

Summary
The aim of the present study was to characterize a patient‐derived in vitro 3D model (ie tumoroid) established from colorectal adenocarcinoma. This study investigated the growth rate of tumoroids and whether the Kirsten rat sarcoma (KRAS) mutations in the parental tumour accelerate this rate. The tumoroids were established from surgical resections of primary and metastatic colorectal adenocarcinoma from 26 patients. The in vitro growth rate of these tumoroids was monitored by automated imaging and recorded as relative growth rate. The KRAS hotspot mutations were investigated on the parental tumours by Ion Torrent™ next‐generation sequencing. The KRAS mutations were detected in 58% of the parental tumours, and a significantly higher growth rate was observed for tumoroids established from the KRAS‐mutated tumours compared to wild‐type tumours (P < 0.0001). The average relative growth rate (log10) on day 10 was 0.360 ± 0.180 (mean ± SD) for the KRAS‐mutated group and 0.098 ± 0.135 (mean ± SD) for the KRAS wild‐type group. These results showed that the presence of KRAS mutations in parental tumours is associated with an acceleration of the growth rate of tumoroids. The future perspective for such a model could be the implementation of chemoassays for personalized medicine.
Keywords: 3D in vitro culture, colorectal cancer, gastrointestinal pathology, Kirsten rat sarcoma mutation, tumoroids
1. INTRODUCTION
Over the past decade, three‐dimensional cultures of cancer cells (tumoroids) have proven to be an important tool in personalized medicine and in vitro investigations of tumours.1, 2 It has been demonstrated that tumoroids can be established in vitro with a high success rate.3, 4 Tumoroids resemble their parental tissue more closely than monolayer cultures, since original cell‐cell and cell‐matrix attachments are maintained during tumoroid formation.3, 5, 6 This similarity is attributed to a major presence of cells positive for epithelial cell adhesion molecules (EpCAM) in tumoroids established from colorectal cancer (CRC).3, 5 Therefore, tumoroids are good candidates in experimental modelling of tumour functionality, disease pathogenesis and therapy response.7, 8 In the present study, tumoroids have been characterized by their growth rate and whether the presence of Kirsten rat sarcoma (KRAS) mutation in the parental tumour has any effect on the growth rate.
More than 70% of colorectal adenocarcinomas show at least one genetic dysregulation in the MAPK/PI3K signalling pathway.9 KRAS protein contributes to the transmission of extracellular growth signals into the nucleus, resulting in increased cellular growth and mitotic activity.10 It has been shown in animal models that KRAS mutations stimulate proliferation of colon epithelial cells.10 KRAS mutations are one of the most common types of mutations in CRC.11 Point mutations in exon 2 (codons 12 and 13) are the most prevalent types of mutations in the KRAS gene.12, 13 Mutations in KRAS exon 2 codon 13 have been shown to be associated with poor prognosis,12 whereas mutations in KRAS exon 2 codon 12 are associated with lymph node metastasis and advanced stages of disease.13
The hyperproliferation of KRAS‐mutated tumours can be attributed to upregulation of the surface receptors involved in MAPK/PI3K signalling pathway. These receptors include but not limited to epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR) and platelet‐derived growth factor receptor (PDGFR).14, 15, 16 The aim of the present study was to investigate whether the parental KRAS status of the tumoroids has a prolonged effect on the growth rate of the tumoroids. For this purpose, the status of KRAS in the parental tumour was determined and the presence of mutations was correlated with the in vitro growth rate of tumoroids.
