

Abstract
Plant γ-glutamylcysteine ligase (GCL), catalyzing the first and tightly regulated step of glutathione (GSH) biosynthesis, is redox-activated via formation of an intramolecular disulfide bond. In vitro, redox-activation of recombinant GCL protein causes formation of homo-dimers. Here, we have investigated whether dimerization occurs in vivo and if so whether it contributes to redox-activation. FPLC analysis indicated that recombinant redox-activated WT (wild type) AtGCL dissociates into monomers at concentrations below 10−6 M, i.e. below the endogenous AtGCL concentration in plastids, which was estimated to be in the micromolar range. Thus, dimerization of redox-activated GCL is expected to occur in vivo. To determine the possible impact of dimerization on redox-activation, AtGCL mutants were generated in which salt bridges or hydrophobic interactions at the dimer interface were interrupted. WT AtGCL and mutant proteins were analyzed by non-reducing SDS–PAGE to address their redox state and probed by FPLC for dimerization status. Furthermore, their substrate kinetics (KM, Vmax) were compared. The results indicate that dimer formation is not required for redox-mediated enzyme activation. Also, crystal structure analysis confirmed that dimer formation does not affect binding of GSH as competitive inhibitor. Whether dimerization affects other enzyme properties, e.g. GCL stability in vivo, remains to be investigated.
Keywords: glutamylcysteine ligase, glutathione biosynthesis, homo-dimerization, redox activation, site-directed mutagenesis
Introduction
Glutathione (GSH) and its oxidized form glutathione disulfide (GSSG) are key players of the cellular redox buffer in plants. Total GSH content and changes in the GSH:GSSG ratio both affect its redox potential. To ensure a stable redox environment within the cell, de novo synthesis of GSH has to be tightly regulated by mechanisms that allow quick response to abiotic and biotic stress factors [1–3].
GSH is synthesized in two ATP-dependent reactions, catalyzed by γ-glutamylcysteine ligase (GCL) and glutathione synthetase (GS), respectively. The first and rate-limiting step is mediated by GCL (EC 6.3.2.2), which forms γ-glutamylcysteine (γ-EC) from glutamate and cysteine. In mammals and Drosophila, GCL is a hetero-dimeric protein, a catalytic subunit (GCLC) interacting with a modifier subunit (GCLM). Upon binding of GCLM to GCLC, Vmax is increased, KM values for glutamate and ATP are decreased and the Ki for GSH-mediated feedback inhibition is increased [4]. GCLM regulates enzyme activity by covalent (formation of inter-subunit disulfides) and non-covalent interactions [5]. Furthermore, activities of mammalian and Drosophila GCL may be modulated via GCL phosphorylation and allosterically by NADPH [6].
In marked contrast, higher plant GCL is encoded by a single gene, the enzyme being exclusively localized in the plastids [7]. In vivo, GSH biosynthesis may be limited by cysteine or glycine availability [8–10]. Furthermore, biochemical and crystal structure analysis has revealed that GCL activity may be post-translationally activated by formation of an intramolecular disulfide bridge [Cys186–Cys406 in AtGCL; 11–14]. In addition, FPLC analysis of recombinant GCL protein has shown that oxidized GCL protein forms homo-dimers in vitro [13,15]. Reducing agents like DTT, which inactivate GCL via disruption of the regulatory disulfide bridge, also cause dissociation of the homo-dimer into monomers [15]. While GCL proteins from plant species of the rosid clade contain a second disulfide bridge (Cys349–Cys364 in AtGCL), forming a β-hairpin motif, the midpoint potential of this second disulfide bridge is not within the range of chloroplast redox control as shown by Hicks et al. [14]. Structural analysis of the homo-dimer interface has revealed many amino acid residues highly conserved among plant GCL sequences [15]. The zipper arrangement at the interface involves several salt bridges and aromatic side chains, and the intramolecular disulfide bridge is positioned in close proximity to the dimer interface [13,15]. While these cited studies were performed with recombinant GCL protein in vitro, Hicks et al. [14] have provided evidence for the physiological relevance of this redox-mediated post-translational regulation in vivo: In Arabidopsis roots, oxidation or depletion of endogenous GSH resulted in increased GCL activity concomitant with a pronounced conformational change of GCL protein, as indicated by a mobility shift in non-reducing gel electrophoresis [14]. These observations demonstrated for the first time that the formation of the oxidized (i.e. redox-activated) GCL protein indeed occurred in vivo. However, it has remained an open question whether homo-dimer formation was an essential part of this activation since the method used (i.e. non-reducing SDS–PAGE) revealed the shift from reduced to oxidized monomer but would have caused disruption of the non-covalently bound homo-dimer.
Thus, while the in vitro analysis of recombinant GCL enzyme indicated that oxidation-mediated enzyme activation was correlated with homo-dimer formation [15], it has remained unresolved, whether (i) homo-dimer formation occurs in vivo and (ii) whether redox-mediated activation is dependent or independent of the homo-dimer formation. To answer both questions, we have first determined in which concentration range recombinant homo-dimer dissociates into monomers and compared the result with an experimental estimation of plastidial GCL concentration. Wild type (WT) GCL from Arabidopsis thaliana was then mutated in amino acid residues involved in the zipper-like arrangement at the dimer interface [13,15]. After disrupting the dimer interface by site-directed mutagenesis, the recombinant mutant GCL proteins were analyzed for enzyme activity and dimerization state. In summary, the results reveal that homo-dimer formation of redox-activated GCL is likely to occur in vivo but is not required for enzyme activation.
