

The more haste, the worse speed. —“The Tortoise and the Hare,” Aesop, 600 bc
MET TYROSINE KINASE, MET INHIBITORS, AND MET PREDICTIVE BIOMARKERS
The hepatocyte growth factor (HGF) ligand and its receptor, MET, are important for tumor cell proliferation, migration, and survival, as first noted in gastroesophageal adenocarcinoma (GEA) cell lines in the 1980s (Appendix Fig A1).1 Notably, most subsequent studies over the next decades suggested that MET inhibitors were most effective when the MET pathway was aberrantly and genomically activated, thus rendering cells addicted to this oncogene. Oncogenic MET activation results from a hijacking of various aspects of the canonical MET pathway otherwise normally active in development and wound healing.2 The canonical pathway consists of HGF ligand-induced dimerization, tyrosine kinase domain activation, and the subsequent activation of a downstream signaling cascade resulting in the MET-driven oncophenotype. In cancer, overactivation most often occurs when MET gene amplification with consequent overexpression leading to constitutive activation of the receptor is present and generally portends a poor prognosis.3,4
The wave of MET inhibitors introduced into the clinic during the next decades came on the tails of the tales of trastuzumab for HER2-amplified breast cancer and GEA and imatinib for BCR/ABL-translocated chronic myeloid leukemia and KIT-mutated GI stromal tumors. A subset of targeted MET-axis inhibitors, ligand-blocking antibodies (LBAbs), came with great promise, hysteria, and early efficacy in one patient with GEA who achieved complete response.5,6 Early efficacy must be qualified, because only one of 43 patients demonstrated a response to the HGF ligand–blocking one-armed monoclonal antibody onartuzumab, with or without bevacizumab therapy, in the phase I study.7 That patient did not have genomically activated MET but did have exceedingly high MET expression in the tumor and also high serum HGF levels.5,8 However, despite these preclinical and early clinical observations pointing toward likely predictive biomarkers, the next steps taken in the development of MET inhibitors (LBAbs or tyrosine kinase inhibitors [TKIs]) in GEA and other tumors was to cast a wider net and include more than the small subset of patients actually expected to derive substantial benefit.9-11 One might ask why this was the case.
TARGETED THERAPIES FOR UNTARGETED POPULATIONS?
A peculiar proposition it is indeed to use targeted therapies for untargeted populations. Shouldn’t a targeted therapy be directed toward a susceptible cancer cell that harbors the target? But what was the biomarker to reliably signify that a tumor was truly MET dependent and would derive substantial benefit from anti-MET therapy? The leading biomarker candidate was, and still is, MET amplification, which has several-fold higher protein expression as observed by mass spectrometry compared with nonamplified tumors.8 Although rare, amounting to only approximately 5% of GEA (the highest incidence of any cancer), MET amplification has been the most consistent and reliable biomarker both of overall poor prognosis and predictive benefit from agents targeting the receptor.3,4,12-14 However, other potential predictive biomarkers have also been proposed, including MET tyrosine kinase phosphorylation (difficult to measure),15 MET activating mutation or fusion (rare), MET exon 14 skipping or deletion (mostly lung cancer),16 high HGF tumor and/or serum levels (not consistently demonstrated), and MET overexpression (in the absence of MET amplification).17 Focus on the latter overexpression ensued, likely because this was generally more prevalent than the more stringent genomic definition of amplification, making this more expedient and lucrative to study. Although not necessarily clear cut, MET gene amplification is a genomic event that generally is reliably discernable from non–MET-amplified cases. In contrast, protein expression is quite complex. What exactly is the definition of MET overexpression? MET expression by immunohistochemistry (IHC) has many descriptions, including cellular distribution (membranous, cytoplasmic, and/or nuclear staining), extensity (the percentage of tumor cells actually staining), and intensity (the semiquantified staining strength from 0 to 3+). Which of these IHC variables or combinations of variables, if any, are significant predictive biomarkers of MET inhibition? As with many IHC biomarkers including programmed death ligand 1 (PD-L1), various alterations in the scoring criteria can change a tumor from MET negative to MET positive, or vice versa, and dramatically alter positivity incidence with slight scoring modifications. Furthermore, does the predictive utility of any putative biomarker change on the basis of specific inhibitor properties (LBAbs v receptor-directed antibodies v TKIs)?