2. MATERIALS AND METHODS
2.1. Tumour samples
Eighteen fresh primary colorectal tumours and eight liver metastasis samples from 26 patients operated for colorectal adenocarcinoma were received from the Digestive Disease Center, Bispebjerg Hospital, and the Department of Gastrointestinal Surgery, Rigshospitalet, Copenhagen, Denmark. Approximately 0.5 cm3 of non‐necrotic tumour was transferred on ice to the culturing laboratory in sterile 50 mL Falcone™ tubes containing transport medium. The transport medium consisted of DPBS (Dulbecco's phosphate‐buffered saline, Sigma‐Aldrich, St. Louis, Missouri, USA) supplemented with 500 U/mL penicillin, 500 U/mL streptomycin (Sigma‐Aldrich), 500 μg/mL gentamicin (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and 12.5 μg/mL amphotericin B (Sigma‐Aldrich). Macroscopic and microscopic descriptions of tumour samples were conducted by independent pathologists at the Department of Pathology, Rigshospitalet. For each patient, the following clinical characteristics were registered from the pathology report: age, gender, TNM stage (local invasion depth (T‐stage), lymph node involvement (N‐stage), presence of distant metastasis (M‐stage)) and vascular invasion.
2.2. Ethical approval
The study protocol was approved by the Science Ethics Committee of The Capital Region of Denmark (No. H‐1‐2011‐125).
2.3. In vitro 3D cultures
Tumoroids were cultured based on the work by Jeppesen et al17 Tumour specimens were cut into 1‐2 mm2 fragments using an ordinary scalpel while keeping the tissue on ice. Tumour fragments were washed with cold DPBS and dissociated partially by 1 mg/mL collagenase type II (Thermo Fisher Scientific) in DPBS at 37°C in several steps. The dissociated fragments were serially filtered to reach a size of 30‐70 μm. These fragments were transferred to a 10‐cm Petri dish containing stem cell medium (StemPro hESC SFM, Thermo Fisher Scientific) coated with 1.5% agarose (Sigma‐Aldrich) by rinsing the filter remnants with 2 mL of StemPro hESC medium (Thermo Fisher Scientific) supplemented with 200 U/mL penicillin, 200 U/mL streptomycin (Sigma‐Aldrich), 200 μg/mL gentamicin (Thermo Fisher Scientific) and 2.5 μg/mL amphotericin B. The Petri dishes were transferred to a 37°C humidified incubator with 5% CO2 and were monitored daily by light microscopy for the formation of tumoroids. Following the formation of tumoroids (integrated and round structures with smooth surface), debris and individual cells were removed by DPBS rinsing and filtered to select tumoroids of 70‐100 μm in diameter. Approximately 15 tumoroids per well were seeded in a 384‐well low volume, non‐treated microtiter plate (Corning Life Sciences, Tewksbury, Massachusetts, USA) containing 1:2 mixture of stem cell medium with Matrigel (Corning Life Sciences).
2.4. Growth monitoring of tumoroids
Tumoroids were cultured in a 37°C humidified incubator with 5% CO2 for 11 days. Microscopy images of the wells were recorded by oCelloScope™ (BioSense Solutions, Farum, Denmark). The growth rate of tumoroids was reported as the total area of tumoroids, defined as regions of interest in each well, relative to the area at day 0. As described by Fredborg et al,18 the oCelloScope™ is a fluid‐samples microscopy technology using tilted imaging planes for 3D image acquisition. By combining the tilted images, a single z‐plane is generated. Focus was set automatically in two equal halves of each well, and subsequently, the recorded images were stitched together. Image processing and analysis were performed using algorithms developed by 2cureX.
2.5. Targeted mutations
The single‐nucleotide polymorphisms on codons 12, 13, 58, 59, 61, 117 and 146 of the KRAS oncogene were investigated in formalin‐fixed, paraffin‐embedded samples of the original tumours. The appropriate section for molecular analysis was selected by microscopy of haematoxylin and eosin‐stained sections following these criteria in each tissue block: more than 400 malignant cells, more than 70% tumour proportion and less than 5% necrosis. Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's recommendations. Targeted Next‐Generation Sequencing (NGS) was carried out on the Ion PGM™ System (Thermo Fisher Scientific) with the Ion Torrent AmpliSeq™ Colon and Lung Cancer Research Panel v2 (Thermo Fisher Scientific) as described by D'Haene et al19 Library preparation was performed from 10 ng of the purified DNA using the Ion AmpliSeq Library kit 2.0 according to the manufacturer's instructions (Thermo Fisher Scientific) and the Ion PGM IC 200 Kit (Thermo Fisher Scientific) as the template kit for the IonChef (Thermo Fisher Scientific). Sequencing was carried out using Ion PGM IC 200 Sequencing Kit (TRS) with the Ion 318TM Chip v2 (Thermo Fisher Scientific). Variant calling was carried out using the Torrent Variant Caller v4.6 of the Torrent SuiteTM Software (Thermo Fisher Scientific). Variants were evaluated according to ACMG guidelines.20
2.6. Statistical methods
Linear mixed‐effects regression model with REML (residual maximum likelihood) estimation and repeated measures ANOVA were performed on Log10‐transformed values of the relative growth rate of tumoroids. Data were handled in R (version 3.0.1) and RStudio (version 0.97.320; RStudio, Boston, Massachusetts, USA).
