

Abstract
Risk for late-onset Alzheimer’s disease (LOAD), the most prevalent dementia, is partially driven by genetics. To identify LOAD risk loci, we performed a large genome-wide association meta-analysis of clinically diagnosed LOAD (94,437 individuals). We confirm 20 previous LOAD risk loci and identify five new genome-wide loci (IQCK, ACE, ADAM10, ADAMTS1, and WWOX), two of which (ADAM10, ACE) were identified in a recent genome-wide association (GWAS)-by-familial-proxy of Alzheimer’s or dementia. Fine-mapping of the human leukocyte antigen (HLA) region confirms the neurological and immune-mediated disease haplotype HLA-DR15 as a risk factor for LOAD. Pathway analysis implicates immunity, lipid metabolism, tau binding proteins, and amyloid precursor protein (APP) metabolism, showing that genetic variants affecting APP and Aβ processing are associated not only with early-onset autosomal dominant Alzheimer’s disease but also with LOAD. Analyses of risk genes and pathways show enrichment for rare variants (P = 1.32 × 10−7), indicating that additional rare variants remain to be identified. We also identify important genetic correlations between LOAD and traits such as family history of dementia and education.
Our previous work identified 19 genome-wide-significant common variant signals in addition to APOE that influence risk for LOAD (onset age > 65 years)1. These signals, combined with ‘subthreshold’ common variant associations, account for ~31% of the genetic variance of LOAD2, leaving the majority of genetic risk uncharacterized3. To search for additional signals, we conducted a GWAS meta-analysis of non-Hispanic Whites (NHW) by using a larger Stage 1 discovery sample (17 new, 46 total datasets; n = 21,982 cases, 41,944 cognitively normal controls) from our group, the International Genomics of Alzheimer’s Project (IGAP) (composed of four consortia: Alzheimer Disease Genetics Consortium (ADGC), Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE), The European Alzheimer’s Disease Initiative (EADI), and Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/ PERADES) (Supplementary Tables 1 and 2, and Supplementary Note). To sample both common and rare variants (minor allele frequency (MAF) ≥ 0.01 and MAF < 0.01, respectively), we imputed the discovery datasets by using a 1,000 Genomes reference panel consisting of 36,648,992 single-nucleotide polymorphisms (SNPs), 1,380,736 insertions/deletions, and 13,805 structural variants. After quality control, 9,456,058 common variants and 2,024,574 rare variants were selected for analysis. Genotype dosages were analyzed within each dataset, and then combined with meta-analysis (Supplementary Fig. 1 and Supplementary Tables 1–3).
Results
Meta-analysis of Alzheimer’s disease GWAS.
The Stage 1 discovery meta-analysis produced 12 loci with genome-wide significance (P ≤ 5 × 10−8) (Table 1), all of which are previously described1,4–11. Genomic inflation factors (λ) were slightly inflated (λ median = 1.05; λ regression = 1.09; see Supplementary Figure 2 for a quantile–quantile (QQ) plot); however, univariate linkage disequilibrium score (LDSC) regression12,13 estimates indicated that the majority of this inflation was due to a polygenic signal, with the intercept being close to 1 (1.026, s.e.m. = 0.006). The observed heritability (h2) of LOAD was estimated at 0.071 (0.011) using LDSC. Stage 1 meta-analysis was first followed by Stage 2, using the I-select chip we previously developed in Lambert et al.1 (including 11,632 variants, n = 18,845; Supplementary Table 4) and finally Stage 3A (n = 11,666) or Stage 3B (n = 30,511) (for variants in regions not well captured in the I-select chip) (see Supplementary Figure 1 for the workflow). The final sample was 35,274 clinical and autopsy-documented Alzheimer’s disease cases and 59,163 controls.
Table 1 |.
Summary of discovery Stage 1, Stage 2 and overall meta-analyses results for identified loci reaching genome-wide significance after Stages 1 and 2
| Stage 1 discovery (n = 63,926) | Stage 2 (n = 18,845) | Overall stage 1 + stage 2 (n = 82,771) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant a | chr. | Positionb | Closest genec | Major/minor alleles | MAFd | OR | 95% CIe | P | OR | 95% CIe | P | OR | 95% CIe | Meta P | I2 (%), Pf |
| Previous genome-wide-significant loci still reaching significance | |||||||||||||||
| rs4844610 | 1 | 207802552 | CR1 | C/A | 0.187 | 1.16 | 1.12–1.20 | 8.2 × 10−16 | 1.20 | 1.13–1.27 | 3.8 × 10−10 | 1.17 | 1.13–1.21 | 3.6 × 10−24 | 0, 8 × 10−1 |
| rs6733839 | 2 | 127892810 | BIN1 | C/T | 0.407 | 1.18 | 1.15–1.22 | 4.0 × 10−28 | 1.23 | 1.18–1.29 | 2.0 × 10−18 | 1.20 | 1.17–1.23 | 2.1 × 10−44 | 15, 2 × 10−1 |
| rs10933431 | 2 | 233981912 | INPP5D | C/G | 0.223 | 0.90 | 0.87–0.94 | 2.6 × 10−7 | 0.92 | 0.87–0.97 | 3.2 × 10−3 | 0.91 | 0.88–0.94 | 3.4 × 10−9 | 0, 8 × 10−1 |
| rs9271058 | 6 | 32575406 | HLA-DRB1 | T/A | 0.270 | 1.10 | 1.06–1.14 | 5.1 × 10−8 | 1.11 | 1.06–1.17 | 5.7 × 10−5 | 1.10 | 1.07–1.13 | 1.4 × 10−11 | 10, 3 × 10−1 |
| rs75932628 | 6 | 41129252 | TREM2 | C/T | 0.008 | 2.01 | 1.65–2.44 | 2.9 × 10−12 | 2.50 | 1.56–4.00 | 1.5 × 10−4 | 2.08 | 1.73–2.49 | 2.7 × 10−15 | 0, 6 × 10−1 |
| rs9473117 | 6 | 47431284 | CD2AP | A/C | 0.280 | 1.09 | 1.05–1.12 | 2.3 × 10−7 | 1.11 | 1.05–1.16 | 1.0 × 10−4 | 1.09 | 1.06–1.12 | 1.2 × 10−10 | 0, 6 × 10−1 |
| rs12539172 | 7 | 100091795 | NYAP1g | C/T | 0.303 | 0.93 | 0.91–0.96 | 2.1 × 10−5 | 0.89 | 0.84–0.93 | 2.1 × 10−6 | 0.92 | 0.90–0.95 | 9.3 × 10−10 | 0, 8 × 10−1 |
| rs10808026 | 7 | 143099133 | EPHA1 | C/A | 0.199 | 0.90 | 0.87–0.94 | 3.1 × 10−8 | 0.91 | 0.86–0.96 | 1.1 × 10−3 | 0.90 | 0.88–0.93 | 1.3 × 10−10 | 0, 5 × 10−1 |
| rs73223431 | 8 | 27219987 | PTK2B | C/T | 0.367 | 1.10 | 1.07–1.13 | 8.3 × 10−10 | 1.11 | 1.06–1.16 | 1.5 × 10−5 | 1.10 | 1.07–1.13 | 6.3 × 10−14 | 0, 6 × 10−1 |
| rs9331896 | 8 | 27467686 | CLU | T/C | 0.387 | 0.88 | 0.85–0.91 | 3.6 × 10−16 | 0.87 | 0.83–0.91 | 1.7 × 10−9 | 0.88 | 0.85–0.90 | 4.6 × 10−24 | 3, 4 × 10−1 |
| rs3740688 | 11 | 47380340 | SPI1h | T/G | 0.448 | 0.91 | 0.89–0.94 | 9.7 × 10−11 | 0.93 | 0.88–0.97 | 1.2 × 10−3 | 0.92 | 0.89–0.94 | 5.4 × 10−13 | 4, 4 × 10−1 |
| rs7933202 | 11 | 59936926 | MS4A2 | A/C | 0.391 | 0.89 | 0.86–0.92 | 2.2 × 10−15 | 0.90 | 0.86–0.95 | 1.6 × 10−5 | 0.89 | 0.87–0.92 | 1.9 × 10−19 | 27, 5 × 10−2 |
| rs3851179 | 11 | 85868640 | PICALM | C/T | 0.356 | 0.89 | 0.86–0.91 | 5.8 × 10−16 | 0.85 | 0.81–0.89 | 6.1 × 10−11 | 0.88 | 0.86–0.90 | 6.0 × 10−25 | 0, 8 × 10−1 |
| rs11218343 | 11 | 121435587 | SORL1 | T/C | 0.040 | 0.81 | 0.76–0.88 | 2.7 × 10−8 | 0.77 | 0.68–0.87 | 1.8 × 10−5 | 0.80 | 0.75–0.85 | 2.9 × 10−12 | 7, 3 × 10−1 |
| rs17125924 | 14 | 53391680 | FERMT2 | A/G | 0.093 | 1.13 | 1.08–1.19 | 6.6 × 10−7 | 1.15 | 1.06–1.25 | 5.0 × 10−4 | 1.14 | 1.09–1.18 | 1.4 × 10−9 | 8, 3 × 10−1 |
| rs12881735 | 14 | 92932828 | SLC24A4 | T/C | 0.221 | 0.92 | 0.88–0.95 | 4.9 × 10−7 | 0.92 | 0.87–0.97 | 4.3 × 10−3 | 0.92 | 0.89–0.94 | 7.4× 10−9 | 0, 6 × 10−1 |
| rs3752246 | 19 | 1056492 | ABCA7 | C/G | 0.182 | 1.13 | 1.09–1.18 | 6.6 × 10−10 | 1.18 | 1.11–1.25 | 4.7 × 10−8 | 1.15 | 1.11–1.18 | 3.1 × 10−16 | 0, 5 × 10−1 |
| rs429358 | 19 | 45411941 | APOE | T/C | 0.216 | 3.32 | 3.20–3.45 | 1.2 × 10−881 | APOE region not carried forward to replication stage | ||||||
| rs6024870 | 20 | 54997568 | CASS4 | G/A | 0.088 | 0.88 | 0.84–0.93 | 1.1 × 10−6 | 0.90 | 0.82–0.97 | 9.0 × 10−3 | 0.88 | 0.85–0.92 | 3.5 × 10−8 | 0, 9 × 10−1 |
| New genome-wide-significant loci reaching significance | |||||||||||||||
| rs7920721 | 10 | 11720308 | ECHDC3 | A/G | 0.389 | 1.08 | 1.05–1.11 | 1.9 × 10−7 | 1.07 | 1.02–1.12 | 3.2 × 10−3 | 1.08 | 1.05–1.11 | 2.3 × 10−9 | 0,8 × 10−1 |
| rs138190086 | 17 | 61538148 | ACE | G/A | 0.020 | 1.29 | 1.15–1.44 | 7.5 × 10−6 | 1.41 | 1.18–1.69 | 1.8 × 10−4 | 1.32 | 1.20–1.45 | 7.5 × 10−9 | 0, 9 × 10−1 |
| Previous genome-wide-significant loci not reaching significance | |||||||||||||||
| rs190982 | 5 | 88223420 | MEF2C | A/G | 0.390 | 0.95 | 0.92–0.97 | 2.8 × 10−4 | 0.93 | 0.89–0.98 | 2.7 × 10−3 | 0.94 | 0.92–0.97 | 2.8 × 10−6 | 0, 6 × 10−1 |
| rs4723711 | 7 | 37844263 | NME8 | A/T | 0.356 | 0.95 | 0.92–0.98 | 2.7 × 10−4 | 0.91 | 0.87–0.95 | 1.0 × 10−4 | 0.94 | 0.91–0.96 | 2.8 × 10−7 | 0, 5 × 10−1 |
Variants showing the best level of association after meta-analysis of Stages 1 and 2.