PHASE III LBAb STUDIES IN GEA: A RACE TO THE FINISH
Only one GEA phase II biomarker-stratified study evaluating an LBAb explored, retrospectively, optimal MET expression IHC criteria before launching a larger phase III study. This phase II study evaluated rilotumumab plus chemotherapy in the first-line setting in all-comers, unselected by any biomarker. Benefit in the MET-high population (64% incidence in that study) hinged on a small retrospective subgroup of 47 and 21 patients receiving rilotumumab and placebo, respectively.18 Unfortunately, the competition to be first to market and, therefore, the rush to initiate phase III studies with rilotumumab in RILOMET-1 (International Phase III Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial of Rilotumumab Plus Epirubicin, Cisplatin, and Capecitabine as First-Line Therapy in Patients With Advanced MET-Positive Gastric or Gastroesophageal Junction Adenocarcinoma) and onartuzumab in METGastric (Fluorouracil, Leucovorin, and Oxaliplatin With or Without Onartuzumab in HER2-Negative, MET-Positive Gastroesophageal Adenocarcinoma), without more attention to these issues, resulted in a fundamental lack of biomarker understanding. Because the questions were unasked, the answers were unknown.
Thus, the market-based engines steamed on ahead with the following two bits of data: one patient who responded to an LBAb monotherapy5 and one retrospective small subgroup analysis of a phase II randomized study.18 Unfortunately, ultimately both LBAb phase III studies were negative.10,11 Regrettably, neither study successfully enriched for a population who would benefit from MET LBAbs. Notably, for the rilotumumab studies, the initial antibody prototype assay and, importantly, the actual scoring system used in the phase II study were changed for the prospective phase III study to officially codevelop a companion diagnostic assay. Indeed, the assay in RILOMET-1 likely inadequately selected any optimal patient population for treatment, with most patients (81%) screened considered positive for MET expression using the lax criteria of greater than or equal to 1+ intensity at 25% or greater extensity staining (hardly selecting) and most patients having low-level expression.11 Prospectively testing freshly cut formalin-fixed paraffin-embedded (FFPE) samples from recently diagnosed patients in RILOMET-1 compared with a retrospective batched archival FFPE analysis in the phase II study may also have contributed to the incidence discrepancy between the studies, because IHC sensitivity is decreased with older FFPE samples and, when still positive in light of this, would represent those samples likely having the highest levels of expression to begin with. Conversely, the METGastric study had only the slightly more stringent criteria of positivity for eligibility of greater than or equal to 1+ intensity at 50% or greater extensity staining (ie, more cells needed to be expressing at least low-level amounts of protein). However, a preplanned coprimary end point of survival in the very high expressing subgroup of greater than or equal to 2+ intensity at 50% or greater extensity was nearly met (pun intended).10 Unfortunately, the phase III METGastric study closed early at only approximately 70% of intended accrual because a parallel, small, randomized phase II trial that was launched simultaneously (ie, not sequentially to plan the phase III trial on the basis of what was found in the phase II trial) with the intention to refine biomarker scoring in the phase III trial in real time (so as to not lose time), “failed to identify an obvious predictive cutoff” in the 62-patient onartuzumab arm.19(p1088) Sadly, the few very high-expressing patients (n = 16) within the 62-patient phase II cohort would lack any real power to identify a true hazard ratio (HR), even less than 0.4, let alone the more likely HR of 0.75 to 0.85. Furthermore, the early phase III closure resulted in loss of the originally calculated power to detect a potential true benefit in the preplanned very high-expressing subgroup of the METGastric study. Notably, in this latter underpowered subgroup analysis, the HR for overall survival was 0.64 in favor of onartuzumab (P = .062), and the median survival increased from 9.7 months to 11 months with onartuzumab.10 Could this have been statistically significant with the originally intended number of patients? Because the question was unasked (study terminated early), the answer is unknown. In the end, the optimal MET biomarker assay, scoring, and positivity criteria of expression for LBAbs, if any, remain undefined. Nevertheless, even