

Abstract
The functionally-tolerated sequence space of proteins can now be explored in an unprecedented way, due to the expansion of genomic databases and the development of high-throughput methods to interrogate protein function. For signaling proteins, several recent studies have shown how the analysis of sequence variation leverages the available protein structure information to provide new insights into specificity and allosteric regulation. In this review, we discuss recent work that illustrates how this emerging approach is providing a deeper understanding of signal transduction mechanisms.
Introduction
Cellular signal transduction involves the transmission of information from the outside to the inside of a cell, evoking a specific response to an extracellular stimulus. The proteins that mediate signal transduction operate under two imperatives. First, they must ensure that signals are relayed in the appropriate direction. This is achieved through adequate interaction specificities. Second, they should transmit signals only when the appropriate cues are received, which requires that they are subject to responsive regulatory control. Foundational insights into the molecular basis of signal transduction have been gained through experimental structure determination, augmented by computer simulations and biochemical investigations of structure and mechanism. Recently, the development of rapid and inexpensive DNA synthesis, coupled with next-generation sequencing, has facilitated new approaches to understand how signaling proteins work.
We have now acquired a remarkably complete atlas of structures of signaling proteins, augmented by powerful modeling approaches that fill in what we do not yet see directly1,2. The principles of signaling through second messengers are now well understood3,4, and the link between cell signaling and transcriptional control is becoming increasingly clear, as exemplified by the structures of nuclear hormone and steroid receptors5. We also have a general understanding of how protein kinases and phosphatases function and are regulated6–9, and the mechanisms by which adapter proteins facilitate signal-induced protein-protein interactions are known10. Ubiquitin ligation, which controls a broad spectrum of cell-biological processes, has been explored in depth11. The structural mechanisms of Ras and related small GTPases have been mapped in detail12. More recently, we have obtained deep insights into how G-protein coupled receptors are activated13. All of these structural efforts have had a tremendous impact on drug discovery.
With this information in place, we are now poised to address questions that pertain to the nuanced architecture and evolution of signaling proteins, and to the complex biological processes that they control. Proteins have arrived at their present state through evolution, filtered by natural selection. Signaling proteins are typically multifunctional, with the ability to parse information from many inputs and to transduce that information to multiple outputs. The evolutionary logic of the design of such devices often does not make immediate sense in terms of how one might design these molecules or pathways from first principles14.
Most signaling proteins are members of large families of homologous proteins, each with a distinct, occasionally overlapping, set of interaction partners. It remains difficult to deduce, from structure alone, why closely-related proteins are biased towards different input and output signals, as differences in binding energies between on- and off-target interactions are often small (that is, comparable to the thermal energy, kBT) 15. As a result, the structures of these proteins often do not reveal how specificity is encoded in these systems. A related challenge comes in trying to understand divergence in the regulation of homologous signaling proteins. Regulation of signaling proteins necessarily involves the adoption of transient conformational states, which are difficult to visualize or probe directly. These transient states may be functionally important, and can be stabilized or destabilized by the forces of evolution. A record of this selection must be imprinted on the sequences of signaling proteins.
We are now gaining insight into the mechanisms of signaling proteins through approaches that examine the impact of sequence variation on structure and function (Figure 1). This new wave of protein science builds upon bioinformatic concepts and functional screens that were developed in parallel with structure determination tools over the past several decades. These approaches have been enhanced by recent advances in DNA synthesis and sequencing techniques, and by the increasing availability of sequence databases derived from the genomes of thousands of organisms. With these improved tools, we are now equipped to explore, in great depth and with great speed, how changes to the amino acid sequences of signaling proteins impact their function. Here, we describe selected examples of recent studies in this area, focusing on animal-cell signaling, and discuss how this work is leading to a new appreciation of the versatile functions of signaling proteins.
Figure 1.
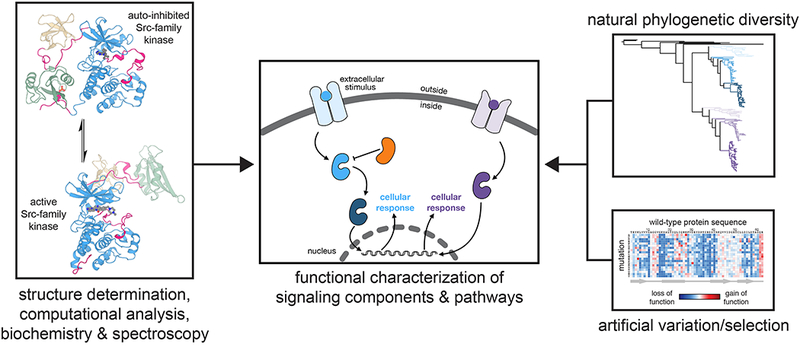
Complementary approaches to elucidate molecular mechanisms of signal transduction. Structures of Src-family kinases are represented by that of Hck in an auto-inhibited conformation (PDB code 1QCF) and c-Src in an active conformation (PDB code 1Y57).
Early explorations of protein sequence space
Natural sequence variation
The diversity of sequences that can map onto a common protein fold has been appreciated since the very beginnings of structural biology. For example, members of the globin family of proteins, which bind to and transport oxygen, can diverge to the point where the sequence identity between proteins is ~15%, while retaining the same overall structure and oxygen-binding mechanism. The analysis of residue conservation in globin sequences has been critical in interpreting how globin structure allosterically controls oxygen binding16, and residue conservation remains an important metric for identifying structurally and functionally important regions of proteins.
More recently, the assessment of co-variation between positions in multiple sequence alignments, rather than simply conservation at a single position, has emerged as a powerful approach to study protein structure17. Residue co-variation across a protein family can be used to identify native contacts in a protein fold, as exemplified by a technique known as Direct Coupling Analysis (DCA), which has been used to predict the structures of many proteins18,19. Residue co-variation can also be used to infer energetic coupling between sites within one protein, using DCA18,19, and by a method called Statistical Coupling Analysis (SCA) 20,21. SCA has been used to identify conserved networks of residues that mediate allosteric regulation, as demonstrated for globins and other protein families22. The optimal use of such methods is a topic of current study23.
Reconstruction of the protein sequences that represent the ancestors of present-day proteins, first proposed by Pauling and Zuckerkandl24, is also a very powerful approach to understanding function. The value of such ancestral sequence reconstructions was first demonstrated by the experimental analysis of predicted ancestral ribonuclease and lysozyme sequences25,26. When compared with sequences from extant organisms, the reconstructed proteins revealed the molecular basis for functional diversification in these protein families.
Artificial sequence variation and selection
An alternative approach to studying natural sequence variation is to generate a collection of related protein sequences artificially, through DNA synthesis, error-prone PCR, or other molecular biology techniques, and to analyze the functions of these proteins using a genetic selection scheme27. In a series of landmark studies, Sauer and co-workers applied this type of screening approach to the λ repressor27–29. By combining new methods for the generation of mutant libraries with an in vivo selection assay in E. coli, the authors demonstrated that these screens could map the remarkable tolerance of proteins to mutations. A critical aspect of this approach is that it provides access to regions of sequence space that have not been sampled in natural evolution.
