture, the size of the treated pancreatic explants was not significantly different from that of the controls (Fig 6E). However, flow cytometry analysis showed that the percentage of both Ins1‐RFP+ and Gcg‐GFP+ cells increased significantly after U0126 treatment (Fig 6F). To examine whether the treatment affected gene expression in the differentiated β cells, we performed sc‐RNA‐seq on the sorted Ins1‐RFP+ cells. PCA and hierarchical clustering analysis revealed that the U0126‐treated and control cells were relatively homogeneous at the transcriptomic level (Fig 6G). Genes related to cell proliferation were not differentially regulated between the control and treated cells, indicating that the increase in pancreatic endocrine cells was due to progenitor specification rather than cell proliferation. Therefore, these results indicate that ERK signaling restrains pancreatic endocrine cell specification.
The EP period involves four stages
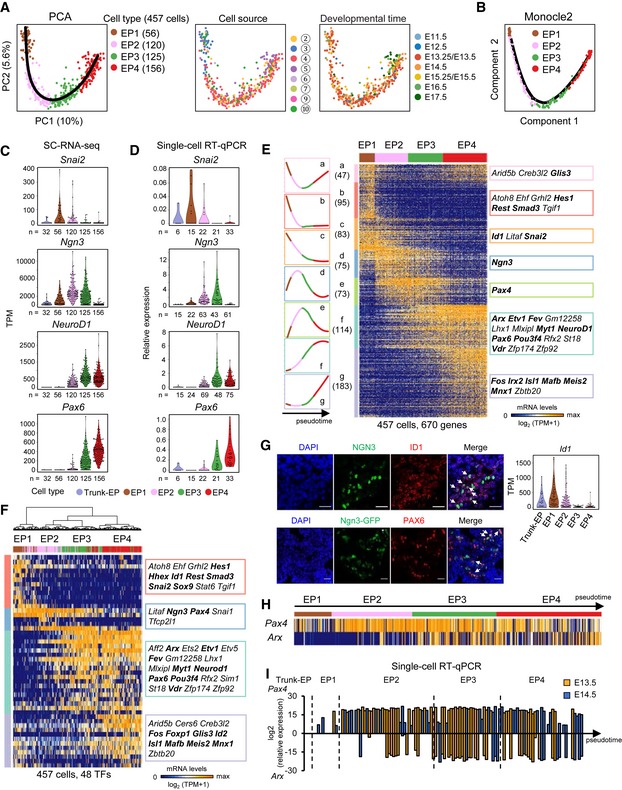
We next focused on the differentiation pathway of EP (EP1–EP4) cells. In the PCA plot, we observed a “V”‐shaped pathway indicating that the principal component 2 (PC2) genes were transiently regulated. These genes returned to their earlier stage of expression after dynamic changes in expression levels during the four‐stage period (Fig 7A). A similar pathway of EP cell differentiation was mapped using Monocle2 (Fig 7B). We examined the dynamics of Ngn3 and NeuroD1 expression during pancreatic endocrine development using the data from sc‐RNA‐seq and verified their expression patterns using a more sensitive approach of single‐cell RT–qPCR (Pijuan‐Sala et al, 2018; Fig 7C and D). Compared with Ngn3‐expressing trunk‐EP cells, we observed that the expression level of Ngn3 increased at EP1, peaked at EP2, was downregulated at EP3 and returned to the background level at EP4 (Fig 7C and D). Due to the persistence of the GFP protein after inhibition of its driven gene, Ngn3, we were able to sort the immediate descendants of EP3 cells. We observed that in EP4 cells, although the GFP signal was high, Ngn3 expression was halted (Fig 7C and D). Notably, in the Ngn3‐GFP reporter mouse strain, the Ngn3 coding sequence was heterozygously knocked in by the GFP gene (Lee et al, 2002). However, PCA showed that the Ngn3‐expressing cells isolated from Ngn3‐GFP knock‐in and from Pdx1‐Cre; Rosa‐RFP embryos were integrated in the same pancreatic developmental path, thus eliminating the concern that heterozygous deletion of the Ngn3 gene could modify the endocrine differentiation pathway (Fig 7A).
Figure 7. Four stages of EP cell development.
-
APCA plots of EP cells. The colors denote cell types (left) and the cell source (middle), the circled number indicating the cell source labeled in Fig 1A, and the developmental time (right). The simultaneous principal curve indicates the developmental pathway of EP cells.
-
BDevelopmental trajectory of EP cells produced by Monocle2 analysis.
-
C, DThe violin plots show the gene expression levels (TPM) in trunk‐EP and four stages of EP cells (C). The expression levels were verified via single‐cell RT–qPCR with normalization to Gapdh expression (D). n: cell counts.
-
EHierarchical clustering of the pseudotemporally ordered cells plotted in the PCA (A) (PC1 and PC2, P < 10−15). The mean relative expression level and TF list of each gene cluster are shown on the left and right of the heat map, respectively. The bolded TFs are known to be important for pancreas development. Gene counts are shown in brackets.
-
FHierarchical clustering of EP cells with TFs. The TFs listed on the right were high‐loading genes (PC1 and PC2, P < 10−13) in the PCA (see Materials and Methods).
-
GImmunofluorescence staining of ID1, Ngn3, and DAPI in paraffin sections of E14.5 WT pancreas (upper) or of PAX6 and DAPI in cryosections of E14.5 Ngn3‐GFP pancreas. Scale bars: 20 μm. The arrows indicate the Ngn3+ID1+ or Ngn3+PAX6+ cells. The violin plots show the Id1 expression levels (TPM) in trunk‐EP and four stages of EP cells. The expression pattern of Pax6 is shown in (C).
-
HArx and Pax4 expression patterns in EP cells. Individual cells are arranged in the same order as in (E).
-
ISingle‐cell RT–qPCR verified the expression levels of Pax4 and Arx in trunk‐EP cells and in four stages of EP cells at E13.5 (yellow) and E14.5 (blue) with normalization to Gapdh expression. Individual cells are arranged by pseudotime. The dashed lines indicate the boundaries of stages.