if we could rewind and do things differently with this hindsight (could we perform prospective, adequately accrued, randomized phase II and/or III studies with only the highest IHC criteria amounting to approximately 20% to 30% of all patients with GEA?), the marginal absolute benefits that could be realistically achieved with LBAb in an optimal expressing subgroup, as exemplified by the METGastric subgroup analysis HR of 0.64, lend pause to question whether this would really provide significant clinical improvement. However, recent approval of ramucirumab in unselected patients with GEA in the second-line setting with HRs of 0.78 to 0.81 makes this an interesting discussion indeed.20 In other words, these types of HRs are how we make incremental progress in the stage IV setting, with many examples of this to date, and therefore, arguably, it would be clinically significant. Despite this, future investigation of LBAbs in GEA with MET overexpression and without genomic activation is certainly unlikely at this point, because the hares have muddied the field and have left an overall perception that MET inhibitors do not work, without full appreciation of the historical details of MET inhibitor development.
MET AMPLIFICATION IN GEA: NOT ENOUGH FOR ACCELERATED APPROVAL
In stark contrast to the feasibility of testing MET LBAbs in MET-overexpressing GEAs as a result of relatively high incidence, MET-amplified GEA is an orphan disease, with only approximately 5% of patients harboring this event, and MET amplification is even less common in other tumor types.14 Even in the large coordinated National Cancer Institute’s Molecular Analysis for Therapy Choice (NCI-MATCH) effort, identifying and enrolling 35 patients of any histology onto the arm C1 cohort has been challenging and unlikely will lead to huge strides toward approval and open availability of MET-targeted therapies for MET-amplified tumors. Yet, frustratingly, MET amplification, now serving as a driver oncogene in this scenario, is the biomarker subset to likely derive the most benefit from targeted MET receptor inhibitors. Despite this, because of the relative infrequency of the event, only small nonrandomized monotherapy efforts in later therapy lines for GEA have been possible. One of the most promising studies evaluated the TKI AMG33721 and was reported to have an unconfirmed response rate of 62% (eight of 13 patients) in a phase I expansion cohort (required to screen approximately 275 to 300 patients to find these 13 patients) for MET-amplified GEA.22 The final report published recently now reports that of 11 evaluable patients with MET-amplified GEA, the confirmed overall response rate was 55% (six of 11 patients) and the best disease control rate was 82% (nine of 11 patients).23 However, the follow-up, single-arm, late-line phase II GEA study demonstrated a lower than desired response rate at an interim analysis and was terminated. In the recent final report, confirmed responses were observed in eight (18%; 95% CI, 8% to 32%) of 45 patients, median duration of response (DOR) was 6 months, disease control rate was 53%, median progression-free survival time was 3.4 months, and median overall survival time was 7.9 months.24 Despite what otherwise are a respectable response rate and outcomes using AMG337 monotherapy for heavily pretreated patients with GEA harboring a negative prognostic biomarker, because of the low incidence of the aberration and inability to reach response rates acceptable for accelerated approval, additional investigation was terminated. In comparison, PD-L1 checkpoint blockade monotherapies, despite the hysteria, ultimately demonstrated an approximate 10% response rate in the third-line or higher setting in GEA all-comers, with a median progression-free survival time of 2 months, and only a 13.3% response rate (19 of 143 patients) in the subset of PD-L1–positive, microsatellite stable (MSS) selected patients, with a median DOR of less than 8 months (only five patients, or five [28%] of 19 responders or five [3%] of 143 PD-L1–positive/MSS patients, had DOR greater than 12 months).25 Yet on the basis of these results, pembrolizumab was recently successfully pursued for accelerated approval for the entire PD-L1–positive/MSS subset by the US Food and Drug Administration.25,26 Similar clinical outcomes are consistently reported in patients with MET-amplified GEA (with comparable incidence of responses lasting greater than 12 months). In the face of being an uncommon genomic event, this discussion still matters for patients with GEA with tumors harboring MET amplification.
