

Abstract
SREBPs are master regulators of lipid homeostasis and undergo sterol‐regulated export from ER to Golgi apparatus for processing and activation via COPII‐coated vesicles. While COPII recognizes SREBP through its escort protein SCAP, factor(s) specifically promoting SREBP/SCAP loading to the COPII machinery remains unknown. Here, we show that the ER/lipid droplet‐associated protein Cideb selectively promotes the loading of SREBP/SCAP into COPII vesicles. Sterol deprivation releases SCAP from Insig and enhances ER export of SREBP/SCAP by inducing SCAP‐Cideb interaction, thereby modulating sterol sensitivity. Moreover, Cideb binds to the guanine nucleotide exchange factor Sec12 to enrich SCAP/SREBP at ER exit sites, where assembling of COPII complex initiates. Loss of Cideb inhibits the cargo loading of SREBP/SCAP, reduces SREBP activation, and alleviates diet‐induced hepatic steatosis. Our data point to a linchpin role of Cideb in regulated ER export of SREBP and lipid homeostasis.
Keywords: Cideb, COPII vesicles, fatty liver, lipid metabolism, SREBP
Subject Categories: Membrane & Intracellular Transport, Metabolism
Introduction
Balanced synthesis, storage, and breakdown of lipids constitute homeostasis of lipid metabolism for the proper development and health of organisms (Shi & Burn, 2004). The liver plays a central and integrative role in lipid homeostasis, whereas a variety of metabolic insults leads to abnormal lipid accumulations in the liver, resulting in hepatic steatosis or fatty liver disease (Browning & Horton, 2004; Cohen et al, 2011; Hooper et al, 2011). Non‐alcoholic fatty liver disease (NAFLD), particularly those associated with obesity, occurs at an epidemic level of ~25% in adults of industrialized countries and often become the forerunner of full‐blown metabolic disorders and other complications (Younossi et al, 2016).
The SREBP family transcription factors (SREBP‐1 and SREBP‐2) are master regulators of lipid metabolism, governing the expression of genes responsible for lipogenesis, sterol production, and lipid uptake (Brown & Goldstein, 1997; Horton et al, 2002). Activation of SREBPs is under the control of a delicate feedback mechanism, ensuring proper response to fluctuations of nutrient levels (Goldstein et al, 2006; Brown & Goldstein, 2009; Goldstein & Brown, 2015). In the presence of sterols, SREBP precursors and SCAP form complex with sterol binding protein Insig‐1/2 (Yabe et al, 2002; Yang et al, 2002) and are retained on the endoplasmic reticulum (ER). Upon sterol deprivation, SREBP/SCAP are exported from ER and relocated to the Golgi apparatus, where proteolytic cleavage of SREBPs takes place, releasing the N‐terminal trans‐activating domain for nuclear localization to activate genes in lipid synthesis and uptake.
The ER‐to‐Golgi transport of SREBP/SCAP is mediated by specialized transport vesicles generated by the COPII complex (Coatomer Complex II) (Zanetti et al, 2011; Miller & Schekman, 2013). The assembly of the COPII complex is initiated at specific exit sites on the ER (ERESs) marked by Sec16 (Connerly et al, 2005; Watson et al, 2006), which anchors guanine nucleotide exchange factor (GEF) Sec12 to activate the small GTPase Sar1 (Barlowe & Schekman, 1993; Weissman et al, 2001). Activated Sar1 localizes to the ER membrane and subsequently recruits the Sec23/24 hetero‐dimer (the inner coat complex), followed by the hetero‐tetramer composed of two copies of Sec13/31 (the outer coat complex). The fully assembled COPII complex thus forms caged vesicles and subsequently inactivates Sar1 to complete scission and release cargo vesicles en route to the Golgi apparatus. Despite our understanding of the fundamental mechanisms of COPII assembly, questions remain concerning the specific function and regulation of COPII‐mediated transport in physiological processes.
Cargo recognition by the COPII complex underlines the specificity of ER‐to‐Golgi transport (Schmidt & Stephens, 2010; Gillon et al, 2012). Previous works have revealed that sterol‐regulated recognition between SCAP and Sec24 constitutes the molecular “switch” that turns on/off the transport of SREBP (Sun et al, 2005, 2007). SCAP contains an MELADL sequence in the 6th cytosolic loop as the COPII sorting signal, which could directly interact with the Sec24C subunit of COPII. Excess sterols lead to SCAP binding with Insig and cause conformational changes of SCAP that block the access of the MELADL sorting signal to COPII proteins, resulting in ER retention of SCAP/SREBP (Sun et al, 2007). Conversely, sterol deprivation displaces Insig from SCAP, thereby allowing COPII recognition and consequently the ER export of SREBP for its activation at the Golgi. Intriguingly, sterol depletion, while activating SREBP ER export, inhibits general COPII‐mediated transport assayed with VSV‐G (Ridsdale et al, 2006; Runz et al, 2006), raising the possibility that unique factor(s) may enhance COPII function to ensure efficient delivery of SREBP/SCAP. Inasmuch the differential roles of various tissues in lipid homeostasis, such factor(s) may promote SREBP activation in tissues with high lipogenic program, thereby effectively meeting physiological demands for lipid synthesis or storage.
We previously reported that cell death‐inducing DFF45‐like effector B (Cideb), an ER and lipid droplet (LD)‐associated protein primarily expressed in the liver, participated in lipid metabolism by regulating lipid droplet fusion and VLDL lipidation (Ye et al, 2009). Here, we report that Cideb regulates hepatic SREBP activation by selectively promoting ER‐to‐Golgi delivery of the SREBP/SCAP complex. Interestingly, sterol depletion induces SCAP to interact with Cideb, which also binds the Sec12, the GEF of Sar1, thereby enriching SCAP/SREBP at ER exit sites and increasing the packaging of SREBP/SCAP into COPII‐coated vesicles. Loss of Cideb, via limiting SREBP activity, protects mice from diet‐induced hepatic steatosis.
Results
Loss of Cideb leads to defective SREBP processing and maturation
During the analysis of hepatic gene expression profiles in Cideb −/− mice and the liver‐specific Scap knockout mice (Horton et al, 2003; Li et al, 2010), we uncovered a significant enrichment of two down‐regulated pathways: the fatty acid biosynthesis pathway and the cholesterol synthesis pathway (Fig 1A). Real‐time PCR revealed reduced mRNA levels for genes in lipogenesis (Srebp‐1c, Fasn, Acaca, Scd1, Acly, and Acss2) and cholesterol metabolism (Srebp‐2, Hmgcr, Hmgcs, Ss, Ldlr, and Pcsk9) under various feeding conditions in Cideb −/− mice comparing to those in controls (Fig 1C). In particular, while the expression of lipogenic genes was robustly induced by re‐feeding (R) after fasting in WT mice (Horton et al, 1998; Rong et al, 2017), the induction was blunted in the Cideb −/− liver under this fasting/re‐feeding paradigm (Fig 1C). The expression of LXR target genes, such as Abcg5, Abcg8, and Lpcat3, which were activated by increased ER cholesterol level (Janowski et al, 1996; Calkin & Tontonoz, 2012), was similar between two genotypes under re‐feeding condition (Fig 1C). The nuclear form of SREBP‐1 (SREBP‐1N) protein was increased by approximately fourfold under fast/re‐feeding condition in WT mice (Fig 1B). In contrast, Cideb −/− mice exhibited approximately 60% reduced SREBP‐1N levels at ad‐lib, as well as ~70% reduced SREBP‐1N and SREBP‐2N levels after re‐feeding (Fig 1B). Re‐introduction of Cideb into Cideb −/− mice liver or isolated hepatocytes rescued the impaired SREBP‐1/2 processing and their downstream genes to a comparable extent as observed in the WT liver (Figs 1D and E, and EV1E). Therefore, deficiency of Cideb in the hepatocytes resulted in a defective SREBP processing and reduced expression of their downstream target genes.
Figure 1. Cideb deficiency leads to reduced SREBP processing and maturation.

- Down‐regulation of SREBP target genes in Cideb‐deficient liver. Microarray heatmap of fatty acids/cholesterol synthesis genes that were down‐regulated in Cideb −/− mice and Scap liver‐specific knockout mice, comparing to the respective WT controls (WT VS Cideb −/−, WT VS L‐Scap −/−, n = 2 per group). The expression level of each gene was linearly normalized to range from −1 to 1 before plotting.
- Decreased SREBP processing and maturation in Cideb‐deficient liver. Immune blotting (IB) of the full‐length, precursor SREBP‐1/2 (SREBP FL) and the cleaved, active SREBP‐1/2 (SREBP N) from WT and Cideb −/− mice liver under ad‐lib (ND), fasting 12 h (F), and re‐feeding (R) conditions. Mice were re‐fed with high‐carbohydrate low‐fat diet for 12 h after 12‐h fasting.
- Defective SREBP transcriptional response to re‐feeding in Cideb‐deficient liver. Transcript levels of lipogenic and cholesterol metabolism genes determined by qPCR from WT and Cideb −/− liver under ad‐lib (ND), fasting 12 h (F), and re‐feeding (R) conditions. Mice were re‐fed with high‐carbohydrate low‐fat diet for 12 h after 12‐h fasting (n = 3 per group).
- Re‐introduction of Cideb rescues SREBP processing in Cideb −/− liver. WT and Cideb −/− mice were injected with AAV‐GFP or AAV‐Cideb, and sacrificed for IB with the indicated antibodies 20 days after the injection.
- Re‐introduction of Cideb rescues the expression of SREBP target genes in Cideb −/− liver. Transcript levels of lipogenic genes determined by qPCR from WT and Cideb −/− liver. WT and Cideb −/− mice were injected with AAV‐GFP or AAV‐Cideb, and sacrificed for qPCR 20 days after the injection (n = 3 per group).
Figure EV1. CIDE family proteins rescue SREBP processing in Cideb −/− liver.

-
A–DRe‐introduction of Cidea or Cidec rescues SREBP processing and the expression of SREBP target genes in Cideb−/− liver. WT and Cideb −/− mice were injected with AAV‐GFP, AAV‐Cidea, or AAV‐Cidec, and sacrificed for IB with the indicated antibodies (A, C) and qPCR (B, D) 20 days after the injection (n = 3 per group).
-
ERe‐introduction of Cideb enhances SREBP processing in Cideb‐deficient primary hepatocytes. Hepatocytes infected with adenovirus expressing GFP or Cideb were cultured in sterol‐deprived condition. When indicated, cells were treated with 10 μg/ml 25‐HC before subjected to IB with the indicated antibodies following SDS–PAGE.
-
F, GRe‐introduction of Cideb‐KRRA rescues SREBP processing and target genes expression in Cideb‐deficient liver. WT and Cideb −/− mice were injected with AAV‐GFP, AAV‐Cideb, or AAV‐Cideb‐KRRA, and sacrificed for IB with the indicated antibodies (F) and qPCR (G) 20 days after the injection (n = 3 per group).
Cidea and Cidec, two other CIDE family members that share high homology with Cideb (Wu et al, 2008), also restored SREBP processing upon introduction into the liver of Cideb −/− mice (Fig EV1A–D), demonstrating a conserved function of CIDE family proteins in regulating SREBP. Interestingly, the Cideb mutant (Cideb‐KRRA) defective for LD fusion (Gong et al, 2011) was able to restore SREBP processing and lipogenic gene expression to the same extent of the WT Cideb (Fig EV1F and G), suggesting that the role of Cideb in regulating SREBP processing is independent of its function in LD fusion and growth.
Loss of Cideb protects mice from lipogenic diet‐induced hepatic steatosis
Hyper‐activation of SREBPs leads to hepatic steatosis (Shimano et al, 1997; Moon et al, 2012). To investigate the physiological function of Cideb‐mediated SREBP processing, we subjected WT and Cideb −/− mice to high‐fructose low‐fat (HFLF) diet feeding, thereby stimulating SREBP processing to induce de novo lipogenesis and eventually hepatic steatosis (Haas et al, 2012; Moon et al, 2012). After HFLF diet feeding for 2 months, WT mice displayed increased fat accumulation in the liver, evidenced by the emergence of enlarged LDs visualized with hematoxylin and eosin staining, oil red‐O staining, and electron microscopy (Fig 2A). However, the liver of Cideb −/− mice only contained small‐sized LDs (Fig 2A). Consistently, Cideb −/− mice on HFLF diet contained ~3 times less hepatic TAG (Fig 2B), ~1.3 times less hepatic cholesterol ester (Fig 2C) compared to those in the WT mice. Similarly, Cideb −/− mice also displayed lower serum free cholesterol, cholesterol ester, and NEFA than those in the WT mice on both chow diet and HFLF diet (Appendix Fig S1F and H). Serum TAG levels were similar between two genotypes under both conditions (Appendix Fig S1G). Cideb −/− mice under HFLF diet showed similar food intake (Appendix Fig S1A), body weight (Appendix Fig S1B), adipose tissue and muscle morphology (Appendix Fig S1C), adiposity index (Appendix Fig S1D), and energy expenditure (Appendix Fig S1I–L) to that of WT mice. Hence, reduced fat accumulation was specific for liver, and no increased ectopic fat deposition was observed in adipose tissue and muscle (Appendix Fig S1E).