Build 37, assembly hg19.
Based on position of top SNP in reference to the RefSeq assembly.
Average in the discovery sample.
Calculated with respect to the minor allele.
Cochran’s Q test
Previously the ZCWPW1 locus.
Previously the CELF1 locus. Chr., chromosome; CI, confidence interval; OR, odds ratio; I2, heterogeneity estimate.
Meta-analysis of Stages 1 and 2 produced 21 genome-wide-significant associations (P ≤ 5 × 10−8) (Table 1 and Fig. 1), 18 of which were previously reported as genome-wide significant in Lambert et al.1. Three other signals were not initially described in the initial IGAP GWAS: the rare R47H TREM2 coding variant previously reported by others7,8,14; ECHDC3 (rs7920721; NC_000010.10: g.11720308A>G), which was recently identified as a potential genome-wide-significant Alzheimer’s disease risk locus in several studies15–17, and ACE (rs138190086; NC_000017.10: g.61538148G>A) (Supplementary Figs. 3 and 4). In addition, seven signals showed suggestive association with P < 5 × 10−7 (closest genes: ADAM10, ADAMTS1, ADAMTS20, IQCK, MIR142/ TSPOAP1-AS1, NDUFAF6, and SPPL2A) (Supplementary Figs. 5–11). Stage 3A and meta-analysis of all three stages for these nine signals (excluding the TREM2 signal; see Supplementary Table 5 for the variant list) identified five genome-wide-significant loci. In addition to ECHDC3, this included four new genome-wide Alzheimer’s disease risk signals at IQCK, ADAMTS1, ACE, and ADAM10 (Table 2). ACE and ADAM10 were previously reported as Alzheimer’s disease candidate genes18–22 but were not replicated in some subsequent studies23–25. A recent GWAS using family history of Alzheimer’s disease or dementia as a proxy26 also identified these two risk loci, suggesting that while use of proxy Alzheimer’s disease/dementia cases introduces less sensitivity and specificity for true Alzheimer’s disease signals overall in comparison to clinically diagnosed Alzheimer’s disease, proxy studies can identify disease-relevant associations. Two of the four other signals approached genome-wide significance: miR142/TSPOAP1-AS1 (P = 5.3 × 10−8) and NDUFAF6 (P = 9.2 × 10−8) (Table 2). Stage 3A also extended the analysis of two loci (NME8 and MEF2C) that were previously genome-wide significant in our 2013 meta-analysis. These loci were not genome-wide significant in our current study and will deserve further investigation (NME8: P = 2.7 × 10−7; MEF2C: P = 9.1 × 10−8; Supplementary Figs. 12 and 13). Of note, GCTA COJO27 conditional analysis of the genome-wide loci indicates that TREM2 and three other loci (BIN1, ABCA7, and PTK2B/CLU) have multiple independent LOAD association signals (Supplementary Table 6), suggesting that the genetic variance associated with some GWAS loci is probably underestimated.
Fig. 1 |. Manhattan plot of meta-analysis of stage 1, 2, and 3 results for genome-wide association with Alzheimer’s disease.
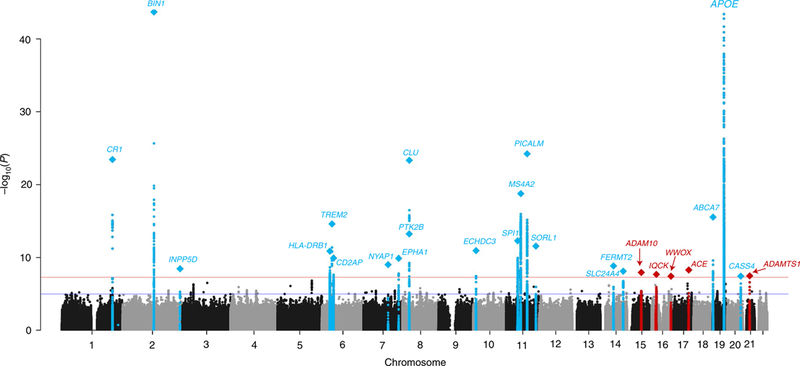
The threshold for genomewide significance (P < 5 × 10−8) is indicated by the red line, while the blue line represents the suggestive threshold (P < 1 × 10−5). Loci previously identified by the Lambert et al.1 IGAP GWAS are shown in blue and newly associated loci are shown in red. Loci are named for the closest gene to the sentinel variant for each locus. Diamonds represent variants with the smallest P values for each genome-wide locus.
Table 2 |.
Summary of discovery Stage 1, Stage 2, Stage 3 (A and B), and overall meta-analysis results of potential novel loci
| Stage 3A | Stage 1 + 2 (n = 82,771) | Stage 3A (n = 11,666) | Overall (n = 94,437) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNPa | Chr. | Positionb | Closest genec | Major/ minor allele | MAFe | OR | 95% CIf | P | OR | 95% CIf | P | OR | 95% CIf | Meta P |
| rs4735340 | 8 | 95976251 | NDUFAF6 | T/A | 0.476 | 0.94 | 0.92–0.96 | 3.4 × 10−7 | 0.92 | 0.83–1.02 | 9.7 × 10−2 | 0.94 | 0.92–0.96 | 9.2 × 10−8 |
| rs7920721g | 10 | 11720308 | ECHDC3 | A/G | 0.390 | 1.08 | 1.05–1.11 | 2.3 × 10−9 | 1.11 | 1.04–1.18 | 1.5 × 10−3 | 1.08 | 1.06–1.11 | 1.8 × 10−11 |
| rs7295246 | 12 | 43967677 | ADAMTS20 | T/G | 0.413 | 1.07 | 1.04–1.09 | 2.7 × 10−7 | 1.02 | 0.96–1.09 | 4.5 × 10−1 | 1.06 | 1.04–1.08 | 3.9 × 10−7 |
| rs10467994 | 15 | 51008687 | SPPL2A | T/C | 0.333 | 0.94 | 0.91–0.96 | 3.9 × 10−7 | 0.97 | 0.87–1.08 | 6.2 × 10−1 | 0.94 | 0.92–0.96 | 4.3 × 10−7 |
| rs593742 | 15 | 59045774 | ADAM10 | A/G | 0.295 | 0.93 | 0.91–0.96 | 1.3 × 10−7 | 0.91 | 0.85–0.98 | 1.5 × 10−2 | 0.93 | 0.91–0.95 | 6.8 × 10−9 |
| rs7185636 | 16 | 19808163 | IQCK | T/C | 0.180 | 0.92 | 0.89–0.95 | 8.4 × 10−8 | 0.94 | 0.86–1.01 | 1.1 × 10−1 | 0.92 | 0.89–0.95 | 2.4 × 10−8 |
| rs2632516 | 17 | 56409089 | MIR142/TSPOAP1-AS1d | G/C | 0.440 | 0.94 | 0.92–0.96 | 2.3 × 10−7 | 0.91 | 0.82–1.01 | 7.5 × 10−2 | 0.94 | 0.91–0.96 | 5.3 × 10−8 |
| rs138190086 | 17 | 61538148 | ACE | G/A | 0.020 | 1.32 | 1.20–1.45 | 7.5 × 10−9 | 1.17 | 0.92–1.48 | 2.1 × 10−1 | 1.30 | 1.19–1.42 | 5.3 × 10−9 |
| rs2830500 | 21 | 28156856 | ADAMTS1 | C/A | 0.308 | 0.93 | 0.91–0.96 | 7.4 × 10−8 | 0.95 | 0.89–1.02 | 1.3 × 10−1 | 0.93 | 0.91–0.96 | 2.6 × 10−8 |
| Stage 3B | Stage 1 (n = 63,926) | Stage 3B (n = 30,511)h | Overall (n = 94,437)h | |||||||||||
| SNPa | Chr. | Positionb | Closest genec | Major/ minor allele | MAFe | OR | 95% CIf | P | OR | 95% CIf | P | OR | 95% CIf | Meta P |
| rs71618613 | 5 | 29005985 | SUCLG2P4 | A/C | 0.010 | 0.68 | 0.57–0.80 | 9.8 × 10−6 | 0.76 | 0.63–0.93 | 6.8 × 10−3 | 0.71 | 0.63–0.81 | 3.3 × 10−7 |
| rs35868327 | 5 | 52665230 | FST T/A | A/C | 0.013 | 0.69 | 0.59–0.80 | 7.8 × 10−7 | 0.58 | 0.29–1.17 | 1.2 × 10−1 | 0.68 | 0.59–0.79 | 2.6 × 10−7 |
| rs114812713 | 6 | 41034000 | OARD1 | G/C | 0.030 | 1.35 | 1.24–1.47 | 4.5 × 10−12 | 1.23 | 1.06–1.42 | 7.2 × 10−3 | 1.32 | 1.22–1.42 | 2.1 × 10−13 |
| rs62039712 | 16 | 79355857 | WWOX | G/A | 0.116 | 1.17 | 1.10–1.23 | 1.2 × 10−7 | 1.14 | 0.96–1.36 | 1.3 × 10−1 | 1.16 | 1.10–1.23 | 3.7 × 10−8 |
Novel loci were defined as loci not reported in Lambert et al.1 with (1) a Stage 1 + 2 meta P < 5 × 10−7 (nine variants after excluding TREM2) (Stage 3A) or (2) a MAF < 0.05 and Stage 1 P < 1 × 10−5 or MAF ≥ 0.05 and Stage 1 P < 5 × 10−6 for genome regions not covered on the Stage 2 custom array (Stage 3B).
SNPs showing the best level of association after meta-analysis of Stages 1, 2 and 3.
Build 37, assembly hg19.
Based on position of top SNP in reference to the RefSeq assembly.
Variant is annotated to both gene features.
Average in the discovery sample.
Calculated with respect to the minor allele.
Recently identified as a LOAD locus in two separate 2017 studies.
Sample sizes for these loci are smaller (overall n = 89,769 for SUCLG2P4, 65,230 for FST, and n = 69,898 for WWOX).
We also selected 33 variants from Stage 1 (28 common and 5 rare variants in loci not well captured in the I-select chip; see Methods for full selection criteria) for genotyping in Stage 3B (including populations of Stage 2 and Stage 3A). We nominally replicated a rare variant (rs71618613; NC_000005.9: g.29005985A>C) within an intergenic region near SUCLG2P4 (MAF = 0.01; P = 6.8 × 10−3; combined P = 3.3 × 10−7) and replicated a low-frequency variant in the TREM2 region (rs114812713; NC_000006.11: g.41034000G>C, MAF = 0.03, P = 7.2 × 10−3; combined P = 2.1 × 10−13) in the gene OARD1 that may represent an independent signal according to our conditional analysis (Table 2, Supplementary Figs. 14 and 15, Supplementary Tables 6 and 7). In addition, rs62039712 (NC_000016.9: g.79355857G>A) in the WWOX locus reached genome-wide significance (P = 3.7 × 10−8), and rs35868327 (NC_000005.9: g.52665230T>A) in the FST locus reached suggestive significance (P = 2.6 × 10−7) (Table 2 and Supplementary Figs. 16 and 17). WWOX may play a role in Alzheimer’s disease through its interaction with tau28,29, and it is worth noting that the sentinel variant (defined as the variant with the lowest P value) is just 2.4 mega-bases from PLCG2, which contains a rare variant that we recently associated with Alzheimer’s disease14. Since both rs62039712 and rs35868327 were only analyzed in a restricted number of samples, these loci deserve further attention.
Candidate gene prioritization at genome-wide loci.