Materials and methods
Cloning and site-directed mutagenesis
WT AtGCL cDNA was amplified by PCR with primers P1 and P2 (see Supplementary Table S1). The sequence encompassed the protein coding region without the predicted plastidial transit peptide (residues 1–73); also for all GCL mutants, the transit peptide was excluded. AtGCL mutants Glu141/Glu403, Lys201/Lys479, Gln141/Met403, and Leu194/Phe483 were generated by site-directed mutagenesis using primers including mutation sites (Table 1): P1/P2/P3/P4 were used for the Glu141/Glu403 mutation, P1/P2/P5/P6 for the Lys201/Lys479 mutation, P1/P2/P7/P8/P9/P10 for the Gln141/Met403 mutation, P1/P2/P11/P12 for the Leu194/Phe483 mutation, and P1/P2/P13/P14 for the Ser186/Cys406 mutation. PCR products were verified by DNA sequencing. All sequences were digested with NcoI and XhoI and cloned into the pETM-20 vector [16] to produce His-tagged proteins fused with thioredoxin to increase solubility.
Table 1. Overview of GCL mutations in A. thaliana.
| Endogenous amino acid residues in pair | Mutated amino acid residues in pair | |
|---|---|---|
| Salt bridges | Glu141/Arg403 | Glu141/Glu403 |
| Gln141/Met403 | ||
| Glu201/Lys479 | Lys201/Lys479 | |
| Hydrophobic interaction | Tyr194/Phe483 | Leu194/Phe483 |
Protein expression and purification
Recombinant fusion proteins were expressed in Escherichia coli Rosetta-gami DE3 (Novagen). Cells grown to an OD of 0.6 (600 nm) were induced with 1 mM isopropyl-β-d-thiogalactopyranoside in lysogeny broth at 37°C for 4 h. Pelleted cells were resuspended in lysis buffer [50 mM Tris (pH 8.0), 250 mM NaCl, 20 mM imidazol]. The suspension was centrifuged at 22 000g for 10 min and recombinant protein was purified by Ni2+ affinity chromatography (Qiagen, Valencia, CA). His-tagged protein was eluted with elution buffer [50 mM Tris (pH 8.0), 250 mM NaCl, 400 mM imidazol]. The eluted protein was dialyzed against buffer [25 mM HEPES (pH 7.5), 5 mM MgCl2, 150 mM NaCl] and cleaved overnight with recombinant tobacco etch virus protease at 4°C. A second Ni2+ affinity step was performed after dialysis to separate GCL protein from 6xHis-tagged protease and thioredoxin. The final protein was concentrated by centrifugal filtration (Amicon Ultra-filter Millipore, 30 kDa MWCO).
Crystallization of GCL protein in the presence of S-methyl-GSH and data collection
S-methyl-GSH (GSM) was added to a final concentration of 10 mM to a 12 mg/ml GCL protein solution; note that for crystallization GCL from Brassica juncea (95.5% protein sequence identity with the Arabidopsis enzyme) was used as previously described by Hothorn et al. [13]. Crystals grew within 3 days in a robotic sitting drop setup, where 100 nl protein solution and 100 nl crystallization buffer [0.2 M sodium acetate, 20% (w/v) PEG 3,350] were mixed and suspended over 70 µl of the latter as reservoir solution. The crystals were transferred to crystallization buffer supplemented by 20% glycerol and flash frozen in liquid N2. Data were collected at beamline BM16 of the European Synchrotron Radiation Facility (ESRF), Grenoble, France.
Structure determination and refinement
Structures were solved using XDS [17] for data processing and PHASER [18] for molecular replacement using l-glutamate bound GCL (PDB code 2GWD) as a search model. The structure was refined through alternating cycles of model building in Coot [19] and refinement with Refmac5 [20]. Refinement statistics are given in Supplementary Table S2. Structure validation was done using Molprobity [21] and Pymol [22] (see Figure 7). Superpositions were made using DaliLite [23] and Superpose [24]. Atomic co-ordinates and structure factors have been deposited in the RCSB Protein Data Bank with accession code 6GMO.
Figure 7. Structure of the GSM–GCL complex confirms that in the dimeric state GSM has free access to the active site.
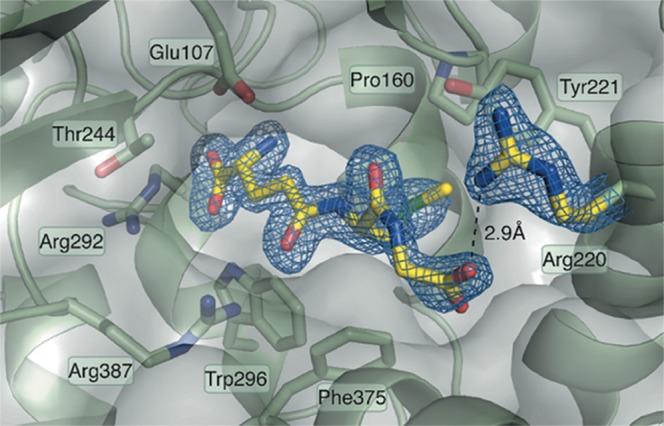
The well-defined density for GSM indicates the binding mode associated with feedback inhibition: the glutamate and cysteine parts of the GSH analog bind deep into the active site pocket, whereas the glycine moiety interacts with the conserved Arg220 at the solvent accessible end of the tunnel. GSM and GSH are therefore competitive inhibitors of GCL.
GCL enzyme activity assay
Specific activities of AtGCL recombinant proteins (WT and mutants) were determined using a coupled enzymatic assay as previously described [25]. A standard reaction mixture (0.5 ml) contained reaction buffer [100 mM MOPSO (pH 7.0), 150 mM NaCl, 20 mM MgCl2], 10 mM l-cysteine, 20 mM sodium glutamate, 5 mM ATP, 2 mM phosphoenolpyruvate, 0.27 mM NADH, 5 units of type II rabbit muscle pyruvate kinase, and 10 units of type II rabbit muscle lactic dehydrogenase. Reactions were started by the addition of WT or mutant GCL protein (50 µg). Enzyme activity was monitored spectrophotometrically at 340 nm. KM values for cysteine were determined at 20 mM ATP and 80 mM glutamate, respectively, for ATP at 20 mM cysteine and 80 mM glutamate, respectively, and for glutamate at 20 mM cysteine and 20 mM ATP, respectively. Kinetic parameters were calculated to fit data to v = [S]/(KM + [S]) using the software GraphPad Prism.