3. RESULTS
Tumoroids were successfully established from all 26 patients with primary colorectal adenocarcinoma or from liver metastasis of colorectal adenocarcinoma. The histology of the tumoroids demonstrated glandular structures that resembled the histological structure of colorectal epithelium. Furthermore, the high nuclear‐cytoplasmic ratio of the cells in the tumoroids indicates the hyperproliferative feature of the tumoroids (Figure 1A‐C). None of the liver metastasis samples were from the same patients as the primary tumours. All the tumours were histologically diagnosed as glandular adenocarcinoma. The clinical characteristics of the tumours are summarized in Table 1. The age of the patients varied from 52 to 85 years old with an average of 70.2 years (± 8.7, SD).
Figure 1.
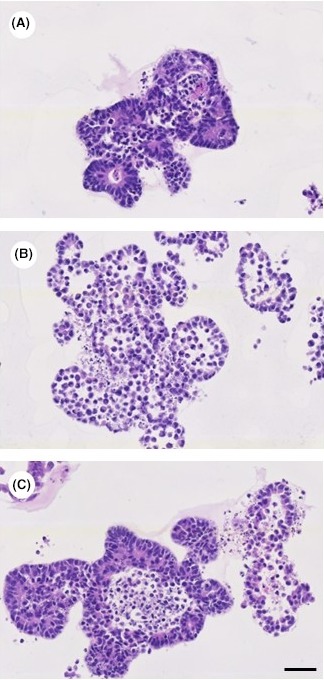
Tumoroids stained with haematoxylin and eosin. Tumoroids recapitulate the glandular structure of the colorectal epithelium. The cells in the tumoroids show a high nuclear‐cytoplasmic ratio, which indicates the hyperproliferative potential of these cells (a: patient no. 8; b: patient no. 10; c: patient no. 14). (bar = 50 μm) [Colour figure can be viewed at wileyonlinelibrary.com]
Table 1.
Overview of the tumours
| Patient no. | Gender | Age (y) | Localization | KRAS status | T‐Stage | N‐Stage | M‐Stage | V‐Stage |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 76 | Colon | c.35G>A; p.G12D | T3 | N0 | M0 | V0 |
| 2 | M | 76 | Colon | wt | T3 | N0 | M0 | V0 |
| 3 | M | 75 | Colon | c.38G>A; p.G13D | T2 | N1 | M0 | V0 |
| 4 | M | 71 | Colon | c.35G>A; p.G12D | T2 | N0 | M0 | V0 |
| 5 | M | 63 | Colon | wt | T3 | N0 | M0 | V0 |
| 6 | M | 53 | Colon | wt | T4 | N1 | M0 | V0 |
| 7 | M | 79 | Colon | c.436G>A; p.A146T | T2 | N0 | M0 | V0 |
| 8 | M | 78 | Colon | wt | T3 | N2 | M0 | V1 |
| 9 | F | 73 | Colon | c.35G>T; p.G12V | T3 | N0 | M0 | V1 |
| 10 | F | 85 | Colon | c.35G>A; p.G12D | T2 | N0 | M0 | V0 |
| 11 | F | 62 | Colon | c.35G>A; p.G12D | T4 | N0 | M0 | V0 |
| 12 | F | 68 | Colon | wt | T1 | N0 | M0 | V0 |
| 13 | F | 73 | Colon | wt | T4 | N0 | M1 | V0 |
| 14 | M | 77 | Colon | c.35G>A; p.G12D and c.34G>A; p.G12S | T3 | N1 | M1 | V0 |
| 15 | F | 61 | Colon | c.35G>T; p.G12V | T3 | N1 | M0 | V2 |
| 16 | F | 67 | Colon | wt | T4 | N2 | M0 | V1 |
| 17 | M | 81 | Rectum | wt | T3 | N2 | M1 | V1 |
| 18 | F | 76 | Rectum | wt | T3 | N0 | M0 | V0 |
| 19 | M | 74 | Liver metastasis | c.35G>T; p.G12V | N/A | N/A | N/A | N/A |
| 20 | F | 52 | Liver metastasis | c.35G>A; p.G12D | N/A | N/A | N/A | N/A |
| 21 | M | 63 | Liver metastasis | c.35G>A; p.G12D | N/A | N/A | N/A | N/A |