To evaluate the biological significance and attempt to identify the underlying risk genes for the newly identified genome-wide signals (IQCK, ACE, ADAM10, ADAMTS1, and WWOX) and those found previously, we pursued five strategies: (1) annotation and gene-based testing for deleterious coding, loss-of-function (LOF) and splicing variants; (2) expression-quantitative trait loci (eQTL) analyses; (3) evaluation of transcriptomic expression in LOAD clinical traits (correlation with the BRAAK stage30 and differential expression in Alzheimer’s disease versus control brains31); (4) evaluation of transcriptomic expression in Alzheimer’s disease–relevant tissues32–34; and (5) gene cluster/pathway analyses. For the 24 signals reported here, other evidence indicates that APOE35,36, ABCA7 (refs. 37–40), BIN1 (ref. 41), TREM2 (refs. 7,8), SORL1 (refs. 42,43), ADAM10 (ref. 44), SPI1 (ref. 45), and CR1 (ref. 46) are the true Alzheimer’s disease risk gene, although there is a possibility that multiple risk genes exist in these regions47. Because many GWAS loci are intergenic, and the closest gene to the sentinel variant may not be the actual risk gene, in these analyses we considered all protein-coding genes within ±500 kilobases (kb) of the sentinel variant linkage disequilibrium (LD) regions (r2 ≥ 0.5) for each locus as a candidate Alzheimer’s disease gene (n = 400 genes) (Supplementary Table 8).
We first annotated all sentinel variants for each locus and variants in LD (r2 > 0.7) with these variants in a search for deleterious coding, LOF or splicing variants. In line with findings that most causal variants for complex disease are non-coding48, only 2% of 1,073 variants across the 24 loci (excluding APOE) were exonic variants, with a majority (58%) being intronic (Supplementary Fig. 18 and Supplementary Table 9). Potentially deleterious variants include the rare R47H missense variant in TREM2, common missense variants in CR1, SPI1, MS4A2, and IQCK, and a relatively common (MAF = 0.16) splicing variant in IQCK. Using results of a large whole-exome-sequencing study conducted in the ADGC and CHARGE sample49 (n = 5,740 LOAD cases and 5,096 controls), we also identified ten genes located in our genome-wide loci as having rare deleterious coding, splicing or LOF burden associations with LOAD (false discovery rate (FDR) P < 0.01), including previously implicated rare-variant signals in ABCA7, TREM2, and SORL1 (refs. 14,49–55), and additional associations with TREML4 in the TREM2 locus, TAP2 and PSMB8 in the HLA-DRB1 locus, PIP in the EPHA1 locus, STYX in the FERMT2 locus, RIN3 in the SLC24A4 locus, and KCNH6 in the ACE locus (Supplementary Table 10).
For eQTL analyses, we searched existing eQTL databases and studies for cis-acting eQTLs in a prioritized set of variants (n = 1,873) with suggestive significance or in LD with the sentinel variant in each locus. Of these variants, 71–99% have regulatory potential when considering all tissues according to RegulomeDB56 and HaploReg57, but restricting to Alzheimer’s disease–relevant tissues (via Ensembl Regulatory Build58 and GWAS4D59) appears to aid in regulatory variant prioritization, with probabilities for functional variants increasing substantially when using GWAS4D cell-dependent analyses with brain or monocytes, for instance (these and other annotations are provided in Supplementary Table 11). Focusing specifically on eQTLs, we found overlapping cis-acting eQTLs for 153 of the 400 protein-coding genes, with 136 eQTL-controlled genes in Alzheimer’s disease–relevant tissues (that is, brain and blood/immune cell types; see Methods for details) (Supplementary Tables 12 and 13). For our newly identified loci, there were significant eQTLs in Alzheimer’s disease–relevant tissue for ADAM10, FAM63B, and SLTM (in the ADAM10 locus); ADAMTS1 (ADAMTS1 locus); and ACSM1, ANKS4B, C16orf62, GDE1, GPRC5B, IQCK, and KNOP1 (IQCK locus). There were no eQTLs in Alzheimer’s disease–relevant tissues in the WWOX or ACE locus, although several eQTLs for PSMC5 in coronary artery tissue were found for the ACE locus. eQTLs for genes in previously identified loci include BIN1 (BIN1 locus), INPP5D (INPP5D locus), CD2AP (CD2AP locus), and SLC24A4 (SLC24A4 locus). Co-localization analysis confirmed evidence of a shared causal variant affecting expression and disease risk in 66 genes over 20 loci, including 31 genes over 13 loci in LOAD-relevant tissue (see Supplementary Table 14 and 15 for complete lists). Genes implicated include CR1 (CR1 locus), ABCA7 (ABCA7 loci), BIN1 (BIN1 locus), SPI1 and MYBPC3 (SPI1 locus), MS4A2, MS4A6A, and MS4A4A (MS4A2 locus), KNOP1 (IQCK locus), and HLA-DRB1 (HLA-DRB1 locus) (Supplementary Table 12).
To study the differential expression of genes in brains of patients with Alzheimer’s disease versus controls, we used 13 expression studies31. We found that 58% of the 400 protein-coding genes within the genome-wide loci had evidence of differential expression in at least one study (Supplementary Table 16). Additional comparisons to Alzheimer’s disease related gene expression sets revealed that 62 genes were correlated with pathogenic stage (BRAAK) in at least one brain tissue30 (44 genes in prefrontal cortex, the most relevant LOAD tissue; 36 in cerebellum and 1 in visual cortex). Finally, 38 genes were present in a set of 1,054 genes preferentially expressed in aged microglial cells, a gene set shown to be enriched for Alzheimer’s disease genes (P = 4.1 × 10−5)34. We also annotated our list of genes with brain RNA-seq data, which showed that 80% were expressed in at least one type of brain cell, and the genes were most highly expressed in fetal astrocytes (26%), followed by microglia/ macrophages (15.8%), neurons (14.8%), astrocytes (11.5%), and oligodendrocytes (6.5%). When not considering fetal astrocytes, mature astrocytes (21%), and microglial cells (20.3%), the resident macrophage cells of the brain thought to play a key role in the pathologic immune response in LOAD8,14,60, became the highest expressed cell types in the genome-wide set of genes, with 5.3% of the 400 genes showing high microglial expression (Supplementary Table 17; see Supplementary Table 18 for the highly expressed gene list by cell type).
We conducted pathway analyses (MAGMA61) separately for common (MAF > 0.01) and rare variants (MAF < 0.01). For common variants, we detected four function clusters including (1) APP metabolism/Aβ formation (regulation of Aβ formation: P = 4.56 × 10−7 and regulation of APP catabolic process: P = 3.54 × 10−6); (2) tau protein binding (P = 3.19 × 10−5); (3) lipid metabolism (four pathways including protein−lipid complex assembly: P = 1.45 × 10−7); and (4) immune response (P = 6.32 × 10−5) (Table 3 and Supplementary Table 19). Enrichment of the four clusters remained after removal of genes in the APOE region. When APOE-region genes and genes near genome-wide-significant genes were removed, tau showed moderate association (P = 0.027), and lipid metabolism and immune-related pathways showed strong associations (P < 0.001) (Supplementary Table 20). Genes driving these enrichments (that is, having a gene-wide P < 0.05) included SCNA, a Parkinson’s risk gene that encodes alpha-synuclein, the main component of Lewy bodies, whch may play a role in tauopathies62,63, for the tau pathway; apolipoprotein genes (APOM, APOA5) and ABCA1, a major regulator of cellular cholesterol, for the lipid metabolism pathways; and 52 immune pathway genes (Supplementary Table 21). While no pathways were significantly enriched for rare variants, lipid and Aβ pathways did reach nominal significance in rare-variant-only analyses. Importantly, we also observed a highly significant correlation between common and rare pathway gene results (P = 1.32 × 10−7), suggesting that risk Alzheimer’s disease genes and pathways are enriched for rare variants. In fact, 50 different genes within tau, lipid, immunity and Aβ pathways showed nominal rare-variant driven associations (P < 0.05) with LOAD.
Table 3 |.
Significant pathways (q value ≤ 0.05) from MAGMA pathway analysis for common and rare variant subsets
| Pathway | No. of genes in the pathway in the dataset | Common variant Pa | Common variant q value | Rare variant Pa | Rare variant q value | Pathway description |
|---|---|---|---|---|---|---|
| GO:65005 | 20 | 1.4 × 10−7a | 9.5 × 10−4 | 6.7 × 10−2 | 8.4 × 10−1 | Protein−lipid complex assembly |
| GO:1902003 | 10 | 4.5 × 10−7a | 1.4 × 10−3 | 4.9 × 10−2 | 8.4 × 10−1 | Regulation of Aβ formation |
| GO:32994 | 39 | 1.1 × 10−6a | 2.5 × 10−3 | 1.7 × 10−2 | 8.1 × 10−1 | Protein−lipid complex |
| GO:1902991 | 12 | 3.5 × 10−6a | 5.8 × 10−3 | 5.6 × 10−2 | 8.4 × 10−1 | Regulation of amyloid precursor protein catabolic process |
| GO:43691 | 17 | 5.5 × 10−6a | 6.7 × 10−3 | 3.0 × 10−2 | 8.1 × 10−1 | Reverse cholesterol transport |
| GO:71825 | 35 | 6.1 × 10−6a | 6.7 × 10−3 | 1.2 × 10−1 | 8.4 × 10−1 | Protein−lipid complex subunit organization |
| GO:34377 | 18 | 1.6 × 10−5a | 1.5 × 10−2 | 1.8 × 10−1 | 8.4 × 10−1 | Plasma lipoprotein particle assembly |
| GO:48156 | 10 | 3.1 × 10−5a | 2.6 × 10−2 | 7.7 × 10−1 | 8.5 × 10−1 | Tau protein binding |
| GO:2253 | 382 | 6.3 × 10−5a | 4.6 × 10−2 | 2.0 × 10−1 | 8.4 × 10−1 | Activation of immune response |
Significant after FDR correction (q value ≤ 0.05).
To further explore the APP/Aβ pathway enrichment, we analyzed a comprehensive set of 335 APP metabolism genes64 curated from the literature. We observed significant enrichment of this gene set in common variants (P = 2.27 × 10−4; P = 3.19 × 10−4 excluding APOE), with both ADAM10 and ACE nominally significant drivers of this result (Table 4 and Supplementary Tables 22 and 23). Several ‘sub-pathways’ were also significantly enriched in the common variants, including ‘clearance and degradation of Aβ’, and ‘aggregation of Aβ’, along with its subcategory ‘microglia’, the latter supporting microglial cells suspected role in response to Aβ in LOAD65. Nominal enrichment for risk from rare variants was found for the pathway ‘aggregation of Aβ: chaperone’ and 23 of the 335 genes.
Table 4 |.