Analytical size-exclusion chromatography
Size-exclusion chromatography was performed with a HiLoad 16/600 Superdex 200 pg column (GE Healthcare). The column was equilibrated in 25 mM HEPES (pH 7.5), 5 mM MgCl2, 150 mM NaCl. For low ionic strength buffer, NaCl concentration was lowered to 10 mM. To determine the impact of elution buffer pH, the equilibration buffer was varied from 7 to 8. One milliliter of recombinant WT or mutant GCL protein solution (equivalent to 50 µg protein if not stated otherwise) was loaded onto the column and eluted at 1 ml/min. Results were detected by ultraviolet absorbance at 280 nm. For in vitro reduction experiments, protein samples were treated with 120 mM dithiothreitol for 1 h before loading on the column. For FPLC profiles (see Figures 1, 4 and 5), representative results from three independent experiments are shown.
Figure 1. FPLC elution profile of endogenous AtGCL in leaf protein extract and recombinant AtGCL protein at different concentrations.

(A) Size-exclusion chromatography of Arabidopsis protein extract from H2O2-treated leaf discs (280 nm absorbance; arrows indicating the elution volumes of recombinant GCL dimer and monomer, respectively). (B) Total leaf protein was extracted from leaf discs pre-treated with 5 mM H2O2 for 1 h, resulting in a shift from reduced (high mobility) to oxidized (low mobility) AtGCL protein (left panel). The FPLC elution profile (right panel) revealed that most of the oxidized AtGCL protein eluted in fraction 5 corresponding to the monomeric state, together with some reduced AtGCL protein (lower band); note that in fraction 3 a weak immunosignal indicated a minor amount of AtGCL dimer. (C–E) 280 nm recordings for FPLC elution profiles for different amounts of recombinant WT AtGCL protein (1 nmol [C], 0.15 nmol [D], 0.044 nmol [E]; corresponding to concentrations of 10−6 M, 1.5 × 10−7 M and 4.4 × 10−8 M, respectively).
Figure 4. Size-exclusion chromatography of WT and mutant AtGCL proteins with or without pre-treatment with DTT reveals dimer disruption in mutant AtGCLs.

After dialysis overnight at 4°C in buffer [25 mM HEPES (pH 7.5); 150 mM NaCl; 5 mM MgCl2], purified GCL proteins were injected on a Superdex-200 FPLC column. Under non-reducing conditions (black) WT AtGCL protein eluted predominantly as dimer, whereas after pre-treatment with 120 mM DTT (orange) it eluted as monomer. According to the retention volume, the estimated Mr values for AtGCL dimer and monomer were 106 and 50 kDa, respectively (Mr of WT AtGCL: 50.9 kDa); for column calibration, see Supplementary Figure S2. Note that all mutant GCL proteins eluted as monomers, irrespective of their oxidized or reduced state, indicating that the mutations disrupted the dimer interface.
Figure 5. Size-exclusion chromatography of WT AtGCL protein at low ionic strength.
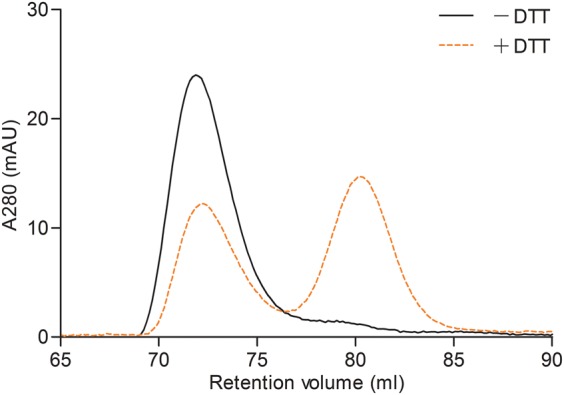
FPLC analysis of WT AtGCL protein in the presence of 10 mM NaCl (instead of 150 mM NaCl; see Figure 4). While without DTT pre-treatment the oxidized protein eluted as a dimer (black), the pre-treatment with DTT did not induce complete conversion to monomeric state (orange), indicative of ionic interactions at the dimer interface that was not completely disrupted by 10 mM NaCl.
SDS–PAGE and immunoblot analysis of native AtGCL protein
Polyclonal antiserum was generated in rabbit against purified recombinant AtGCL protein. A. thaliana (Col-0) seedlings were grown in a growth chamber at 22°C under long-day conditions (16 h light/8 h dark) for 4 weeks. Leaf discs from rosette leaves were treated with or without 5 mM H2O2 for 1 h, homogenized under liquid nitrogen, mixed with extraction buffer [50 mM HEPES (pH 7.5), 1 mM EDTA, 2 mM MgCl2, 10 mM ascorbate, 10 mM KCl, 10 mM N-ethylmaleimide, 1 mM phenylmethanesulfonyl fluoride] and centrifuged at 6000g. Supernatant protein samples were separated by SDS–PAGE on 12% gels. Immunoblot analysis was performed as described by Han et al. [26].
Isothermal titration calorimetry
Protein samples were dialyzed overnight in ITC (isothermal titration calorimetry) buffer containing 25 mM HEPES (pH 7.5), 150 mM NaCl, and 5 mM MgCl2 at 4°C. Concentrations of proteins were determined by measuring the absorption at 280 nm using a NanoPhotometer NP80 (Implen). ITC experiments were performed in triplicate on a MicroCal PEAQ-ITC machine (Malvern Instruments) at 20°C. For determination of the dissociation constant (KD) the syringe was loaded with Arabidopsis WT GCL protein (450 µM) and titrated into the sample cell filled with ITC buffer. ITC data were processed using the MicroCal PEAQ-ITC Analysis Software (Malvern Instruments) and thermodynamic parameters were obtained by fitting the data to a dissociation model.