In an important investigation of the mechanisms of resistance to the cancer drug imatinib (Gleevec), Daley and co-workers screened a random mutant library of the oncogenic kinase Bcr-Abl, the target of imatinib, to identify mutations that overcome drug inhibition30. They found that a collection of residues, many of which are far from the imatinib binding site, can allosterically perturb drug binding and kinase activity when mutated. Remarkably, these mutations predicted that the auto-inhibited structure of Abl kinases, not known at that time, would resemble the known structures of Src-family kinases30. They also predicted the presence of an allosteric site in the kinase domain, unique to Abl kinases, which could not have been inferred from the Src structures. This site was later shown to bind a lipid molecule that allosterically modulates kinase activity31,32. These findings were in accordance with the results of X-ray crystallographic and biochemical studies of the structure and regulation of Abl that were carried out independently31,32. Related screens analyzing resistance to an allosteric inhibitor of Abl and activating mutations in its proto-oncogenic form, c-Abl, have also revealed insights into kinase regulation33. The concordance of these studies with classical structural approaches testifies to the power of deep-mutagenesis methods to reveal new principles of molecular regulation.
New DNA technologies to enhance studies of sequence variation
DNA sequencing has now become fast and cheap, enabling the sequencing of thousands of genomes, the collection of metagenomic datasets, and the dense population of databases cataloguing natural variation in gene and protein sequences34. DNA sequencing has also become quantitative, due to the advent of next-generation sequencing methods that allow the rapid analysis of complex mixtures of DNA to obtain accurate counts of each sequence in a mixture35. Methods to synthesize and manipulate DNA have also become streamlined, enabling the easy construction of large DNA libraries encoding protein variants that can be functionally characterized in high-throughput selection and sequencing assays36. These innovations in DNA technology have led to the development or enhancement of methods such as directed evolution37, ancestral sequence reconstruction38, and deep mutational scanning39 (Box 1).
Box 1. Experimental approaches that utilize protein sequence variation.
In this review, we primarily focus on three strategies to experimentally analyze the impact of sequence variation on protein function. Directed evolution is an established protein engineering strategy, in which the goal is to isolate a protein with new or optimized functionality through multiple rounds of relatively unbiased sequence variation and functional selection37. These efforts can also yield insights into the fitness landscapes of proteins, through the isolation and characterization of intermediates along an evolutionary trajectory96.
Ancestral sequence reconstruction allows the identification of plausible evolutionary paths between two states, and is often used to identify sequence features that confer functional divergence between paralogous protein families38. Protein sequences are aligned and used to generate a phylogenetic tree, and the sequences of internal nodes in the tree, the ancestors, are predicted using an evolutionary model for amino acid substitutions. The value of this approach is rooted in the ability to readily synthesize gene sequences encoding the predicted ancestors, and to experimentally characterize those proteins.
In deep mutational scanning experiments, defined DNA libraries are subject to expression and functional selection, followed by deep sequencing39. Here, the power lies in the ability to quantitatively compare the abundance of variants in the DNA library before and after selection using modern deep sequencing methods. This comparison yields an “enrichment score” for each variant in a population. These scores have been shown to correlate with biophysical and biochemical parameters of proteins, including fold stability, binding affinity, and catalytic activity.
Interaction specificity during cell signaling
Evolution of nuclear hormone receptors
A common feature of many signaling proteins is that they exist within large families of paralogous members that are the products of gene duplication events followed by specialization40–43. How do new or specialized functions arise in these protein families? With the compilation of large sequence databases and the means to analyze numerous protein variants rapidly, these questions can now be addressed.
Thornton and co-workers have combined ancestral sequence reconstruction, directed evolution, and deep sequencing to analyze possible trajectories for the evolution of new steroid and DNA binding preferences in steroid-hormone receptors. Early work focused on the analysis of reconstructed sequences that provided an evolutionary path between vertebrate glucocorticoid and mineralocorticoid receptors. These investigations revealed that ancient corticoid receptors likely had steroid specificity resembling that of mineralocorticoid receptors (Figure 2A, left panel). The acquisition of binding preferences akin to that of the glucocorticoid receptor required the introduction of “permissive” background mutations that facilitated tolerance to other mutations that alter ligand binding preferences (Figure 2A, middle and right panels) 44–46. This epistasis was highly specific46, likely reflecting the fact that mutations along an evolutionary path must not substantially destabilize a protein or dramatically alter the energetic balance between all of its functionally important conformations.
Figure 2.
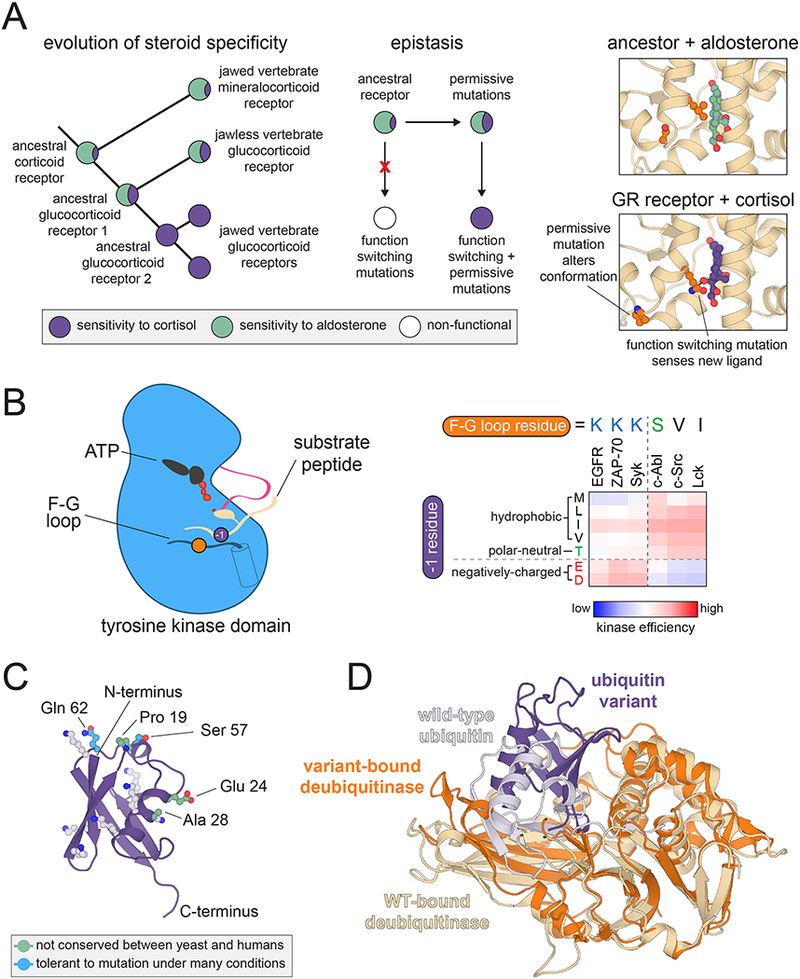
Insights into the interaction specificity of signaling proteins. (A) Epistasis in nuclear hormone receptor evolution. Left: Diagram depicting the divergence of glucocorticoid and mineralocorticoid receptors, highlighting measured binding preferences at various nodes. Middle: Diagram depicting epistasis between permissive and functional mutations. Right: Structural changes induced by permissive mutations that allow for binding to cortisol (PDB codes 2Q1H and 4P6X). (B) Tyrosine kinase substrate recognition. Left: Schematic diagram of a peptide substrate bound to a tyrosine kinase domain, highlighting the −1 residue on the substrate and a key specificity-determining position on the F-G loop of the kinase. Right: −1 residue preferences for six tyrosine kinases measured using a high-throughput bacterial surface-display and deep sequencing assay, with the identity of the key F-G loop residue given above each kinase. (C) Structure and conservation of ubiquitin. A cartoon diagram of ubiquitin is shown, highlighting the lysine residues and chain termini that are involved in ubiquitin ligation, along with residues that diverge between yeast and humans, and those residues that are completely tolerant to mutation in yeast selection assays (PDB code 1UBQ). (D) Alternative binding mode of a designed ubiquitin variant. Overlaid cartoon diagrams are shown of the deubiquitylating enzyme USP21 bound to wild-type ubiquitin (light purple, bound to beige enzyme) and an engineered variant that inhibits USP8 (dark purple, bound to orange enzyme). Structures are superimposed using only the coordinates for the deubiquitinase, and the designed variant binds with a ~90º rotation and 5 Å translation relative to wild-type ubiquitin (PDB codes 3I3T and 3N3K).