Top results of pathway analysis of the Aβ-centered biological network from Campion et al.64 (see Supplementary Table 12 for full results)
| Category | Subcategory | No. of genes | Common variant P 0 kb | Common variant P 35–10 kb | Rare variant P 0 kb | Rare variant P 35–10 kb |
|---|---|---|---|---|---|---|
| Aβ-centered biological network (all genes) | – | 331 | 2.2 × 10−4a | 1.5 × 10−4a | 8.2 × 10−1 | 5.1 × 10−1 |
| Clearance and degradation of Aβ | – | 74 | 2.1 × 10−4a | 3.2 × 10−3 | 3.1 × 10−1 | 5.1 × 10−1 |
| Clearance and degradation of Aβ | Microglia | 47 | 2.2 × 10−4a | 1.8 × 10−2 | 2.4 × 10−1 | 6.8 × 10−1 |
| Aggregation of Aβ | – | 35 | 7.0 × 10−4a | 9.9 × 10−3 | 9.0 × 10−2 | 1.6 × 10−1 |
| Aggregation of Aβ | Miscellaneous | 21 | 1.0 × 10−3a | 3.3 × 10−2 | 9.5 × 10−2 | 1.9 × 10−1 |
| APP processing and trafficking | Clathrin/caveolin-dependent endocytosis | 10 | 1.1 × 10−3 | 1.1 × 10−2 | 3.6 × 10−1 | 1.8 × 10−1 |
| Mediator of Aβ toxicity | – | 51 | 3.8 × 10−2 | 4.6 × 10−2 | 5.8 × 10−1 | 5.7 × 10−1 |
| Mediator of Aβ toxicity | Calcium homeostasis | 6 | 6.9 × 10−2 | 1.2 × 10−1 | 3.9 × 10−1 | 2.5 × 10−1 |
| Mediator of Aβ toxicity | Miscellaneous | 3 | 7.6 × 10−2 | 2.3 × 10−2 | 9.7 × 10−1 | 7.6 × 10−1 |
| Clearance and degradation of Aβ | Enzymatic degradation of Aβ | 15 | 7.7 × 10−2 | 2.6 × 10−2 | 6.1 × 10−1 | 2.9 × 10−1 |
| Mediator of Aβ toxicity | Tau toxicity | 20 | 9.0 × 10−2 | 3.4 × 10−1 | 7.1 × 10−1 | 6.8 × 10−1 |
| Aggregation of Aβ | Chaperone | 9 | 1.5 × 10−1 | 3.0 × 10−1 | 1.9 × 10−1 | 1.1 × 10−2 |
Significant after Bonferroni correction for 33 pathway sets tested.
To identify candidate genes for our novel loci, we combined results from our five prioritization strategies in a priority ranking method similar to that of Fritsche et al.66 (Fig. 2 and Supplementary Table 24). ADAM10 was the top ranked gene of the 11 genes within the ADAM10 locus. ADAM10, the most important α-secretase in the brain, is a component of the non-amyloidogenic pathway of APP metabolism67 and sheds TREM2 (ref. 68), an innate immunity receptor expressed selectively in microglia. Overexpression of ADAM10 in mouse models can halt Aβ production and subsequent aggregation69. In addition, two rare ADAM10 alterations segregating with disease in LOAD families increased Aβ plaque load in ‘Alzheimer-like’ mice, with diminished α-secretase activity from the alterations probably the causal mechanism19,44. For the IQCK signal, which is also an obesity locus70,71, IQCK, a relatively uncharacterized gene, was ranked top, although four of the other 11 genes in the locus have a priority rank ≥ 4, including KNOP1 and GPRC5B, the latter being a regulator of neurogenesis72,73 and inflammatory signaling in obesity74. Of the 22 genes in the ACE locus, PSMC5, a key regulator of major histocompatibility complex (MHC)75,76, has a top score of 4, while DDX42, MAP3K3, an important regulator of macrophages and innate immunity77,78, and CD79B, a B lymphocyte antigen receptor subunit, each have a score of 3. Candidate gene studies have associated ACE variants with Alzheimer’s disease risk20,22,79, including a strong association in the Wadi Ara, an Israeli Arab community with high risk of Alzheimer’s disease21. However, these studies yielded inconsistent results23, and our work reports a clear genome-wide association in NHW at this locus. While ACE was not prioritized, it should not be rejected as a candidate gene, as its expression in Alzheimer’s disease brain tissue is associated with Aβ load and Alzheimer’s disease severity80. Furthermore, cerebrospinal fluid (CSF) levels of the angiotensin-converting enzyme (ACE) are associated with Aβ levels81 and LOAD risk82, and studies show ACE can inhibit Aβ toxicity and aggregation83. Finally, angiotensin II, a product of ACE function, mediates a number of neuropathological processes in Alzheimer’s disease84 and is now a target for intervention in phase II clinical trials of Alzheimer’s disease85. Another novel genome-wide locus reported here, ADAMTS1, is within 665 kb of APP on chromosome 21. Of three genes at this locus, our analyses nominate ADAMTS1 as the likely risk gene, although we cannot rule out that this signal is a regulatory element for APP. ADAMTS1 is elevated in Down’s syndrome with neurodegeneration and Alzheimer’s disease86, and it is a potential neuroprotective gene87–89 or a neuroinflammatory gene important to microglial response90. Finally, WWOX and MAF, which surround an intergenic signal in an obesity associated locus91, were both prioritized for the WWOX locus, with MAF, another important regulator of macrophages92,93, being highly expressed in microglia in the Brain RNA-seq database, and WWOX, a high-density-lipoprotein cholesterol and triglyceride–associated gene94,95, being expressed most highly in astrocytes and neurons. WWOX has been implicated in several neurological phenotypes96; in addition, it binds tau and may play a critical role in regulating tau hyper-phosphorylation, neurofibrillary formation and Aβ aggregation28,29. Intriguingly, treatment of mice with its binding partner restores memory deficits97, hinting at its potential in neurotherapy.
Fig. 2 |. Top prioritized genes of 400 genes located in genome-wide-significant loci.

The criteria include: (1) deleterious coding, LOF or splicing variant in the gene; (2) significant gene-based tests; (3) expression in a tissue relevant to Alzheimer’s disease (astrocytes, neurons, microglia/macrophages, oligodendrocytes); (4) a HuMi microglial-enriched gene; (5) having an eQTL effect on the gene in any tissue, in Alzheimer’s disease–relevant tissue, and/ or a co-localized eQTL; (6) being involved in a biological pathway enriched in Alzheimer’s disease (from the current study); (7) expression correlated with the BRAAK stage; and (8) differential expression in a 1 + Alzheimer’s disease (AD) study. Novel genome-wide loci from the current study are listed first, followed by known genome-wide loci. Each category is assigned an equal weight of 1, with the priority score equaling the sum of all categories. Colored fields indicate that the gene meets the criteria. Genes with a priority score ≥ 4 are listed for each locus. If no gene reached a score of ≥ 5 in a locus, then the top ranked gene(s) is listed.
For previously reported loci, applying the same prioritization approach highlights several genes, as described in Fig. 2, some of which are involved in APP metabolism (FERMT2, PICALM) or tau toxicity (BIN1, CD2AP, FERMT2, CASS4, PTK2B)98–101. Pathway, tissue and disease trait enrichment analyses support the utility of our prioritization method, as the 53 prioritized genes with a score ≥ 5 are (1) enriched in substantially more Alzheimer’s disease–relevant pathways, processes and dementia-related traits; (2) enriched in candidate Alzheimer’s disease cell types such as monocytes (adjusted P = 9.0 × 10−6) and macrophages (adjusted P = 5.6 × 10−3); and (3) more strongly associated with dementia-related traits and Alzheimer’s disease–relevant pathways (Supplementary Table 25 and 26; see Supplementary Fig. 19 for the interaction network of these prioritized genes). To further investigate the cell types and tissues the prioritized genes are expressed in, we performed differentially expressed gene (DEG) set enrichment analysis of the prioritized genes by using GTEx102 tissues, and we identified significant differential expression in several potentially relevant Alzheimer’s disease tissues including immune-related tissues (upregulation in blood and spleen), obesity-related tissue (upregulation in adipose), heart tissues (upregulation in left ventricle and atrial appendage), and brain tissues (dowregulation in cortex, cerebellum, hippo-campus, basal ganglia, and amygdala). Furthermore, the 53 genes are overexpressed in ‘adolescence’ and ‘young adult’ brain tissues in BrainSpan103, a transcriptomic atlas of the developing human brain, which is consistent with accumulating evidence suggesting Alzheimer’s disease may start decades before the onset of disease104,105 (Supplementary Fig. 20; see Supplementary Fig. 21 for a tissue expression heat map for the 53 genes).
Fine-mapping of the HLA region.
The above approach prioritized HLA-DRB1 as the top candidate gene in the MHC locus, known for its complex genetic organization and highly polymorphic nature (see Supplementary Fig. 22 for a plot of the region of the Stage 1 results). Previous analyses in the ADGC (5,728 Alzheimer’s disease cases and 5,653 controls) have linked both HLA class I and II haplotypes with Alzheimer’s disease risk106. In order to further investigate this locus in a much larger sample, we used a robust imputation method and fine-mapping association analysis of alleles and haplotypes of HLA class I and II genes in 14,776 cases and 23,047 controls from our datasets (Supplementary Table 27). We found risk effects of HLA-DQA1*01:02 (FDR P = 0.014), HLA-DRB1*15:01 (FDR P = 0.083), and HLA-DQB1*06:02 (FDR P = 0.010) (Supplementary Table 28). After conditioning on the sentinel meta-analysis variant in this region (rs78738018), association signals were lost for the three alleles, suggesting that the signal observed at the variant level is due to the association of these three alleles. These alleles form the HLA-DQA1*01:02~HLA-DQB1*06:02~HLA-DRB1*15:01 (DR15) haplotype, which is also associated with Alzheimer’s disease in our sample (FDR P = 0.013) (Supplementary Table 29). Taken together, these results suggest a central role of the DR15 haplotype in Alzheimer’s disease risk, a finding originally discovered in a small study in the Tunisian population107 and more recently in a large ADGC analysis106. Intriguingly, the DR15 haplotype and its component alleles also associate with protection against diabetes108, a high risk for multiple sclerosis109,110, and risk or protective effects with many other immune-mediated diseases (Supplementary Table 30). Moreover, the associated diseases include a large number of traits queried from an HLA-specific Phewas111, including neurological diseases (for example, Parkinson’s disease112,113) and diseases with risk factors for Alzheimer’s disease (for example, hyperthyroidism114), pointing to potential shared and/or interacting mechanisms and co-morbidities, a common paradigm in the MHC locus115. Two additional alleles, HLA-DQA1*03:01 and HLA-DQB1*03:02, belonging to another haplotype, show a protective effect on Alzheimer’s disease, but their signal was lost after conditioning on HLA-DQA1*01:02, and the HLA-DQA1*03:01~HLA-DQB1*03:02 haplotype is not associated with Alzheimer’s disease (FDR P = 0.651).
Genetic correlations with Alzheimer’s disease.
As described above, several of our genome-wide loci have potentially interesting co-morbid or pleiotropic associations with traits that may be relevant to the pathology of Alzheimer’s disease. To investigate the extent of LOAD’s shared genetic architecture with other traits, we performed LD-score regression to estimate the genetic correlation between LOAD and 792 human diseases, traits and behaviors12,116 (Supplementary Table 31). The common variant genetic architecture of LOAD was positively correlated with a maternal family history of Alzheimer’s disease/dementia (rg for the genetic correlation of two traits = 0.81; FDR P = 2.79 × 10−7), similar to the Marioni et al. family proxy analyses26, which found maternal genetic correlation with Alzheimer’s disease to be higher than that for paternal Alzheimer’s disease (rg = 0.91 and 0.66, respectively). There is substantial overlap between these estimates, as the Marioni et al. analyses include the 2013 IGAP summary statistics and employed the same UK Biobank variable that we used for rg estimates with maternal history of dementia. We also find significant negative correlation between Alzheimer’s disease and multiple measures of educational attainment (for example, college completion, rg = −0.24; years of schooling, rg range = −0.19 to −0.24; cognitive scores, rg = −0.24 and −0.25) (FDR P < 0.05), supporting the theory that a greater cognitive reserve could help protect against development of LOAD117. The extent to which socioeconomic, environmental, or cultural factors contribute to the correlation between educational attainment and risk for Alzheimer’s disease is unknown, but research shows dementia risk to be associated with lower socio-economic status, independently of education status118,119. We also found negative correlations at P < 0.05 with multiple measures of cardiovascular health (that is, family history of high blood pressure and heart disease and vascular/heart problems) and diabetes (that is, fasting proinsulin, basal metabolic rate and fasting insulin), supporting previous research suggesting that use of blood pressure and diabetic medications may reduce the risk of Alzheimer’s disease120. In fact, use of blood pressure medication does show a negative genetic correlation with Alzheimer’s disease in our study (rg = −0.12; P = 0.035), although this result does not survive FDR correction. These and other top results from this analysis (for example, body mass index, height; see Supplementary Table 31 for a full list of other nominally significant correlations) have been linked to Alzheimer’s disease previously116,120–127, either through suggestive or significant genetic or epidemiological associations (see Kuzma et al.128 for a recent review), but the multiple measures here support and emphasize their genetic correlation with LOAD and highlight the possible genetic pleiotropy or co-morbidity of these traits with pathology of LOAD.