Hierarchical clustering analysis of PC1‐ and PC2‐related genes identified seven groups comprising a total of 670 genes that presented distinct expression patterns during EP cell development (Fig 7E and Dataset EV8). This result indicates that EP cell differentiation proceeds through a series of transient stages. Notably, the TF Fev is highly expressed in EP3 and EP4 cells (Fig 7E and Appendix Fig S2E). A recent study using genetic tracing demonstrated that all α and β cells are derived from Fev‐expressing cells (Byrnes et al, 2018), which indicates that Fev+ cells are at a stage before islet lineage allocation. In addition to Fev, these gene clusters included 36 other TFs (Fig 7E, Appendix Fig S2A–F, and Dataset EV8). To examine whether TFs alone can distinguish the four developmental stages, we employed all of the highly variously expressed TFs for PCA and performed hierarchical clustering analysis. We identified 48 TFs, including the 36 TFs (except Irx2) identified in Fig 7E, that sufficiently defined the developmental stages of EP cell differentiation (Fig 7F and Dataset EV8). This finding suggests that the developmental process of EP cell differentiation is driven by these TFs. In addition, these variously expressed TFs displayed a cascade expression pattern along with EP cell differentiation (Fig 7E and F) that allowed us to speculate the functions of these TFs. The TFs expressed at early stages may play roles in promoting endocrine fate commitment by avoiding the alternative “trunk” progenitor fate; TF expression in middle stages may regulate EP cell specification, while TF expression in late stages might be necessary for the allocation of islet subtypes. Accordingly, genes with known functions, such as Hes1 and Snai2 (EP fate choice; Rukstalis & Habener, 2007; Gouzi et al, 2011; Kopinke et al, 2011); Ngn3 and NeuroD1 (EP specification; Naya et al, 1997; Petri et al, 2006); and Pax6, Arx, Isl1, and Mafb (islet lineage specification; Pan & Wright, 2011), were expressed at the early, middle, and late stages, respectively (Fig 7C–F and Appendix Fig S2A–F). Notably, a fraction of TFs for the second wave of endocrine fate transition, such as Ngn3 and Pax4, also transiently expressed during the first wave of endocrine fate transition (Fig 3E and H). We used immunofluorescence to validate the heterogeneous expression pattern of two TFs, Id1 and Pax6, which were expressed in the early and late EP stages, respectively. We found that ID1 labeled Ngn3‐GFPlow cells and a fraction of Ngn3‐GFPhigh cells, whereas PAX6 was detected in part of the Ngn3‐GFPhigh cells (Fig 7G). The TFs Arx and Pax4 play crucial roles in α‐ and β‐lineage allocation, respectively (Collombat et al, 2003). Curiously, the sequencing data, which were validated by single‐cell RT–qPCR, showed that the expression of Pax4 preceded the expression of Arx because many cells at EP1 and the early stage of EP2 started to express Pax4 but not Arx, whereas in EP3 and EP4 cells, Arx + cells generally co‐expressed Pax4 (Fig 7H and I). Notably, the co‐expression of Pax4 and Arx was also observed in pre‐α‐1st cell population (Fig 3G and H). This finding suggests that before pancreatic islet lineage allocation, EP cells present heterogeneity in the expression of important TFs. Taken together, our studies identified distinct TF clusters associated with the establishment of transient cell subtypes during EP cell differentiation.
We also examined the E14.5 single‐cell datasets generated from 10X Genomics to define the developmental stages of pancreatic endocrine development. t‐SNE analysis revealed three EP subpopulations (EP110X‐310X) with 85 differentially expressed genes (Fig EV3L and M). The stage‐specific gene expression patterns indicated that stages EP1–EP3 identified by Smart‐seq2 technology roughly correspond to stages EP110X to EP210X based on the expression patterns of Ngn3 (Figs 7E and EV3L and M, and Dataset EV8). EP4 and EP310X likely represent cells of the same EP stage because they commonly expressed the TFs Isl1 and Mafb (Figs 7E and EV3L and M, and Dataset EV8).
Identification of the earliest events of islet lineage allocation
At the end of the EP cell differentiation course, the EP4 cells began to differentiate into islet lineages (Fig 2A). Due to an insufficient cell number for high‐quality analysis, we excluded the cells expressing Sst, Ghrl, and Ppy (Fig EV2I), and only focused on the islet lineages of α and β cells. To increase the significance of the analysis, we included previously published sc‐RNA‐seq datasets for α and β cells at E17.5 (Qiu et al, 2017a). PCA and Monocle2 analysis clearly mapped α and β divisions from the branching node of the EP4 cells (Fig 8A and B). By comparing the gene expression profiles between α and EP4 cells or β and EP4 cells, we identified hundreds of genes that were differentially expressed during islet lineage allocation (Fig 8C and D, and Dataset EV9); these genes could be divided into α‐cell‐specific, β‐cell‐specific, and overlapped differentially expressed genes. The overlapped genes may be associated with the general process of islet differentiation, whereas the α‐cell‐ and β‐cell‐specific differentially expressed genes were related to the lineage allocation process. However, among the differentially expressed genes, only a small fraction overlapped between these two lineages. This finding suggests that α and β cells primarily use different strategies for early differentiation. Among the groups of genes that were differentially and specifically expressed in α cells and β cells, we identified many group‐specific TFs (Fig 8C and D, Appendix Fig S3A–D, and Dataset EV9). Interestingly, during islet lineage allocation, Pax4 was downregulated in both α cells and β cells, while Arx was only downregulated in β cells (Fig 8D, Appendix Fig S3D and E). Whether these group‐specific TFs regulate islet general differentiation or lineage‐specific differentiation requires further investigation. Surprisingly, GO analysis did not indicate any significantly enriched cell signaling pathway terms (Dataset EV9), suggesting that the differentiation of α‐ and β‐lineages is primarily dependent on TFs or other regulatory factors. Therefore, this analysis provides many candidate regulators of islet lineage differentiation.
Figure 8. Allocation of α‐cell and β‐cell lineages.

-
APCA plots of EP4, α‐2nd, and β cells. The colors denote cell types (left) and the cell source (middle), the circled number indicating the cell source labeled in Fig 1A, and the developmental time (right). The simultaneous principal curves indicate the developmental pathways of EP4, α‐2nd, and β cells. * and ** indicate the E17.5 Ins1‐RFP+ and Gcg‐Cre;Rosa‐RFP+ cells, respectively, from our previously published work (Qiu et al, 2017a).
-
BDevelopmental trajectory of EP4 cell differentiation produced by Monocle2 analysis.
-
C, DUpregulated (C) and downregulated (D) genes in β cells and α‐2nd cells compared with EP4 cells. The numbers indicate the gene counts. TFs are listed next to the Venn diagrams. The bolded TFs are known to be important for pancreas development.
-
E3D PCA plot of EP4, β, and α cells. The simultaneous principal curves indicate the developmental pathways of these cells. Data on E17.5‐P60 quiescent β and α cells from our published study (Qiu et al, 2017a) were integrated into this PCA.
-
FThe heat map shows the transcriptomic differences between α‐1st and α‐2nd cells. TFs are listed on the right. The bolded TFs are known to be important for pancreas development. Gene counts are labeled in brackets.
-
GSummary of pancreatic lineage developmental pathways.
Our previous study deciphered the mature pathways of both the α‐ and β‐lineages from the embryonic stage to the mature stage (Qiu et al, 2017a). Here, when we combined the analyses of the current data with previous data, we defined the entire α‐ and β‐lineage developmental pathways (Fig 8E).