Statistical treatment of data
All data are presented as the mean ± SD of three or more independent experiments. Statistical analyses, including two-way ANOVA, the Holm-–Šídák method and Welch's ANOVA, were performed using the software package IBM SPSS Statistics. A P-value of <0.05 indicated in the figure legends was considered statistically significant.
Results
Evidence for homo-dimer formation of redox-activated GCL in plastids
To assess the dimerization state of endogenous AtGCL protein, leaf discs from 4-week-old Arabidopsis plants (rosette stage) were treated with 5 mM H2O2 for 1 h. This treatment causes a shift from the reduced (high mobility form) to the oxidized state (low mobility form) in non-reducing SDS–PAGE analysis as previously shown by Hicks et al. [14]. A total protein extract was then analyzed by FPLC to determine the homo-dimer/monomer ratio by analyzing elution fractions via non-reducing SDS–PAGE and subsequent immunoblot detection. In contrast with redox-activated recombinant GCL protein which previously had been shown to form homo-dimers [15], the endogenous GCL protein, while being predominantly in its oxidized state [i.e. showing lower mobility in non-reducing SDS–PAGE (Figure 1B)] eluted in fractions corresponding to monomeric GCL protein (Figure 1A,B). As protein extraction causes dilution of GCL protein when compared with its in vivo concentration in the chloroplasts, it was important to estimate dimerization kinetics for homo-dimer formation and to compare it with the in vivo GCL concentration. First, different concentrations of recombinant WT AtGCL protein were subjected to FPLC analysis (Figure 1C–E). The elution profiles revealed dissociation of AtGCL protein into monomers within the concentration range of 10−6 M to 10−7 M. To estimate endogenous GCL protein concentration in plastids, a dilution series of Arabidopsis root extracts was analyzed for its GCL content, in comparison with a dilutions series of recombinant AtGCL protein (for quantification, root extracts were preferred since in leaf extracts Rubisco Large SU tends to distort GCL immune signals). The estimated endogenous GCL concentration was ∼0.5 µmol kg−1 fresh weight (i.e. 5 × 10−7 M; Figure 2). Based on a conserved estimate, total plastidial volume in roots corresponds to <10% of total tissue volume. Thus, plastidial GCL concentration is expected to be ≥ 5 × 10−6 M. Since in leaf extracts GCL protein is substantially more abundant than in root extracts (Figure 2), GCL concentration in the chloroplasts is expected to be even higher than in root plastids. Together, these results indicate that in plastids (including chloroplasts) a stress-mediated redox-activation of GCL enzyme will lead to homo-dimer formation. To explore whether this homo-dimer formation contributes to the observed redox-activation, the following experiments were performed with recombinant AtGCL mutant proteins compromised in homo-dimer formation.
Figure 2. Quantification of AtGCL protein in Arabidopsis root extract by immunoblot analysis.
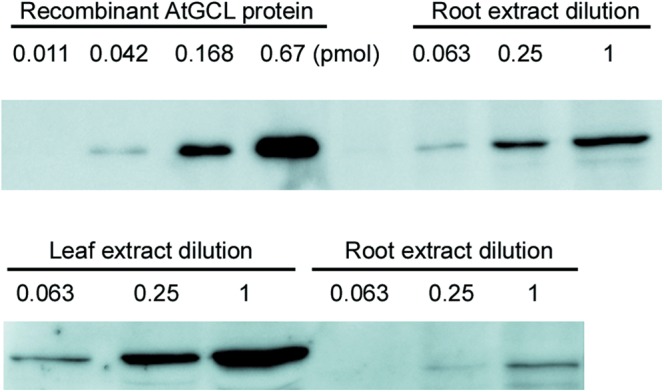
Upper panel: Recombinant AtGCL protein at different dilutions (0.011, 0.042, 0.168 and 0.67 pmol) when compared with root protein extract dilutions. Diluted extract (0.063) is equivalent to 0.078 mg fresh weight. The comparison indicates that the amount of endogenous AtGCL protein in 0.078 mg of root tissue approximately equals 0.042 pmol of recombinant AtGCL protein, corresponding to 538 nmol endogenous AtGCL protein per kilogram fresh weight. Lower panel: Comparison of leaf and root extracts at different dilutions. As in leaf extracts, Rubisco Large SU tends to distort GCL immune signals, the root part was used for quantification. Numbers indicate the dilution factors, factor 1 corresponds to 90 mg fresh weight equivalent.
Generation of recombinant AtGCL proteins mutagenized at the homo-dimer interface
In the GCL enzyme of B. juncea, a species closely related to A. thaliana, eleven amino acids contribute to the zipper arrangement of the dimer interface (Glu133, Phe135, Glu136, Gln176, Asn182, Tyr186, Glu193, Trp394, Arg395, Lys471, Phe475; numbering according to Hothorn et al. [13]). For the present study, the GCL enzyme of A. thaliana (AtGCL) was chosen, which displays the same amino acid residues at corresponding positions. Based on structural considerations, AtGCL mutants for amino acids forming the salt bridges (Glu141/Arg403 and Glu201/Lys479), and providing hydrophobic interaction (Tyr194/Phe483) were selected for analysis (Table 1 and Figure 3). The salt bridge Glu141/Arg403 was mutated to Glu141/Glu403 or to Gln141/Met403, the salt bridge Glu201/Lys479 to Lys201/Lys479, and the hydrophobic contact Tyr194/Phe483 was mutated to Leu194/Phe483. WT and mutant AtGCL proteins (without plastidial transit peptide) were expressed as 6His:TrxA:GCL fusions in E. coli, as described in the Materials and methods section. All mutant proteins were obtained as soluble proteins, yields being highest for WT protein and the mutants Glu141/Glu403 and Lys201/Lys479 (Supplementary Figure S1).