The same research group has used ancestral sequence reconstruction to examine the evolution of specificity in the DNA binding domain of nuclear steroid receptors47–49. Within this family of transcription factors, receptors that bind estrogen-like ligands with aromatized rings have one DNA-binding specificity, while those that bind ligands lacking aromatized rings interact with different DNA sequences. The predicted common ancestor of these families has DNA-binding specificity similar to that of estrogen receptors. Biochemical characterization of plausible evolutionary intermediates along these two lineages again revealed permissive mutations that were neutral on their own, but facilitated the ability of other mutations to create new DNA-binding specificity. A critical factor in the evolution of new DNA-binding specificity was not the introduction of new favorable interactions, but rather the introduction of mutations that negatively affected binding to the ancestral recognition sequence and relief of stereochemical clashes with the new recognition sequence47.
Ancestral sequence reconstruction and deep mutational scanning were also used to analyze all possible combinations of amino acid residues at the four sites in the DNA-binding domain of the ancestral steroid receptor that confer DNA sequence specificity49. This revealed numerous alternative paths to generate the specificity switch. This study highlighted how multiple solutions could arise through evolution to achieve the same functional property, and how the background sequence (i.e., the sequence containing existing permissive substitutions) impacts the evolutionary outcome.
Protein kinase substrate specificity
Protein kinases represent one of the largest classes of eukaryotic signaling enzymes, with ~500 human protein kinase genes41. Despite having a common fold, individual protein kinases phosphorylate distinct sets of substrates in cells. Box 2 provides a brief discussion of the structurally distinct bacterial histidine kinases. The substrate specificity of eukaryotic kinases is dictated by differential expression patterns and subcellular localization, but the sequence preferences of the catalytic domains also play an important role in controlling cell signaling50. The sequence preferences of protein kinase domains have been defined by using degenerate peptide libraries to extract sequence motifs that are preferred by individual kinases51,52. The advent of deep mutational scanning and new bioinformatic approaches has provided complementary strategies to further investigate kinase specificity.
Box 2. Insights into bacterial two-component signaling from sequence co-variation.
Bacterial histidine kinases are structurally distinct from eukaryotic serine/threonine and tyrosine kinases, but present the same challenges in understanding their interaction specificity. In an early landmark study, Laub and co-workers analyzed the sequences of ~1000 pairs of histidine kinases and their substrates, the response regulator proteins, and identified sets of covarying residues between the proteins97. They established that these residues confer specificity, and demonstrated that mutations at these positions could rewire histidine kinase specificity. A larger-scale analysis was carried out on bacterial histidine kinases using Direct Coupling Analysis. This approach recapitulated known interfacial determinants of specificity and also allowed the prediction of previously unknown histidine kinase-response regulator pairs98.
Recently, a method was developed that couples bacterial surface-display of genetically-encoded peptide libraries with cell sorting and deep sequencing to compare the phosphorylation of hundreds-to-thousands of discrete sequences by individual tyrosine kinases53,54. This platform was used to analyze comprehensive point-mutant libraries derived from key phosphorylation sites in T cell receptor and epidermal growth factor receptor (EGFR) signaling. The sequence-activity relationships extracted from these screens revealed an electrostatic selection mechanism in the T cell kinase ZAP-70 that controls ordered signaling upon T cell activation and likely contributes to the accuracy of the T cell response53,54. The screens also revealed functional trade-offs in the evolution of EGFR substrates. Specifically, the data suggest that the sequences of phosphorylation sites in the EGFR tail have been tuned to suppress phosphorylation by cytoplasmic tyrosine kinases, such as c-Src and c-Abl, at the expense of tight binding to downstream effectors55.
One observation that emerged from these investigations is that the successful coordination of the actions of multiple tyrosine kinases in a pathway often hinges on strong exclusionary rules at the residue immediately preceding the tyrosine phosphosite (the “−1” position). A comparison of specificity preferences at the −1 position across several kinases, coupled with sequence and structural analysis, has identified a specific residue in the F-G loop of the tyrosine kinase domain that can tune −1 preferences dramatically to direct substrate specificity (Figure 2B) 54,55.
Ancestral sequence reconstruction has been used by Holt, Turk, and co-workers to pinpoint the molecular determinants of kinase specificity, as illustrated for the divergence in specificity within the CMGC family of serine/threonine kinases56. Biochemical characterization of predicted ancestral sequences revealed that the identity of a single residue adjacent to the conserved Asp-Phe-Gly (DFG) motif in the activation loop of these kinases controls preferences for the residue at the +1 position in substrates. These experiments also showed that the specificity switch from a preferred +1 Pro to +1 Arg residue in one branch of the CMGC tree likely occurred through a promiscuous intermediate with dual specificity at this position.
Creixell, Linding, and co-workers developed a machine-learning strategy that integrates the full complement of human kinase sequences with experimentally-derived position-specific scoring matrices for hundreds of kinases57 to predict residues that impact sequence preferences across the “kinome” 58. This strategy recapitulated known specificity determinants obtained through studies on individual kinases, but also revealed a larger, sparse network of residues that control kinase specificity. Notably, many of these residues are distinct from those important for catalytic activity and regulation59,60, and mutations at some of these positions are found in cancers, where they may rewire the topology of signaling networks61.