Our single‐cell transcriptomic study successfully distinguished α‐1st cells from α‐2nd cells (Fig 1B). Comparative transcriptomic analysis identified 684 genes that were differentially expressed between the α‐1st and fetal α‐2nd populations (Fig 8F and Dataset EV10). The key TF genes for α‐cell differentiation, Arx, Pou3f4, and Mafb (Pan & Wright, 2011), were expressed homogeneously in these two waves of α cells (Appendix Fig S3F). Curiously, the TF genes Nkx6.2 and Gata4, which play roles in early pancreatic development (Schaffer et al, 2010; Pan & Wright, 2011), were expressed in α‐1st cells but downregulated in α‐2nd cells (Appendix Fig S3G). We also identified a set of TF genes, including Meis2 and Zbtb20, that were highly expressed in α‐2nd cells (Appendix Fig S3H). Therefore, our studies comprehensively identified the developmental pathways and many lineage‐specific genes of α and β cells.
Discussion
Understanding the lineage differentiation pathway in solid organogenesis is challenging. Although traditional approaches, such as genetic tracing and immunostaining of limited protein markers, have been employed to explore the developmental relationships between progenitors and their descendant cells, these methods cannot discover precise pathways, especially at the branch points of cell fate choices. To resolve this issue, in this study, we performed single‐cell RNA transcriptomic analyses of purified pancreatic lineages from various genetically modified reporter mouse strains across the E9.5–E17.5 developmental stages. Compared with droplet‐based microfluidics methods, the Smart‐seq2 protocol used in this study has the advantages of higher transcript coverage (Figs EV2C and EV3C) and lower transcript 3′‐end bias and is considered the gold standard in the single‐cell transcriptomic field (Pijuan‐Sala et al, 2018). To comprehensively map the lineage trajectory, we used 2,282 high‐quality single‐cell transcriptomic datasets. For most cell types, we used at least two different reporter mouse strains to enrich the target cells, which prevented the omission of cell types due to imperfect labeling efficiency of a sole reporter. The cells of a specific cell type but with different genetic backgrounds were clustered together in the t‐SNE, which indicated that the genetic background had a negligible impact on our single‐cell transcriptomic analyses for the identification of cell types and developmental pathways. An advantage of using reporter mice is that they provide genetic information to reconstruct lineage relationships among cell populations. Benefiting from the high data quality and rich mouse resources, we comprehensively depicted the differentiation paths of both pancreatic exocrine and endocrine α and β cells.
Our studies defined four main branching lineage choices that cascade along pancreatic development. The first branching node is MP‐early cells, which differentiate into tip cells via the MP‐late stage and α‐1st cells. The second node is the tip cells, which differentiate into acinar cells and trunk cells. The trunk cells are heterogeneous. Trunk‐early cells can be considered the third node, which branches into ductal and EP cells via the trunk‐duct and trunk‐EP transient stages, respectively. Strikingly, we found that rapid EP cell differentiation involved four stages (EP1–EP4). The EP4 cells are the fourth branching node, from which islet lineage allocation begins. Combined with our previous data (Qiu et al, 2017a), we mapped the entire α‐ and β‐cell developmental pathways from the EP stage to the mature stage. Therefore, in addition to the precise fate map defined in this study, our analyses identified many intermediate cell populations along the developmental roadmap (Fig 8G).
Identification of the key lineage branching points is extremely important for understanding the regulatory strategies and logic during stepwise cell fate choices in pancreatic lineage differentiation. Therefore, in this study, we carefully investigated the dynamics of gene expression patterns at each step of cell fate choice and identified many cell population‐specific TFs and cell signaling pathways as well as other genes related to the establishment of cell identity. Based on these analyses, we hypothesized that repression of ERK signaling is necessary for endocrine cell specification, which was confirmed in a pancreas explant system and through single‐cell transcriptomic analyses. However, our data cannot exclude the possibility that treatment with the ERK signaling inhibitor U0126 affects endocrine function, which could not be detected using single‐cell RNA‐seq. It has been suggested that MAPK/ERK signaling restrained endocrine differentiation in an in vitro study of human embryonic stem cell differentiation (Mfopou et al, 2010). Moreover, in in vivo research, αE‐catenin mutant mice showed impaired islet formation due to increased Sox9+ pancreatic progenitors and enhanced MAPK signaling (Jimenez‐Caliani et al, 2017). However, our work discovered the precise time during pancreas development at which MAPK/ERK regulates endocrine progenitor specification. Therefore, this study provides a rich resource for further understanding the regulatory mechanisms underlying cell lineage differentiation throughout pancreatic development.
One of the most striking findings of this work is that the rapid differentiation of EP cells includes four stages that are accompanied by an expression cascade of specific groups of TFs. A rapid change in gene expression activity indicates the involvement of positive feedback or negative feedback regulation of gene expression. In support of this viewpoint, Ngn3 expression is discovered to positively regulate the expression of key TFs, such as NeuroD1. The expression of NeuroD1 in turn promotes its own expression (Huang et al, 2000). Based on these findings, we infer that these stage‐specific TFs form a complicated genetic network that governs pancreatic endocrine differentiation. A recent study generated 15,228 single‐cell datasets from E14.5 pancreas using a droplet‐based method and defined four subpopulations. The heterogeneity of EP cells was verified using the differentially expressed genes (Scavuzzo et al, 2018). However, the second subpopulation (a small population marked by “N14_2”), which expressed the mesenchymal cell marker gene Col3a1, deviated from the developmental trajectory of other three subpopulations (Scavuzzo et al, 2018). Considering that the droplet‐based method inevitably produces doublet contamination, the existence of this population needs to be carefully verified.
An Ngn3‐Timer mouse model has been utilized to identify early, middle, and late Ngn3‐expressing cell populations (Miyatsuka et al, 2009). We speculate that if we perform single‐cell transcriptomic analyses in different Ngn3‐expressing cell populations from Ngn3‐Timer mice, we could understand how EP1‐4 and the early islet lineage cells classified in our study correspond to the early, middle, and late Ngn3‐expressing cells identified using the Ngn3‐Timer model.
Our work provides new insights into cell fate choice and the key features of both exocrine and endocrine lineage differentiation. A comprehensive roadmap for pancreas development in vivo would guide the in vitro generation of functional β cells and other cell lineages from either embryonic stem cells or induced pluripotent stem cells in a stepwise manner. Single‐cell transcriptomic analyses during in vitro induction would contribute to the evaluation of the efficiency of a specific procedure and to the assessment of the potential risks caused by improper cell differentiation pathways or abnormal expression of key genes.