Figure 3. Alignment of AtGCL and BjGCL protein sequences.

The amino acid residues responsible for the salt bridges at the dimer interface of AtGCL are marked in red and blue for Glu141/Arg403 and Glu201/Lys479, respectively. The amino acid residues responsible for the hydrophobic interaction at the dimer interface of AtGCL are marked in green. The cysteine residues forming the two disulfide bridges of AtGCL (Cys186/Cys406 (CC2) and Cys349/Cys364 (CC1)) are marked in yellow.
Comparison of WT and mutant GCL proteins reveals that single amino acid mutations at the dimer interface prevent dimer formation
To first determine whether recombinant WT and mutant proteins expressed in bacteria were all recovered in their oxidized redox-activated state, SDS–PAGE analysis was performed under non-reducing conditions to estimate the ratio of oxidized (low mobility) to reduced protein (high mobility) (Supplementary Figure S1). Note that non-reducing SDS–PAGE does not differentiate between monomer and homo-dimer since SDS disrupts homo-dimers (see Introduction). Under non-reducing conditions all recombinant GCL proteins (WT and mutants) displayed two separate bands, reflecting the oxidized (upper band) and reduced (lower band) state as evident from comparison with gels run under reducing conditions, however, only WT GCL and mutants Glu141/Glu403 and Lys201/Lys479 were predominantly in their oxidized state.
Size-exclusion chromatography by FPLC was then performed to determine whether dimer formation was disrupted in mutated GCL proteins. In agreement with a previous study [15], WT AtGCL protein eluted predominantly as homo-dimer at a volume corresponding to a molecular mass of 106 kDa under non-reducing conditions (Figure 4), whereas after pre-treatment with 120 mM DTT it eluted as a 50 kDa species, reflecting dissociation into the monomeric state. In marked contrast, all four GCL mutants eluted at a volume corresponding to the monomer size, independent of pre-treatment with reducing agent, indicating that mutation in only one of the contact sites of the zipper-like interface [13] was sufficient to prevent in vitro dimerization.
As protein–protein interactions primarily based on ionic interactions are dependent on the ionic strength of the incubation medium [27], the elution behavior of WT AtGCL protein was compared at different NaCl concentrations. Under low ionic strength (10 mM NaCl; Figure 5) the active and dimerized (i.e. oxidized) WT AtGCL protein eluted with the same elution profile as in the presence of 150 mM NaCl. However, when performing the same experiment with reduced WT AtGCL protein (i.e. after DTT treatment) the elution profile differed between 10 mM and 150 mM NaCl; both dimer and monomer were observed at low ionic strength, in marked contrast with exclusive monomer formation at high ionic strength. Apart from ionic strength, the pH may also impact on dimer-to-monomer transition [28]. Due to its localization in the chloroplast stroma, the GCL enzyme will be exposed to in situ pH shifts between pH 7 (dark phase) and pH 8 (light phase). Therefore, the elution behavior of WT AtGCL protein was determined at pH 7 and pH 8. The comparison did not reveal a significant change in the elution profile (data not shown), indicating that the GCL homo-dimer is stable within this pH range.
GCL dimer formation is not required for redox-mediated enzyme activation
Having confirmed that the dimer interface had been disrupted in the mutant proteins, their enzyme activities were compared in the absence or presence of DTT. This analysis was restricted to WT protein and the two mutants Glu141/Glu403 and Lys201/Lys479, as they reproducibly provided the highest yields of recombinant protein and were predominantly obtained in their oxidized (i.e. redox-activated) state (Supplementary Figure S1). Note that under the chosen assay conditions (i.e. AtGCL concentration 2 µM), oxidized WT AtGCL is in its homodimeric state (Supplementary Figure S1 and Figures 1 and 4), whereas the mutants Glu141/Glu403 and Lys201/Lys479, while also being predominantly in the oxidized state are monomeric (Supplementary Figure S1 and Figure 4). In the absence of DTT, WT AtGCL displayed a specific activity of 112 nmol min−1 mg−1 (Figure 6), which in the presence of DTT was reduced to 38% of this value. The statistical analysis indicated that considering the enzyme activities in the presence/absence of DTT, the dimer interface mutants Glu141/Glu403 and Lys201/Lys479 were not significantly different when compared with WT GCL, although the oxidized mutant proteins did not dimerize (see Figure 4). Likewise, KM values for the substrates glutamate, cysteine and ATP did not differ significantly between WT AtGCL and mutant proteins (Table 2). Thus, GCL dimer formation does not appear to be required for redox-mediated enzyme activation. As expected, the effect of DTT on a mutant AtGCL protein with disrupted regulatory disulfide bridge (Ser186/Cys406) was negligible.
Figure 6. Disruption of homo-dimer does not significantly affect the enzyme activity of AtGCL.