GPCR interactions with G-proteins and ligands
Like kinases, G protein coupled receptors (GPCRs) make up a large fraction (~4%) of the genes in the human genome43. Unlike receptor kinases, which always have distinct extracellular sensory domains separated from intracellular catalytic domains, GPCRs are capable of binding a ligand and generating a signal transduction output using a single integrated transmembrane module13. Despite the growing number of crystal structures of GPCRs bound to extracellular and intracellular ligands, our understanding of the molecular determinants of ligand selectivity, and how distinct ligands transduce stimulatory or inhibitory signals in the same receptor, remains incomplete.
A recent investigation by Procko and co-workers utilized deep mutational scanning to examine cell-surface expression and ligand binding for two important human GPCRs, the chemokine receptors CXCR4 and CCR562. There is also a conceptually related study by Garcia and co-workers on a different GPCR63. Both CXCR4 and CXCR5 engage the HIV-1 envelope glycoprotein during infection, and they are activated by distinct sets of endogenous ligands to regulate the trafficking of white blood cells. The mutational screens revealed a previously unknown asymmetric mode of binding for the chemokine CXCL12 to CXCR4. These screens also identified mutations within the cores of these GPCRs, not in direct contact with ligands, that enhance ligand affinity62. Such residues are likely to underlie the allosteric network in these GPCRs that couples ligand binding to conformational changes in the intracellular region to engage downstream effectors.
The activation of GPCRs is coupled to the activation of heterotrimeric G proteins complexes (made up of α, β, and γ subunits). Each of the ~800 human GPCRs engages a distinct subset of the 16 different Gα proteins in these heterotrimeric complexes. Upon GPCR stimulation, the receptor facilitates release of GDP from the Gα protein in exchange for GTP, which leads to the dissociation of the αβγ complex and propagation of the signal.
Babu and co-workers took an evolutionary approach to understand the specificity of GPCR-G protein interactions by analyzing conservation across paralogs and orthologs of all 16 human Gα proteins in 66 diverse genomes64. The 16 human Gα proteins fall into four families (subtypes) of paralogous GTPases that engage similar sets of GPCRs, and there is an analogous distribution of Gα proteins in organisms ranging from animals to unicellular eukaryotes. The authors analyzed hundreds of Gα sequences and identified residues that were conserved in orthologs of the same Gα protein across different organisms, but that diverged between paralogous families within the same organism.
When mapped onto structures of GPCR-Gα complexes, subtype-specific residues at the interface were found that surrounded a core set of residues that were highly conserved across all Gα proteins64. The conserved residues at the core of the interface are required for the activation of Gα proteins by GPCRs (discussed below) 65. The surrounding subtype-specific residues comprise a selectivity “barcode”, where the combination of residue identities at these key positions defines which GPCRs can effectively engage a particular G protein64. Since the Gα residues that make up this “barcode” to selectively engage GPCRs are distinct from the conserved residues that form the core of the interaction, GPCRs can readily evolve to be either promiscuous or highly-specific. In GPCRs, segregation of molecular determinants for extracellular ligand-selectivity and residues important for allosteric activation of Gα proteins from those that control GPCR-Gα interaction specificity has allowed for rampant diversification of these receptors64.
Control of ubiquitylation pathways
Ubiquitin is one of the most highly conserved proteins in eukaryotic genomes, with human and yeast ubiquitin sequences differing in only three out of 76 residues (Figure 2C). The linking of single ubiquitin molecules, or of ubiquitin chains, to other proteins can impact the stability of those proteins, their localization, or their engagement in protein-protein interactions66. Virtually every surface of ubiquitin is involved in protein-protein interactions, and the numerous binding partners of ubiquitin often have overlapping footprints on its surface67.
The diversity of interactions made by ubiquitin is one likely explanation for its strict conservation. This hypothesis was tested in a series of studies using deep mutational scans of ubiquitin in yeast68–70. Under particular selection conditions, most positions in ubiquitin are remarkably tolerant to amino acid substitution. By conducting screens in the presence of different chemical additives, however, Fraser and co-workers showed that virtually every residue in ubiquitin, with the exception of two, can be sensitized to mutation under a particular selection condition (Figure 2C). These results demonstrate that the evolutionary trajectory of ubiquitin is shaped by a necessity to function under different environmental conditions, in which ubiquitin may engage in distinct sets of interactions69,70.
The numerous interactions that ubiquitin participates in has made it challenging to dissect the importance of individual ubiquitylation events in the cell. To tackle this problem, Sidhu and co-workers generated a phage-display library encoding billions of ubiquitin variants, and selected for tight binding to particular ubiquitin ligases or deubiquitinases71,72. The screens yielded potent and selective inhibitors of ubiquitin-modifying enzymes, which were used in cell-based experiments to identify new ubiquitin-mediated signaling events. In a related study, this screening approach was combined with computational protein design to identify subtle conformational fluctuations in the ubiquitin fold that impact binding specificity73.
These screens not only yielded potent and selective inhibitors of ubiquitin signaling, they also revealed surprising features of ubiquitin. Cellular experiments with variant ubiquitin molecules showed that sequence changes that promote tight binding to individual ubiquitin-binding proteins are incompatible with the dynamic nature of ubiquitin-based signaling71. Thus, the sequence of ubiquitin may be constrained by the need to maintain numerous weak interactions. Structural analysis of the ubiquitin variants showed that small changes in the sequence of ubiquitin could result in dramatic changes in the orientation of variants when bound to the same target protein, by as much as a ~90º rotation and 5 Å translation (Figure 2D) 71. This finding is intriguing when one considers that protein-protein interactions occur through the formation of encounter complexes that can involve quite different interacting surfaces than those seen in the final stable complex74. We speculate that the ubiquitin variants that bound preferentially to a particular target were selected from distinct configurations that were sampled during the initial stages of intermolecular encounter. Selection on transient intermediates may also be at play in the natural evolution of signaling proteins, and could be an important driving force in the evolution of new protein-protein interactions14.
Allosteric regulation of signaling proteins
Determinants of protein kinase regulation
The underlying conformational landscape of eukaryotic protein kinase domains that allows for dynamic regulation is slightly different in each kinase, and thus provides unique structural targets for the design of selective inhibitors. Kornev, Taylor, and co-workers analyzed a number of protein kinases that had been crystallized in both active and inactive states, and they identified two clusters of physically contiguous residues, termed “spines”, whose arrangement is likely to impact kinase activation (Figure 3A, left panel) 59,60. The identity of residues in these spines, and of the residues that contact them, affect the stability of either the active or inactive conformations of the kinase domain. These spines have been used to predict the intrinsic activities of kinases, and to understand kinase dysregulation and drug resistance (Figure 3A, right panel) 6,75. Box 3 highlights a comparative biochemistry study examining the evolution of allostery in protein kinases.