| 22 | F | 64 | Liver metastasis | c.35G>A; p.G12D | N/A | N/A | N/A | N/A |
| 23 | F | 75 | Liver metastasis | c.35G>T; p.G12V | N/A | N/A | N/A | N/A |
| 24 | M | 81 | Liver metastasis | 36‐37insGGT; G12‐G13insG | N/A | N/A | N/A | N/A |
| 25 | M | 70 | Liver metastasis | wt | N/A | N/A | N/A | N/A |
| 26 | M | 54 | Liver metastasis | wt | N/A | N/A | N/A | N/A |
A, adenine; A, alanine; D, aspartic acid; F, female; G, glycine; G, guanine; ins, insertion; M, distant metastasis; M, Male; N, lymph node metastasis; N/A, not applicable; S, serine; T, threonine; T, thymine; T, tumour stage; V, valine; V, vascular invasion; wt, wild‐type.
3.1. KRAS mutations in the parental tumours enhanced growth rate of tumoroids established from colorectal adenocarcinomas
The tumoroids were categorized into those established from KRAS wild‐type parental tumours and those from KRAS‐mutated parental tumours. The KRAS mutations were detected in 58% of the parental tumours. The investigated subtypes of KRAS mutations included G12D, G13D, A146T, G12V, G12S and G12‐G13insG (Table 1). The average relative growth rate (log10) for tumoroids differed for each patient (Figure 2). Overall, the growth rate on day 10 was 0.360 ± 0.180 (mean ± SD) for the KRAS‐mutated group and 0.098 ± 0.135 (mean ± SD) for the KRAS wild‐type group. The linear regression analysis with mixed effects on the whole period of cultures from day 1 to day 11 showed that the log10 relative growth rate of tumoroids in the KRAS‐mutated group was significantly higher than the growth rate for tumoroids in the KRAS wild‐type group (P < 0.0001). Figure 2 shows that the growth rate of tumoroids in the KRAS‐mutated and in the wild‐type group differed from day 7. Subtypes of KRAS mutation in the parental tumours did not show any significant effect on the relative growth rate of tumoroids established from these tumours (Table S1).
Figure 2.
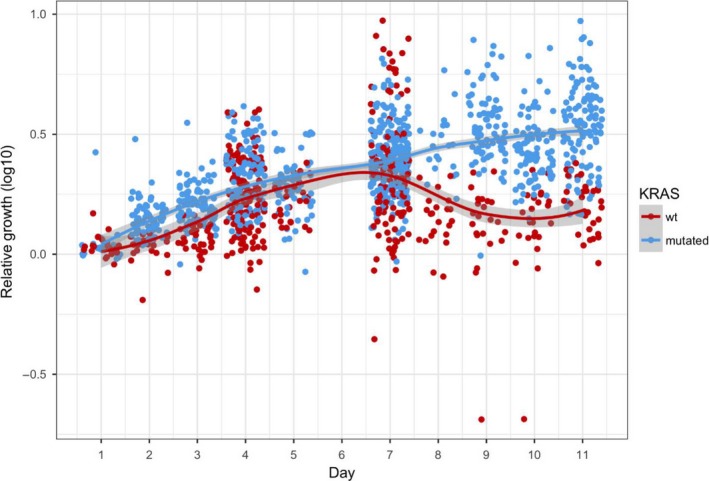
Scatter plot with fitted regression line showing log10 values of different relative rate of growth area for tumoroids. Each dot represents the relative rate of growth area in a specific culture‐well for a specific patient. The growth rate of tumoroids established from the parental tumours with mutated KRAS was higher that the growth rate for tumoroids established from the tumours with wild‐type (wt) KRAS ( P < 0.0001) [Colour figure can be viewed at wileyonlinelibrary.com]
Table S2 shows distribution of KRAS mutations correlated to staging of the primary tumours. The stage of primary parental tumours was not correlated to the growth rate of tumoroids.