Figure 2. Loss of Cideb protects mice from diet‐induced fatty liver diseases.
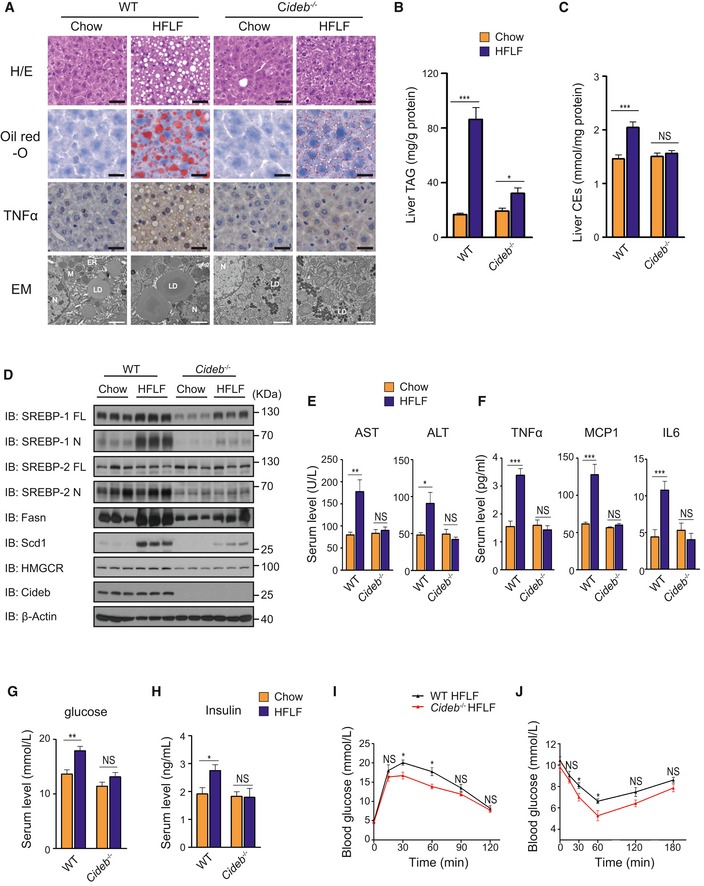
-
ALiver morphology of WT and Cideb −/− liver under chow diet and HFLF diet. H/E staining, oil red‐O staining, immunohistochemistry, and ultrastructure (Electron Microscope) were performed. Scale bar represented 50 μm in the upper three rows of images and 2 μm in the bottom row of images. LD: lipid droplet; ER: endoplasmic reticulum; M: mitochondrial; N: nucleus.
-
B, CTotal liver TAG levels (B) and liver cholesterol ester levels (C) of WT and Cideb −/− mice under chow diet and HFLF diet (n = 8 per group).
-
DLoss of Cideb inhibits diet‐induced SREBP activation. IB of hepatic SREBP processing and lipogenic enzyme levels (Fasn, Scd1) of WT and Cideb −/− mice under chow diet and HFLF diet.
-
ESerum AST and ALT levels of WT and Cideb −/− mice under chow diet and HFLF diet (n = 8 per group).
-
FCideb deficiency alleviates the whole body inflammation response. Serum TNFα, MCP1, and IL6 levels of WT and Cideb −/− mice under chow diet and HFLF diet (n = 8 per group).
-
G, HSerum fed glucose levels (G) and insulin levels (H) of WT and Cideb −/− mice under chow diet and HFLF diet (n = 8 per group).
-
IGlucose tolerance test (GTT) of WT and Cideb −/− mice under HFLF diet (n = 5 per group).
-
JInsulin tolerance test (ITT) of WT and Cideb −/− mice under HFLF diet (n = 5 per group).
To further verify the role of SREBP in the protection of liver steatosis in Cideb −/− mice, we analyzed protein expression in liver lysates. In the WT mice, the cleaved nuclear form of SREBP‐1 was elevated up to ~3.5‐fold after HFLF diet feeding. Consequently, mRNA and protein levels of lipogenic enzymes downstream of SREBP, such as Fasn, Acaca, and Scd1, were also significantly increased (Fig 2D and Appendix Table S1). In contrast, the activation of SREBP‐1 and expression of downstream lipogenic genes in the Cideb −/− liver under HFLF diet were significantly lower compared with the WT liver (Fig 2D and Appendix Table S1). The SREBP‐2 processing and its downstream gene expression in Cideb −/− mice were lower than WT mice under HFLF diet feeding but were not significantly different from those fed with normal diets (Fig 2D and Appendix Table S1). Expression levels of genes related to fatty acid oxidation (Cpt1, Cpt2, and Cox4) and LXR target genes (Abcg5, Abca1, and Lpcat3) were similar between two genotypes (Appendix Table S1).
Blood chemistry analysis revealed that HFLF diet feeding caused ~2‐fold induction of serum AST and ALT level in WT, but not in Cideb −/− mice (Fig 2E), indicating that Cideb inactivation alleviated liver injury associated with steatosis. Consistently, hepatic tissues in WT mice displayed elevated inflammation, manifested by the increased TNFα staining (Fig 2A), higher expression of inflammatory genes such as F4/80, Tnfα, and Mcp1 (Appendix Table S1). These changes were largely absent in Cideb −/− liver. Furthermore, WT mice exhibited ~two‐fold induction in serum levels of cytokines such as TNFα, MCP1, and IL6 after HFLF feeding (Fig 2F). Cideb −/− mice displayed no changes in the systemic levels in these pro‐inflammatory factors. Consequently, Cideb −/− mice had lower levels of blood glucose and insulin (Fig 2G and H) and a slightly improved glucose clearance and higher insulin response than WT control mice after HFLF diet treatment (Fig 2I and J). Taken together, these data suggested that loss of Cideb, via limiting SREBP activation, protected mice from lipogenic diet‐induced hepatic steatosis and inflammation.
Loss of Cideb inhibits loading of SREBP/SCAP to COPII vesicles
The physiological role of Cideb in regulating SREBP activation prompted us to investigate the underlying molecular mechanism. ER‐localized SREBP precursors are delivered to the Golgi apparatus for processing, in a manner requiring the escort protein SCAP for packaging into the COPII‐coated transport vesicles (Nohturfft et al, 2000). We thus assessed the subcellular distribution of SREBP and SCAP in the liver of WT and Cideb −/− mice. In the ER fractions, the protein levels of SREBP‐1/2 and SCAP were similar between Cideb −/− and WT mice. In contrast, Golgi‐localized SREBP‐1 showed near 80% reduction, and SREBP‐2 and SCAP displayed ~60% reduction in the liver of Cideb −/− mice comparing to those in WT mice (Fig 3A). Consequently, nuclear SREBP‐1/2 were also reduced by ~70% by Cideb deficiency, pointing to a role of Cideb in regulating ER–Golgi transport of SREBP.
Figure 3. Loss of Cideb specifically inhibits loading of SREBP/SCAP into COPII vesicles.
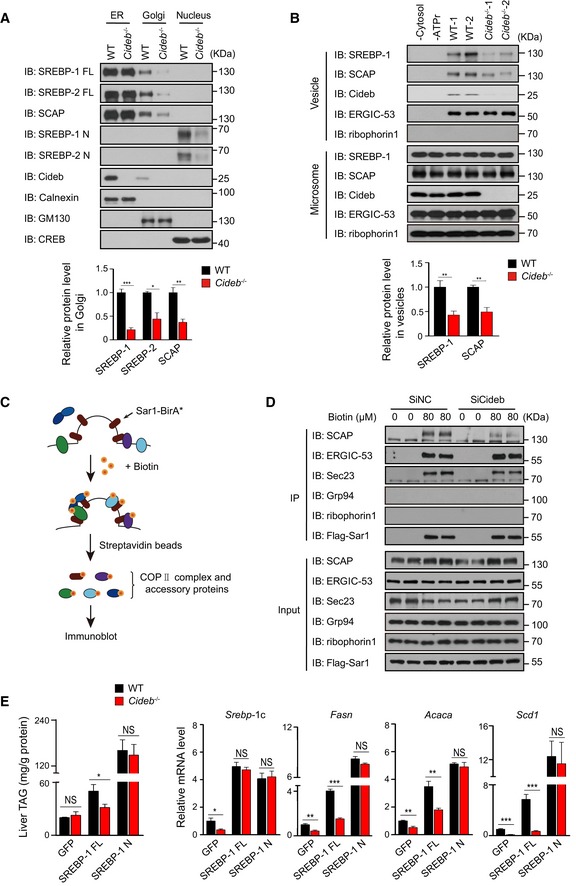
- Decreased SREBP‐1/2 and SCAP levels in Golgi apparatus in Cideb −/− mice. IB of organelle fractions of the indicated proteins and quantification of IB data from three independent experiments.
- Cideb deficiency impairs the packaging of SCAP/SREBP into COPII vesicles. Protein present in the vesicle or microsome fractions obtained from WT or Cideb −/− liver was detected by IB. Quantification of IB data from three independent experiments.
- Schematic of the BioID assay with Sar1A‐BirA*.
- Cideb depletion decreases the recruitment of SCAP to the COPII machinery in cells. Primary hepatocytes transfected with control or Cideb siRNA were infected with adenovirus expressing Sar1A‐Flag‐BirA*. Cells were cultured in sterol‐depleted medium and treated with 80 μM biotin, prior to pull down with streptavidin‐conjugated beads. The biotinylated proteins were detected by IB with the indicated antibodies following SDS–PAGE.
- Cleaved SREBP bypasses Cideb deficiency. WT and Cideb −/− mice were injected with adenovirus expressing GFP, SREBP‐1c full length (FL), or SREBP‐1c active form (N). Mice were sacrificed 7 days after injection, and liver TAG levels and hepatic expression SREBP‐1 target genes in the indicated groups were determined (n = 5 per group).
Next, we directly examined the efficiency of SREBP/SCAP packaging into COPII vesicles with an in vitro budding assay (Fig EV2A), using microsomes isolated from WT or Cideb −/− liver (Nohturfft et al, 2000; Schindler & Schekman, 2009). The addition of cytosol and ATP regeneration system (ATPr) led to efficient recruitment of SCAP/SREBP into the vesicle fractions from WT microsomes (Figs 3B and EV2B). However, this packaging process was substantially reduced, as evidenced by decreased levels of SREBP‐1 and SCAP in the vesicles budded from the microsome of Cideb −/− liver (Fig 3B). In contrast, the packaging of ERGIC‐53, one classical COPII cargos (Nichols et al, 1998), was unaffected by Cideb deficiency (Fig 3B). The ER resident protein ribophorin I was not recovered in the vesicle fractions. Interestingly, Cideb was also detected in the vesicle fractions, implying that Cideb may be a component of COPII vesicles. Since the ER export of SREBP/SCAP was regulated by sterols (Nohturfft et al, 2000), we tested the incorporation of SCAP into COPII vesicles in sterol depletion or 25‐hydroxycholesterol (25‐HC)‐replenished conditions. The addition of sterol blocked the packaging of SCAP into COPII vesicles as reported before (Nohturfft et al, 2000), and Cideb deficiency inhibited the recruitment of SCAP into COPII vesicles under sterol depletion condition. Meanwhile, other COPII cargos such as ERGIC‐53 and Sec22B were not affected by either 25‐HC or Cideb deficiency (Fig EV2C).
Figure EV2. Loss of Cideb specifically inhibits loading of SREBP/SCAP into COPII vesicles.