Materials and Methods
Mice
Pdx1‐Cre (Hingorani et al, 2003), Ngn3‐Cre (Schonhoff et al, 2004), Sox9‐CreER (Kopp et al, 2011), Ptf1a‐CreER (Kopinke et al, 2012), Rosa‐RFP, Pdx1‐GFP (Gu et al, 2004), Ngn3‐GFP (Lee et al, 2002), and Ins1‐RFP (Piccand et al, 2014) genetically modified reporter mouse strains were used to obtain pancreatic cells at various developmental stages. To activate Cre expression in Ptf1a‐CreER and Sox9‐CreER embryos, tamoxifen (Sigma T5648, stock solution of 16 mg/ml in corn oil) was intraperitoneally injected (0.2 mg/g body weight) into pregnant females 1.5–2 days before sacrifice. The day on which a vaginal plug appeared was counted as E0.5. All animals were maintained under specific pathogen‐free conditions in the animal facility at Peking University. All animal experiments were conducted following the rules of the ethics committee for animal care.
To generate Gcg‐P2A‐GFP transgenic reporter mice, the P2A‐GFP cassette was inserted upstream of the TAG translation stop codon of the Gcg gene using the CRISPR/Cas9‐mediated system and expressed under the control of endogenous Gcg expression. Constitutive Cas9 mRNA, sgRNA (TGAAATACCTATTTCCTACG), and the P2A‐Gcg‐EGFP donor fragment were co‐injected into C57BL/6 mouse zygotes, and the zygotes were then transplanted into the uterus of a C57BL/6 mouse. The pups were genotyped using PCR primers (forward: 5′‐GCCTATGTGGGGTCCCTAACTCAGTC‐3′; reverse: 5′‐ACCATAGGACCGGGGTTTTCTTCC‐3′). The sgRNAs were designed using the CRISPR tool (http://crispr.mit.edu/). The sgRNA expression plasmids were cloned by inserting annealed oligos into a pGK1.1 linear vector and were employed as templates for in vitro transcription of sgRNA using a MEGAshortscript Kit (Ambion, AM1354). The Cas9 expression plasmid (Addgene: 44758) was used as the template for in vitro transcription of Cas9 mRNA using a T7 Ultra Kit (Ambion, AM1345).
Cell suspension preparation and FACS
E9.5–E14.5 dorsal pancreata were dissected and digested with 0.25% trypsin (Sigma, T4799) for 5 min at 37°C. Digestion was terminated with 0.4 volumes of fetal bovine serum (FBS). E15.5–E17.5 pancreata were dissected and digested with 0.5 mg/ml collagenase P (Roche, 11213873001) for 2 min at 37°C, followed by 0.25% trypsin treatment as described above. The cell solutions were filtered through a polystyrene test tube with a cell strainer snap cap (Corning, 352235), and the cells were sorted via FACS using a BD FACSAria II Cell Sorter.
Single‐cell RNA‐sequencing and single‐cell RT–qPCR
For the Smart‐seq2 method, single cells were mouth‐pipetted from FACS‐purified cells into a PCR tube or were directly sorted into a 96‐well plate containing lysis buffer. The lysis buffer for each cell contained 0.1 μl of ERCC spike‐in RNA (1:1,000,000 dilution, Life Technologies, 4456740). The cell lysis and cDNA synthesis (18 cycles for PCR pre‐amplification step) methods followed the Smart‐seq2 protocol (Picelli et al, 2014). The cDNA was purified twice with 1X VAHTS DNA Clean Beads (Vazyme, N411‐03). A library was then prepared using 2 ng of cDNA for eight cycles of PCR amplification with a TruePrep DNA Library Prep Kit (Vazyme, TD502). The cDNA libraries had an average size of 350 bp and were sequenced as 51‐bp single‐end reads using an Illumina HiSeq 2500 System.
For the droplet‐based method, sc‐RNA‐seq was performed using the Single Cell 3′ Reagent Kit v2 of 10X Genomics following the user's guide. Briefly, the digested E14.5 pancreatic cells were passed through a 35‐μm filter to remove undigested pieces and diluted to a concentration of 700 cells/μl in FACS buffer (HBSS containing 1% FBS, pH 7.4). We loaded approximately 10,000 cells into one channel of the chromium system. We then employed 11 amplification cycles for cDNA amplification and 12 cycles for cDNA library construction. The cDNA library was sequenced as 150‐bp paired‐end reads using the Illumina HiSeq 4000 system.
A portion of the cDNA of individual single cells was diluted for qPCR using AceQ qPCR SYBR Green Master Mix (Vazyme, Q121‐02) on a Roche LightCycler 480 Instrument II. The PCR primers were as follows:
| Gapdh | forward, 5′‐ ATGGTGAAGGTCGGTGTGAAC‐3′ |
| reverse, 5′‐ GCCTTGACTGTGCCGTTGAAT‐3′ | |
| Arx | forward, 5′‐ TCCGGATACCCCACTTAGCTT‐3′ |
| reverse, 5′‐ GACGCCCCTTTCCTTTAAGTG‐3′ | |
| Pax4 | forward, 5′‐ ACCTCATCCCAGGCCTATCT‐3′ |
| reverse, 5′‐ AGGCCTCTTATGGCCAGTTT‐3′ | |
| Ngn3 | forward, 5′‐ AGCTCTTGGCCCATAGATGATG‐3′ |
| reverse, 5′‐ AAGAAGGCAGATCACCTTCGTG‐3′ | |
| NeuroD1 | forward, 5′‐ GCCCAGCTTAATGCCATCTTT‐3′ |
| reverse, 5′‐ CAAAAGGGCTGCCTTCTGTAA‐3′ | |
| Snai2 | forward, 5′‐ TCTGCAGACCCACTCTGATG‐3′ |
| reverse, 5′‐ AGCAGCCAGACTCCTCATGT‐3′ | |
| Pax6 | forward, 5′‐ AGTGAATGGGCGGAGTTATGA‐3′ |
| reverse, 5′‐ AACTTGGACGGGAACTGACA‐3′ |
Immunofluorescence and microscopy
Gcg‐P2A‐GFP pancreatic tissues were from 8‐week‐old mice, and E9.5–E14.5 pancreatic tissues were from WT or Ngn3‐GFP mice. The tissues were fixed in 4% paraformaldehyde for 6–8 h at 4°C, then cryoprotected in 30% sucrose overnight at 4°C, or dehydrated with 30–100% ethanol. The tissues were then embedded in OCT (Thermo Fisher, 6502) and frozen on dry ice or embedded in paraffin. For antigen retrieval of paraffin sections, slides were boiled in antigen unmasking solution (Vector, H‐3300) at high pressure for 10 min. Sections (5 μm thick) were probed with primary antibodies against Glucagon (1:200; Santa Cruz Biotechnology, sc‐7779), PDX1 (1:5,000, Abcam, ab47383), NR2F2 (1:500, Abcam, ab41859), ANXA2 (1:100, Santa Cruz Biotechnology, sc‐28385), MUC1 (1:200, Abcam, ab15481), Amylase (1:200, Santa Cruz Biotechnology, sc‐46657), ID1 (1:200, Proteintech, 18475‐1‐AP), PAX6 (1:100, Thermo Fisher, 42‐6600), and Ngn3 (1:20, DSHB, F25A1B3; Zahn et al, 2004), followed by incubation with donkey anti‐goat conjugated to Alexa Fluor 488 (Thermo Fisher, A11055) or to Alexa Fluor 594 (Thermo Fisher, A11058), donkey anti‐rabbit conjugated to Alexa Fluor 594 (Thermo Fisher, A21207), or donkey anti‐mouse conjugated to Alexa Fluor 488 (Thermo Fisher, A21202) as the secondary antibodies. In addition to the manufacturer's validation, the specificity of NR2F2, ANXA2, ID1, and PAX6 was also validated by Western blot (Appendix Fig S4). Fluorescence images were obtained using a ZEISS AxioImager M2 or a ZEISS Axio Scan Z1.