Figure 3.
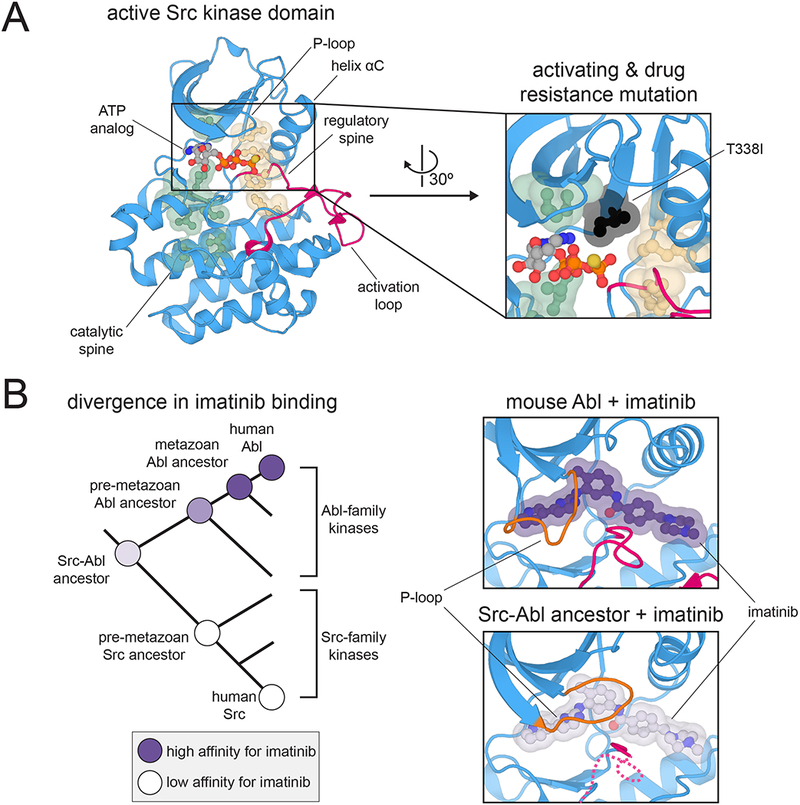
Sequence and structural features that control kinase dynamics and allostery. (A) Hydrophobic spines that control kinase activity and regulation. Left: Structure of the kinase domain of c-Src kinase in an active conformation, highlighting key structural elements and the hydrophobic spines (PDB code 3DQW). Right: Position of the T338I mutation (chicken c-Src numbering) relative to the hydrophobic spines. Mutations at this “gatekeeper” position in many kinases are often activating and confer resistance to ATP-competitive inhibitors (PDB code 3DQW). (B) Divergence of imatinib binding between the Src- and Abl-family kinases. Left: Phylogenetic tree depicting the divergence of Src- and Abl-family kinases, highlighting measured preferences at various nodes for binding to imatinib. Right: Closing of the P-loop over imatinib when bound to Abl but not a Src-Abl ancestral kinase (PDB codes 1OPJ and 4CSV).
Box 3. Evolution of allostery in yeast kinases.
The identification of hydrophobic spines and sectors comprised of co-evolving residues in all protein kinases has provided a useful framework for understanding kinase regulation by reinforcing the concept that all protein kinases share commonalities in their regulatory mechanisms. Given this observation, it is interesting to consider how different kinases have evolved to respond to distinct allosteric perturbations, while relying on a conserved structural scaffold to convert these perturbations into the same biochemical activity.
In an elegant study by Lim and co-workers, mitogen-activated protein (MAP) kinases from yeast were analyzed from an evolutionary perspective to understand how two closely-related kinases, Fus3 and Kss1, could have evolved from a single common ancestor to be either allosterically-regulated by the scaffold protein Ste5, or to be scaffold-independent, respectively99. In this study, these two paralogous proteins and their orthologs across 13 different yeast species were compared biochemically for their ability to be activated by Ste5 orthologs from those same species, when a discernable Ste5-like scaffold was present. The authors found that MAP kinases in organisms that diverged before the emergence of Ste5 and before duplication to yield distinct Fus3 and Kss1 proteins, could still be activated by Ste599. This suggests that allosteric regulation is a latent feature of protein kinases that can be exploited or suppressed by just a few amino acid substitutions. Importantly, this work used sequences from just a few diverse yeast genomes, and it foreshadowed current-day high-throughput approaches.
The implementation of Statistical Coupling Analysis to protein kinases has identified three, roughly independent, sectors of co-evolving residues in the kinase fold that control intrinsic catalytic activity, confer substrate specificity, and coordinate the reception of allosteric perturbations76. The sectors that control catalytic activity and allosteric regulation contain the two hydrophobic spines that are implicated in the control of these two functions59,60. The majority of somatic cancer mutations found in kinase genes map to the catalytic sector, and residues that coordinate conformation-selective kinase inhibitors typically map to the regulatory sector76.
Ancestral sequence reconstruction has been implemented to identify specific sequence changes that impact the dynamics of protein kinases. One investigation by Kern and colleagues used this approach to examine the structural basis for the 3000-fold tighter binding of the cancer drug imatinib to Abl-family kinases over the closely-related Src-family kinases77. Imatinib binds tightly to relatively few kinases in the human kinome. This was first thought to be due to the ability of only a few kinases, including Abl, to adopt an inactive conformation in which the conserved catalytic site DFG motif was flipped relative to most kinases, such as Src-family kinases78. The identification of compounds that can bind Src- and Abl-family kinases equipotently in the DFG-flipped conformation indicated that this explanation was not correct79,80.
To identify the origins of imatinib selectivity, sequences of the common ancestors of the Abl and Src lineages were predicted and experimentally characterized (Figure 3B, left panel) 77. By tracing steps in the lineages leading from the common ancestor of the Src and Abl kinases to each family of extant kinases, a series of sequence changes were identified that strongly correlate with drug selectivity. The authors identified a set of residues that form a hydrogen bond network in Src-like kinases that is lacking in Abl-like kinases. These hydrogen bonds prevent the P-loop, a structural element near the drug binding site, from closing over the drug in the case of Src, but not in Abl (Figure 3B, right panel). As a consequence, although imatinib can bind both Src and Abl in the DFG-flipped state, it dissociates more slowly from Abl than from Src77,78. The principle that conformational changes that lock down the inhibitor occur after initial binding may be quite general81, and could guide future drug design efforts.