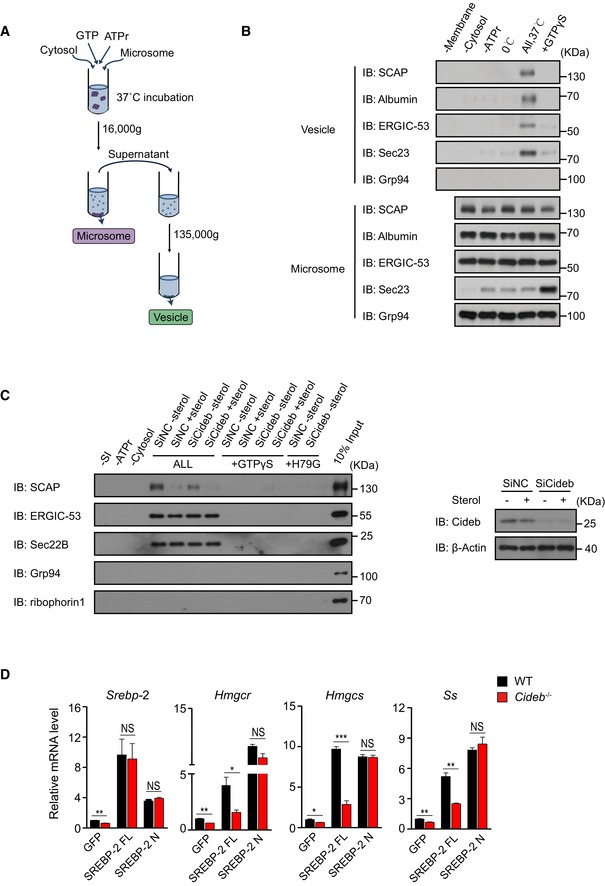
- Schematic of the in vitro reconstitution assay.
- Controls of in vitro reconstitution assay. Hepatic microsomes from WT mice were incubated in the indicated conditions, and the resulting vesicle fractions were analyzed to demonstrate the specificity of the vesicle production from the in vitro reconstitution system.
- Cideb deficiency impairs the recruitment of SCAP into COPII vesicles under sterol depletion condition. WT or Cideb‐deficient primary hepatocytes were treated with or without 25‐HC, and protein presented in the vesicle fractions was detected by IB. 10% input was showed in the last lane to indicate the budding efficiency (left). Cideb knockdown efficiency in the WT hepatocytes (right).
- Cleaved SREBP bypasses Cideb deficiency. WT and Cideb −/− mice were injected with adenovirus expressing GFP, SREBP‐2 full length (FL), or SREBP‐2 active form (N). Mice were sacrificed 7 days after injection, and hepatic expression of SREBP‐2 target genes in the indicated four groups was determined (n = 3 per group). Data represent the Mean ± SEM; NS: not significant; *P < 0.05; **P < 0.01; ***P < 0.001, by 2‐tailed Student's t‐test.
Source data are available online for this figure.
We further employed the BioID assay to assess the dynamic recruitment of COPII machinery and cargo proteins in cells, by fusing the promiscuous biotin ligase BirA* (R118G) to the C‐terminus of Sar1A (Fig 3C) (Nie et al, 2018). In WT hepatocytes, the COPII subunit Sec23, as well as cargos such as SCAP and ERGIC‐53, was biotinylated by Sar1A‐BirA* and captured by streptavidin‐conjugated beads, as a result of COPII assembly and cargo recruitment initiated by the activated Sar1 (Fig 3D). The ER luminal protein Grp94 or ER transmembrane protein ribophorin I was not biotinylated by Sar1A‐BirA* (Fig 3D). Notably, Cideb knockdown decreased the biotinylated SCAP without affecting other proteins such as Sec23 and ERGIC‐53, further confirming a selective effect of Cideb on the recruitment of SCAP into COPII vesicles in cells (Fig 3D).
To further ascertain that Cideb selectively regulates SREBP processing to control lipid metabolism, we tested whether the processed SREBP could bypass Cideb deficiency. To this end, we introduced full‐length SREBP‐1c (FL) or the N‐terminal, cleaved form of SREBP‐1c (N) into WT and Cideb −/− liver, respectively (Figs 3E and EV2D). The expression of FL SREBP‐1c in WT liver caused ~two‐fold increase in hepatic triglyceride (TAG) levels, but failed to achieve so in Cideb −/− livers. On the contrary, modest expression of nuclear form of SREBP‐1c bypassed the Cideb deficiency and resulted in a similar induction of liver TAG in both the WT and Cideb −/− liver (Fig 3E). Similarly, the expression of lipogenic genes including Fasn, Acaca, and Scd1 was robustly induced to the same extent by the processed SREBP in WT and Cideb −/− liver, contrasting the cases of full‐length SREBP (Figs 3E and EV2D). Taken together, our data suggested that Cideb selectively regulates SREBP processing by promoting the packaging of SCAP/SREBP complex into COPII vesicles from the ER.
Sterol‐controlled Cideb/SCAP interaction sensitizes SREBP processing
To dissect the mechanism by which Cideb selectively promotes SCAP/SREBP transport, we screened interactions between Cideb and major components in SREBP transport by co‐immunoprecipitation. SCAP, but not SREBP‐1/2 or Insig, was recovered in the Flag‐Cideb immune‐precipitated complex (Fig 4A). Furthermore, endogenous Cideb was detected in the immuno‐complexes isolated by an anti‐SCAP antibody, as did SREBP‐1 (Fig 4B). Consistent with their functional role in restoring Cideb activity, co‐immunoprecipitation of Cidea and Cidec with SCAP was also observed (Fig EV3A). Molecular mapping showed that Cideb interacted with the region containing a.a.416–800 of SCAP, covering the previously reported motif that interacts with COPII (Fig 4C and D; Sun et al, 2005), while Insig interacted with the transmembrane regions a.a.1–446 of SCAP (Fig EV3B).
Figure 4. Sterol‐controlled SCAP/Cideb interaction sensitizes SREBP processing.
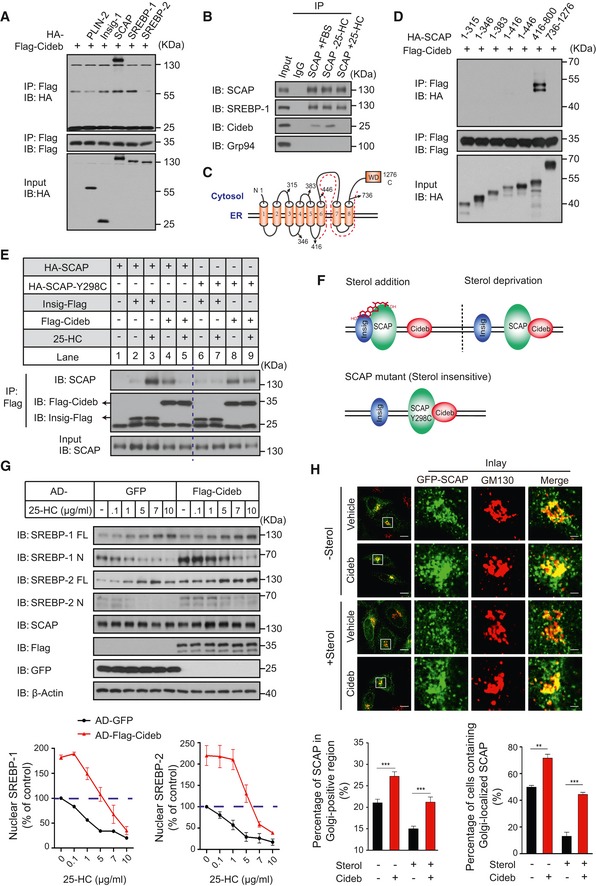
- Cideb co‐precipitates with SCAP. Flag‐tagged Cideb was co‐expressed with different HA‐tagged proteins (PLIN2, Insig, SCAP, SREBP‐1, or SREBP‐2) in 293T cells and immunoprecipitated with an anti‐Flag antibody, and the co‐immunoprecipitated protein was detected by an anti‐HA antibody following SDS–PAGE.
- Interaction between the endogenous SCAP and Cideb. Endogenous SCAP was immunoprecipitated from primary hepatocytes under normal medium, 25‐HC depletion, or addition conditions. And the presence of Cideb in the immune complex was detected by IB. SREBP‐1 was detected as the positive control. Grp94 was detected as the negative control.
- Membrane topology of SCAP. The dashed line labels the portion of SCAP (a.a.416–800) that interacts with Cideb.
- Cideb interacts with the 416–800 portion of SCAP. Flag‐tagged Cideb was co‐expressed with the indicated HA‐tagged SCAP truncation mutants in 293T cells and immunoprecipitated with an anti‐Flag antibody. Levels of the co‐immunoprecipitated protein were detected by an anti‐HA antibody following SDS–PAGE.
- Sterol‐dependent interaction switch of SCAP. SCAP interacts with Insig in the presence of sterol, while switches to Cideb binding upon sterol deprivation. Flag‐tagged Insig‐1 or Cideb was co‐expressed with HA‐tagged SREBP‐2 and SCAP or SCAP‐Y298C in CHO‐K1 cells in sterol‐depleted medium. When indicated, 10 μg/ml 25‐HC was supplemented before cells were immunoprecipitated with an anti‐Flag antibody, and the levels of co‐immunoprecipitated proteins were detected by IB following SDS–PAGE. The dotted blue vertical line was drawn to separate the result between SCAP‐WT and SCAP‐Y298C.
- A “Ping‐Pong” scheme of sterol‐regulated SCAP/Insig and SCAP/Cideb interactions. SCAP switches from Insig binding to Cideb binding in the absence of sterol, while the sterol‐insensitive SCAP‐Y298C consistently interacts with Cideb.
- Cideb relieves sterol‐dependent suppression of SREBP processing. Hepatocytes infected with adenovirus expressing GFP or Flag‐Cideb were cultured in sterol‐deprived condition. When indicated, cells were treated with different doses of 25‐HC before subjected to IB with the indicated antibodies following SDS–PAGE. Quantification of IB data from three independent experiments. Data represent Mean ± SEM.
- Cideb promotes Golgi localization of SCAP. HepG2 cells transfected with GFP‐SCAP alone or with HA‐Cideb were cultured in sterol‐deprived conditions or treated with 1 μg/ml 25‐HC. Cells were fixed and subjected to confocal microscopy following staining with antibodies against GM130 or HA. Scale bars represent 10 μm (Merge) and 2 μm (Inlay). Bottom: quantification of the percentage of SCAP localized in Golgi‐positive region per cell (left) and quantification of the percentage of cells containing Golgi‐localized GFP‐SCAP (right) under indicated conditions, from three independent experiments; Data represent Mean ± SEM; **P < 0.01; ***P < 0.001, by 2‐tailed Student's t‐test.
Source data are available online for this figure.
Figure EV3. Sterol‐controlled SCAP/Cideb interaction.

- CIDE family proteins co‐precipitate with SCAP. Flag‐tagged SCAP was co‐expressed with different HA‐tagged proteins (Cidea, Cideb, or Cidec) in 293T cells and immunoprecipitated with an anti‐Flag antibody, and the co‐immunoprecipitated proteins were detected by an anti‐HA antibody.
- Insig interacts with the transmembrane portion of SCAP. Flag‐tagged Insig was co‐expressed with the indicated HA‐tagged SCAP truncations in 293T cells and immunoprecipitated with an anti‐Flag antibody. Levels of the co‐immunoprecipitated protein were detected by IB following SDS–PAGE.
- Sterol inhibits SCAP/Cideb interaction in a dose‐dependent manner. Flag‐tagged SCAP was co‐expressed with Cideb‐HA and Insig‐HA in CHO‐K1 cells in sterol‐depleted medium. When indicated, different doses of 25‐HC were supplemented before cells were immunoprecipitated with an anti‐Flag antibody, and the levels of co‐immunoprecipitated proteins were detected by IB following SDS–PAGE.
- High level of sterol arrests SCAP on the ER. HepG2 cells transfected with GFP‐SCAP alone or with HA‐Cideb were treated with 10 μg/ml 25‐HC. Cells were fixed and subjected to confocal microscopy following staining with antibodies against GM130 or HA. Scale bars represent 10 μm (Merge) and 2 μm (Inlay). Bottom: quantification of the percentage of SCAP in Golgi‐positive region under indicated conditions; Data represent Mean ± SEM; NS: not significant, by 2‐tailed Student's t‐test. Number of cells used for quantification was listed below the graph.
Source data are available online for this figure.