The sorted cells were attached to Poly‐Prep slides (Sigma, P0425‐72EA) for 30 min at room temperature (RT) and fixed with 4% paraformaldehyde for 10 min at RT. The cells were subsequently stained with primary antibodies and with secondary antibodies conjugated to fluorophores. Fluorescence images were obtained using a ZEISS AxioImager M2 or a ZEISS Axio Scan Z1. The primary antibodies used were rabbit anti‐pERK1/2 (1:200; CST, 9101s), goat anti‐Glucagon (1:200; Santa Cruz Biotechnology, sc‐7779), and guinea pig anti‐Insulin (1:500, Abcam, ab7842). The secondary antibodies used were donkey anti‐rabbit antibody conjugated to Alexa Fluor 594 (Thermo Fisher, A21207), donkey anti‐goat conjugated to Alexa Fluor 594 (Thermo Fisher, A11058), and goat anti‐guinea pig conjugated to Alexa Fluor 594 (Thermo Fisher, A11076).
Pancreas explant cultures
E13.0 dorsal pancreata were dissected from Ins1‐RFP and Gcg‐GFP transgenic mice. Three pancreata were cultured on one Whatman® Nuclepore Track‐Etched Membrane (110414) in a 12‐well plate with 2 ml of DMEM (Gibco, 11885) per well containing 10% FBS and 1% penicillin/streptomycin. U0126 (Sigma, U120), an inhibitor of MEK1 and MEK2, was added to the medium at a final concentration of 10 μM. The tissues were subsequently cultured at 37°C in a humidified incubator with 5% CO2 for 48 h. The tissues were then harvested and digested with 0.5 mg/ml collagenase P for 1 min at 37°C, followed by 0.25% trypsin for 4 min at 37°C. After digestion was complete, the cells were sorted into 96‐well plates containing 4.1 μl of lysis buffer and analyzed using a BD FACSAria II Cell Sorter.
Processing of Smart‐seq2 data
Sequencing reads were mapped to the mouse genome (mm10) using TopHat (v2.1.0; Kim et al, 2013) with the parameters “‐o out_dir ‐G gtf –transcriptome‐index trans_index bowtie2_index input_fastq”. The reads mapped to each gene were quantified with HTSeq (Anders et al, 2015; v0.6.0) using the parameters “htseq‐count ‐f bam ‐r pos ‐s no ‐a 30 input_bam gtf”. Gene expression levels were calculated as transcripts‐per‐million (TPM) values (Wagner et al, 2012). Cells were filtered out if they exhibited fewer than 0.2 million mapped reads or if fewer than 4,000 genes were detected (TPM > 1). For CD45−;Tie2− cells (GSE89798), the quality control criteria were (i) mapped read count > 0.2 million; (ii) detected gene count > 4,000; and (iii) unique mapped read % > 35%. Batch effects between CD45−;Tie2− data and other Smart‐seq2 data were corrected with the MNN algorithm (Haghverdi et al, 2018).
Identification of cell types of Smart‐seq2 data
The Seurat package (v2.3.1; Satija et al, 2015) was used to identify cell types based on log2(TPM + 1) values. PCA was conducted using the RunPCA function employing significantly highly variably expressed genes, which were identified based on ERCC spike‐in as described previously (FDR < 0.1; Brennecke et al, 2013). The “RunTSNE” function was used to perform t‐SNE. To identify cell types, “FindClusters” or “DBClustDimension” was applied. Marker genes for each subgroup were obtained using the “FindAllMarkers” function with the parameters “logfc.threshold = 1, return.thresh = 0.7, only.pos = T, test.use = roc”. Genes highly expressed in multiple subgroups were excluded. 2D and 3D graphs were generated with ggplot2 (v2.2.1; Hadley, 2009) and rgl (v0.96.0; Adler et al, 2016), respectively.
Identification of proliferative cells
Hierarchical clustering was performed on significantly highly variably expressed genes among pancreatic lineage cells. Then, a subset of genes enriched for cell cycle‐related annotation terms in GO enrichment analysis was extracted as cell cycle‐related genes (Dataset EV2). A second round of hierarchical clustering was performed with cell cycle‐related genes, and cells expressing these cell cycle‐related genes were considered proliferative cells.
Developmental trajectory analysis
We used both DDRTree in Monocle2 (v2.64; Qiu et al, 2017b) and slingshot (v0.1.2‐3; Street et al, 2018) for developmental trajectory analyses. In the Monocle2 pipeline, “ordering genes” were set as significantly highly variably expressed genes (excluding the cell cycle‐related genes identified above). Then, a DDRTree was constructed with the “reduceDimension” function. Finally, cells were ordered along the trajectory with the “orderCells” function. On the trunk differentiation pathway, trunk cells were divided into three subgroups according to the Monocle2 results. The trunk cells in the branch toward EP and ductal cells were defined as trunk‐EP and trunk‐duct cells, respectively. The rest of the trunk cells were considered trunk‐early cells (Fig 5B).
We also employed slingshot (v0.1.2‐3) for developmental trajectory analyses according to the PCA results. PCA was performed using FactoMineR (v1.31.4; Le et al, 2008) employing the log2 (TPM + 1) value of significantly highly variably expressed genes (excluding the cell cycle‐related genes identified above). Slingshot analyses were performed as previously described (Street et al, 2018). Briefly, the lineage structure was inferred with the cluster‐based MST (minimum spanning tree), and then, developmental trajectories were fit as simultaneous principal curves. To define the developmental trajectories precisely, we set the “start cluster” and “end cluster” according to our biological knowledge.
The genes used for hierarchical clustering were also identified according to the PCA results. We identified genes with the highest principal component (PC) loadings using the dimdesc function of FactoMineR (v1.31.4; PCs and P‐values are indicated in the figure legends). To obtain more heterogeneously expressed genes, we excluded genes with a TPM ≥ 1 in ≥ 90% of the total cells (Figs 3B and E, 6G and 7E). In Fig 7F, PCA was performed with significantly highly variably expressed TFs, and 48 TFs related to PC1 and PC2 (P < 10−13) were identified for hierarchical clustering analysis.