Discussion
In conclusion, our work identifies five new genome-wide associations for LOAD and shows that GWAS data combined with high-quality imputation panels can reveal rare disease risk variants (for example, TREM2). The enrichment of rare variants in pathways associated with Alzheimer’s disease indicates that additional rare variants remain to be identified, and larger samples and better imputation panels will facilitate identifying them. While these rare variants may not contribute substantially to the predictive value of genetic findings, they will enhance the understanding of disease mechanisms and potential drug targets. Discovery of the risk genes at genome-wide loci remains challenging, but we demonstrate that converging evidence from existing and new analyses can prioritize risk genes. We also show that APP metabolism is associated with not only early-onset Alzheimer’s disease but also LOAD, suggesting that therapies developed by studying early-onset families could also be applicable to the more common late-onset form of the disease. Pathway analysis showing that tau is involved in LOAD supports recent evidence that tau may play an early pathological role in Alzheimer’s disease129–131 and confirms that therapies targeting tangle formation/degradation could potentially affect LOAD. Finally, our fine-mapping analyses of HLA and genetic correlation results point to LOAD’s shared genetic architecture with many immunemediated and cognitive traits, suggesting that research and interventions that elucidate mechanisms behind these relationships could also yield fruitful therapeutic strategies for LOAD.
Methods
Samples.
All Stage 1 meta-analysis samples are from four consortia: ADGC, CHARGE, EADI, and GERAD/PERADES. Summary demographics of all 46 case-control studies from the four consortia are described in Supplementary Tables 1 and 2. Written informed consent was obtained from study participants or, for those with substantial cognitive impairment, from a caregiver, legal guardian, or other proxy. Study protocols for all cohorts were reviewed and approved by the appropriate institutional review boards. Further details of all cohorts can be found in the Supplementary Note.
Pre-imputation genotype chip quality control.
Standard quality control was performed on all datasets individually, including exclusion of individuals with low call rate, individuals with a high degree of relatedness, and variants with low call rate. Individuals with non-European ancestry according to principal components analysis of ancestry-informative markers were excluded from the further analysis.
Imputation and pre-analysis quality control.
Following genotype chip quality control, each dataset was phased and imputed to the 1,000 Genomes Project (phase 1 integrated release 3, March 2012)132 using SHAPEIT/IMPUTE2133,134 or MaCH/Minimac135,136 software (Supplementary Table 3). All reference population haplotypes were used for the imputation, as this method improves accuracy of imputation for low-frequency variants137. Common variants (MAF ≥ 0.01%) with an r2 or an information measure <0.40 from MaCH and IMPUTE2 were excluded from further analyses. Rare variants (MAF < 0.01%) with a ‘global’ weighted imputation quality score of <0.70 were also excluded from analyses. This score was calculated by weighting each variant’s MACH/IMPUTE2 imputation quality score by study sample size and combining these weighted scores for use as a post-analysis filter. We also required the presence of each variant in 30% of cases and 30% of controls across all datasets.
Stage 1 association analysis and meta-analysis.
Stage 1 single variant-based association analysis employed an additive genotype model adjusting for age (defined as age-at-onset for cases and age-at-last exam for controls), sex, and population substructure using principal components138. The score test was implemented on all case-control datasets. This test is optimal for meta-analysis of rare variants due to its balance between power and control of type 1 error139. Family datasets were tested using GWAF140, with generalized estimating equations (GEE) implemented for common variants (MAF ≥ 0.01), and a general linear mixed effects model (GLMM) implemented for rare variants (MAF < 0.01), per our preliminary data showing that the behavior of the test statistics for GEE was fine for common variants but inflated for rare variants, while GLMM controlled this rare-variant inflation. Variants with regression coefficient |β| > 5 or P value equal to 0 or 1 were excluded from further analysis.
Within-study results for Stage 1 were meta-analyzed in METAL141 using an inverse-variance-based model with genomic control. The meta-analysis was split into two separate analyses according to the study sample size, with all studies being included in the analysis of common variants (MAF ≥ 0.01), and only studies with a total sample size of 400 or greater being included in the rare-variant (MAF < 0.01) analysis. See the Supplementary Note for further details of the meta-analyses methods.
Stage 1 summary statistics quality control and analysis.
Genomic inflation was calculated for lambda (λ) in the GenABEL package142. In addition, we performed LDSC regression via LD Hub v.1.9.0 (refs. 12,13) to calculate the LD score regression intercept and derive a heritability estimate for the inverse-variance weighted meta-analysis summary statistics. The APOE region (Chr19:45,116,911–46,318,605) was removed to calculate the intercept. Removal of the APOE region reduced the heritability estimate slightly from 0.071 (s.e.m. = 0.011) to 0.0637 (s.e.m. = 0.009).
LDSC was also employed via the LD Hub web server to obtain genetic correlation estimates (rg)116 between LOAD and a wide range of other disorders, diseases and human traits, including 518 UK BioBank traits143. UK BioBank is a large long-term study (~500,000 volunteers aged 40 to 69) begun in 2006 in the United Kingdom, which is investigating the contributions of genetic predisposition and environmental exposure (that is, nutrition, lifestyle, and medications) to the development of disease. While volunteers in the study are generally healthier than the overall United Kingdom population144, its large size and comprehensive data collection make the study an invaluable resource for researchers looking to interrogate the combined effect of genetics and environmental factors on disease. Before analyses in LD Hub, we removed all SNPs with extremely large effect sizes including the MHC (Chr6:26,000,000–34,000,000) and APOE region, as outliers can overly influence the regression analyses. A total of 1,180,989 variants were used in the correlation analyses. Statistical significance of the genetic correlations was estimated using 5% Benjamini−Hochberg FDR corrected P values.
GCTA COJO27 was used to conduct conditional analysis of the Stage 1 summary statistics, with 28,730 unrelated individuals from the ADGC as a reference panel for calculation of LD. See URLs for methods for creation of the ‘ADGC reference dataset’.
Stage 2 and 3 genotyping, quality control, and analysis.
Stage 2 genotypes were determined for 8,362 cases and 10,483 controls (Supplementary Table 4). 1,633 variants from the I-select chip were located in the 24 genome-wide loci (defined by the LD blocks of the sentinel variants; excluding the APOE region), with an average of 68 variants per locus. The most well-covered loci were HLA-DRB1, M24A2, and PICALM (763, 202, and 156 variants available, respectively); the least were MAF, ADAMTS1, and INPP5D (0, 4, and 5 variants, respectively).
Stage 3A was conducted for variants selected as novel loci from meta-analyses of Stages 1 and 2 with P < 5 × 10−7 (9 variants) and variants that were previously significant (P < 5 × 10−8) that were not genome-wide significant after Stages 1 and 2 (2 variants) (4,930 cases and 6,736 controls) (Supplementary Table 5). Variants were genotyped using Taqman.
Stage 3B, which combined samples from Stage 2 and 3A, included variants with MAF < 0.05 and P < 1 × 10−5 or variants with MAF ≥ 0.05 and P < 5 × 10−6 in novel loci not covered in the 2013 iSelect genotyping1 (13,292 cases and 17,219 controls) (Supplementary Table 7). See the Supplementary Note for details on selection of variants for Stage 3B follow-up genotyping. For Stages 1, 2, and 3, samples did not overlap.
Per-sample quality checks for genetic sex and relatedness were performed in PLINK. Sex mismatches or individuals showing a high degree of relatedness (identical-by-descent value of 0.98 or greater) were removed from the analysis. A panel of ancestry-informative markers was used to perform principal component analysis with SMARTPCA from EIGENSOFT 4.2 software145, and individuals with non-European ancestry were excluded. Variant quality control was also performed separately in each country including removal of variants missing in more than 10% of individuals, having a Hardy−Weinberg P value in controls lower than 1 × 10−6 or a P value for missingness between cases and controls lower than 1 × 10−6.
Per-study analysis for Stage 2 and Stage 3 followed the same analysis procedures described for Stage 1, except for covariate adjustments per cohort, where all analyses were adjusted on sex and age apart from the Italian, Swedish, and Gr@ACE cohorts, which were also adjusted for principal components. Within-study results were meta-analyzed in METAL141 using an inverse-variance-based model.
Characterization of gene(s) and non-coding features in associated loci.
We determined the base-pair boundaries of the search space for potential gene(s) and non-coding features in each of the 24 associated loci (excluding APOE) using the ‘proxy search’ mechanism in LDLink146. LDLink uses 1,000 genomes genotypes to calculate LD for a selected population; in our case all five European populations were selected (population codes CEU, TSI, FIN, GBR, and IBS). The boundaries for all variants in LD (r2 ≥ 0.5) with the top associated variant from the Stage 2 meta-analysis for each region ±500 kb of the ends of the LD blocks (as eQTL controlled genes are typically less than 500 kb from their controlling variant147) were input into the UCSC genome browser’s ‘Table Browser’ for RefSeq148 and GENCODEv24lift37149 genes at each associated locus. The average size of the LD blocks was 123 kb.
Identification of potentially causal coding or splicing variants.
To identify deleterious coding or splicing variants that may represent causal variants for our genome-wide loci, we first used SNIPA150 to identify variants in high LD (defined as r2 > 0.7) with the sentinel variants of the 24 genome-wide loci (excluding APOE) (n = 1,073). The sentinel variants were defined as the variants with the lowest P in each genome-wide locus. We then used Ensembl VEP151 for annotation of the set of sentinel variants and their proxies. We used BLOSUM62 (ref. 152), SIFT153, Polyphen-2 (ref. 154), CADD155, Condel156, MPC157 and Eigen158 to predict the pathogenicity of protein-altering exonic variants and MaxEntScan to predict the splicing potential of variants. Splicing variants with high splicing potential according to MaxEntScan159 and protein-coding variants predicted to be deleterious by two or more programs were considered to be potentially causal variants for a locus. It should be noted that while we do include rare variants from imputation in our analyses, we may be missing many rare causal variants in this study.
Identification of genes with rare-variant burden via gene-based testing.
We used the summary statistics results of a large whole-exome sequencing (WES) study of LOAD, the Alzheimer’s Disease Sequencing Project (ADSP) case-control study (n = 5,740 LOAD cases and 5,096 cognitively normal controls of NHW ancestry) to identify genes within our genome-wide loci that may contribute to the association signal through rare deleterious coding, splicing or LOF variants. The individuals in the ADSP study largely overlap with individuals in the ADGC and CHARGE cohorts included in our Stage 1 meta-analysis. All 400 protein-coding genes within our LD-defined genome-wide loci were annotated with the gene-based results from this study, and the results were corrected using a 1% FDR P as a cutoff for significance. Complete details of the analysis can be found in Bis et al.49 and the Supplementary Note.
Regulatory variant and eQTL analysis.