SREBP processing is suppressed by sterols, which promote the interaction between Insig and SCAP (Yang et al, 2002). Surprisingly, we found an opposite regulatory pattern of SCAP/Cideb interaction by sterols. When assayed by co‐immunoprecipitation (Fig 4E), Insig formed little interaction with SCAP in the absence of sterol (lane 2), while 25‐HC (Hydroxycholesterol) drastically increased interaction between SCAP‐Insig (lane 3) as expected. However, Cideb interacted with SCAP only in the absence of sterol and their interaction was disrupted by 25‐HC (lane 4, 5). Consistently, the addition of 25‐HC blocked the interaction between endogenous SCAP and Cideb in WT hepatocytes (Fig 4B). SCAP‐Y298C, a sterol‐resistant mutant that constantly activated SREBP (Nohturfft et al, 1998), showed little interaction with Insig even in the presence of 25‐HC (lane 6, 7). However, SCAP‐Y298C formed a strong interaction with Cideb in either sterol‐deprived or present conditions (lane 8, 9). Consistently, an increased dose of 25‐HC gradually decreased the interaction between SCAP and Cideb, along with the increased interaction between SCAP and Insig (Fig EV3C). These data pointed to a “Ping‐Pong” scheme in which SCAP switched binding partners, departing Insig and forming interactions with Cideb when SREBP is being activated upon sterol deprivation (Fig 4F).
We further tested whether Cideb could decrease the sensitivity of SREBP processing to sterol levels, a quantitative trait defined by SCAP freed from Insig (Yang et al, 2002; Engelking et al, 2004, 2005). Addition of 25‐HC to GFP‐expressing control cells inhibited SREBP processing in primary hepatocytes, with more than 50% inhibition of SREBP‐1/2 processing at 1 μg/ml sterol. Cideb expression caused ~2‐fold increase of levels of nuclear form of SREBP‐1/2, comparing to control cells in the sterol‐depleted state. The increased SREBP‐1/2 processing persisted in the presence of 1 μg/ml sterol and was only reduced to that observed in sterol‐depleted cells by the addition of 5 μg/ml sterol (Fig 4G). In the addition of 10 μg/ml sterol, SREBP‐1/2 cleavage was reduced to an undetected level in both control cells and Cideb expression cells.
Next, we examined the subcellular localization of SCAP under various sterol concentrations. When quantified by fluorescent signals, ~20% of SCAP signals were localized to the Golgi region in sterol‐deprived control HepG2 cells, and Cideb expression increased SCAP signals in the Golgi to ~27%. Sterol treatment decreased Golgi localization of SCAP to ~15% in control cells; however, the presence of Cideb retained ~20% of SCAP in the Golgi, similar to the control cells in the absence of sterol (Fig 4H). We further quantified the percentage of cells with Golgi‐localized SCAP, by using SCAP signals restricted at the Golgi region in the presence of 10 μg/ml of 25‐HC as baseline (Fig EV3D). In the absence of sterol, ~50% of control cells displayed Golgi localization of SCAP, whereas 72% of Cideb‐expressing cells contained Golgi‐localized SCAP. After 1 μg/ml of 25‐HC treatment, only ~13% of control cells displayed Golgi localization of SCAP. Notably, the presence of Cideb maintained Golgi localization of SCAP in ~44% of cells (Fig 4H). Taken together, the biochemical and imaging data revealed that Cideb, via a sterol‐dependent interaction with SCAP, promoted the transportation of SCAP/SREBP from the ER to the Golgi to enhance SREBP processing.
Cideb facilitates SREBP/SCAP loading to COPII complex by interacting with Sec12
The sensitizing effects on the ER‐to‐Golgi delivery of SCAP/SREBP by Cideb prompted us to further examine whether Cideb may also interact with proteins involved in the production of COPII vesicles. Interestingly, Sec12, the GEF for Sar1 GTPase, was selectively recovered from the Flag‐Cideb immuno‐complex, whereas Sar1, Sec23, Sec13, Sec31, or Sec16 was not detected (Figs 5A and EV4A and B). As Sec12 initiates the assembly of the COPII complex at the ER exit sites (ERESs) that is also marked by Sec16, we examined whether SCAP and Cideb are being enriched at ERESs prior to COPII‐mediated ER export (Presley et al, 1997; Budnik & Stephens, 2009). Indeed, triple staining showed that Cideb and SCAP were localized at the ERESs marked by Sec16A or Sec12 (Figs 5B and EV4C). We further verified the localization of endogenous Cideb by knocking in a GFP tag into the C‐terminus of Cideb with the CRISPR/Cas9 system (Fig EV4D). The endogenous Cideb also exhibited precise co‐localization with a subset of Sec12‐positive ER exit sites (Fig 5C), supporting the notion that Cideb is localized to the ER exit sites.
Figure 5. Cideb enhances SREBP processing by interacting with Sec12.
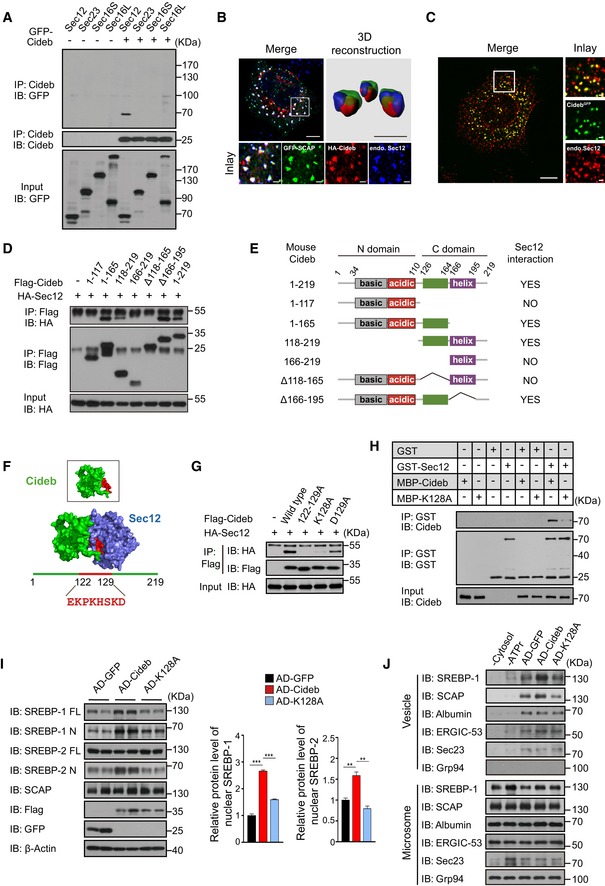
- Cideb interacts with Sec12. 293T cells transfected with Cideb and GFP‐tagged Sec12, Sec23, Sec16S, or Sec16L were subjected to immunoprecipitation with an anti‐Cideb antibody, and levels of the co‐immunoprecipitated protein were detected by an anti‐GFP antibody.
- Co‐localization of SCAP and Cideb at the ER exit sites. HepG2 cells transfected with GFP‐SCAP and HA‐Cideb were arrested with 10 μM BFA and 1 μM nocodazole for 2 h, then fixed with methanol, and stained with antibodies against Sec12 or HA. 3D reconstruction was obtained by processing 3D‐original image with Imaris. Scale bars represent 10 μm (Merge) and 1 μm (Inlay and 3D reconstruction).
- Localization of Cideb at the ER exit sites. AML12 cells knocked‐in with a GFP tag at the C‐terminus of Cideb genome were treated with 10 μM BFA for 1 h, then fixed with methanol, and stained with antibodies against Sec12. Scale bars represent 10 μm (Merge) and 1 μm (Inlay).
- Sec12 interacts with the 118–165 portion of Cideb. HA‐tagged Sec12 was co‐expressed with the indicated Flag‐tagged Cideb truncation mutants in 293T cells and immunoprecipitated with an anti‐Flag antibody. Levels of the co‐immunoprecipitated protein were detected by an anti‐HA antibody following SDS–PAGE.
- The schematic diagram of Cideb and its truncations.
- Sec12/Cideb interaction modeled by molecular dynamic simulation. The red portion highlighted the predicted domain of Cideb interacting with Sec12.
- Lysine 128 of Cideb is required for the interaction with Sec12. 293T cells transfected with HA‐Sec12 and the indicated Flag‐tagged Cideb constructs were subjected to immunoprecipitation with an anti‐Flag antibody, and levels of the co‐immunoprecipitated HA‐Sec12 were detected with an anti‐HA antibody.
- Recombinant Cideb interacts with Sec12. GST‐Sec12 and MBP‐tagged Cideb or Cideb‐K128A were purified from E. coli and subjected to in vitro binding.
- Cideb‐K128A fails to promote SREBP processing. Cideb −/− mice were injected with adenovirus expressing GFP, Flag‐Cideb, or Flag‐Cideb‐K128A, and sacrificed 7 days after injection. Liver lysates were subjected to IB with the indicated antibodies following SDS–PAGE and quantification of IB data from three independent experiments. Data represent Mean ± SEM; **P < 0.01; ***P < 0.001, by 2‐tailed Student's t‐test.
- Cideb, but not Cideb‐K128A, promotes SCAP/SREBP incorporation into COPII vesicles. Liver microsomes were isolated from mice expressing the indicated proteins and subjected to in vitro budding assay. Resulting vesicles and the donor microsomes were subject to IB with the indicated antibodies following SDS–PAGE.
Source data are available online for this figure.
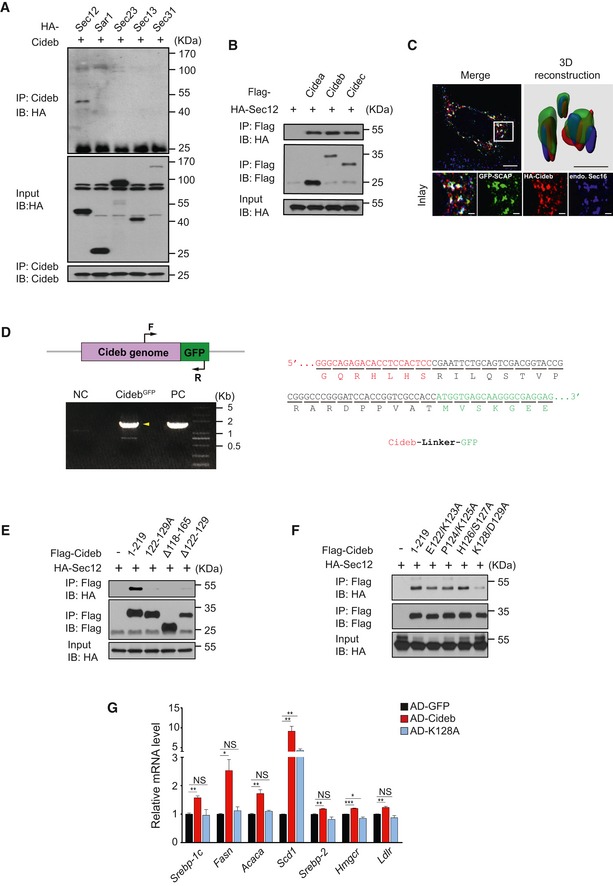
Figure EV4. Cideb enhances SREBP processing by interacting with Sec12.
-
ACideb interacts with Sec12. 293T cells transfected with Cideb and HA‐tagged Sar1, Sec23, Sec13, or Sec31 were subjected to immunoprecipitation with an anti‐Cideb antibody, and levels of the co‐immunoprecipitated protein were detected by an anti‐HA antibody following SDS–PAGE.
-
BCIDE family protein co‐precipitates with Sec12. HA‐tagged Sec12 was co‐expressed with different Flag‐tagged proteins (Cidea, Cideb, or Cidec) in 293T cells and immunoprecipitated with an anti‐Flag antibody, and the co‐immunoprecipitated protein was detected by an anti‐HA antibody following SDS–PAGE.
-
CCo‐localization of SCAP and Cideb at the ER exit sites. HepG2 cells transfected with GFP‐SCAP and HA‐Cideb were arrested with 10 μM BFA and 1 μM nocodazole for 2 h, then fixed with methanol, and stained with antibodies against Sec16 or HA. 3D reconstruction was obtained by processing 3D‐original image with Imaris. Scale bars represent 10 μm (Merge) and 1 μm (Inlay and 3D reconstruction).
-
DCharacterization of CidebGFP knock‐in cells. Left: schematic representation of GFP fused to the C‐terminus of Cideb. Arrows indicated PCR primers designed for genotyping (top). Genotyping analysis. Normal AML12 cells were used as negative control (NC), and plasmid containing Cideb genome and GFP fusion sequence was used as positive control (PC). Yellow arrowhead indicated the PCR product containing Cideb genome and GFP (bottom). Right: sequencing analysis of the CidebGFP genome.
-
E, FHA‐tagged Sec12 was co‐expressed with the indicated Flag‐tagged Cideb truncation or mutation in 293T cells and immunoprecipitated with an anti‐Flag antibody. Levels of the co‐immunoprecipitated protein were detected by an anti‐HA antibody following SDS–PAGE.