Differential gene expression analysis
Differential expression analysis between the two groups of cells was performed with scde (v1.99.1; Kharchenko et al, 2014). Genes with a FDR < 0.05 and ce (conservative estimate of expression‐fold change) > 1 were identified as differentially expressed genes. Genes were identified as TFs using data from the Animal Transcription Factor Database (AnimalTFDB; Zhang et al, 2011). Enriched GO annotation categories were identified with GOstats (v2.46.0; Falcon & Gentleman, 2007).
Processing of 10X Genomics data
Sequencing reads were pre‐processed with cellranger count (v2.0.2; https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger). The UMI (unique molecular identifiers) matrix generated by cellranger count was normalized as the log2 value (“transcripts‐per‐million”/10+1). The batch effect was corrected with the MNN algorithm (Haghverdi et al, 2018). The variably expressed genes of each batch were obtained using “FindVariableGenes” in the Seurat package with the parameters “mean.function = ExpMean, dispersion.function = LogVMR, x.low.cutoff = 0.0125, x.high.cutoff = 3, y.cutoff = 0.5”. Principal component analysis was performed using the RunPCA function with the union gene set of variably expressed genes in each batch. Cell types and marker genes identification were performed as the relevant part of the Smart‐seq2 data process. To eliminate the influence of doublet cells in 10X Genomics data, we performed an additional clustering analysis on pancreatic epithelial cells, and the group of cells expressing both the pancreatic markers Pdx1/Prox1 and the mesenchymal cell marker Col3a1 was excluded.
Statistical analysis
SEM and P‐value of unpaired two‐tailed t‐test were calculated for immunofluorescence staining and FACS analyses with at least three independent biological replicates (P‐value < 0.05 was considered significant). Other statistical criteria for single‐cell transcriptomic analysis were detailed in the data analysis sections.
Author contributions
C‐RX conceived the project; C‐RX, X‐XY, and W‐LQ designed the research; X‐XY, LY, YZ, M‐YH, and L‐CL performed the research; W‐LQ, X‐XY, and C‐RX analyzed the data; and C‐RX, X‐XY, and W‐LQ wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Review Process File
Acknowledgements
We thank Dr. Fuchou Tang for sc‐RNA‐seq advice; Ms. Si Si for technical assistance; members of the Xu laboratory for advice and comments; and the High Performance Computing Platform of Peking‐Tsinghua Center for Life Sciences. We thank Ms. Fei Wang for assistance with FACS and Mr. Guopeng Wang from the Core Facility of the School of Life Sciences, Peking University, for fluorescent image acquisition with the ZEISS Axio Scan Z1. This work was supported by the National Basic Research Program of China (973 Program 2015CB942800) and the National Natural Science Foundation of China (31521004, 31471358, 31522036, 91753138) to C.‐R.X.
The EMBO Journal (2019) 38: e100164
See also: Z Liu & JB Sneddon (April 2019)
Data availability
The RNA‐seq data from this publication have been deposited to the Gene Expression Omnibus (GEO) and assigned the identifier GSE115931 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115931).
References
- Adler D, Murdoch D, Nenadic O, Urbanek S, Chen M, Gebhardt A, Bolker B, Csardi G, Strzelecki A, Senger A (2016) rgl: 3D visualization using OpenGL. R package version 096. https://CRAN.R-project.org/package=rgl
- Anders S, Pyl PT, Huber W (2015) HTSeq‐a Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de Angelis M, Lendahl U, Edlund H (1999) Notch signalling controls pancreatic cell differentiation. Nature 400: 877–881 [DOI] [PubMed] [Google Scholar]
- Azzarelli R, Hurley C, Sznurkowska MK, Rulands S, Hardwick L, Gamper I, Ali F, McCracken L, Hindley C, McDuff F, Nestorowa S, Kemp R, Jones K, Gottgens B, Huch M, Evan G, Simons BD, Winton D, Philpott A (2017) Multi‐site Neurogenin3 phosphorylation controls pancreatic endocrine differentiation. Dev Cell 41: 274–286.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastidas‐Ponce A, Scheibner K, Lickert H, Bakhti M (2017) Cellular and molecular mechanisms coordinating pancreas development. Development 144: 2873–2888 [DOI] [PubMed] [Google Scholar]
- Baumgartner BK, Cash G, Hansen H, Ostler S, Murtaugh LC (2014) Distinct requirements for beta‐catenin in pancreatic epithelial growth and patterning. Dev Biol 391: 89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhushan A, Itoh N, Kato S, Thiery JP, Czernichow P, Bellusci S, Scharfmann R (2001) Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development 128: 5109–5117 [DOI] [PubMed] [Google Scholar]
- Brennecke P, Anders S, Kim JK, Kołodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, Heisler MG (2013) Accounting for technical noise in single‐cell RNA‐seq experiments. Nat Methods 10: 1093–1095 [DOI] [PubMed] [Google Scholar]
- Burlison JS, Long Q, Fujitani Y, Wright CV, Magnuson MA (2008) Pdx‐1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev Biol 316: 74–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes LE, Wong DM, Subramaniam M, Meyer NP, Gilchrist CL, Knox SM, Tward AD, Ye CJ, Sneddon JB (2018) Lineage dynamics of murine pancreatic development at single‐cell resolution. Nat Commun 9: 3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P, Mansouri A, Hecksher‐Sørensen J, Serup P, Krull J, Gradwohl G, Gruss P (2003) Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev 17: 2591–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgraz R, Herrera PL (2009) Pancreatic neurogenin 3‐expressing cells are unipotent islet precursors. Development 136: 3567–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R (2007) Using GOstats to test gene lists for GO term association. Bioinformatics 23: 257–258 [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM (1998) Identification of a novel inhibitor of mitogen‐activated protein kinase kinase. J Biol Chem 273: 18623–18632 [DOI] [PubMed] [Google Scholar]
- Fletcher RB, Das D, Gadye L, Street KN, Baudhuin A, Wagner A, Cole MB, Flores Q, Choi YG, Yosef N, Purdom E, Dudoit S, Risso D, Ngai J (2017) Deconstructing olfactory stem cell trajectories at single‐cell resolution. Cell Stem Cell 20: 817–830.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH (2008) Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev 22: 3435–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma'ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ, Immunological Genome C (2012) Gene‐expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13: 1118–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittes GK (2009) Developmental biology of the pancreas: a comprehensive review. Developmental biology 326: 4–35 [DOI] [PubMed] [Google Scholar]
- Gouzi M, Kim YH, Katsumoto K, Johansson K, Grapin‐Botton A (2011) Neurogenin3 initiates stepwise delamination of differentiating endocrine cells during pancreas development. Dev Dyn 240: 589–604 [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F (2000) Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA 97: 1607–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA (2002) Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129: 2447–2457 [DOI] [PubMed] [Google Scholar]