To identify potential functional risk variants and genes at each associated locus, we first annotated a list of prioritized variants from the 24 associated loci (excluding APOE) (n = 1,873). This variant list combined variants in LD with the sentinel variants (r2 ≥ 0.5) using INFERNO160 LD expansion (n = 1,339) and variants with suggestive significance (P < 10−5) and LD (r2 ≥ 0.5) with the sentinel variants for the 24 associated loci (excluding APOE) (n = 1,421 variants). We then identified variants with regulatory potential in this set of variants using four programs that incorporate various annotations to identify likely regulatory variants: RegulomeDB56, HaploReg v.4.1 (refs. 57,161), GWAS4D59, and the Ensembl Regulatory Build58. We used the ChromHMM (core 15-state model) as ‘source epigenomes’ for the HaploReg analyses. We used immune (Monocytes-CD14+, GM12878 lymphoblastoid, HSMM myoblast) and brain (NH-A astroctyes) for the Ensembl Regulatory Build analyses. We then used the list of 1,873 prioritized variants to search for genes functionally linked via eQTLs in LOAD relevant tissues including various brain and blood tissue types, including all immune-related cell types, most specifically myeloid cells (macrophages and monocytes) and B-lymphoid cells, which are cell types implicated in LOAD and neurodegeneration by a number of recent studies14,45,162,163. While their specificity may be lower for identifying Alzheimer’s disease risk eQTLs, we included whole blood cell studies in our Alzheimer’s disease–relevant tissue class due to their high correlation of eQTLs with Alzheimer’s disease–relevant tissues (70% with brain164; 51–70% for monocytes and lymphoblastoid cell lines165) and their large sample sizes that allow for increased discovery power. See the Supplementary Note for details on the eQTL databases and studies searched, and Supplementary Table 13 for sample sizes of each database/study.
Formal co-localization testing of our summary Stage 1 results was conducted using (1) COLOC166 via INFERNO and (2) Summary Mendelian Randomization (SMR)-Heidi analysis167. The approximate Bayes factor (ABF), which was used to assess significance in the INFERNO COLOC analysis, is a summary measure that provides an alternative to the P value for the identification of associations as significant. SMR-Heidi analysis, which employs a heterogeneity test (HEIDI test) to distinguish pleiotropy or causality (a single genetic variant affecting both gene expression and the trait) from linkage (two distinct genetic variants in LD, one affecting gene expression and one affecting trait), was also employed for co-localization analysis. Genes located less than 1 Mb from the GWAS sentinel variants that pass a 5% Benjamini–Hochberg FDR-corrected SMR P-value significance threshold and a HEIDI P-value > 0.05 threshold were considered significant. The Westra eQTL168 summary data and Consortium for the Architecture of Gene Expression (CAGE) eQTL summary data were used for analysis. These datasets, conducted in whole blood, are large eQTL studies (Westra: discovery phase n = 5,311, replication phase n = 2,775; CAGE: n = 2,765), and while there is some overlap in samples between the two datasets, CAGE provides finer coverage. The ADGC reference panel dataset referenced above for GCTA COJO analysis was used for LD calculations.
Human brain gene expression analyses.
We also evaluated gene expression of all candidate genes in the associated loci (see Supplementary Table 8 for a complete list of genes searched), using differential Alzheimer’s disease gene expression results from AlzBase31, brain tissue expression from the Brain RNA-seq Database32,33 (see URLs), and the HuMi_Aged gene set34, a set of genes preferentially expressed in aged human microglia established through RNA-seq expression analysis of aged human microglial cells from ten post-mortem brains. AlzBase includes transcription data from brain and blood from aging, non-dementia, mild cognitive impairment, early-stage Alzheimer’s disease, and late-stage Alzheimer’s disease. See ALZBase (see URLs) for a complete list of studies included in the search. Correlation values for the BRAAK stage expression were taken from the Zhang et al.30 study of 1,647 post-mortem brain tissues from LOAD patients and non-demented subjects.
Pathway analysis.
Pathway analyses were performed with MAGMA61, which performs SNP-wise gene analysis of summary statistics with correction for LD between variants and genes to test whether sets of genes are jointly associated with a phenotype (that is, LOAD), compared to other genes across the genome. Adaptive permutation was used to produce an empirical P value and an FDR-corrected q value. Gene sets used in the analyses were from GO169,170, KEGG171,172, REACTOME173,174, BIOCARTA, and MGI175 pathways. Analyses were restricted to gene sets containing between 10 and 500 genes, a total of 10,861 sets. Variants were restricted to common variants (MAF ≥ 0.01) and rare variants (MAF < 0.01) only for each analysis, and separate analyses for each model included and excluded the APOE region. Analyses were also performed after removal of all genome-wide-significant genes. Primary analyses used a 35-kb upstream/10-kb downstream window around each gene in order to capture potential regulatory variants for each gene, while secondary analyses were run using a 0-kb window176. To test for significant correlation between common and rare-variant gene results, we performed a gene property analysis in MAGMA, regressing the gene-wide association statistics from rare variants on the corresponding statistics from common variants, correcting for LD between variants and genes using the ADGC reference panel. The Aβ-centered network pathway analysis used a curated list of 32 Aβ-related gene sets and all 335 genes combined (see Campion et al.64 for details). The combined dataset of 28,730 unrelated individuals from the ADGC referenced in the GCTA COJO analysis was used as a reference set for LD calculations in these analyses.
Validation of prioritization method.
Evaluation of the prioritization of the risk genes in genome-wide loci was done using STRING177, and Jensen Diseases178, Jensen Tissues179, dbGAP gene sets, and the ARCHS4180 resource via the EnrichR181 tool. We evaluated both the 400 genes set list and a list of 53 genes with priority score ≥ 5 (adding in APOE to both lists as the top gene in the APOE locus) using the standard settings for both STRING and EnrichR. We used the q value, which is the adjusted P value using the Benjamini–Hochberg FDR method with a 5% cutoff for correction for multiple hypotheses testing. We also performed ‘differentially expressed gene (DEG)’ sets analysis via FUMA182. These analyses were performed in order to assess whether our 53 prioritized genes were significantly differentially expressed in certain GTEx v.7 (ref. 102; 30 general tissues and 53 specific tissues) or BrainSpan tissues (11 tissue developmental periods with distinct DEG sets ranging from early prenatal to middle adulthood)103. FUMA defines DEG sets by calculating a two-sided t-test per tissue versus all remaining tissue types or developmental periods. Genes with a Bonferonni-corrected P < 0.05 and absolute log(fold change) ≥ 0.58 were considered DEGs. Input genes were tested against each of the DEG sets using the hypergeometric test. Significant enrichment was defined by Bonferonni-corrected P ≤ 0.05.
HLA region analysis.
Non-familial datasets from the ADGC, EADI and GERAD consortiums were used for HLA analysis. After imputation quality control, a total of 14,776 cases and 23,047 controls were available for analysis (Supplementary Table 27). Within ADGC, GenADA, ROSMAP, TARC1, TGEN2, and a subset of the UMCWRMSSM datasets were not imputed as Affymetrix genotyping arrays are not supported by the imputation software.
Imputation of HLA alleles.
Two-field resolution HLA alleles were imputed using the R package HIBAG v.1.4 (ref. 183) and the NHW-specific training set. This software uses specific combinations of variants to predict HLA alleles. Alleles with an imputation posterior probability lower than 0.5 were considered as undetermined as recommended by HIBAG developers. HLA-A, HLA-B, HLA-C class I genes, and HLA-DPB1, HLA-DQA1, HLA-DQB1, and HLA-DRB1 class II genes were imputed. Individuals with more than two undetermined HLA alleles were excluded.
Statistical analysis.
All analyses were performed in R184. Associations of HLA alleles with disease were tested using logistic regressions, adjusting for age, sex, and principal components as specified above for single variant association analysis. Only HLA alleles with a frequency higher than 1% were analyzed. Haplotype estimations and association analyses with disease were performed using the ‘haplo.glm’ function from the haplo.stats R package185 with age, sex, and principal components as covariates. Analysis was performed on two-loci and three-loci haplotypes of HLA-DQA1, HLA-DQB1, and HLA-DRB1 genes. Haplotypes with a frequency below 1% were excluded from the analysis. Considering the high LD in the MHC region, only haplotypes predicted with posterior probabilities higher than 0.2 were considered for analysis. Meta-analysis P values were computed using an inverse-variance-based model as implemented in METAL software141. For haplotypes analysis, only individuals with no undetermined HLA alleles and only datasets with more than 100 cases or controls were included. Adjustments on HLA significant variants and HLA alleles were performed by introducing the variant or alleles as covariates in the regression models. Adjusted P values were computed using the FDR method and the R ‘p.adjust’ function, and applied to the meta-analysis P values. The FDR threshold was set to 10%.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank all the participants of this study for their contributions. Additional acknowledgements and detailed acknowledgments of funding sources for the study are provided in the Supplementary Note.
Footnotes
URLs. ADGC Reference Dataset: https://kauwelab.byu.edu/Portals/22/adgc_combined_1000G_09192014.pdf; AlzBase: http://alz.big.ac.cn/alzBase/; Brain RNA-seq Database: http://www.brainrnaseq.org/; Enrichr: http://amp.pharm.mssm.edu/Enrichr/; exSNP: http://www.exsnp.org/; NESDA eQTL catalog: https://eqtl.onderzoek.io/index.php?page=info; FUMA: http://fuma.ctglab.nl/; HLA-PheWas catalog: https://phewascatalog.org/hla; INFERNO: http://inferno.lisanwanglab.org/index.php; LD Hub: http://ldsc.broadinstitute.org/ldhub/; STRING: https://string-db.org/.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41588-019-0358-2.