This AMG337 example, along with other MET inhibitors, has well-elucidated reasons for perceived failure. These include underappreciating the general poor prognosis of MET-amplified tumors in single-arm, late-line studies, intrapatient heterogeneity with regions harboring and not harboring MET amplification, and the presence of concurrent genomic aberrations in some MET-amplified cells. All of these factors could lead to relatively quick progression on monotherapy either outright or shortly after an initial response in most, but importantly not all, responding patients. However, the ideal randomized and combination therapy studies have been elusive to date because of the infeasibility of classic clinical study designs and the requirement of extraordinarily high numbers of patients screened to identify the MET-amplified subset, with the consequent abandonment of additional investigation by pharma because of this apparently high-risk and low-yield scenario. Thus, there is true market failure here, not unlike other situations in oncology, including evaluating second-line anti–human epidermal growth factor receptor 2 (HER2) therapy in the small subset of persistently HER2-amplified tumors after HER2 conversion to negative status, occurring in anywhere between 30% and 70% of patients after first-line anti-HER2 therapy.27 This provides a strong argument in favor of cooperative groups stepping in and leading in a new direction and new initiatives to design patient-centered next-generation studies, with necessary pharma and regulatory support.28,29
LESSONS LEARNED FROM MET INHIBITOR DEVELOPMENT AND FUTURE DIRECTIONS
The example here on rushing MET inhibitor development in GEA and its consequences is not unique and has been observed in a number of other instances and scenarios across various tumor types, from the hurried choice of drug dosing30 to similar lack of effective biomarker strategy rushing from early phase I and II studies to phase III,31-33 and therefore, the lessons learned are broadly applicable. Hasty Hares often ultimately fail to move the field forward without due diligence before embarking on the pivotal study and also hinder appropriate additional development because of added baggage. With MET, supportive evidence abounds that there are patients who derive benefit from MET inhibitors, particularly those with MET-amplified tumors, yet to date, no MET-specific inhibitor is approved by the US Food and Drug Administration in any MET-specific indication for any tumor. For LBAbs and TKIs alike, there has been failure to focus with appropriate studies on the respective subset of patients with GEA most likely to derive benefit, which are relatively small subsets of patients. Lack of progress can be attributed to a number of issues including inadequate validation of biomarker cut-offs, along with the inherent hurdles to developing drugs in low-incidence biomarker populations, coupled with complex intrapatient heterogeneous biology rendering monotherapy in late-line therapy less likely to impress,34 and certainly so for accelerated approval paths. Newer trial designs implementing MET inhibitors for MET-amplified tumors into earlier lines of therapy combined with chemotherapy and/or other targeted agents and using novel molecular profiling technology such as cell-free DNA profiling may help to overcome several obstacles that have served as barriers to implementing matched targeted MET inhibitors.28,34,35 For the successful implementation of MET-directed therapy into the clinic for the appropriate patients, we await the Tortoise.
Appendix
FIG A1.