- Gu G, Wells JM, Dombkowski D, Preffer F, Aronow B, Melton DA (2004) Global expression analysis of gene regulatory pathways during endocrine pancreatic development. Development 131: 165–179 [DOI] [PubMed] [Google Scholar]
- Hadley W (2009) ggplot2: elegant graphics for data analysis. New York, NY: Springer Science & Business Media; [Google Scholar]
- Haghverdi L, Lun ATL, Morgan MD, Marioni JC (2018) Batch effects in single‐cell RNA‐sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36: 421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hald J, Hjorth JP, German MS, Madsen OD, Serup P, Jensen J (2003) Activated Notch1 prevents differentiation of pancreatic acinar cells and attenuate endocrine development. Dev Biol 260: 426–437 [DOI] [PubMed] [Google Scholar]
- Haumaitre C, Barbacci E, Jenny M, Ott MO, Gradwohl G, Cereghini S (2005) Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc Natl Acad Sci USA 102: 1490–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA (2003) Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437–450 [DOI] [PubMed] [Google Scholar]
- Horn S, Kobberup S, Jorgensen MC, Kalisz M, Klein T, Kageyama R, Gegg M, Lickert H, Lindner J, Magnuson MA, Kong YY, Serup P, Ahnfelt‐Ronne J, Jensen JN (2012) Mind bomb 1 is required for pancreatic beta‐cell formation. Proc Natl Acad Sci USA 109: 7356–7361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HP, Liu M, El‐Hodiri HM, Chu K, Jamrich M, Tsai MJ (2000) Regulation of the pancreatic islet‐specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol 20: 3292–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, Madsen OD, Carmeliet P, Dewerchin M, Collen D, Rousseau GG, Lemaigre FP (2000) Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol Cell Biol 20: 4445–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez‐Caliani AJ, Pillich R, Yang W, Diaferia GR, Meda P, Crisa L, Cirulli V (2017) AlphaE‐catenin is a positive regulator of pancreatic islet cell lineage differentiation. Cell Rep 20: 1295–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HS, Takeda Y, Jeon K, Jetten AM (2016) The spatiotemporal pattern of Glis3 expression indicates a regulatory function in bipotent and endocrine progenitors during early pancreatic development and in beta, PP and ductal cells. PLoS One 11: e0157138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Silberstein L, Scadden DT (2014) Bayesian approach to single‐cell differential expression analysis. Nat Methods 11: 740–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopinke D, Brailsford M, Shea JE, Leavitt R, Scaife CL, Murtaugh LC (2011) Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development 138: 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopinke D, Brailsford M, Pan FC, Magnuson MA, Wright CV, Murtaugh LC (2012) Ongoing Notch signaling maintains phenotypic fidelity in the adult exocrine pancreas. Dev Biol 362: 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, Ma J, Sander M (2011) Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 138: 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krentz NAJ, van Hoof D, Li Z, Watanabe A, Tang M, Nian C, German MS, Lynn FC (2017) Phosphorylation of NEUROG3 links endocrine differentiation to the cell cycle in pancreatic progenitors. Dev Cell 41: 129–142.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen HL, Grapin‐Botton A (2017) The molecular and morphogenetic basis of pancreas organogenesis. Semin Cell Dev Biol 66: 51–68 [DOI] [PubMed] [Google Scholar]
- Larsen HL, Martin‐Coll L, Nielsen AV, Wright CVE, Trusina A, Kim YH, Grapin‐Botton A (2017) Stochastic priming and spatial cues orchestrate heterogeneous clonal contribution to mouse pancreas organogenesis. Nat Commun 8: 605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson LI (1998) On the development of the islets of Langerhans. Microsc Res Tech 43: 284–291 [DOI] [PubMed] [Google Scholar]
- Le S, Josse J, Husson F (2008) FactoMineR: an R package for multivariate analysis. J Stat Softw 25: 1–18 [Google Scholar]
- Lee CS, Perreault N, Brestelli JE, Kaestner KH (2002) Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev 16: 1488–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Zhai WJ, Teng CB (2015) Notch signaling in pancreatic development. Int J Mol Sci 17: 48–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LC, Qiu WL, Zhang YW, Xu ZR, Xiao YN, Hou C, Lamaoqiezhong, Yu P, Cheng X, Xu CR (2018) Single‐cell transcriptomic analyses reveal distinct dorsal/ventral pancreatic programs. EMBO Rep 19: e46148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lof‐Ohlin ZM, Nyeng P, Bechard ME, Hess K, Bankaitis E, Greiner TU, Ameri J, Wright CV, Semb H (2017) EGFR signalling controls cellular fate and pancreatic organogenesis by regulating apicobasal polarity. Nat Cell Biol 19: 1313–1325 [DOI] [PubMed] [Google Scholar]
- Lynn FC, Smith SB, Wilson ME, Yang KY, Nekrep N, German MS (2007) Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc Natl Acad Sci USA 104: 10500–10505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastracci TL, Sussel L (2012) The endocrine pancreas: insights into development, differentiation, and diabetes. Wiley Interdiscip Rev Dev Biol 1: 609–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui T, Swift GH, Deering T, Shen C, Coats WS, Long Q, Elsasser HP, Magnuson MA, MacDonald RJ (2010) Replacement of Rbpj with Rbpjl in the PTF1 complex controls the final maturation of pancreatic acinar cells. Gastroenterology 139: 270–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memic F, Knoflach V, Sadler R, Tegerstedt G, Sundstrom E, Guillemot F, Pachnis V, Marklund U (2016) Ascl1 is required for the development of specific neuronal subtypes in the enteric nervous system. J Neurosci 36: 4339–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mfopou JK, Chen B, Sui L, Sermon K, Bouwens L (2010) Recent advances and prospects in the differentiation of pancreatic cells from human embryonic stem cells. Diabetes 59: 2094–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatsuka T, Li Z, German MS (2009) Chronology of islet differentiation revealed by temporal cell labeling. Diabetes 58: 1863–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatsuka T, Kosaka Y, Kim H, German MS (2011) Neurogenin3 inhibits proliferation in endocrine progenitors by inducing Cdkn1a. Proc Natl Acad Sci USA 108: 185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh LC, Stanger BZ, Kwan KM, Melton DA (2003) Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci USA 100: 14920–14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ (1997) Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD‐deficient mice. Genes Dev 11: 2323–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SB, Schaffer AE, Sander M (2007) The transcription factors Nkx6.1 and Nkx6.2 possess equivalent activities in promoting beta‐cell fate specification in Pdx1+ pancreatic progenitor cells. Development 134: 2491–2500 [DOI] [PubMed] [Google Scholar]