Allosteric regulation of G proteins
The activation of all G proteins from their signaling-inactive GDP-bound state typically requires that a guanine nucleotide exchange factor (GEF) binds to the G protein and facilitates the dissociation of GDP, allowing for binding of the more abundant nucleotide, GTP. For small GTPases, such as Ras, GEFs are a diverse set of cytoplasmic proteins. For heterotrimeric G proteins, the GEF that acts on the Gα subunit is the GPCR. Numerous structures of Ras-like small GTPases bound to their corresponding GEFs have been determined, all of which show related molecular mechanisms for nucleotide exchange82,83. By contrast, until recently, very few structures of heterotrimeric G proteins bound to GPCRs had been determined, and these structures reveal a very different mechanism of activation13. Given that the 16 distinct Gα proteins in humans can potentially be activated by hundreds of different GPCRs, it had remained unclear whether all heterotrimeric G proteins would be activated through the same allosteric mechanism.
This question was addressed by Babu and colleauges through a structure-guided bioinformatic approach, which integrated residue contact information from crystal structures of Gα proteins with conservation scores from an alignment of ~600 sequences65. This analysis revealed that many of the highly conserved residues at GPCR-G protein interfaces lie on a single helix, H5, which extends out of the core GTPase domain, away from the guanine nucleotide binding site. These residues represent a unified integration point for signal transduction from all GPCRs to all heterotrimeric G proteins. Helix H5 undergoes a large conformational change upon GPCR binding, and bioinformatic analysis suggested that this conformational change in H5 would reduce the structural integrity of the adjacent helix, H1, by disrupting conserved interactions between the two helices. As H1 leads into the nucleotide binding pocket, an ordered H1 is critical for tight nucleotide binding. Thus, direct propagation of conserved structural changes from the GPCR to H5 to H1 drives a universal allosteric activation mechanism in heterotrimeric G proteins65.
Amino acid substitutions at a handful of positions in the Ras family of small GTPases, particularly at residues 12, 13, and 61, are among the most common missense mutations in human cancers84. They act by slowing the rate of GTP hydrolysis and by biasing Ras towards its signaling-active conformation (Figure 4A). The three major Ras isoforms, H-Ras, K-Ras, and N-Ras, are virtually identical in their GTPase domains and are highly conserved throughout vertebrate evolution40. To better understand Ras activation and evolution, H-Ras activity was analyzed by deep mutational scanning, using a bacterial “two-hybrid” system that could sense the ability of H-Ras to interact with a downstream binding partner85. All possible single-amino acid substitutions in the GTPase domain of H-Ras were analyzed in the presence and absence of a GEF and a negative-regulatory GTPase activating protein (GAP), which accelerates the rate of GTP hydrolysis.
Figure 4.
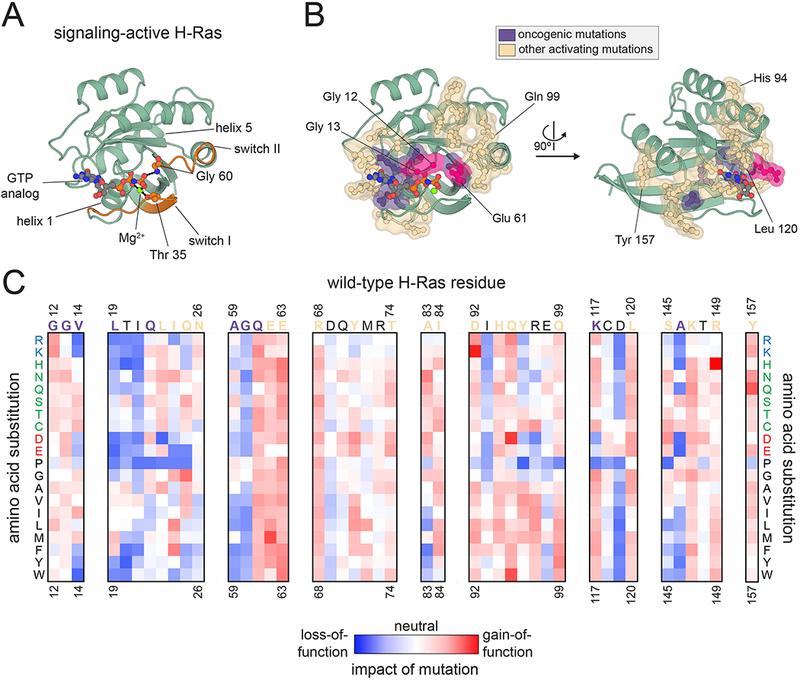
Allosteric activation of Ras. (A) Structural features of Ras. A cartoon representation of H-Ras is shown, highlighting several key structural elements and showing how the switch regions are anchored to the γ phosphate of GTP (PDB code 5P21). (B) Hotspots of activation in Ras. The major sites of oncogenic activating mutations in Ras proteins are highlighted in purple and pink, and sites of activating second-shell hotspot residues, identified through deep mutational scanning of H-Ras, are shown in beige (PDB code 5P21). The residues colored in purple and pink are found to be mutated in the COSMIC cancer genome database (https://cancer.sanger.ac.uk/cosmic), but of these, mutations are found with high frequency at only three sites (Glycine 12, Glycine 13, and Glutamine 61, colored in pink). (C) Deep mutational scanning of H-Ras. The effects of all possible point mutations at several positions in H-Ras, measured in a deep mutational scanning experiment in bacteria, are shown as a heatmap. The wild-type amino acid residue is shown above each column of the heatmap, and each row represents a different amino acid substitution. The wild-type residue label is colored according to whether it is a site of oncogenic mutations in human cancers (purple) or deemed a second-shell hotspot residue (beige).
These experiments resulted in two important findings. First, the necessity to cycle between on and off states, and to respond to the presence of a GAP and GEF, constrains the accessible sequence space of Ras. This may partly explain the high conservation of Ras proteins across evolution. Second, although only a few sites are commonly mutated in human cancers to activate Ras, many more “hotspot” residues, where numerous amino acid substitutions are activating, were observed in the screens (Figure 4B,C) 85. These residues form a contiguous shell surrounding the sites of the canonical oncogenic Ras mutations (Figure 4B). Molecular dynamics simulations suggest that mutations at these positions cause a general ‘loosening’ of Ras structure. Given the large number of sites of activating mutations found in this study, it is surprising that most cancer mutations are found at only three positions in Ras. Understanding the origins of this inconsistency requires further investigation.
Concluding remarks
It has long been appreciated that analysis of natural sequence variation can provide insights into protein structure and function, and mutational analysis has been the common currency of biochemistry since the advent of modern molecular biology techniques. These classical approaches to explore the sequence space of proteins have seen a recent increase in their power, facilitated by the development of new DNA sequencing and synthesis methods.
It is now possible to reconstruct plausible evolutionary trajectories for a family of proteins and to functionally characterize all members of that protein family simultaneously. Deep mutational scans are providing data that both corroborate findings from structural studies and inspire new ways of thinking about protein structure and dynamics. Further insights into protein allostery are likely to be unveiled as we develop strategies to generate and sequence higher-order mutational libraries, which will allow for an experimental assessment of energetic coupling between sites in a protein86. A challenge lies in the development of robust selection assays for proteins of interest. For signaling proteins, many of which are proto-oncogenes that promote cell proliferation, high-throughput screens that report on oncogene-dependent cell growth in a native context can be readily developed. Such screens are particularly informative when correlated with the growing information available in cancer genome databases87–89.