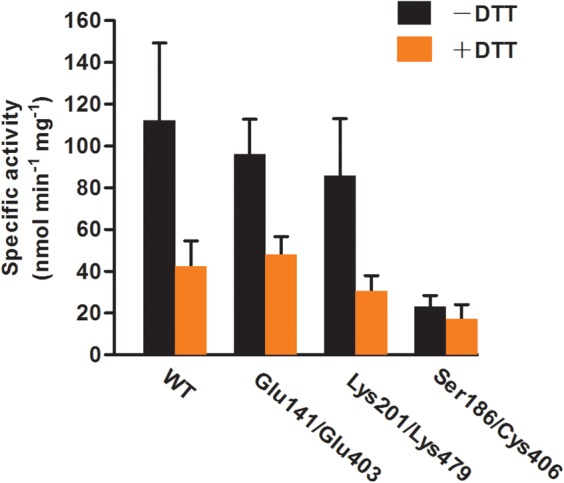
The statistical analysis (two-way ANOVA followed by the Holm–Šídák method) indicates that both factors, DTT treatment and enzyme type, have an impact on enzyme activities, but there is no significant interaction between two sources of variation (DTT treatment and enzyme type). With respect to the activity in the presence/absence of DTT, the dimer interface mutants Glu141/Glu403 and Lys201/Lys479 are not significantly different when compared with WT GCL, although oxidized mutant proteins do not dimerize (see Figure 4). The activity of the disulfide bridge mutant Ser186/Cys406 is not significantly affected by DTT as expected. Finteraction (3, 23) = 2.90, Pinteraction = 0.06; FDTT(1, 23) = 35.33, PDTT < 0.001; Fenzyme type(3, 23) = 10.57, Penzyme type < 0.001. Enzyme reactions were performed as described under Materials and methods section. Values for specific activities (nmol min−1 mg−1) are expressed as means ± SD (n > 3).
Table 2. KM values of WT and mutant AtGCL enzymes for cysteine, ATP and glutamate.
Enzyme assays were performed as described under Materials and methods section. Values for KM (mM) are expressed as a mean ± SD (n > 3). Statistical analysis by Welch's ANOVA indicates that GCL variants (WT and mutants) do not differ significantly in their affinity for substrates. [FCys(2, 7.32) = 0.65, PCys = 0.55; FGlu(2, 9.10) = 3.48, PGlu = 0.08; FATP(2, 7.02) = 2.87, PATP = 0.12].
| KM values | |||
|---|---|---|---|
| Cysteine | Glutamate | ATP | |
| AtGCL WT | 0.34 ± 0.12 | 6.64 ± 1.02 | 1.25 ± 0.18 |
| Glu141/Glu403 | 0.33 ± 0.26 | 7.56 ± 0.58 | 1.16 ± 0.04 |
| Lys201/Lys479 | 0.28 ± 0.04 | 8.33 ± 1.15 | 1.45 ± 0.30 |
Structure of a GSM–GCL complex confirms that binding of GSH to the active site is not affected by the dimeric state of GCL
Since dimerization was shown to be dispensable for enzyme activation, we speculated that dimer formation could possibly interfere with competitive feedback inhibition by GSH [12,15,29]. Thus, to address the question whether GCL dimerization affects interaction with GSH at the active site, recombinant GCL protein was co-crystallized with the non-reducing GSH analog GSM. GSM was used to uncouple redox activity from competitive binding, thus avoiding any sample heterogeneity that might arise due to partially reduced protein or oxidized GSH (like GSH, GSM inhibits GCL activity, however, slightly less efficiently [Table 3]). Note that during crystallization GCL protein concentration was 0.24 × 10−3 M (i.e. the oxidized GCL was in its dimeric state). The structure of the GCL–GSM complex was solved at 1.75 Å (see Material and methods) and shows the GSM derived protein interactions in detail (Figure 7 and Supplementary Table S2). One GCL homo-dimer was found per asymmetric unit and the electron density allowed clear localization of GSM in the active site of both chains of the dimer. The GSM–GCL complex displayed the glutamate-cysteine moiety well defined in a position equivalent to that in the previously characterized BSO-binding site [13] and similar to GSH binding in yeast GCL [30]. In the cysteine binding pocket, the glycine moiety of GSH exhibits less defined electron density compared with the other parts of the ligand, indicating local flexibility. However, the density indicates that the carboxy terminus of the GSH-glycine residue interacts with the conserved Arg 220 (Figure 6). GSM binding induces a rearrangement of Arg 220 protruding towards the shallower end of the active site tunnel. The close similarity of the GSM–GCL complex with BSO and glutamate bound GCL [13] suggests that there is hardly any conformation change resulting in ligand-induced cross-talk between the two subunits upon GSH binding. Thus, it can be assumed that if in the oxidized dimer only one active site is blocked by GSH the second monomer will keep its activity.
Table 3. Inhibition of AtGCL activity by GSH and GSM.
| mM | GSH | GSM |
|---|---|---|
| 0 | 100 | 100 |
| 1 | 79 ± 3 | 97 ± 4 |
| 2 | 49 ± 4 | 85 ± 4 |
| 3 | 30 ± 6 | 73 ± 3 |
| 4 | 22 ± 4 | 64 ± 2 |
| 5 | 18 ± 1 | 55 ± 3 |
Discussion
With the goal to explore the possible link between the formation of the regulatory intramolecular disulfide bridge in plant GCL [13,14] and the simultaneous formation of homo-dimers [15], the present study has revealed that homo-dimer formation can be assumed to occur in vivo, however, it does not appear to be required for redox-mediated activation via the formation of the regulatory intramolecular disulfide bridge (Cys186/Cys406).
In particular, the recombinant mutant proteins Glu141/Glu403 and Lys201/Lys479 (Table 1), both being compromised in homo-dimer formation (Figure 4), displayed a response to DTT-treatment comparable to the WT AtGCL protein (Figure 6) with no significant change in affinity for the three substrates cysteine, glutamate and ATP (Table 2). Note that GCL protein concentration in the enzyme assay was always 2 × 10−6 M, i.e. well above the KD value of WT AtGCL, thus assuring that WT AtGCL enzyme activity was determined under conditions causing homo-dimer formation (Figure 1). Under in vivo conditions, we expect GCL to rapidly dissociate into monomers after reduction in its disulfide bridge since (i) the high ionic strength of chloroplast stroma (∼100–200 mM) is favorable to dissociation (Figure 5), and (ii) in the mutants the elimination of a single salt bridge already prevents dimerization (Figure 4).