-
GCideb‐K128A fails to increase the expression of SREBP target genes. Cideb −/− mice were injected with adenovirus expressing GFP, Flag‐Cideb, or Flag‐Cideb‐K128A. Mice were sacrificed 7 days after injection, and hepatic expression of SREBP target genes in the indicated groups was determined by qPCR (n = 3 per group). Data represent the Mean ± SEM; NS: not significant; *P < 0.05; **P < 0.01; ***P < 0.001, by 2‐tailed Student's t‐test.
Source data are available online for this figure.
Domain mapping showed that a.a.118‐165 of Cideb was responsible for interacting with Sec12 (Fig 5D and E). Molecular dynamic simulation based on individual structures of Sec12p and Cideb (Lugovskoy et al, 1999; McMahon et al, 2012) indicated that a peptide covering a.a.122–129 of Cideb may contribute to Sec12/Cideb interaction (Fig 5F). Indeed, deletion of this peptide or mutating all amino acids in this region to alanines (122–129A) in Cideb completely disrupted its interaction with Sec12 (Fig EV4E). Further mutagenesis in this region pinpointed to a single point mutation of Cideb at K128A (Cideb‐K128A), which abolished the interaction between Cideb and Sec12 (Figs 5G and EV4F). Furthermore, recombinant GST‐Sec12 protein displayed a specific interaction with MBP‐Cideb, but not MBP‐Cideb‐K128A, revealing a direct binding between these two proteins (Fig 5H).
To assess whether Cideb/Sec12 interaction was required for the increased SREBP processing by Cideb, we introduced WT Cideb or Cideb‐K128A to the Cideb −/− liver. While modest expression of Cideb‐WT restored the cleavage of both SREBP‐1 and SREBP‐2, Cideb‐K128A expression failed to rescue SREBP processing (Fig 5I). Concomitantly, the mRNA levels of SREBPs and their target genes exhibited a similar pattern (Fig EV4G). In vitro budding assay showed that Cideb, but not Cideb‐K128A, increased the packaging of SCAP/SREBP into COPII vesicles synthesized in the reconstitution system. Other proteins, such as albumin, ERGIC‐53, and Sec23, were not affected by the over‐expression of either Cideb or Cideb‐K128A (Fig 5J).
Since both ApoB (mediated VLDL secretion) and Sec12 (mediated SREBP activation) bound to the similar region (a.a.118–165) of Cideb (Ye et al, 2009), we further dissected the relationship between the role of Cideb in VLDL secretion and SREBP/SCAP trafficking. Co‐immunoprecipitation revealed that increasing dose of Sec12 potentiated Sec12/Cideb interaction, but did not affect ApoB/Cideb interaction (Fig EV5A), indicating that ApoB and Sec12 bound to Cideb in a non‐competitive manner. In addition, Cideb‐K128A, a mutant failed to interact with Sec12, maintained similar interaction with ApoB (Fig EV5B), indicating that Cideb differentially interacted with ApoB and Sec12. Further, we found that both wild type and Cideb‐K128A were able to rescue VLDL lipidation defects in Cideb −/− liver (Fig EV5C–G). These results suggested that Cideb‐mediated VLDL lipidation and SREBP trafficking are independent of each other.
Figure EV5. Cideb plays independent roles during SREBP transport and VLDL lipidation.
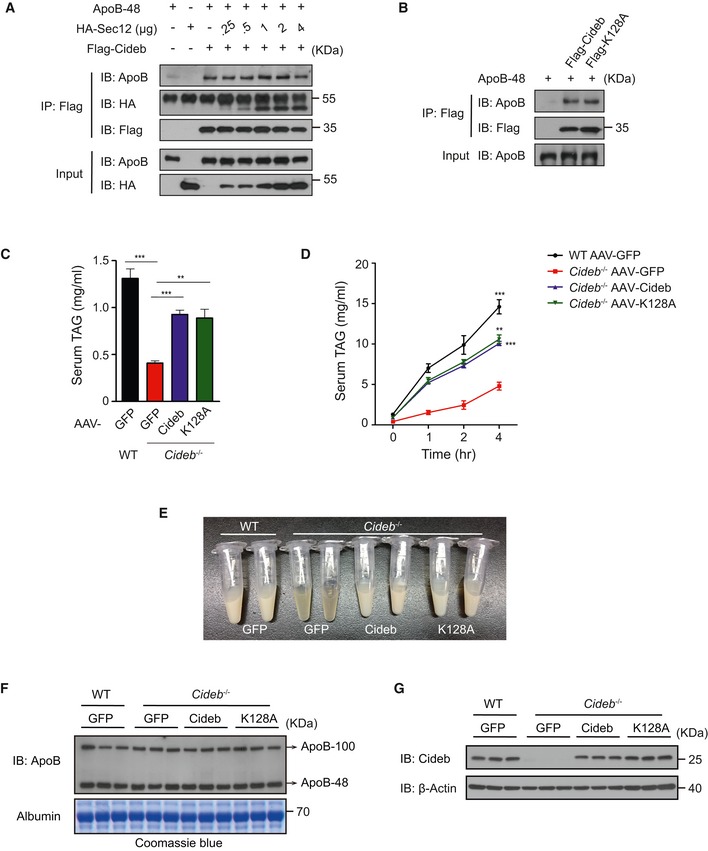
- The interaction between ApoB/Cideb and Sec12/Cideb is not competitive. Flag‐tagged Cideb was co‐expressed with apoB‐48 and an increased dose of HA‐Sec12 in 293T cells. Cells were immunoprecipitated with an anti‐Flag antibody, and the levels of co‐immunoprecipitated proteins were detected by IB following SDS–PAGE.
- Cideb‐WT and Cideb‐K128A show similar interaction with ApoB. Flag‐tagged Cideb or Cideb‐K128A was co‐expressed with apoB‐48 in 293T cells. Cells were immuno‐precipitated with an anti‐Flag antibody, and the levels of co‐immunoprecipitated ApoB‐48 were detected by IB following SDS–PAGE.
- Levels of serum TAG in WT and Cideb −/− mice injected with AAV containing GFP, Cideb‐WT, or Cideb‐K128A (n = 4 per group) under 16‐h fasting condition.
- In vivo VLDL‐TAG secretion. Mice mentioned in (C) were injected with Triton WR‐1339 (500 mg/Kg in PBS), and serum samples at indicated time point were collected for TAG analysis.
- Represented photograph image of serum samples from mice treated with Triton WR‐1339 for 4 h.
- Serum ApoB levels. Levels of serum ApoB‐48/100 in WT and Cideb −/− mice expressed GFP, Cideb, or Cideb‐K128A after 16‐h fasting and 4‐h Triton WR‐1339 injection. Albumin stained with Coomassie Blue G‐250 was used as loading control.
- Expression levels of Cideb in WT and Cideb −/− liver.
Cideb enhances SREBP/SCAP loading at ER exit site
To further understand the role of Sec12/Cideb interaction in promoting the ER export of SREBP/SCAP, we employed quantitative imaging to evaluate the spatial recruitment of SCAP to ERESs prior to COPII‐mediated export in the presence of Cideb or Cideb‐K128A (Fig 6A). As previously reported (Presley et al, 1997), nocodazole treatment resulted in the localization of SCAP to dispersed ERESs that is positive for Sec12. Expression of Cideb, but not Cideb‐K128A, significantly increased both the number of SCAP‐positive ERESs and the intensity of SCAP at each ERES (Fig 6A–D and Appendix Fig S2A). Overall, Cideb expression led to ~50% increase of the total amount of SCAP at all ERESs in a cell, representing a significant enrichment of SCAP at ERESs (Fig 6E). Expression of Cideb or Cideb‐K128A did not affect the total number of ERESs that were positive for Sec12 (Fig 6F and Appendix Fig S2B). Taken together with the biochemical data, these results suggested that Cideb, via interacting with Sec12, enriched SCAP/SREBP to the active budding COPII machinery at the ERESs, therefore enhancing the transport of SREBP to the Golgi for processing (Fig 6G).
Figure 6. Cideb enhances the targeting of SREBP/SCAP to ER exit sites.
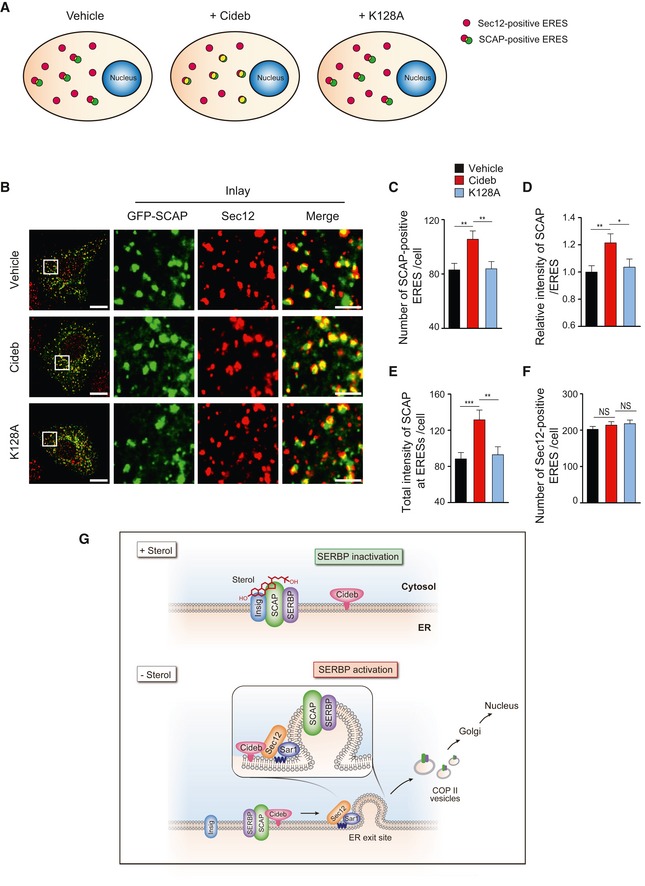
- Schematic of SCAP targeting to ER exit sites under different conditions. The red puncta represent the Sec12‐positive ER exit sites, and the yellow puncta represent the SCAP‐positive ER exit sites.
- Cideb enriches SCAP to ERESs via interacting with Sec12. CHO‐K1 cells transfected with GFP‐SCAP, HA‐Cideb, or HA‐Cideb‐K128A were arrested with 5 μM nocodazole for 30 min, then fixed with methanol, and stained with antibodies against Sec12 and HA. Scale bars represent 10 μm (left) and 2.5 μm (Inlay).
- Number of SCAP‐positive ER exit sites per cell.
- Normalized fluorescent intensity of SCAP at each individual ER exit site.
- Total fluorescent intensity of SCAP at ER exit sites per cell.
- Number of Sec12‐positive ER exit sites per cell.
- Model depicting the role of Cideb in enhancing SREBP processing. When cellular sterol level is high, SCAP binds to Insig, resulting in the retention of SREBP/SCAP on ER and low SREBP activation. The absence of sterol frees SCAP from Insig and induces the interaction between Cideb and SCAP, leading to the targeting of SREBP/SCAP to the ERESs via the interaction between Cideb and Sec12 and the enrichment of SREBP/SCAP in Golgi. Cleaved SREBP enters nucleus and activates the expression of fatty acids/cholesterol synthesis genes.
Discussion
Precise regulation of SREBP processing and activation is pivotal to lipid homeostasis (Brown & Goldstein, 2009). Here, we demonstrate that Cideb acts as a critical factor to promote SREBP processing by interacting with SCAP and Sec12 in response to deprivation of dietary sterol. These interactions of Cideb efficiently tether the SREBP/SCAP complex to the assembling COPII complex at the ER exit sites, thereby enhancing the loading of SREBP/SCAP into COPII vesicles for their delivery to the Golgi apparatus in response to sterol deprivation. Consistently, increased Cideb expression causes accelerated processing of SREBP, leading to resistance to sterol inhibition of SREBP processing, whereas loss of Cideb blunted SREBP activation and protected mice from lipogenic diet‐induced hepatic steatosis.