Looking forward, we anticipate substantial synergy between the analyses of sequence variation and other methods to interrogate signaling proteins. For example, cryo-electron microscopy is providing structural insights into multi-component signaling machines of increasing size and complexity90, and the interpretation of these structures will undoubtedly be guided by an assessment of tolerated sequence space. Where structure determination remains challenging, co-evolutionary information is already aiding in the prediction of protein-protein interactions91,92 and is shedding light on the organization of macromolecular complexes93,94. Long time-scale molecular dynamics simulations are revealing the detailed motions of signaling proteins95. In conjunction with information about sequence variation, these simulations will help elucidate how closely-related proteins have diverged and specialized through nuanced changes in their conformational dynamics. Like these other recent advances in protein science, we envision that the strategies described in this review will become commonplace in the toolkit of biologists studying cell signaling, and will help to guide the development of therapeutics that modulate cellular signal transduction.
Acknowledgements
We thank Pradeep Bandaru and Rama Ranganathan for insights and stimulating discussions. N.H.S. is a funded by a Damon Runyon-Dale F. Frey Award for Breakthrough Scientists from the Damon Runyon Cancer Research Foundation. J.K. is funded by NIH grant P01 A1091580.
Footnotes
Competing interests The authors declare no competing interests.
References
- 1.Song Y et al. High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Webb B & Sali A Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics 47, 5.6.1–32 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Clapham DE Calcium signaling. Cell 131, 1047–1058 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Berman HM et al. The cAMP binding domain: an ancient signaling module. Proc. Natl. Acad. Sci. USA 102, 45–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang P, Chandra V & Rastinejad F Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu. Rev. Physiol. 72, 247–272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kornev AP & Taylor SS Dynamics-Driven Allostery in Protein Kinases. Trends Biochem. Sci. 40, 628–647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Tonks NK Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Shah NH, Amacher JF, Nocka LM & Kuriyan J The Src module: an ancient scaffold in the evolution of cytoplasmic tyrosine kinases. Crit Rev Biochem Mol Biol 53, 535–563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pawson T & Scott JD Signaling through scaffold, anchoring, and adaptor proteins. Science 278, 2075–2080 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Lorenz S, Cantor AJ, Rape M & Kuriyan J Macromolecular juggling by ubiquitylation enzymes. BMC Biol. 11, 65 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wittinghofer A & Vetter IR Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 80, 943–971 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Hilger D, Masureel M & Kobilka BK Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuriyan J & Eisenberg D The origin of protein interactions and allostery in colocalization. Nature 450, 983–990 (2007). [DOI] [PubMed] [Google Scholar]
- 15.O’Rourke L & Ladbury JE Specificity is complex and time consuming: mutual exclusivity in tyrosine kinase-mediated signaling. Acc. Chem. Res. 36, 410–416 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Kapp OH, Moens L, Vanfleteren J, Trotman CN, Suzuki T and Vinogradov SN Alignment of 700 globin sequences: extent of amino acid substitution and its correlation with variation in volume. Protein Science 4, 2179–2190 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Juan D, Pazos F & Valencia A Emerging methods in protein co-evolution. Nat. Rev. Genet. 14, 249–261 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Marks DS et al. Protein 3D structure computed from evolutionary sequence variation. PLoS One 6, e28766 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morcos F et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 108, E1293–301 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lockless SW & Ranganathan R Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286, 295–299 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Halabi N, Rivoire O, Leibler S & Ranganathan R Protein sectors: evolutionary units of three-dimensional structure. Cell 138, 774–786 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Süel GM, Lockless SW, Wall MA & Ranganathan R Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 10, 59–69 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Teşileanu T, Colwell LJ & Leibler S Protein sectors: statistical coupling analysis versus conservation. PLoS Comput. Biol. 11, e1004091 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pauling L, Zuckerkandl E, Henriksen T & Lövstad R Chemical Paleogenetics. Molecular “Restoration Studies” of Extinct Forms of Life. Acta Chem. Scand. 17 supl, 9–16 (1963). [Google Scholar]
- 25.Malcolm BA, Wilson KP, Matthews BW, Kirsch JF & Wilson AC Ancestral lysozymes reconstructed, neutrality tested, and thermostability linked to hydrocarbon packing. Nature 345, 86–89 (1990). [DOI] [PubMed] [Google Scholar]
- 26.Stackhouse J, Presnell SR, McGeehan GM, Nambiar KP & Benner SA The ribonuclease from an extinct bovid ruminant. FEBS Lett. 262, 104–106 (1990). [DOI] [PubMed] [Google Scholar]
- 27.Bowie JU, Reidhaar-Olson JF, Lim WA & Sauer RT Deciphering the message in protein sequences: tolerance to amino acid substitutions. Science 247, 1306–1310 (1990). [DOI] [PubMed] [Google Scholar]
- 28.Reidhaar-Olson JF & Sauer RT Combinatorial cassette mutagenesis as a probe of the informational content of protein sequences. Science 241, 53–57 (1988). [DOI] [PubMed] [Google Scholar]
- 29.Lim WA & Sauer RT Alternative packing arrangements in the hydrophobic core of lambda repressor. Nature 339, 31–36 (1989). [DOI] [PubMed] [Google Scholar]
- 30.Azam M, Latek RR & Daley GQ Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 112, 831–843 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Nagar B et al. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112, 859–871 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Hantschel O et al. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 112, 845–857 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Lee BJ & Shah NP Identification and characterization of activating ABL1 1b kinase mutations: impact on sensitivity to ATP-competitive and allosteric ABL1 inhibitors. Leukemia 31, 1096–1107 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Mukherjee S et al. Genomes OnLine Database (GOLD) v.6: data updates and feature enhancements. Nucleic Acids Res. 45, D446–D456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodwin S, McPherson JD & McCombie WR Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kosuri S & Church GM Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 11, 499–507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Packer MS & Liu DR Methods for the directed evolution of proteins. Nat. Rev. Genet. 16, 379–394 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Hochberg GKA & Thornton JW Reconstructing ancient proteins to understand the causes of structure and function. Annu. Rev. Biophys. 46, 247–269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fowler DM & Fields S Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rojas AM, Fuentes G, Rausell A & Valencia A The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J. Cell Biol. 196, 189–201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manning G, Plowman GD, Hunter T & Sudarsanam S Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 27, 514–520 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Laudet V, Hänni C, Coll J, Catzeflis F & Stéhelin D Evolution of the nuclear receptor gene superfamily. EMBO J. 11, 1003–1013 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fredriksson R, Lagerström MC, Lundin L-G & Schiöth HB The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63, 1256–1272 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Ortlund EA, Bridgham JT, Redinbo MR & Thornton JW Crystal structure of an ancient protein: evolution by conformational epistasis. Science 317, 1544–1548 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bridgham JT, Ortlund EA & Thornton JW An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature 461, 515–519 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harms MJ & Thornton JW Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature 512, 203–207 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKeown AN et al. Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell 159, 58–68 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson DW, McKeown AN & Thornton JW Intermolecular epistasis shaped the function and evolution of an ancient transcription factor and its DNA binding sites. Elife 4, e07864 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Starr TN, Picton LK & Thornton JW Alternative evolutionary histories in the sequence space of an ancient protein. Nature 549, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller CJ & Turk BE Homing in: mechanisms of substrate targeting by protein kinases. Trends Biochem. Sci. 43, 380–394 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Songyang Z et al. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature 373, 536–539 (1995). [DOI] [PubMed] [Google Scholar]