To address the question whether oxidized GCL protein dimerizes in situ in the chloroplast required a reliable assessment of its concentration in plastids, since plant GCL is exclusively localized in this compartment [7]. Based on the conservative assumption that plastid volume represents not more than 10% of total tissue volume, the immunological quantification of GCL protein (Figure 2) predicted a plastidial concentration of ≥5 × 10−6 M. At this concentration, oxidized GCL protein is expected to be entirely dimerized (Figure 1C–E). Due to lack of sensitivity, it was not possible to estimate the KD value of the monomer-dimer transition by ITC. However, ITC experiments at higher GCL concentration indicated that dimers may associate to form tetramers, albeit with much lower affinity (KD: 9 × 10−5 M; Supplementary Figure S3).
Since GCL protein concentration in plastids is high enough to induce in vivo homo-dimerization upon redox-activation, but in vitro studies indicate that homo-dimerization does not show significant impact on enzyme activation, it remains an intriguing question whether GCL homo-dimerization conveys another function in vivo. Interestingly, dimerization of oxidized active GCL proteins is a feature specific to plants, while the structurally related GCL proteins from Agrobacterium tumefaciens (AtuGCL, α-proteobacteria) and Xanthomonas campestris (XcaGCL, γ-proteobacteria) do not dimerize, and are not redox-regulated, suggesting that the combination of redox-activation and dimerization might be a special adaptation of plant GCL to its plastidial environment [15].
Since GSH acts as a competitive inhibitor for GCL [12,15 and literature cited therein], we addressed the question whether GCL dimer formation impacts on access of GSH to its active site (Figure 7). However, based on the structural analysis of the GCL–GSM complex it can be excluded that GCL homo-dimer formation interferes with feedback inhibition by GSH. As GCL homo-dimerization in plants does not significantly affect KM values for the substrates cysteine, glutamate and ATP (Table 2), and does not interfere with the physiologically important feedback inhibition by GSH, it may be speculated that dimerization impacts on GCL protein stability in vivo. Stabilization via homo-dimer formation has been previously reported for other proteins [31,32]. Noteworthy, a shift of reduced (i.e. less active) to activated GCL is generally observed under different stress exposures such as heavy metal treatment or oxidative stress [14], conditions which also cause an increased proteolytic activity in plastids [33,34]. Whether homo-dimerization of redox-activated GCL in plastids confers a higher stability by possibly shielding certain domains remains to be shown.
Acknowledgements
We thank Jeremy Sloan for the technical support with ITC experiments.
Abbreviations
- ESRF
European Synchrotron Radiation Facility
- GCL
γ-glutamylcysteine ligase
- GS
glutathione synthetase
- GSM
S-methyl-GSH
- GSSG
glutathione disulfide
- ITC
isothermal titration calorimetry
Author Contribution
Y.Y., E.D.L., R.G., S.W. M.W. and K.S. contributed to experimental design (Figures 1–7; Tables 2 and 3); R.H. contributed to the concept of study; T.P.-B. contributed to discussion part and statistics; T.R. was involved in the concept of study and manuscript writing.
Funding
This work was supported by the DFG (SPP1710; grants to Thomas Rausch, Markus Wirtz and Rüdiger Hell). Y.Y. was supported by a scholarship from the China Scholarship Council. Part of this study was funded by DFG (CRC 1063, TP13; grant to MW and RH).
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
Supplementary Material
References
- 1.Rausch T., Gromes R., Liedschulte V., Müller I., Bogs J., Galovic V. et al. (2007) Novel insight into the regulation of GSH biosynthesis in higher plants. Plant Biol. 9, 565–572 10.1055/s-2007-965580 [DOI] [PubMed] [Google Scholar]
- 2.Meyer A.J. and Rausch T. (2008) Biosynthesis, compartmentation and cellular functions of glutathione in plant cells In Sulfur Metabolism in Phototrophic Organisms. Advances in Photosynthesis and Respiration (Hell R., Dahl C., Knaff D. and Leustek T., eds), pp. 161–184, Springer, Dordrecht [Google Scholar]
- 3.Galant A., Preuss M.L., Cameron J. and Jez J.M. (2011) Plant glutathione biosynthesis: diversity in biochemical regulation and reaction products. Front. Plant Sci. 2, 45 10.3389/fpls.2011.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franklin C.C., Backos D.S., Mohar I., White C.C., Forman H.J. and Kavanagh T.J. (2009) Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Aspects Med. 30, 86–98 10.1016/j.mam.2008.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fraser J.A., Kansagra P., Kotecki C., Saunders R.D.C. and McLellan L.I. (2003) The modifier subunit of Drosophila glutamate-cysteine ligase regulates catalytic activity by covalent and noncovalent interactions and influences glutathione homeostasis in vivo. J. Biol. Chem. 278, 46369–46377 10.1074/jbc.M308035200 [DOI] [PubMed] [Google Scholar]
- 6.Toroser D., Yarian C.S., Orr W.C. and Sohal R.S. (2006) Mechanisms of gamma-glutamylcysteine ligase regulation. Biochim. Biophys. Acta 1760, 233–244 10.1016/j.bbagen.2005.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wachter A., Wolf S., Steininger H., Bogs J. and Rausch T. (2005) Differential targeting of GSH1 and GSH2 is achieved by multiple transcription initiation: implications for the compartmentation of glutathione biosynthesis in the Brassicaceae. Plant J. 41, 15–30 10.1111/j.1365-313X.2004.02269.x [DOI] [PubMed] [Google Scholar]