The recognition of different cargos by the COPII complex is a prerequisite step in ensuring specificity of cargo delivery at the entrance of the secretory pathway (Gillon et al, 2012). Our work demonstrates that Cideb is triggered to form complex with SCAP in response to falling supply of exogenous sterol. Intriguingly, the domain of SCAP (a.a.416–800) mediating its interaction with Cideb coincidentally contains the MELADL motif that interacts with the COPII subunit Sec24C (Sun et al, 2005). It is possible that conformational changes of SCAP in the absence of sterol allow the exposure of the regions that interact with Cideb and COPII. Direct interaction between SCAP and the COPII subunit Sec24C may allow a constitutive yet less efficient SREBP processing (Sun et al, 2007). However, the formation of the SCAP/Cideb/Sec12 complex may generate a spatially oriented “molecular crowding” effect, enriching the reactants to enhance the specificity and overall efficiency of SREBP/SCAP cargo loading into COPII vesicles at ER exit sites. In fact, ER‐to‐Golgi transport represents a central node among the different levels of SREBP regulation. In previous reports, Insulin modulates SREBP pathway by promoting the association of SCAP/SREBP with COPII components (Yellaturu et al, 2009). The transcriptional coactivator CRTC2 decreases the ER export of SREBP by disrupting COPII vesicles formation (Han et al, 2015). Hence, the recognition and incorporation of SREBP/SCAP by COPII represent a plausible target to control SREBP activity and lipogenesis.
The role of Cideb in regulating SREBP/SCAP cargo loading may be analogous to other factors, such as TANGO1 and Sedlin, which have been shown to load unique cargos like pro‐collagen to specialized COPII transport vesicles for their ER export (Saito et al, 2009; Venditti et al, 2012; Liu et al, 2017). It is particularly noteworthy that unlike TANGO1 and Sedlin, Cideb may be part of the COPII vesicles. The conclusion is supported by the detection of endogenous Cideb in Golgi apparatus following a biochemical subcellular fractionation and the detection of Cideb in the in vitro reconstituted COPII vesicle fractions. However, compared with other cargo proteins such as SCAP, ERGIC‐53, and Sec22B, the incorporation of Cideb into COPII vesicles only involved a smaller percentage of total Cideb. This might reflect the nature of Cideb as a multi‐functional protein or potentially additional regulation for Cideb trafficking which we failed to recapitulate with our current in vitro reconstitution assay.
We have previously shown that Cideb −/− mice contained reduced plasma cholesterol and NEFA, and Cideb −/− liver shows 50% reduction in fatty acid synthesis rates and ~60% reduction in cholesterol synthesis (Li et al, 2007, 2010). Here, we demonstrate that mature forms of both SREBP‐1/2 and the expression of their downstream target genes are significantly decreased in Cideb −/− liver under both normal diet and the pro‐lipogenic diet (HFLF). These phenotypes are strikingly similar to those of liver‐specific knockout of Scap (Matsuda et al, 2001). Consistently, when SREBP activity was induced by the pro‐lipogenic HFLF diet, Cideb −/− mice were protected from the diet‐induced pathologies including liver steatosis and inflammation. These data further support the notion that Cideb acts as a physiological regulator of the SCAP/SREBP axis.
We found that Cidea and Cidec, two Cideb‐related proteins (Wu et al, 2008), also interact with SCAP and Sec12 and are able to functionally replace Cideb in SREBP processing and activation. Therefore, besides liver, CIDE proteins may also regulate SREBP processing in adipose tissues, mammary gland, and skin sebocytes where high levels of Cidea or Cidec expression are observed and active lipogenesis is required to maintain their function in lipid homeostasis (Tontonoz et al, 1993; Gao et al, 2017).
As Cideb has also been shown to enhance VLDL lipidation (Ye et al, 2009), the question then arises whether SREBP inhibition in Cideb‐deficient condition is due to an indirect effect of lipid accumulation. Though defective VLDL secretion or lipidation leads to hepatic lipid accumulation in the form of lipid droplets, these defects may not proportionally result in SREBP inhibition (Lin et al, 2002; Smagris et al, 2016). Indeed, SREBP is fine‐tuned by sterol levels on ER membrane, but not total lipid contents in cells, likely reflecting the nature of SCAP as a sensor of membrane sterols (Radhakrishnan et al, 2008). In addition, our current study pointed that Cideb −/− liver exhibited defective SREBP processing and downstream genes expression with similar (under normal diet) or even lower (under HFLF diet) hepatic TAG levels compared with the control mice, indicating that there was no correlation between increased lipid accumulation and inhibition of SREBP activity in Cideb −/− liver. Further, our result pointed that the role of Cideb on SREBP activation and VLDL secretion was independent of each other. These points mentioned above, combined with the extensive evidence showing the regulation of Cideb on SREBP ER export in the current study, strongly suggested that Cideb controls the ER export of SREBP/SCAP by facilitating their cargo loading at ERESs.
Cideb also promotes LD fusion and lipid storage (Xu et al, 2016), representing the third aspect of Cideb's function that may affect hepatic lipid levels. Under normal diet condition, the absence of Cideb reduced SREBP activity and LD fusion and led to lower hepatic lipid accumulation. Meanwhile, Cideb deficiency reduced VLDL lipidation and resulted in an increased hepatic lipid accumulation. The combinatory effects of these processes resulted in the similar lipid levels in WT and Cideb −/− mice under normal diet condition. However, when mice were treated with pro‐lipogenic high‐fructose low‐fat diet, the SREBP pathway would be activated and the deficiency of Cideb would significantly decrease the level of hepatic TAG. Therefore, we postulated that different dietary nutrients may play important roles in exerting the role of Cideb in controlling different lipid metabolic pathways. For instance, under fasting condition, Cideb induces VLDL lipidation and secretion when fatty acids are supplied by the adipose tissue. Under high‐fat diet and fatty acid abundant condition when TAG synthesis is active, Cideb promotes the fusion of LDs and increases lipid storage. In response to low‐fat or low‐cholesterol conditions, Cideb promotes the ER export of SREBP and lipid synthesis. De novo synthesized lipids could then be re‐esterified and stored in LDs, providing a cell‐autonomous positive loop for Cideb‐mediated lipid synthesis and storage. Overall, Cideb acts as a critical regulatory factor to coordinate hepatic lipid homeostasis by modulating multiple aspects of lipid metabolism including LD fusion, lipid synthesis, and lipid secretion in different metabolic conditions. Conceivably, approaches leading to Cideb inhibition would consequently down‐regulate lipid synthesis and accumulation, either in the liver or in extra‐hepatic tissues, thereby providing beneficial effects in combating lipotoxicity associated with obesity and fatty liver diseases.
Materials and Methods
Plasmids, antibodies, chemical reagents, and recombinant protein
GFP‐Sec16S, GFP‐Sec16L, AAV‐CMV‐GFP, AAV‐TBG‐GFP, Rep/Cap 2/8, and adenoviral helper plasmid constructs were obtained from Addgene. HA‐Sec13 and HA‐Sec31 constructs were gifts from Dr. Y.G. Wang (Tsinghua University, Beijing, China). Point mutations of Cideb and SCAP were generated by a PCR‐based site‐directed mutagenesis method (StrataGene). Each plasmid was verified by DNA sequencing.
Antibodies against SREBP‐1 (sc‐13551, sc‐8984), GFP (sc‐8334), albumin (sc‐46293), Sec22B (sc‐101267), ribophorin I (sc‐48367), Grp94 (sc‐32249), and HA (sc‐7392) were purchased from Santa Cruz Biotechnology (USA). SREBP‐2 antibody (04‐479) was purchased from Merck Millipore (USA). Antibodies against SCAP (13102), CREB (9197), Sec23 (8162), and Scd‐1 (2438) were purchased from Cell Signaling Technology (USA). Antibody against GM130 (bd610823) was purchased from BD (USA). Antibodies against Sec12 (ab3422) for IB were purchased from Abcam (USA), and (6B3) for IF was gift from Kota Saito (Tokyo University, Japan). Antibodies against Fasn (ab128870) and HMGCR (ab174830) were purchased from Abcam (USA). Antibody against Sec16 (A300‐648A) was purchased from Bethyl Laboratories, Inc. (USA). Antibodies against ERGIC‐53 (E1031), β‐actin (A5441), calnexin (C4731), Flag (F1804), anti‐Flag M2 agarose beads (A2220), and EZview™ Red Streptavidin Affinity Gel (E5529) were purchased from Sigma‐Aldrich (USA). Antibodies against GST (AB101) were purchased from Tiangen (China). Western blots were developed using HRP‐conjugated anti‐mouse (NA‐931), anti‐rabbit (NA‐934) secondary antibodies from GE Healthcare (USA). The rabbit polyclonal antibodies against mouse Cidea, Cideb, and Cidec were generated by the injection of rabbits with His‐tagged truncated mouse Cidea (a.a.1–195), mouse Cideb (a.a.1–176), and mouse Cidec (a.a.1–190) recombinant proteins that were expressed in and purified in Escherichia coli.
Brefeldin A (S7046) and nocodazole (S2775) were purchased from Selleckchem. GST‐Sec12, MBP‐Cideb, and MBP‐Cideb‐K128A were purified from bacteria using a standard protocol.
Animal procedures
Cideb −/− mice were generated and maintained as previously described (Li et al, 2007). All mice were housed in an environmentally controlled animal facility (SPF) of the Center of Biomedical Analysis, Tsinghua University, Beijing, China. The laboratory animal facility in Tsinghua University was accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) and the Institutional Animal Care and Use Committee (IACUC). Mice had access to food and autoclaved water ad libitum. The mice were handled according to the Responsible Care and Use of Laboratory Animals guideline set by Tsinghua University, and the animal protocols were approved by the animal ethics committee of Tsinghua University. For the re‐feeding experiment, mice were fed with high‐carbohydrate diet (TD.98090, Harlan Teklad Diets, with small modification: 72.6% carbohydrate, 2.6% fat, and 17.7% protein by weight). For the HFLF feeding experiment, mice were fed ad libitum to high‐fructose low‐fat diet (TD.89827, Harlan Teklad Diets, with small modification: 63% fructose, 2.6% fat, and 18.3% protein by weight) for 2 months.
Glucose tolerance tests of WT and Cideb −/− mice were performed in overnight‐fasted mice (16 h) following an intraperitoneal injection of glucose (1.0 g/kg body weight), and insulin tolerance tests were carried out in 4‐h‐fasted mice following an intraperitoneal injection of recombinant human insulin (0.5 units/kg body weight). The tail blood sample was collected, and blood glucose levels were measured at indicated time point by using Accu‐Chek Performa (Roche). Plasma TAG levels were measured by free glycerol and TAG reagent (F5428, T2449, Sigma). Plasma cholesterol level was measured by using cholesterol quantification kit (MAK043, Sigma) following the manufacturer's instructions. Plasma NEFA level was quantified by enzymatic assay kit (294‐63601, Wako). Plasma insulin level (ab100578, Abcam), MCP1 level (ab100721, Abcam), TNFα level (430907, BioLegend), and IL6 (431307, BioLegend) were quantified by ELISA kit.
Primary hepatocyte isolation
Primary hepatocytes were isolated from livers of 12‐week‐old mice as follows. Mice were anesthetized with 1% Pelltobarbitalum Natricum (AMRESCO, USA) and dissected. The liver was perfused through the portal vein to remove the residual blood and followed by perfusion with type IV collagenase (C5138, Sigma) until the liver became soft. The liver was immediately removed, cut into pieces, and filtered through a 70‐mm membrane (Millipore, USA) to remove tissue debris. After washing twice with cold DMEM and centrifuging at 50 g for 5 min at 4°C, the isolated hepatocytes were suspended in DMEM containing 10% FBS and seeded at 1 × 106 cells/dish in 6‐cm dishes. Cells were maintained in 5% CO2 at 37°C.
TAG measurement
The extraction of TAG from liver, adipocytes, and muscle was performed as described previously (Folch et al, 1957). Briefly, liver was homogenized in PBS, and the homogenates were rapidly mixed with a chloroform/methanol (2:1, v/v) solution by vortex and centrifuged at 3,000 g for 10 min to separate the phases. The lower lipid‐containing phase was carefully aspirated and dried in a 70°C metal bath with nitrogen steam. The dried lipids were reconstituted in methylbenzene and loaded onto a TLC plate. The lipids were separated in a hexane/diethyl ether/acetic acid (70:30:1, by volume) solution. The TLC plates were sprayed with 10% CuSO4 in 10% phosphoric acid and developed by drying in a 120°C oven. TAG spots of samples and standard on TLC plates (Sigma‐Aldrich) were scanned and quantified using Quantity One software (Bio‐Rad). TAG levels were normalized with the protein concentration of each sample.
Cell culture
293T cells, AD293 cells, and HepG2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, USA) containing 10% FBS (Invitrogen). CHO‐K1 cells were cultured in DMEM/F‐12 (Invitrogen) containing 5% FBS, and AML12 cells were cultured in DMEM/F‐12 containing 10% FBS, 10 μg/ml insulin, 5.5 μg/ml transferrin, 5 ng/ml selenium (Invitrogen), and 40 ng/ml dexamethasone. All medium contained 2 mM l‐glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were cultured at 37°C in a humidified incubator containing 5% CO2.