Competing interests
D. Blacker is a consultant for Biogen, Inc. R.C.P. is a consultant for Roche, Inc.; Merck, Inc.; Genentech, Inc.; Biogen, Inc.; GE Healthcare; and Eisai, Inc. A.R.W. is a former employee and stockholder of Pfizer, Inc., and a current employee of the Perelman School of Medicine at the University of Pennsylvania Orphan Disease Center in partnership with the Loulou. A.M.G. is a member of the scientific advisory board for Denali Therapeutics. N.E.-T. is a consultant for Cytox. J.Hardy holds a collaborative grant with Cytox cofunded by the Department of Business (Biz). F.J. acts as a consultant for Novartis, Eli Lilly, Nutricia, MSD, Roche and Piramal. Neither J.M. nor his family owns stock or has equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company. J.M. is currently participating in clinical trials of antidementia drugs from Eli Lilly and Company, Biogen and Janssen. J.M. serves as a consultant for Lilly USA. He receives research support from Eli Lilly/Avid Radiopharmaceuticals and is funded by NIH grant nos. P50AG005681, P01AG003991, P01AG026276 and UF01AG032438. C.Cruchaga receives research support from Biogen, EISAI, Alector and Parabon. The funders of the study had no role in the collection, analysis or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. C.Cruchaga is a member of the advisory board of ADx Healthcare. M.R.F. receives grant/research support from AbbVie, Accera, ADCS Posiphen, Biogen, Eisai, Eli Lilly, Genentech, Novartis and Suven Life Sciences, Ltd. He is a consultant/advisory board/DSMB board member for Accera, Avanir, AZTherapies, Cognition Therapeutics, Cortexyme, Eli Lilly & Company, Longeveron, Medavante, Merck and Co. Inc., Otsuka Pharmaceutical, Proclara (formerly Neurophage Pharmaceuticals), Neurotrope Biosciences, Takeda, vTv Therapeutics and Zhejian Hisun Pharmaceuticals. He has a transgenic mouse model patent that is licensed to Elan. R.A.S. receives consulting fees as a member of the Alzheimer’s Disease Advisory Board, Biogen; and as member of the Executive Committee for AZD3293 Alzheimer’s Disease Studies, Eli Lilly. R.B.L. receives consulting fees from Merch, Inc. E.M.R. receives grant funding from several NIH grant and research contracts with Genentech/Roche, Novartis/ Amgen and Avid/Lilly. He is a compensated scientific advisor to Alkahest, Alzheon, Aural Analytics, Denali, Takeda and Zinfandel. He is an advisor to Roche and Roche Diagnostics, which reimburse his expenses only. T.G.B. has research support/contracts from the National Institutes of Health, State of Arizona, Michael J Fox Foundation, Avid Radiopharmaceuticals, Navida Biopharmaceuticals and Aprinoia Therapeutics. He is an advisory board member with Vivid Genomics and has consultancy work with Roche Diagnostics. A.G.S. conducts multiple industry-funded clinical trials, but all funds go to her academic institution. They have current (within last 12 months) research contracts with Eli Lilly, Novartis, Roche, Janssen, AbbVie, Biogen, NeuroEM, Suven and Merck. She does not receive personal compensations from these organizations. G.D.S. is a consultant for Biogen, Inc. J.M.B. is participating in clinical trials of antidementia drugs for Eli Lilly, Toyama Chemical Company, Merck, Biogen, AbbVie, vTv Therapeutics, Janssen and Roche. He has received research grants from Eli Lilly, Avid Radiopharmaceuticals and Astra Zeneca. He is a consultant for Stage 2 Innovations. L.F. is a consultant for Allergan, Eli Lilly, Avraham Pharmaceuticals, Axon Neuroscience, Axovant, Biogen, Boehringer Ingelheim, Eisai, Functional Neuromodulation, Lundbeck, MerckSharpe & Dohme, Novartis, Pfizer, Pharnext, Roche and Schwabe Pharma. M.B has consulted as an advisory board member for Araclon, Grifols, Lilly, Nutricia, Roche and Servier. She received fees for lectures and funds for research from Araclon, Grifols, Nutricia, Roche and Servier. She has not received personal compensations from these organizations. A.Ruiz has consulted for Grifols and Landsteiner Genmed. He received fees for lectures or funds for research and/or reimbursement of expenses for congresses attendance from Araclon and Grifols. He has not received personal compensations from these organizations. O.P. acts as a consultant for Roche and Biogen, Inc. He is currently participating in clinical trials of antidementia drugs from Novartis, Genentech, Roche and Pharmatrophix. B.T.H. is a consultant for Aztherapy, Biogen, Calico, Ceregene, Genentech, Lilly, Neurophage, Novartis and Takeda, and receives research support from Abbvie, Amgen, Deanli, Fidelity Biosciences, General Electric, Lilly, Merck, Sangamo and Spark therapeutics. BTH owns Novartis stock. H.Hampel serves as Senior Associate Editor for the Journal Alzheimer’s & Dementia; he received lecture fees from Biogen and Roche, research grants from Pfizer, Avid and MSD AVENIR (paid to the institution), travel funding from Functional Neuromodulation, Axovant, Eli Lilly and company, Takeda and Zinfandel, GE Healthcare and Oryzon Genomics, consultancy fees from Jung Diagnostics, Cytox Ltd., Axovant, Anavex, Takeda and Zinfandel, GE Healthcare, Oryzon Genomics and Functional Neuromodulation, and participated in scientific advisory boards of Functional Neuromodulation, Axovant, Eli Lilly and company, Cytox Ltd., GE Healthcare, Takeda and Zinfandel, Oryzon Genomics and Roche Diagnostica. Harald Hampel is a co-inventor on numerous patents relating to biomarker measurement but has received no royalties from these patents. A.A.-C. has consultancies for GSK, Cytokinetics, Biogen Idec, Treeway Inc, Chronos Therapeutics, OrionPharma and Mitsubishi-Tanabe Pharma, and was Chief Investigator for commercial clinical trials run by OrionPharma and Cytokinetics.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41588-019-0358-2.
Publisher's Disclaimer: Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data availability
Genome-wide summary statistics for the Stage 1 discovery have been deposited in The National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS)—a NIA/NIH-sanctioned qualified-access data repository, under accession NG00075. Stage 1 data (individual level) for the GERAD cohort canbe accessed by applying directly to Cardiff University. Stage 1 ADGC data are deposited in NIAGADS. Stage 1 CHARGE data are accessible by applying to dbGaP for all US cohorts and to Erasmus University for Rotterdam data. AGES primary data are not available owing to Icelandic laws. Stage 2 and Stage 3 primary data are available upon request.
References
- 1.Lambert JC et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams PM et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol. Aging 41, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gatz M et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Naj AC et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet 43, 436–441 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seshadri S et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollingworth P et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet 43, 429–435 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jonsson T et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med 368, 107–116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guerreiro R et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368, 117–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jun G et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol 67, 1473–1484 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harold D et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet 41, 1088–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambert JC et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet 41, 1094–1099 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Zheng J et al. LD Hub: A centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 051094 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulik-Sullivan BK et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sims RC et al. Novel rare coding variants in PLCG2, ABI3 and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet 49, 1373–1387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu JZ et al. Case-control association mapping by proxy using family history of disease. Nat. Genet 49, 325–331 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Desikan RS et al. Polygenic overlap between c-reactive protein, plasma lipids, and Alzheimer’s disease. Circulation 131, 2061–2069 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jun GR et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement 13, 727–738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vassar R ADAM10 prodomain mutations cause late-onset Alzheimer’s disease: not just the latest FAD. Neuron 80, 250–253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim M et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate alpha-secretase activity. Hum. Mol. Genet 18, 3987–3996 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kehoe PG et al. Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nat. Genet 21, 71–72 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Meng Y et al. Association of polymorphisms in the Angiotensin-converting enzyme gene with Alzheimer disease in an Israeli Arab community. Am. J. Hum. Genet 78, 871–877 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehmann DJ et al. Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of Alzheimer’s disease. Am. J. Epidemiol 162, 305–317 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Wang X-B et al. Angiotensin-converting enzyme insertion/deletion polymorphism is not a major determining factor in the development of sporadic Alzheimer disease: evidence from an updated meta-analysis. PLoS ONE 9, e111406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai G et al. Evidence against a role for rare ADAM10 mutations in sporadic Alzheimer disease. Neurobiol. Aging 33, 416–417.e3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belbin O et al. A multi-center study of ACE and the risk of late-onset Alzheimer’s disease. J. Alzheimers. Dis 24, 587–597 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marioni RE et al. GWAS on family history of Alzheimeras disease. Transl. Psychiatry 8, 99 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet 44, 369–375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang J-Y & Chang N-S WWOX dysfunction induces sequential aggregation of TRAPPC6AΔ, TIAF1, tau and amyloid β, and causes apoptosis. Cell Death Discov 1, 15003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sze CI et al. Down-regulation of WW domain-containing oxidoreductase induces tau phosphorylation in vitro: a potential role in Alzheimer’s disease. J. Biol. Chem 279, 30498–30506 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Zhang B et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai Z et al. AlzBase: an integrative database for gene dysregulation in Alzheimer’s disease. Mol. Neurobiol 53, 310–319 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olah M et al. A transcriptomic atlas of aged human microglia. Nat. Commun 9, 539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corder EH et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet 7, 180–184 (1994). [DOI] [PubMed] [Google Scholar]
- 36.Kim J, Basak JM & Holtzman DM The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinberg S et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat. Genet 47, 445–447 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Vasquez JB, Fardo DW & Estus S ABCA7 expression is associated with Alzheimer’s disease polymorphism and disease status. Neurosci. Lett 556, 58–62 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Roeck A et al. An intronic VNTR affects splicing of ABCA7 and increases risk of Alzheimer’s disease. Acta Neuropathol 135, 827–837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Roeck A et al. Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer’s disease. Acta Neuropathol 134, 475–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chapuis J et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogaeva E et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet 39, 168–177 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vardarajan BN et al. Coding mutations in SORL 1 and Alzheimer disease. Ann. Neurol 77, 215–227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suh J et al. ADAM10 missense mutations potentiate beta-amyloid accumulation by impairing prodomain chaperone function. Neuron 80, 385–401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang K et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci 20, 1052–1061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brouwers N et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol. Psychiatry 17, 223–233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flister MJ et al. Identifying multiple causative genes at a single GWAS locus. Genome Res 23, 1996–2002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farh KK-H et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bis JC et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-associated variants involved in immune response and transcriptional regulation. Mol. Psychiatry 10.1038/s41380-018-0112-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vardarajan BN et al. Coding mutations in SORL1 and Alzheimer disease. Ann. Neurol 77, 215–227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verheijen J et al. A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol 132, 213–224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bellenguez C et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 59, 220.e1–220.e9 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Kunkle BW et al. Targeted sequencing of ABCA7 identifies splicing, stop-gain and intronic risk variants for Alzheimer disease. Neurosci. Lett 649, 124–129 (2017). [DOI] [PubMed] [Google Scholar]
- 54.May P et al. Rare ABCA7 variants in 2 German families with Alzheimer disease. Neurol. Genet 4, e224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guennec K. Le et al. ABCA7 rare variants and Alzheimer disease risk. Neurology 86, 1–4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyle AP et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22, 1790–1797 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ward LD & Kellis M HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 40, D930–D934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zerbino DR, Wilder SP, Johnson N, Juettemann T & Flicek PR The Ensembl Regulatory Build. Genome. Biol 16, 56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang D et al. GWAS4D: multidimensional analysis of context-specific regulatory variant for human complex diseases and traits. Nucleic Acids Res 46, W114–W120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gjoneska E et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol 11, 1–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stefanis L alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med 2, 1–23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takeda A et al. C-terminal alpha-synuclein immunoreactivity in structures other than Lewy bodies in neurodegenerative disorders. Acta Neuropathol 99, 296–304 (2000). [DOI] [PubMed] [Google Scholar]
- 64.Campion D, Pottier C, Nicolas G, Le Guennec K & Rovelet-Lecrux A Alzheimer disease: modeling an Aβ-centered biological network. Mol. Psychiatry 7, 861–871 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Yeh FL, Wang Y, Tom I, Gonzalez LC & Sheng M TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Fritsche LG et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet 48, 134–143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haass C, Kaether C, Thinakaran G & Sisodia S Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med 2, a006270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kleinberger G et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med 6, 243ra86 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Postina R et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest 113, 1456–1464 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hinney A et al. Genetic variation at the CELF1 (CUGBP, elav-like family member 1 gene) locus is genome-wide associated with Alzheimer’s disease and obesity. Am. J. Med. Genet. B 165B, 283–293 (2014). [DOI] [PubMed] [Google Scholar]