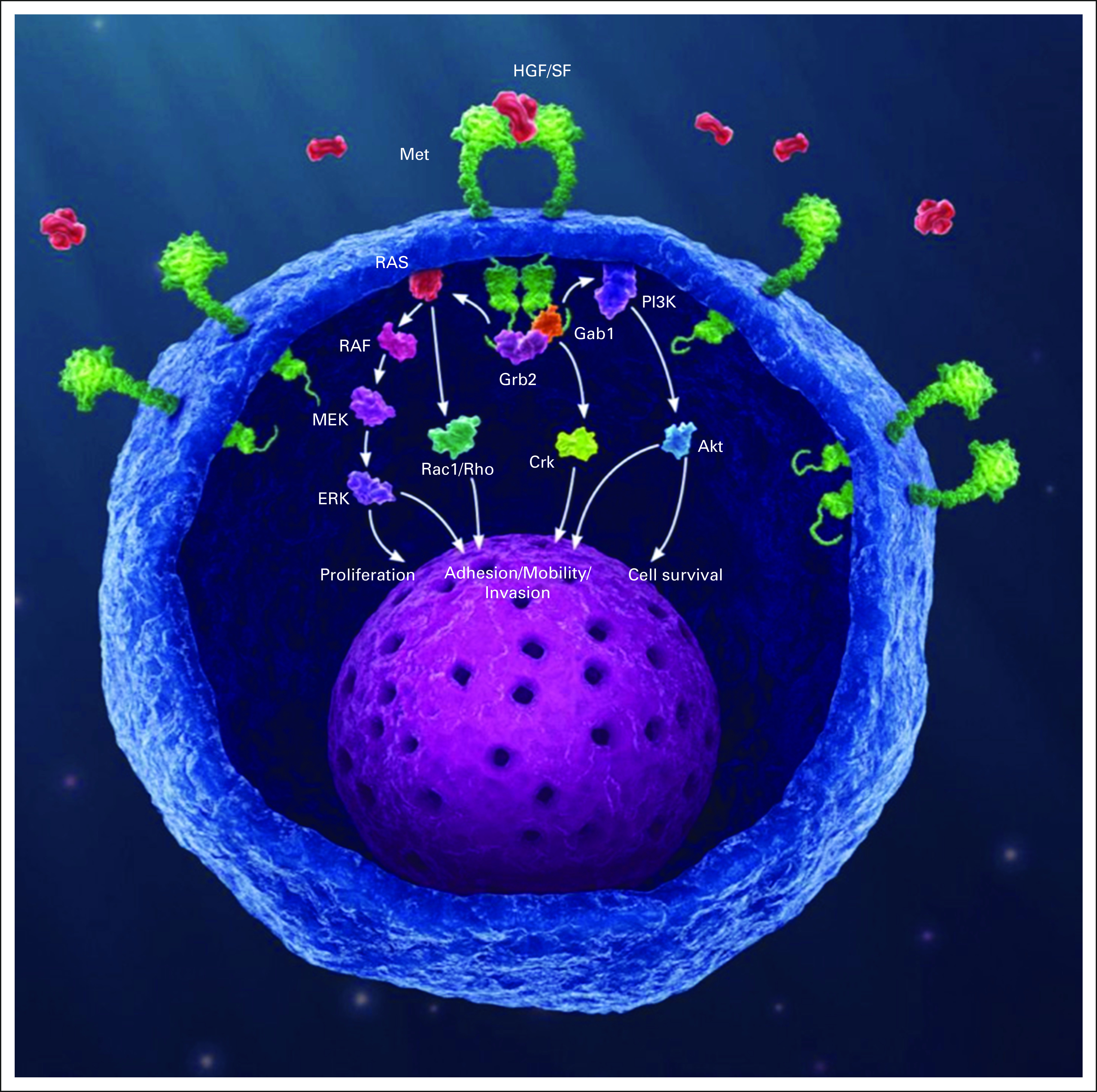
The HGF/MET signaling pathway. Gene amplification, or increased gene dosage, leads to increased messenger RNA expression, which leads to exponential increase in the amount of MET protein receptor (green) expression at the membrane. This in turn yields constitutive activation of the MET receptors through autodimerization, independent of ligand presence or absence, and consequent downstream signaling cascade resulting in oncogenic phenotype. HGF/SF, hepatocyte growth factor/scatter factor (the only known ligand for the MET receptor).