Preparation of lipoprotein‐deficient serum (LPDS)
LPDS was prepared as described before (Goldstein et al, 1983). Briefly, fetal bovine serum was adjusted to a final density of 1.215 g/ml with KBr and centrifuged at 180,000 g for 24 h at 10°C. Following centrifugation, the top fraction containing lipoproteins was separated from the bottom fraction containing the LPDS. LPDS were pooled together and then dialyzed against 150 mM NaCl for 48–72 h at 4°C. After dialysis, LPDS was adjusted to a protein concentration of 50 mg/ml by dilution with 150 mM NaCl, filter sterilized, and kept frozen until use.
Sterol treatment
Sterol treatment of the cells was performed as previously described (Nohturfft et al, 2000). For HepG2 or hepatocytes, the medium was changed to DMEM containing 10% LPDS, 1% (w/v) hydroxypropyl‐β‐cyclodextrin, 50 μM sodium mevalonate, and 50 μM compactin and incubated for 1 h. Then, the cells were incubated for 3 h with DMEM containing 10% LPDS, 50 μM sodium mevalonate, and 50 μM compactin in the absence or presence of 25‐HC at indicated concentration.
Isolation of ER, Golgi, and nucleus fractions
Subcellular fractionation was performed essentially as previously described (Gusarova et al, 2003). Livers were removed and rapidly homogenized with a loose‐fitted Dounce homogenizer in 5 ml of ice‐cold solution containing 10 mM HEPES, pH 7.4, 150 mM sucrose, 0.5 mM DTT, and 1× EDTA‐free protease inhibitors cocktails (Roche). The homogenates were centrifuged at 1,900 g for 10 min at 4°C, the subsequent pellets were prepared for further nucleus isolation (re‐suspended with buffer containing 20 mM HEPES, PH 7.6, 0.42 M NaCl, 1.5 mM MgCl2, 2.5 w/v glycerol, 1 mM EDTA, 1 mM EGTA, rotate for 1 h at 4°C, and centrifuged at 185,000 g for 1 h at 4°C in a Beckman SW41 rotor. The supernatants were collected as nucleus fraction), and the supernatants were centrifuged at 100,000 g for 90 min at 4°C in a Beckman SW60 rotor. The pellets were re‐suspended in 2.3 ml of an 8.58% (w/v) sucrose solution and loaded on top of a sucrose density gradient containing the following layers (from the bottom): 56% (0.46 ml), 50% (0.92 ml), 45% (1.38 ml), 40% (2.3 ml), 35% (2.3 ml), 30% (1.38 ml), and 20% (0.46 ml) sucrose. After ultracentrifugation at 39,000 rpm for 18 h at 4°C in a Beckman SW41 rotor, 22 fractions were unloaded from top to bottom of each centrifuge tube. The distribution patterns of the subcellular compartment markers were determined by IB (ER marker: calnexin; Golgi apparatus marker: GM130) and pooled together as ER and Golgi fraction. All procedures were performed at 4°C, and protease inhibitors cocktails were added.
In vitro budding using isolated microsomes
In vitro budding experiments using isolated microsomes were performed as previously described with slight modifications (Nohturfft et al, 2000; Schindler & Schekman, 2009). Liver from 2‐month mice was perfused with 0.9% (w/v) NaCl and rapidly homogenized in 3 ml of ice‐cold solution A (50 mM HEPES, PH 7.2, 250 mM Sorbitol, 70 mM KOAc, 2.5 mM Mg(OAc)2) plus 1 mM DTT. The homogenates were centrifuged at 1,900 g for 10 min, and the supernatants were sequentially centrifuged at 20,000 g for 20 min, 200,000 g for 1 h, and 200,000 g for 1 h. The cytosol fraction was carefully collected to get rid of the fat layer and dialyzed with solution A plus 1 mM DTT for 3–4 h. The final supernatant was divided into multiple aliquots and stored at −80°C. Microsomes were isolated as “Isolation of ER and Golgi” mentioned above, except that after centrifuged at 1,900 g for 10 min, the supernatant was centrifuged at 20,000 g for 30 min, and the spin pellet was re‐suspended with solution A and divided into multiple aliquots.
Budding reactions (100 μl) were assembled in non‐stick Eppendorf tubes on ice with 80 μg microsomes as donor membranes, ATP regeneration system (1.5 mM ATP, 10 mM creatine phosphate, and 4 units/ml creatine phosphokinase), 0.5 mM GTP, and 700 μg liver cytosol. Reactions were performed at 37°C for 1 h and stopped by centrifuging at 16,000 g for 10 min at 4°C. 75 μl supernatant was centrifuged in a TLA‐100 rotor at 135,000 g for 10 min at 4°C. The supernatants were discarded by pipetting with gel loading tips, and the high‐speed pellet fractions (include COPII vesicles) were thoroughly re‐suspended in 20 μl of 1× SDS sample buffer and heated at 55°C for 20 min before SDS–PAGE.
Immunoprecipitation and Western blot
Immunoprecipitation (IP) and Western blot (WB) were carried out as described previously (Ye et al, 2009; Nie et al, 2018). For IP of endogenous proteins, cells were collected and lysed in IP buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X‐100) by sonication. Antibodies or pre‐immune serum were conjugated to protein A/G beads and used for IP. WB was blocked with 5% fat‐free milk and secondary antibodies.
Immunofluorescent staining and confocal microscopy
For sterol deprivation or addition condition, cells were rinsed twice in PBS, fixed with 4% PFA for 20 min, permeabilized with 0.4% Triton X‐100 in PBS for 20 min, blocked with 5% BSA in PBS for 30 min, followed by incubation with primary antibody for 2 h, washed three times with PBS, and incubated with fluorescently labeled secondary antibody (1:500 dilution) for 1 h. For ERESs localization, cells were pre‐treated with Brefeldin A and nocodazole for the indicated time and then fixed with ice‐cold methanol for 6 min. Coverslips were mounted after three times washing with PBS. Images for protein localization were acquired under A1 confocal microscope (Nikon) with 100× oil immersion objective.
To quantify the Golgi localization of SCAP, the images of 1,024 × 1,024 pixels were processed by ImageJ. The total intensity gray levels of SCAP on Golgi‐positive region (defined by GM130, IG) and in the whole cell region (IC) were measured, and the ratio (RG) was calculated by IG divided to IC. In the condition of 10 μg/ml 25‐HC, SREBP processing was inhibited, and the RG of all cells was lower than 25%. Therefore, we set the ratio larger than 25% as a criterion to identify that SCAP could localize on Golgi.
To measure the recruitment of SCAP to the ERESs, the images were processed by NIS‐Elements program (Nikon). The region of each ERES was used to measure the intensity gray level of SCAP per ERES, and the region of a whole cell was used to measure the SCAP‐positive ERESs number and Sec12‐positive number per cell. The total intensity gray level of SCAP at ERESs per cell was the sum of intensity gray level of SCAP at each ERES in a whole cell.
Production of adenovirus and AAV (adeno‐associated virus)
The recombinant adenoviruses were constructed by using the AdEasy‐1 System (StrataGene, USA). AD‐Cideb, AD‐Cideb‐K128, AD‐SREBP‐1c‐FL, AD‐SREBP‐1c‐N (1‐451), AD‐SREBP‐2‐FL, and AD‐SREPB‐2‐N (1‐410) were constructed by PCR, the fragment from pCMV5‐Flag‐Cideb, pCMV5‐Flag‐Cideb‐K128A, pCMV5‐HA‐SREBP‐1c, and pCMV5‐HA‐SREBP‐2. Recombinant adenoviruses were amplified in AD293 cells in large scale and purified by CsCl density gradient ultracentrifugation according to manufacturer's description. Adenoviruses were injected via the tail veins at a dosage of 1 × 109 pfu/mouse.
AAV‐TBG‐Cideb, AAV‐TBG‐Cideb‐KRRA, AAV‐CMV‐Flag‐Cidea, and AAV‐CMV‐Flag‐Cidec plasmids were constructed by PCR, the fragment from pCMV5‐Cideb, pCMV5‐Cideb‐KRRA, pCMV5‐Flag‐Cidea, and pCMV5‐Flag‐Cidec. Not1 and BamH1, or EcoR1, were used as the restriction sites for subcloning. The cloning primers were used as follows:
Cideb: forward 5′‐ATAAGAATGCGGCCGCATGGAGTACCTTTCA‐3′ and reverse 5′‐CGGGATCCTTAGGAGTGGAGGT‐3′. Cidea: forward 5′‐CGGGATCCATGGACTACAAAGA‐3′ and reverse 5′‐CGGAATTCTTACATGAACCAG‐3′. Cidec: forward 5′‐CGGGATCCATGGACTACAAAGA‐3′ and reverse 5′‐CGGAATTCTCATTGCAGCATCTT‐3′. AAV was produced in HEK293T cells by the three plasmid‐based methods as previously described (Huang et al, 2013). Briefly, subconfluent cells were co‐transfected with the three plasmids by using PEI. At 60 h post‐transfection, cells were collected and lysed by treating with benzonase. AAV was purified via iodixanol gradient centrifugation, at 50,000 rpm for 1.5 h at 16°C. Fractions of the virus were collected and diluted with 1× PBS, further concentrated in ultracentrifuge tube (Amicon 100k columns, UFC‐910008). The purified and concentrated virus were aliquoted and stored at −80°C. Virus titers were determined by qPCR with primers corresponding to the insert sequences.
RNA extraction and real‐time PCR
Total RNA was extracted from mice tissues or cells with TRIzol reagent (Invitrogen). The first‐strand cDNA synthesis was performed with Superscript III RT kit and oligo‐dT primers (Invitrogen). Real‐time PCR was performed with the SYBR Green PCR system (Applied Biosystems) in an ABI 7500 thermal cycler (Applied Biosystems). β‐Actin was used as a reference gene. The primer sequences used are listed in Appendix Table S2.
Molecular dynamic simulation and structural analysis
Monomer structures of mouse Sec12 and Cideb were first generated using the homology modeling methods I‐TASSER (Roy et al, 2010), based on the published crystal structure of Sec12p and Cideb. A total of 20,000 models were generated to obtain the I‐TASSER scores and fit experimental data, resulting in the best‐fit individual structures. Molecular docking of the two proteins was performed with the docking method HoDock (Gong et al, 2010), which incorporates an initial rigid docking and a refined semi‐flexible docking. A total of 12,000 complex structures were generated and scored to produce the initial model of Sec12/Cideb complex. Molecular dynamic simulation package Gromacs 4.5 (Pronk et al, 2013) with OPLS force field was used to minimize, relax, and equilibrate the structure in solution, producing the final model with minimal atom clashes and best fitting with stereo‐chemical restraints.
Quantification and statistical analysis
Statistical analysis was performed with GraphPad Prism 5 (GraphPad Software, USA). Results represent the Mean ± SEM of at least three independent experiments as indicated in figure legends. Two‐tailed Student's t‐test based on the analysis of variance was used for two‐group comparisons. Statistical difference was considered significant at P < 0.05 and indicated in figures as *P < 0.05, **P < 0.01, and ***P < 0.001. NS represents no significant difference.
Microarray data were analyzed with DAVID Bioinformatics Resources 6.8 (Huang da et al, 2009) to transform the Affymetrix probe ID to gene Entrez ID. For the gene mapped with multiple probe IDs, the average intensity value of probes was used as its expression level. The R package of Limma was used to identify the differentially up‐regulated and down‐regulated genes between wide types and two treatments. The genes with P‐value smaller than 0.05 were considered as statistically significant. The heatmaps were plotted using the R package of pheatmap.
Author contributions
LS, LZ, XC, and PL designed the study. LS and LZ performed most experiments and analyzed data. F‐JC helped with image analysis. HW contributed to the plasmid construction and AAV package. HQ helped with the animal experiments. YS contributed to the protein purification. YZ performed EM analysis. HY and XY helped with the microarray analysis. XG helped with the molecular dynamic simulation. LS, LZ, LC, T‐JZ, JZL, LX, XC, and PL participated in result discussion. LS, XC, and PL wrote the manuscript. All authors discussed and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Dr Kota Saito for Sec12 antibody and thank Drs. Rob Yang, Shengcai Lin, and members of the P.L. laboratory for critical reading of the manuscript and helpful discussion. This work was supported by grants from National Key R&D Program of China (2018YFA0506900 to P.L.), from National Natural Science Foundation of China (31430040, 31690103, and 31621063 to P.L. 31571213 and 31521062 to X.W.C.), from National Key R&D Program of China (2016YFA0502002 to P.L.), and from the Young 1000 Talent Plan (to X.W.C.).