- 8.Meyer A.J. and Fricker M.D. (2002) Control of demand-driven biosynthesis of glutathione in green Arabidopsis suspension culture cells. Plant Physiol. 130, 1927–1937 10.1104/pp.008243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noctor G., Gomez L., Vanacker H. and Foyer C.H. (2002) Interactions between biosynthesis, compartmentation and transport in the control of glutathione homeostasis and signalling. J. Exp. Bot. 53, 1283–1304 10.1093/jexbot/53.372.1283 [DOI] [PubMed] [Google Scholar]
- 10.Kopriva S. and Rennenberg H. (2004) Control of sulphate assimilation and glutathione synthesis: interaction with N and C metabolism. J. Exp. Bot. 55, 1831–1842 10.1093/jxb/erh203 [DOI] [PubMed] [Google Scholar]
- 11.May M.J., Vernoux T., Leaver C., Montagu M.V. and Inze D. (1998) Glutathione homeostasis in plants: implications for environmental sensing and plant development. J. Exp. Bot. 49, 649–667 10.1093/jexbot/49.321.649 [DOI] [Google Scholar]
- 12.Jez J.M., Cahoon R.E. and Chen S. (2004) Arabidopsis thaliana Glutamate-Cysteine Ligase functional properties, kinetic mechanism, and regulation of activity. J. Biol. Chem. 279, 33463–33470 10.1074/jbc.M405127200 [DOI] [PubMed] [Google Scholar]
- 13.Hothorn M., Wachter A., Gromes R., Stuwe T., Rausch T. and Scheffzek K. (2006) Structural basis for the redox control of plant glutamate cysteine ligase. J. Biol. Chem. 281, 27557–27565 10.1074/jbc.M602770200 [DOI] [PubMed] [Google Scholar]
- 14.Hicks L.M., Cahoon R.E., Bonner E.R., Rivard R.S., Sheffield J. and Jez J.M. (2007) Thiol-based regulation of redox-active glutamate-cysteine ligase from Arabidopsis thaliana. Plant Cell 19, 2653–2661 10.1105/tpc.107.052597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gromes R., Hothorn M., Lenherr E.D., Rybin V., Scheffzek K. and Rausch T. (2008) The redox switch of γ-glutamylcysteine ligase via a reversible monomer-dimer transition is a mechanism unique to plants. Plant J. 54, 1063–1075 10.1111/j.1365-313X.2008.03477.x [DOI] [PubMed] [Google Scholar]
- 16.Hothorn M., Bonneau F., Stier G., Greiner S. and Scheffzek K. (2003) Bacterial expression, purification and preliminary X-ray crystallographic characterisation of the invertase inhibitor Nt-CIF from tobacco. Acta Crystallogr. D Biol. Crystallogr. 59, 2279–2282 10.1107/S0907444903021036 [DOI] [PubMed] [Google Scholar]
- 17.Kabsch W. (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800 10.1107/S0021889893005588 [DOI] [Google Scholar]
- 18.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C. and Read R.J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emsley P. and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 20.Murshudov G.N., Vagin A.A. and Dodson E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 21.Davis I.W., Leaver-Fay A., Chen V.B., Block J.N., Kapral G.J., Wang X. et al. (2007) Molprobity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeLano W. (2008) The PyMOL Molecular Graphics System, DeLano Scientific LLC, Palo Alto, CA [Google Scholar]
- 23.Holm L. and Park J. (2000) Dalilite workbench for protein structure comparison. Bioinformatics 16, 566–567 10.1093/bioinformatics/16.6.566 [DOI] [PubMed] [Google Scholar]
- 24.Krissinel E. and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 25.Abbott J.J., Pei J., Ford J.L., Qi Y., Grishin V.N., Pitcher L.A. et al. (2001) Structure prediction and active site analysis of the metal binding determinants in γ-glutamylcysteine synthetase. J. Biol. Chem. 276, 42099–42107 10.1074/jbc.M104672200 [DOI] [PubMed] [Google Scholar]
- 26.Han M., Heppel S.C., Su T., Bogs J., Zu Y., An Z. et al. (2013) Enzyme inhibitor studies reveal complex control of methyl-D-erythritol 4-phosphate (MEP) pathway enzyme expression in Catharanthus roseus. PLoS ONE 8, e62467 10.1371/journal.pone.0062467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aymard P., Durand D. and Nicolai T. (1996) The effect of temperature and ionic strength on the dimerisation of β-lactoglobulin. Int. J. Biol. Macromol. 19, 213–221 10.1016/0141-8130(96)01130-0 [DOI] [PubMed] [Google Scholar]
- 28.Mohan P., Barve M., Chatterjee A. and Hosur R.V. (2006) Ph driven conformational dynamics and dimer-to-monomer transition in DLC8. Protein Sci. 15, 335–342 10.1110/ps.051854906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hell R. and Bergmann L. (1990) λ-Glutamylcysteine synthetase in higher plants: catalytic properties and subcellular localization. Planta 180, 603 10.1007/BF02411460 [DOI] [PubMed] [Google Scholar]
- 30.Biterova E.I. and Barycki J.J. (2010) Structural basis for feedback and pharmacological inhibition of Saccharomyces cerevisiae glutamate cysteine ligase. J. Biol. Chem. 285, 14459–14466 10.1074/jbc.M110.104802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Messaritou G., Grammenoudi S. and Skoulakis E.M. (2010) Dimerization is essential for 14-3-3ζ stability and function in vivo. J. Biol. Chem. 285, 1692–1700 10.1074/jbc.M109.045989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans E.L., Saxton J., Shelton S.J., Begitt A., Holliday N.D., Hipskind R.A. et al. (2011) Dimer formation and conformational flexibility ensure cytoplasmic stability and nuclear accumulation of Elk-1. Nucleic Acids Res. 39, 6390–6402 10.1093/nar/gkr266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishimura K. and van Wijk K.J. (2015) Organization, function and substrates of the essential Clp protease system in plastids. Biochim. Biophys. Acta, Bioenerg. 1847, 915–930 10.1016/j.bbabio.2014.11.012 [DOI] [PubMed] [Google Scholar]
- 34.Pullido P., Llamas E. and Rodriguez-Concepcion M. (2017) Both Hsp70 chaperone and Clp protease plastidial systems are required for protection against oxidative stress. Plant Signal. Behav. 12, e1290039 10.1080/15592324.2017.1290039 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.