AUTHOR’S DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Daniel Catenacci
Honoraria: Genentech, Eli Lilly, Amgen, OncoPlex Diagnostics, Foundation Medicine, Taiho Pharmaceutical, Genmab, NantOmics, Guardant Health, Merck, Bristol-Myers Squibb, Gritstone Oncology, Five Prime Therapeutics, Astellas Pharma
Consulting or Advisory Role: Genentech, Amgen, Merck, Eli Lilly, Taiho Pharmaceutical, Bristol-Myers Squibb, Astellas Pharma
Speakers' Bureau: Guardant Health, Foundation Medicine, Genentech, Eli Lilly, Merck
REFERENCES
- 1.Giordano S, Di Renzo MF, Ferracini R, et al. p145, a protein with associated tyrosine kinase activity in a human gastric carcinoma cell line Mol Cell Biol 83510–35171988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trusolino L, Bertotti A, Comoglio PM.MET signalling: Principles and functions in development, organ regeneration and cancer Nat Rev Mol Cell Biol 11834–8482010 [DOI] [PubMed] [Google Scholar]
- 3.Graziano F, Galluccio N, Lorenzini P, et al. Genetic activation of the MET pathway and prognosis of patients with high-risk, radically resected gastric cancer J Clin Oncol 294789–47952011 [DOI] [PubMed] [Google Scholar]
- 4.Catenacci DV, Ang A, Liao WL, et al. MET tyrosine kinase receptor expression and amplification as prognostic biomarkers of survival in gastroesophageal adenocarcinoma Cancer 1231061–10702017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catenacci DV, Henderson L, Xiao SY, et al. Durable complete response of metastatic gastric cancer with anti-Met therapy followed by resistance at recurrence Cancer Discov 1573–5792011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng Y, Ma PC.Anti-MET targeted therapy has come of age: The first durable complete response with MetMAb in metastatic gastric cancer Cancer Discov 1550–5542011 [DOI] [PubMed] [Google Scholar]
- 7.Salgia R, Patel P, Bothos J, et al. Phase I dose-escalation study of onartuzumab as a single agent and in combination with bevacizumab in patients with advanced solid malignancies Clin Cancer Res 201666–16752014 [DOI] [PubMed] [Google Scholar]
- 8.Catenacci DV, Liao WL, Thyparambil S, et al. Absolute quantitation of Met using mass spectrometry for clinical application: Assay precision, stability, and correlation with MET gene amplification in FFPE tumor tissue. PLoS One. 2014;9:e100586. doi: 10.1371/journal.pone.0100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah MA, Wainberg ZA, Catenacci DV, et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One. 2013;8:e54014. doi: 10.1371/journal.pone.0054014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah MA, Bang YJ, Lordick F, et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-negative, MET-positive gastroesophageal adenocarcinoma: The METGastric randomized clinical trial JAMA Oncol 3620–6272017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Catenacci DVT, Tebbutt NC, Davidenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): A randomised, double-blind, placebo-controlled, phase 3 trial Lancet Oncol 181467–14822017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smolen GA, Sordella R, Muir B, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752 Proc Natl Acad Sci USA 1032316–23212006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lennerz JK, Kwak EL, Ackerman A, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib J Clin Oncol 294803–48102011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jardim DLF, Tang C, Gagliato DDM, et al. Analysis of 1,115 patients tested for MET amplification and therapy response in the MD Anderson phase I clinic Clin Cancer Res 206336–63452014 [DOI] [PubMed] [Google Scholar]
- 15.Raghav KP, Wang W, Liu S, et al. cMET and phospho-cMET protein levels in breast cancers and survival outcomes Clin Cancer Res 182269–22772012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Awad MM, Oxnard GR, Jackman DM, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression J Clin Oncol 34721–7302016 [DOI] [PubMed] [Google Scholar]