The EMBO Journal (2019) 38: e100156
Contributor Information
Xiao‐Wei Chen, Email: xiaowei_chen@pku.edu.cn.
Peng Li, Email: li-peng@mail.tsinghua.edu.cn.
References
- Barlowe C, Schekman R (1993) Sec12 encodes a guanine‐nucleotide‐exchange factor essential for transport vesicle budding from the ER. Nature 365: 347–349 [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (1997) The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane‐bound transcription factor. Cell 89: 331–340 [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (2009) Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J Lipid Res 50(Suppl): S15–S27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JD, Horton JD (2004) Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 114: 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik A, Stephens DJ (2009) ER exit sites–localization and control of COPII vesicle formation. FEBS Lett 583: 3796–3803 [DOI] [PubMed] [Google Scholar]
- Calkin AC, Tontonoz P (2012) Transcriptional integration of metabolism by the nuclear sterol‐activated receptors LXR and FXR. Nat Rev Mol Cell Biol 13: 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, Hobbs HH (2011) Human fatty liver disease: old questions and new insights. Science 332: 1519–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connerly PL, Esaki M, Montegna EA, Strongin DE, Levi S, Soderholm J, Glick BS (2005) Sec16 is a determinant of transitional ER organization. Curr Biol 15: 1439–1447 [DOI] [PubMed] [Google Scholar]
- Engelking LJ, Kuriyama H, Hammer RE, Horton JD, Brown MS, Goldstein JL, Liang G (2004) Overexpression of Insig‐1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin‐stimulated lipogenesis. J Clin Invest 113: 1168–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelking LJ, Liang G, Hammer RE, Takaishi K, Kuriyama H, Evers BM, Li WP, Horton JD, Goldstein JL, Brown MS (2005) Schoenheimer effect explained–feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J Clin Invest 115: 2489–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: 497–509 [PubMed] [Google Scholar]
- Gao G, Chen FJ, Zhou L, Su L, Xu D, Xu L, Li P (2017) Control of lipid droplet fusion and growth by CIDE family proteins. Biochim Biophys Acta 1862: 1197–1204 [DOI] [PubMed] [Google Scholar]
- Gillon AD, Latham CF, Miller EA (2012) Vesicle‐mediated ER export of proteins and lipids. Biochim Biophys Acta 1821: 1040–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Basu SK, Brown MS (1983) Receptor‐mediated endocytosis of low‐density lipoprotein in cultured cells. Methods Enzymol 98: 241–260 [DOI] [PubMed] [Google Scholar]
- Goldstein JL, DeBose‐Boyd RA, Brown MS (2006) Protein sensors for membrane sterols. Cell 124: 35–46 [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS (2015) A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161: 161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Wang P, Yang F, Chang S, Liu B, He H, Cao L, Xu X, Li C, Chen W, Wang C (2010) Protein‐protein docking with binding site patch prediction and network‐based terms enhanced combinatorial scoring. Proteins 78: 3150–3155 [DOI] [PubMed] [Google Scholar]
- Gong J, Sun Z, Wu L, Xu W, Schieber N, Xu D, Shui G, Yang H, Parton RG, Li P (2011) Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J Cell Biol 195: 953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarova V, Brodsky JL, Fisher EA (2003) Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII‐dependent, and its lipidation to very low density lipoprotein occurs post‐ER. J Biol Chem 278: 48051–48058 [DOI] [PubMed] [Google Scholar]
- Haas JT, Miao J, Chanda D, Wang Y, Zhao E, Haas ME, Hirschey M, Vaitheesvaran B, Farese RV Jr, Kurland IJ, Graham M, Crooke R, Foufelle F, Biddinger SB (2012) Hepatic insulin signaling is required for obesity‐dependent expression of SREBP‐1c mRNA but not for feeding‐dependent expression. Cell Metab 15: 873–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Li E, Chen L, Zhang Y, Wei F, Liu J, Deng H, Wang Y (2015) The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature 524: 243–246 [DOI] [PubMed] [Google Scholar]
- Hooper AJ, Adams LA, Burnett JR (2011) Genetic determinants of hepatic steatosis in man. J Lipid Res 52: 593–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Bashmakov Y, Shimomura I, Shimano H (1998) Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc Natl Acad Sci USA 95: 5987–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109: 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA 100: 12027–12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Huang X, Hartley AV, Yin Y, Herskowitz JH, Lah JJ, Ressler KJ (2013) AAV2 production with optimized N/P ratio and PEI‐mediated transfection results in low toxicity and high titer for in vitro and in vivo applications. J Virol Methods 193: 270–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ (1996) An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 383: 728–731 [DOI] [PubMed] [Google Scholar]
- Li JZ, Ye J, Xue B, Qi J, Zhang J, Zhou Z, Li Q, Wen Z, Li P (2007) Cideb regulates diet‐induced obesity, liver steatosis, and insulin sensitivity by controlling lipogenesis and fatty acid oxidation. Diabetes 56: 2523–2532 [DOI] [PubMed] [Google Scholar]
- Li JZ, Lei Y, Wang Y, Zhang Y, Ye J, Xia X, Pan X, Li P (2010) Control of cholesterol biosynthesis, uptake and storage in hepatocytes by Cideb. Biochim Biophys Acta 1801: 577–586 [DOI] [PubMed] [Google Scholar]
- Lin X, Schonfeld G, Yue P, Chen Z (2002) Hepatic fatty acid synthesis is suppressed in mice with fatty livers due to targeted apolipoprotein B38.9 mutation. Arterioscler Thromb Vasc Biol 22: 476–482 [DOI] [PubMed] [Google Scholar]
- Liu M, Feng Z, Ke H, Liu Y, Sun T, Dai J, Cui W, Pastor‐Pareja JC (2017) Tango1 spatially organizes ER exit sites to control ER export. J Cell Biol 216: 1035–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugovskoy AA, Zhou P, Chou JJ, McCarty JS, Li P, Wagner G (1999) Solution structure of the CIDE‐N domain of CIDE‐B and a model for CIDE‐N/CIDE‐N interactions in the DNA fragmentation pathway of apoptosis. Cell 99: 747–755 [DOI] [PubMed] [Google Scholar]
- Matsuda M, Korn BS, Hammer RE, Moon YA, Komuro R, Horton JD, Goldstein JL, Brown MS, Shimomura I (2001) SREBP cleavage‐activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev 15: 1206–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon C, Studer SM, Clendinen C, Dann GP, Jeffrey PD, Hughson FM (2012) The structure of Sec12 implicates potassium ion coordination in Sar1 activation. J Biol Chem 287: 43599–43606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EA, Schekman R (2013) COPII ‐ a flexible vesicle formation system. Curr Opin Cell Biol 25: 420–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon YA, Liang G, Xie X, Frank‐Kamenetsky M, Fitzgerald K, Koteliansky V, Brown MS, Goldstein JL, Horton JD (2012) The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate‐induced hypertriglyceridemia in animals. Cell Metab 15: 240–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols WC, Seligsohn U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, Moussalli MJ, Hauri HP, Ciavarella N, Kaufman RJ, Ginsburg D (1998) Mutations in the ER‐Golgi intermediate compartment protein ERGIC‐53 cause combined deficiency of coagulation factors V and VIII. Cell 93: 61–70 [DOI] [PubMed] [Google Scholar]
- Nie C, Wang H, Wang R, Ginsburg D, Chen XW (2018) Dimeric sorting code for concentrative cargo selection by the COPII coat. Proc Natl Acad Sci USA 115: E3155–E3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohturfft A, Brown MS, Goldstein JL (1998) Sterols regulate processing of carbohydrate chains of wild‐type SREBP cleavage‐activating protein (SCAP), but not sterol‐resistant mutants Y298C or D443N. Proc Natl Acad Sci USA 95: 12848–12853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohturfft A, Yabe D, Goldstein JL, Brown MS, Espenshade PJ (2000) Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell 102: 315–323 [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott‐Schwartz J (1997) ER‐to‐Golgi transport visualized in living cells. Nature 389: 81–85 [DOI] [PubMed] [Google Scholar]
- Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high‐throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29: 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS (2008) Switch‐like control of SREBP‐2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab 8: 512–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridsdale A, Denis M, Gougeon PY, Ngsee JK, Presley JF, Zha X (2006) Cholesterol is required for efficient endoplasmic reticulum‐to‐Golgi transport of secretory membrane proteins. Mol Biol Cell 17: 1593–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X, Wang B, Palladino EN, de Aguiar Vallim TQ, Ford DA, Tontonoz P (2017) ER phospholipid composition modulates lipogenesis during feeding and in obesity. J Clin Invest 127: 3640–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y (2010) I‐TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz H, Miura K, Weiss M, Pepperkok R (2006) Sterols regulate ER‐export dynamics of secretory cargo protein ts‐O45‐G. EMBO J 25: 2953–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Chen M, Bard F, Chen S, Zhou H, Woodley D, Polischuk R, Schekman R, Malhotra V (2009) TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell 136: 891–902 [DOI] [PubMed] [Google Scholar]
- Schindler AJ, Schekman R (2009) In vitro reconstitution of ER‐stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci USA 106: 17775–17780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Stephens DJ (2010) Cargo loading at the ER. Mol Membr Biol 27: 398–411 [DOI] [PubMed] [Google Scholar]
- Shi Y, Burn P (2004) Lipid metabolic enzymes: emerging drug targets for the treatment of obesity. Nat Rev Drug Discov 3: 695–710 [DOI] [PubMed] [Google Scholar]
- Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL (1997) Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 99: 846–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH (2016) Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem 291: 10659–10676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LP, Li L, Goldstein JL, Brown MS (2005) Insig required for sterol‐mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem 280: 26483–26490 [DOI] [PubMed] [Google Scholar]
- Sun LP, Seemann J, Goldstein JL, Brown MS (2007) Sterol‐regulated transport of SREBPs from endoplasmic reticulum to Golgi: insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci USA 104: 6519–6526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Kim JB, Graves RA, Spiegelman BM (1993) ADD1: a novel helix‐loop‐helix transcription factor associated with adipocyte determination and differentiation. Mol Cell Biol 13: 4753–4759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venditti R, Scanu T, Santoro M, Di Tullio G, Spaar A, Gaibisso R, Beznoussenko GV, Mironov AA, Mironov A Jr, Zelante L, Piemontese MR, Notarangelo A, Malhotra V, Vertel BM, Wilson C, De Matteis MA (2012) Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science 337: 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P, Townley AK, Koka P, Palmer KJ, Stephens DJ (2006) Sec16 defines endoplasmic reticulum exit sites and is required for secretory cargo export in mammalian cells. Traffic 7: 1678–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman JT, Plutner H, Balch WE (2001) The mammalian guanine nucleotide exchange factor mSec12 is essential for activation of the Sar1 GTPase directing endoplasmic reticulum export. Traffic 2: 465–475 [DOI] [PubMed] [Google Scholar]
- Wu C, Zhang Y, Sun Z, Li P (2008) Molecular evolution of Cide family proteins: novel domain formation in early vertebrates and the subsequent divergence. BMC Evol Biol 8: 159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wu L, Yu M, Chen FJ, Arshad M, Xia X, Ren H, Yu J, Xu L, Xu D, Li JZ, Li P, Zhou L (2016) Differential roles of cell death‐inducing DNA fragmentation factor‐alpha‐like effector (CIDE) proteins in promoting lipid droplet fusion and growth in subpopulations of hepatocytes. J Biol Chem 291: 4282–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe D, Brown MS, Goldstein JL (2002) Insig‐2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element‐binding proteins. Proc Natl Acad Sci USA 99: 12753–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS (2002) Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to Insig‐1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110: 489–500 [DOI] [PubMed] [Google Scholar]
- Ye J, Li JZ, Liu Y, Li X, Yang T, Ma X, Li Q, Yao Z, Li P (2009) Cideb, an ER‐ and lipid droplet‐associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab 9: 177–190 [DOI] [PubMed] [Google Scholar]
- Yellaturu CR, Deng X, Cagen LM, Wilcox HG, Mansbach CM II, Siddiqi SA, Park EA, Raghow R, Elam MB (2009) Insulin enhances post‐translational processing of nascent SREBP‐1c by promoting its phosphorylation and association with COPII vesicles. J Biol Chem 284: 7518–7532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M (2016) Global epidemiology of nonalcoholic fatty liver disease‐Meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 64: 73–84 [DOI] [PubMed] [Google Scholar]
- Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R (2011) COPII and the regulation of protein sorting in mammals. Nat Cell Biol 14: 20–28 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5