- Norgaard GA, Jensen JN, Jensen J (2003) FGF10 signaling maintains the pancreatic progenitor cell state revealing a novel role of Notch in organ development. Dev Biol 264: 323–338 [DOI] [PubMed] [Google Scholar]
- Pan FC, Wright C (2011) Pancreas organogenesis: from bud to plexus to gland. Dev Dyn 240: 530–565 [DOI] [PubMed] [Google Scholar]
- Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, Heimberg H, Wright CV (2013) Spatiotemporal patterns of multipotentiality in Ptf1a‐expressing cells during pancreas organogenesis and injury‐induced facultative restoration. Development 140: 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri A, Ahnfelt‐Ronne J, Frederiksen KS, Edwards DG, Madsen D, Serup P, Fleckner J, Heller RS (2006) The effect of neurogenin3 deficiency on pancreatic gene expression in embryonic mice. J Mol Endocrinol 37: 301–316 [DOI] [PubMed] [Google Scholar]
- Piccand J, Meunier A, Merle C, Jia Z, Barnier JV, Gradwohl G (2014) Pak3 promotes cell cycle exit and differentiation of beta‐cells in the embryonic pancreas and is necessary to maintain glucose homeostasis in adult mice. Diabetes 63: 203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, Sandberg R (2014) Full‐length RNA‐seq from single cells using Smart‐seq2. Nat Protoc 9: 171–181 [DOI] [PubMed] [Google Scholar]
- Pijuan‐Sala B, Guibentif C, Gottgens B (2018) Single‐cell transcriptional profiling: a window into embryonic cell‐type specification. Nat Rev Mol Cell Biol 19: 399–412 [DOI] [PubMed] [Google Scholar]
- Pin CL, Rukstalis JM, Johnson C, Konieczny SF (2001) The bHLH transcription factor Mist1 is required to maintain exocrine pancreas cell organization and acinar cell identity. J Cell Biol 155: 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu WL, Zhang YW, Feng Y, Li LC, Yang L, Xu CR (2017a) Deciphering pancreatic islet beta cell and alpha cell maturation pathways and characteristic features at the single‐cell level. Cell Metab 25: 1194–1205.e4 [DOI] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, Trapnell C (2017b) Reversed graph embedding resolves complex single‐cell trajectories. Nat Methods 14: 979–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukstalis JM, Habener JF (2007) Snail2, a mediator of epithelial‐mesenchymal transitions, expressed in progenitor cells of the developing endocrine pancreas. Gene Expr Patterns 7: 471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelens W, Cannoodt R, Todorov H, Saeys Y (2018) A comparison of single‐cell trajectory inference methods: towards more accurate and robust tools. bioRxiv 10.1101/276907 [PREPRINT] [DOI] [PubMed]
- Satija R, Farrell JA, Gennert D, Schier AF, Regev A (2015) Spatial reconstruction of single‐cell gene expression data. Nat Biotechnol 33: 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scavuzzo MA, Hill MC, Chmielowiec J, Yang D, Teaw J, Sheng K, Kong Y, Bettini M, Zong C, Martin JF, Borowiak M (2018) Endocrine lineage biases arise in temporally distinct endocrine progenitors during pancreatic morphogenesis. Nat Commun 9: 3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer AE, Freude KK, Nelson SB, Sander M (2010) Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell 18: 1022–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhoff SE, Giel‐Moloney M, Leiter AB (2004) Neurogenin 3‐expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non‐endocrine cell types. Dev Biol 270: 443–454 [DOI] [PubMed] [Google Scholar]
- Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, German MS (2000) Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 127: 3533–3542 [DOI] [PubMed] [Google Scholar]
- Serafimidis I, Rodriguez‐Aznar E, Lesche M, Yoshioka K, Takuwa Y, Dahl A, Pan D, Gavalas A (2017) Pancreas lineage allocation and specification are regulated by sphingosine‐1‐phosphate signalling. PLoS Biol 15: e2000949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HP, Wang A, Sander M (2013) Pancreas organogenesis: from lineage determination to morphogenesis. Annu Rev Cell Dev Biol 29: 81–105 [DOI] [PubMed] [Google Scholar]
- Smith SB, Qu HQ, Taleb N, Kishimoto NY, Scheel DW, Lu Y, Patch AM, Grabs R, Wang J, Lynn FC, Miyatsuka T, Mitchell J, Seerke R, Desir J, Vanden Eijnden S, Abramowicz M, Kacet N, Weill J, Renard ME, Gentile M et al (2010) Rfx6 directs islet formation and insulin production in mice and humans. Nature 463: 775–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solar M, Cardalda C, Houbracken I, Martin M, Maestro MA, De Medts N, Xu X, Grau V, Heimberg H, Bouwens L, Ferrer J (2009) Pancreatic exocrine duct cells give rise to insulin‐producing beta cells during embryogenesis but not after birth. Dev Cell 17: 849–860 [DOI] [PubMed] [Google Scholar]
- Spence JR, Lange AW, Lin S‐CJ, Kaestner KH, Lowy AM, Kim I, Whitsett JA, Wells JM (2009) Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Dev Cell 17: 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S (2018) Slingshot: cell lineage and pseudotime inference for single‐cell transcriptomics. BMC Genomics 19: 477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznurkowska MK, Hannezo E, Azzarelli R, Rulands S, Nestorowa S, Hindley CJ, Nichols J, Gottgens B, Huch M, Philpott A, Simons BD (2018) Defining lineage potential and fate behavior of precursors during pancreas development. Dev Cell 46: 360–375.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiso N, Filippi A, Pauls S, Bortolussi M, Argenton F (2002) BMP signalling regulates anteroposterior endoderm patterning in zebrafish. Mech Dev 118: 29–37 [DOI] [PubMed] [Google Scholar]
- Wagner GP, Kin K, Lynch VJ (2012) Measurement of mRNA abundance using RNA‐seq data: RPKM measure is inconsistent among samples. Theor Biosci 131: 281–285 [DOI] [PubMed] [Google Scholar]
- Yu XX, Qiu WL, Yang L, Li LC, Zhang YW, Xu CR (2018) Dynamics of chromatin marks and the role of JMJD3 during pancreatic endocrine cell fate commitment. Development 145: dev163162 [DOI] [PubMed] [Google Scholar]
- Zahn S, Hecksher‐Sorensen J, Pedersen IL, Serup P, Madsen O (2004) Generation of monoclonal antibodies against mouse neurogenin 3: a new immunocytochemical tool to study the pancreatic endocrine progenitor cell. Hybrid Hybridomics 23: 385–388 [DOI] [PubMed] [Google Scholar]
- Zhang HM, Chen H, Liu W, Liu H, Gong J, Wang H, Guo AY (2011) AnimalTFDB: a comprehensive animal transcription factor database. Nucleic Acids Res 40: D144–D149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, Melton DA (2007) A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell 13: 103–114 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Review Process File
Data Availability Statement
The RNA‐seq data from this publication have been deposited to the Gene Expression Omnibus (GEO) and assigned the identifier GSE115931 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115931).