- 17.Herrera LJ, El-Hefnawy T, Queiroz de Oliveira PE, et al. The HGF receptor c-Met is overexpressed in esophageal adenocarcinoma Neoplasia 775–842005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iveson T, Donehower RC, Davidenko I, et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: An open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study Lancet Oncol 151007–10182014 [DOI] [PubMed] [Google Scholar]
- 19.Shah MA, Cho JY, Tan IB, et al. A randomized phase II study of FOLFOX with or without the MET inhibitor onartuzumab in advanced adenocarcinoma of the stomach and gastroesophageal junction Oncologist 211085–10902016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial Lancet Oncol 151224–12352014 [DOI] [PubMed] [Google Scholar]
- 21.Du Z, Caenepeel S, Shen Y, et al. Preclinical evaluation of AMG 337, a highly selective small molecule MET inhibitor, in hepatocellular carcinoma Mol Cancer Ther 151227–12372016 [DOI] [PubMed] [Google Scholar]
- 22.Kwak EL, LoRusso P, Hamid O, et al. Clinical activity of AMG 337, an oral MET kinase inhibitor, in adult patients (pts) with MET-amplified gastroesophageal junction (GEJ), gastric (G), or esophageal (E) cancer. J Clin Oncol. 2015;33(suppl; abstr 1-1) [Google Scholar]
- 23.Hong DS, LoRusso PM, Hamid O, et al. Phase 1 study of AMG 337, a highly selective small-molecule MET inhibitor, in patients with advanced solid tumors. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-18-1341. [epub ahead of print on November 13, 2018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Cutsem E, Karaszewska B, Kang YK, et al. A multicenter phase 2 study of AMG 337 in patients with MET-amplified gastric/gastroesophageal junction/esophageal adenocarcinoma and other solid tumors. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-18-1337. [epub ahead of print on October 26, 2018] [DOI] [PubMed] [Google Scholar]
- 25.US Food and Drug Administration FDA grants accelerated approval to pembrolizumab for advanced gastric cancer. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm577093.htm
- 26.Fuchs CS, Doi T, Jang RW, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: Phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 2018;4:e180013. doi: 10.1001/jamaoncol.2018.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Catenacci DVT, Park H, Uronis HE, et al. Margetuximab (M) plus pembrolizumab (P) in ERBB2-amplified PD-L1+ gastroesophageal adenocarcinoma (GEA) post trastuzumab (T) J Clin Oncol. 2018;36(suppl; abstr 4030) [Google Scholar]
- 28.Catenacci DVT.Expansion platform type II: Testing a treatment strategy Lancet Oncol 161276–12782015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joshi SS, Maron SB, Lomnicki S, et al. Personalized antibodies for gastroesophageal adenocarcinoma (PANGEA): A phase II precision medicine trial ( NCT02213289) J Clin Oncol. 2018;36(suppl; abstr TPS198) [Google Scholar]
- 30.Twombly R.Failing survival advantage in crucial trial, future of Iressa is in jeopardy J Natl Cancer Inst 97249–2502005 [DOI] [PubMed] [Google Scholar]
- 31.Jardim DL, Groves ES, Breitfeld PP, et al. Factors associated with failure of oncology drugs in late-stage clinical development: A systematic review Cancer Treat Rev 5212–212017 [DOI] [PubMed] [Google Scholar]
- 32.Hamid O, Gajewski TF, Frankel AE, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma: Phase 1 and 2 efficacy and safety results from ECHO-202/KEYNOTE-037 Ann Oncol 28v428–v4482017 [Google Scholar]
- 33. Long GV, Dummer R, Hamid O, et al: Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: Results of the phase 3 ECHO-301/KEYNOTE-252 study. J Clin Oncol 36, 2018.
- 34.Catenacci DV.Next-generation clinical trials: Novel strategies to address the challenge of tumor molecular heterogeneity Mol Oncol 9967–9962015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pectasides E, Stachler MD, Derks S, et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma Cancer Discov 837–482018 [DOI] [PMC free article] [PubMed] [Google Scholar]