- 71.Speliotes EK et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet 42, 937–948 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kurabayashi N, Nguyen MD & Sanada K The G protein-coupled receptor GPRC5B contributes to neurogenesis in the developing mouse neocortex. Development 140, 4335–4346 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Cool BH et al. A flanking gene problem leads to the discovery of a Gprc5b splice variant predominantly expressed in C57BL/6J mouse brain and in maturing neurons. PLoS ONE 5, e10351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim Y-J, Sano T, Nabetani T, Asano Y & Hirabayashi Y GPRC5B activates obesity-associated inflammatory signaling in adipocytes. Sci. Signal 5, ra85–ra85 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Bhat K et al. The 19S proteasome ATPase Sug1 plays a critical role in regulating MHC class II transcription. Mol. Immunol 45, 2214–2224 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Inostroza-Nieves Y, Venkatraman P & Zavala-Ruiz Z Role of Sug1, a 19S proteasome ATPase, in the transcription of MHC I and the atypical MHC II molecules, HLA-DM and HLA-DO. Immunol. Lett 147, 67–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim K, Duramad O, Qin XF & Su B MEKK3 is essential for lipopolysaccharide-induced interleukin-6 and granulocyte-macrophage colony-stimulating factor production in macrophages. Immunology 120, 242–250 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamazaki K et al. Two mechanistically and temporally distinct NF-κB activation pathways in IL-1 signaling. Sci. Signal 2, 1–12 (2009). [DOI] [PubMed] [Google Scholar]
- 79.Farrer LA et al. Association between angiotensin-converting enzyme and Alzheimer disease. New Engl. J. Med 57, 210–214 (2000). [DOI] [PubMed] [Google Scholar]
- 80.Miners JS et al. Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: differences in brain and CSF ACE and association with ACE1 genotypes. Am. J. Transl. Res 1, 163–177 (2009). [PMC free article] [PubMed] [Google Scholar]
- 81.Jochemsen HM et al. The association of angiotensin-converting enzyme with biomarkers for Alzheimer’s disease. Alzheimers Res. Ther 6, 1–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kauwe JSK et al. Genome-wide association study of CSFl Levels of 59 Alzheimer’s disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet 10, e1004758 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baranello RJ et al. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr. Alzheimers Res 12, 32–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kehoe PG The coming of age of the angiotensin hypothesis in Alzheimer’s disease: progress toward disease prevention and treatment? J. Alzheimers. Dis 62, 1443–1466 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kehoe PG et al. The rationale and design of the reducing pathology in Alzheimer’s disease through Angiotensin TaRgeting (RADAR) Trial. J. Alzheimers. Dis 61, 803–814 (2017). [DOI] [PubMed] [Google Scholar]
- 86.Miguel RF, Pollak A & Lubec G Metalloproteinase ADAMTS-1 but not ADAMTS-5 is manifold overexpressed in neurodegenerative disorders as Down syndrome, Alzheimer’s and Pick’s disease. Brain. Res. Mol. Brain. Res 133, 1–5 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Suttkus A et al. Aggrecan, link protein and tenascin-R are essential components of the perineuronal net to protect neurons against iron-induced oxidative stress. Cell Death Dis 5, e1119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Végh MJ et al. Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun 2, 76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morawski M, Filippov M, Tzinia A, Tsilibary E & Vargova L ECM in brain aging and dementia. Prog. Brain. Res 214, 207–227 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Wilcock DM Neuroinflammation in the aging down syndrome brain; lessons from Alzheimer’s disease. Curr. Gerontol. Geriatr. Res 2012, 170276 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang K et al. A genome-wide association study on obesity and obesity-related traits. PLoS ONE 6, 3–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kang K et al. Interferon-γ represses M2 gene expression in human macrophages by disassembling enhancers bound by the transcription factor MAF. Immunity 47, 235–250.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cao S, Liu J, Song L & Ma X The protooncogene c-Maf Is an essential transcription factor for IL-10 gene expression in macrophages. J. Immunol 174, 3484–3492 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee JC et al. WW-domain-containing oxidoreductase is associated with low plasma HDL-C levels. Am. J. Hum. Genet 83, 180–192 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saez ME et al. WWOX gene is associated with HDL cholesterol and triglyceride levels. BMC. Med. Genet 11, 148 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chang HT et al. WW domain-containing oxidoreductase in neuronal injury and neurological diseases. Oncotarget 5, 11792–11799 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee MH et al. Zfra restores memory deficits in Alzheimer’s disease triple-transgenic mice by blocking aggregation of TRAPPC6AΔ, SH3GLB2, tau, and amyloid β, and inflammatory NF-κB activation. Alzheimers Dement. Transl. Res. Clin. Interv 3, 189–204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dourlen P et al. Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of Tau pathology. Mol. Psychiatry 22, 874–883 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chapuis J et al. Genome-wide, high-content siRNA screening identifies the Alzheimer’s genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol 133, 955–966 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shulman JM et al. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates tau-mediated mechanisms. Hum. Mol. Genet 23, 870–877 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhao Z et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci 18, 978–987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aguet F et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Miller JA et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Knickmeyer RC & Ross ME Imaging and rare APOE alleles. Neurology 87, 558–559 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Douaud G et al. A common brain network links development, aging, and vulnerability to disease. Proc. Natl Acad. Sci. USA 111, 17648–17653 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Steele NZ et al. Fine-mapping of the human leukocyte antigen locus as a risk factor for Alzheimer disease: a case-control study. PLoS Med 14, 1–25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fekih Mrissa N et al. Association of HLA-DR-DQ polymorphisms with diabetes in Tunisian patients. Transfus. Apher. Sci 49, 200–204 (2013). [DOI] [PubMed] [Google Scholar]
- 108.Pugliese A et al. HLA-DRB1 15:01-DQA1 01:02-DQB1 06:02 haplotype protects autoantibody-positive relatives from type 1 diabetes throughout the stages of disease progression. Diabetes 65, 1109–1119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Patsopoulos Na et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet 9, e1003926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schmidt H, Williamson D & Ashley-Koch A HLA-DR15 haplotype and multiple sclerosis: a HuGE review. Am. J. Epidemiol 165, 1097–1109 (2007). [DOI] [PubMed] [Google Scholar]
- 111.Karnes JH et al. Phenome-wide scanning identifies multiple diseases and disease severity phenotypes associated with HLA variants. Sci. Transl. Med 9, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wissemann WT et al. Association of Parkinson disease with structural and regulatory variants in the HLA region. Am. J. Hum. Genet 93, 984–993 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Misra MK, Damotte V & Hollenbach JA The immunogenetics of neurological disease. Immunology 153, 399–414 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tan ZS Thyroid function and the risk of Alzheimer disease: the Framingham study. Arch. Intern. Med 168, 1514 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dendrou CA, Petersen J, Rossjohn J & Fugger L HLA variation and disease. Nat. Rev. Immunol 18, 325–339 (2018). [DOI] [PubMed] [Google Scholar]
- 116.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stern Y Cognitive reserve in ageing and Alzheimer’s disease. Lancet. Neurol 11, 1006–1012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cadar D et al. Individual and area-based socioeconomic factors associated with dementia incidence in England: evidencefrom a 12-year follow-up in the English longitudinal study of ageing. JAMA Psychiatry 75, 723–732 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marden JR, Tchetgen Tchetgen EJ, Kawachi I & Glymour MM Contribution of socioeconomic status at 3 life-course periods to late-life memory function and decline: early and late predictors of dementia risk. Am. J. Epidemiol 186, 805–814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Østergaard SDSD et al. Associations between potentially modifiable risk factors and Alzheimer disease: a Mendelian randomization study. PLoS Med 12, e1001841 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhu Z et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun 9, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Baumgart M et al. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population-based perspective. Alzheimers Dement 11, 1–9 (2015). [DOI] [PubMed] [Google Scholar]
- 123.Larsson SC, Traylor M, Burgess S & Markus HS Genetically-predicted adult height and Alzheimer’s disease. J. Alzheimers. Dis 60, 691–698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Helzner EP et al. Contribution of vascular risk factors to the progression in Alzheimer disease. Arch. Neurol 66, 343–348 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Reitz C et al. Association of higher levels of high-density lipoprotein cholesterol in elderly individuals and lower risk of late-onset Alzheimer disease. Arch. Neurol 67, 1491–1497 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mukherjee S et al. Genetically predicted body mass index and Alzheimer’s disease-related phenotypes in three large samples: Mendelian randomization analyses. Alzheimers Dement 11, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Arvanitakis Z et al. Late-life blood pressure association with cerebrovascular and Alzheimer disease pathology. Neurology 91, e517–e525 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kuźma E et al. Which risk factors causally influence dementia? A systematic review of mendelian randomization studies. J. Alzheimers. Dis 36, 215–221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murray ME et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain 138, 1370–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shi Y et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Brier MRMR et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med 8, 338ra66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Genomes Project, C.. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Howie BN, Donnelly P & Marchini J A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Delaneau O, Marchini J & Zagury JF A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012). [DOI] [PubMed] [Google Scholar]
- 135.Li Y, Willer CJ, Ding J, Scheet P & Abecasis GR MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes.. Genet. Epidemiol 34, 816–834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Howie B, Fuchsberger C, Stephens M, Marchini J & Abecasis GR Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet 44, 955–959 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Howie B, Marchini J & Stephens M Genotype imputation with thousands of genomes. G3 1, 457–470 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 139.Ma C et al. Recommended joint and meta-analysis strategies for case-control association testing of single low-count variants. Genet. Epidemiol 37, 539–550 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen M-HH & Yang Q GWAF: an R package for genome-wide association analyses with family data. Bioinformatics 26, 580–581 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Aulchenko YS, Ripke S, Isaacs A & van Duijn CM GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296 (2007). [DOI] [PubMed] [Google Scholar]
- 143.Sudlow C et al. UK Biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fry A et al. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am. J. Epidemiol 186, 1026–1034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Patterson N, Price AL & Reich D Population structure and eigenanalysis. PLoS Genet 2, e190 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Machiela MJ & Chanock SJ LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang X et al. Synthesis of 53 tissue and cell line expression QTL datasets reveals master eQTLs. BMC Genomics 15, 532 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pruitt KD, Tatusova T, Brown GR & Maglott DR NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res 40, D130–D135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Harrow J et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Arnold M, Raffler J, Pfeufer a, Suhre K & Kastenmuller G. SNiPA: an interactive, genetic variant-centered annotation browser. Bioinformatics 31, 1334–1336 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McLaren W et al. The Ensembl variant effect predictor. Genome Biol 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cargill M et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet 22, 231–238 (1999). [DOI] [PubMed] [Google Scholar]
- 153.Ng PC & Henikoff S SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31, 3812–3814 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Adzhubei IA et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gonzalez-Perez A & Lopez-Bigas N Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet 88, 440–449 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Samocha KE et al. Regional missense constraint improves variant deleteriousness prediction Preprint at 10.1101/148353 (2017). [DOI] [Google Scholar]
- 158.Ionita-Laza I, McCallum K, Xu B & Buxbaum JD A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet 48, 214–220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yeo G & Burge CB Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol 11, 377–394 (2004). [DOI] [PubMed] [Google Scholar]
- 160.Amlie-Wolf A et al. INFERNO—INFERring the molecular mechanisms of NOncoding genetic variants. Nucleic Acids Res 46, 8740–8753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ward LD & Kellis M HaploRegv4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res 44, D877–D881 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Thériault P, ElAli A & Rivest S The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res. Ther 7, 41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Raj T et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Qi T et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat. Commun 9, 2282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schramm K et al. Mapping the genetic architecture of gene regulation in whole blood. PLoS ONE 9, e93844 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Giambartolomei C et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet 48, 481–489 (2016). [DOI] [PubMed] [Google Scholar]
- 168.Westra H-J et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet 45, 1238–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Blake JA et al. Gene ontology consortium: going forward. Nucleic Acids Res 43, D1049–D1056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kanehisa M, Sato Y, Kawashima M, Furumichi M & Tanabe M KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–D462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ogata H et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 27, 29–34 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fabregat A et al. The reactome pathway knowledgebase. Nucleic Acids Res 44, D481–D487 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Croft D et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 39, D691–D697 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Eppig JT, Blake Ja, Bult CJ, Kadin Ja & Richardson JE. The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res 43, D726–D736 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.O’Dushlaine C et al. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci 18, 199–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Szklarczyk D et al. STRINGv10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–D452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pletscher-Frankild S, Pallejà A, Tsafou K, Binder JX & Jensen LJ DISEASES: text mining and data integration of disease-gene associations. Methods 74, 83–89 (2015). [DOI] [PubMed] [Google Scholar]
- 179.Santos A et al. Comprehensive comparison of large-scale tissue expression datasets. PeerJ 3, e1054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lachmann A et al. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun 9, 1366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Watanabe K, Taskesen E, Van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zheng X et al. HIBAG—HLA genotype imputation with attribute bagging. Pharmacogenomics. J 14, 192–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.R v.3.4.3 (R Development Core Team, 2017). 185.
- 185.haplo.stats v.1.7.9 (2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.