- 52.Hutti JE et al. A rapid method for determining protein kinase phosphorylation specificity. Nat. Methods 1, 27–29 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Shah NH et al. An electrostatic selection mechanism controls sequential kinase signaling downstream of the T cell receptor. Elife 5, e20105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shah NH, Löbel M, Weiss A & Kuriyan J Fine-tuning of substrate preferences of the Src-family kinase Lck revealed through a high-throughput specificity screen. Elife 7, e35190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cantor AJ, Shah NH & Kuriyan J Deep mutational analysis reveals functional trade-offs in the sequences of EGFR autophosphorylation sites. Proc. Natl. Acad. Sci. USA (2018). doi: 10.1073/pnas.1803598115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Howard CJ et al. Ancestral resurrection reveals evolutionary mechanisms of kinase plasticity. Elife 3, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller ML et al. Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal 1, ra2 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Creixell P et al. Unmasking determinants of specificity in the human kinome. Cell 163, 187–201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kornev AP, Haste NM, Taylor SS & Eyck LFT Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 103, 17783–17788 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kornev AP, Taylor SS & Ten Eyck LF A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. USA 105, 14377–14382 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Creixell P et al. Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell 163, 202–217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heredia JD et al. Mapping interaction sites on human chemokine receptors by deep mutational scanning. J. Immunol. 200, 3825–3839 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miles TF et al. Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flock T et al. Selectivity determinants of GPCR-G-protein binding. Nature 545, 317–322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flock T et al. Universal allosteric mechanism for Gα activation by GPCRs. Nature 524, 173–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pickart CM & Eddins MJ Ubiquitin: structures, functions, mechanisms. Biochim. Biophys. Acta 1695, 55–72 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Winget JM & Mayor T The diversity of ubiquitin recognition: hot spots and varied specificity. Mol. Cell 38, 627–635 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Roscoe BP, Thayer KM, Zeldovich KB, Fushman D & Bolon DNA Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J. Mol. Biol. 425, 1363–1377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mavor D et al. Determination of ubiquitin fitness landscapes under different chemical stresses in a classroom setting. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mavor D et al. Extending chemical perturbations of the ubiquitin fitness landscape in a classroom setting reveals new constraints on sequence tolerance. Biol. Open 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ernst A et al. A strategy for modulation of enzymes in the ubiquitin system. Science 339, 590–595 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang W et al. System-Wide Modulation of HECT E3 Ligases with Selective Ubiquitin Variant Probes. Mol. Cell 62, 121–136 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y et al. Conformational stabilization of ubiquitin yields potent and selective inhibitors of USP7. Nat. Chem. Biol. 9, 51–58 (2013). [DOI] [PubMed] [Google Scholar]
- 74.Tang C, Iwahara J & Clore GM Visualization of transient encounter complexes in protein-protein association. Nature 444, 383–386 (2006). [DOI] [PubMed] [Google Scholar]
- 75.Hu J et al. Kinase regulation by hydrophobic spine assembly in cancer. Mol. Cell. Biol. 35, 264–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Creixell P et al. Hierarchical Organization Endows the Kinase Domain with Regulatory Plasticity. Cell Syst. 7, 371–383.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilson C et al. Kinase dynamics. Using ancient protein kinases to unravel a modern cancer drug’s mechanism. Science 347, 882–886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seeliger MA et al. c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure 15, 299–311 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Dar AC, Lopez MS & Shokat KM Small molecule recognition of c-Src via the Imatinib-binding conformation. Chem. Biol. 15, 1015–1022 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Seeliger MA et al. Equally potent inhibition of c-Src and Abl by compounds that recognize inactive kinase conformations. Cancer Res. 69, 2384–2392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pitsawong W et al. Dynamics of human protein kinase Aurora A linked to drug selectivity. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cherfils J & Zeghouf M Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 93, 269–309 (2013). [DOI] [PubMed] [Google Scholar]
- 83.Bandaru P, Kondo Y & Kuriyan J The Interdependent Activation of Son-of-Sevenless and Ras. Cold Spring Harb. Perspect. Med. (2018). doi: 10.1101/cshperspect.a031534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prior IA, Lewis PD & Mattos C A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bandaru P et al. Deconstruction of the Ras switching cycle through saturation mutagenesis. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Salinas VH & Ranganathan R Coevolution-based inference of amino acid interactions underlying protein function. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Findlay GM et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pahuja KB et al. Actionable activating oncogenic ERRB2/HER2 transmembrane and juxtamembrane domain mutations. Cancer Cell (2018). doi: 10.1016/j.ccell.2018.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma L et al. CRISPR-Cas9-mediated saturated mutagenesis screen predicts clinical drug resistance with improved accuracy. Proc. Natl. Acad. Sci. USA 114, 11751–11756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Baretić D et al. Structures of closed and open conformations of dimeric human ATM. Sci. Adv. 3, e1700933 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bitbol A-F, Dwyer RS, Colwell LJ & Wingreen NS Inferring interaction partners from protein sequences. Proc. Natl. Acad. Sci. USA 113, 12180–12185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gueudré T, Baldassi C, Zamparo M, Weigt M & Pagnani A Simultaneous identification of specifically interacting paralogs and interprotein contacts by direct coupling analysis. Proc. Natl. Acad. Sci. USA 113, 12186–12191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hopf TA et al. Sequence co-evolution gives 3D contacts and structures of protein complexes. Elife 3, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ovchinnikov S, Kamisetty H & Baker D Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. Elife 3, e02030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dror RO et al. SIGNAL TRANSDUCTION. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science 348, 1361–1365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Romero PA & Arnold FH Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Skerker JM et al. Rewiring the specificity of two-component signal transduction systems. Cell 133, 1043–1054 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Procaccini A, Lunt B, Szurmant H, Hwa T & Weigt M Dissecting the specificity of protein-protein interaction in bacterial two-component signaling: orphans and crosstalks. PLoS One 6, e19729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coyle SM, Flores J & Lim WA Exploitation of latent allostery enables the evolution of new modes of MAP kinase regulation. Cell 154, 875–887 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]