4. DISCUSSION
Our group has previously shown that tumoroids established from colorectal adenocarcinoma consisted almost purely of the EpCAM‐positive epithelial cells with very little fibroblast contamination. These results have shown that the cellular composition of the tumoroids reflects the variation in cellular composition of the parental tumours. Furthermore, the average expression of cytokeratin 20 and carcinoembryonic antigen (CEA) in the tumoroids was comparable to the expression rate of these markers in the parental tumours and remained unchanged over a 10‐day period in the culture.17
The hallmarks of cancer biology presented by Hanahan and Weinberg show that different factors in the tumour microenvironment play significant roles in tumour development and progression.21 One of these hallmarks is the hyperproliferative character of the invasive tumours. The KRAS mutation makes tumours prone to such a hyperproliferative state.10 In the present study, the prolonged effect of the parental KRAS status on growth rate of the tumoroids was investigated. The incidence of KRAS mutations in primary tumours was 58%, which is similar to previous reports.9, 22, 23, 24 The previous studies on KRAS‐mutated mouse models showed significantly more proliferative cells in the bottom of colonic crypts 10 and significantly higher tumour size than non‐mutated models.25 The present study showed that tumoroids established from tumours with KRAS mutations grew faster than tumoroids established from KRAS wild‐type tumours.
The most frequent alterations in the KRAS are G > A transition as well as G > T and G > C transversions in codon 12 and codon 13.9, 26 Haigis et al10 have shown that expression of the G12D subtype of KRAS in engineered mice stimulates hyperproliferation of colon epithelium. Furthermore, Collado et al27 showed in a mouse model that activating KRAS mutation triggers development of adenocarcinomas. This study showed that the KRAS status in the parental tumour functionally influences the in vitro growth rate of tumoroids. To the best of our knowledge, this is the first study to demonstrate that in vitro patient‐derived 3D tumoroid cultures of CRC with KRAS mutations in their parental tumours grow faster than tumoroids established from KRAS wild‐type tumours.
The results of this study show that patient‐derived tumoroids originated from CRC grow in laboratory conditions and that automated image analysis can be implemented to monitor their growth rate. This model can be implemented in high‐throughput assays to study the in vitro growth of solid tumours. Furthermore, this in vitro model may be used in chemoassays in relation to personalized medicine. In such an assay, different combinations of chemotherapeutic agents can be tested in laboratory conditions prior to the selection of chemotherapy regimen. This tool can help oncologists choose the most effective chemotherapy regimen with the minimum side effects.
In summary, the present study characterized the in vitro growth of the patient‐derived tumoroids established from colorectal adenocarcinoma. The results showed that KRAS mutations in the parental tumour correlate with higher growth rate of tumoroids in vitro. The specific subtypes of KRAS mutation did not show any different growth rate. These findings shed light on an important factor affecting the in vitro model of patient‐derived tumoroids from colorectal adenocarcinoma. The future perspectives for this model could be the implementation of chemo‐screening assays for personalized chemotherapy or investigations of different functional aspects of the solid tumours.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
Supporting information
ACKNOWLEDGEMENTS
This study was supported by Innovation Fund Denmark.
Mousavi N, Truelsen SLB, Hagel G, et al. KRAS mutations in the parental tumour accelerate in vitro growth of tumoroids established from colorectal adenocarcinoma. Int. J. Exp. Path. 2019;100:12–18. 10.1111/iep.12308
REFERENCES
- 1. Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller‐Klieser W, Kunz‐Schughart LA. Multicellular tumor spheroids: an underestimated tool is catching up again. J Biotechnol 2010;148:3‐15. [DOI] [PubMed] [Google Scholar]
- 2. Smalley KS, Lioni M, Herlyn M. Life isn't flat: taking cancer biology to the next dimension. In Vitro Cell Dev Biol Anim 2006;42:242‐247. [DOI] [PubMed] [Google Scholar]
- 3. Kondo J, Endo H, Okuyama H, et al. Retaining cell‐cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc Natl Acad Sci USA 2011;108:6235‐6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van dWM, Francies HE, Francis JM, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015;161:933‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee SH, Hong JH, Park HK, et al. Colorectal cancer‐derived tumor spheroids retain the characteristics of original tumors. Cancer Lett 2015;367:34‐42. [DOI] [PubMed] [Google Scholar]
- 6. Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum‐cultured cell lines. Cancer Cell 2006;9:391‐403. [DOI] [PubMed] [Google Scholar]
- 7. Vlachogiannis G, Hedayat S, Vatsiou A, et al. Patient‐derived organoids model treatment response of metastatic gastrointestinal cancers. Science (New York, NY) 2018;359:920‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weiswald LB, Richon S, Validire P, et al. Newly characterised ex vivo colospheres as a three‐dimensional colon cancer cell model of tumour aggressiveness. Br J Cancer 2009;101:473‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brink M. K‐ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis 2003;24:703‐710. [DOI] [PubMed] [Google Scholar]
- 10. Haigis KM, Kendall KR, Wang Y, et al. Differential effects of oncogenic K‐Ras and N‐Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet 2008;40:600‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marmol I, Sanchez‐de‐Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ. Colorectal carcinoma: a general overview and future perspectives in colorectal cancer. Int J Mol Sci 2017;18:E197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J, Guo F, Shi X, et al. BRAF V600E mutation and KRAS codon 13 mutations predict poor survival in Chinese colorectal cancer patients. BMC Cancer 2014;14:802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li W, Qiu T, Zhi W, et al. Colorectal carcinomas with KRAS codon 12 mutation are associated with more advanced tumor stages. BMC Cancer 2015;15:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diersch S, Wirth M, Schneeweis C, et al. Kras(G12D) induces EGFR‐MYC cross signaling in murine primary pancreatic ductal epithelial cells. Oncogene 2016;35:3880‐3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kitai H, Ebi H. Key roles of EMT for adaptive resistance to MEK inhibitor in KRAS mutant lung cancer. Small GTPases 2017;8:172‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B 2015;5:390‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeppesen M, Hagel G, Glenthoj A, et al. Short‐term spheroid culture of primary colorectal cancer cells as an in vitro model for personalizing cancer medicine. PLoS ONE 2017;12:e0183074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fredborg M, Andersen KR, Jorgensen E, et al. Real‐time optical antimicrobial susceptibility testing. J Clin Microbiol 2013;51:2047‐2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. D'Haene N, Le MM, De NN, et al. Clinical Validation of Targeted Next Generation Sequencing for Colon and Lung Cancers. PLoS ONE 2015;10:e0138245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Green RC, Berg JS, Grody WW, et al. CORRIGENDUM: ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2017;19(5):606. [DOI] [PubMed] [Google Scholar]
- 21. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 22. Brink M, Weijenberg MP, de GA, et al. Meat consumption and K‐ras mutations in sporadic colon and rectal cancer in The Netherlands Cohort Study. Br J Cancer 2005;92:1310‐1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cancer GAN. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch‐repair status. Nature 2002;418:934. [DOI] [PubMed] [Google Scholar]
- 25. Ju HL, Calvisi DF, Moon H, et al. Transgenic mouse model expressing P53(R172H), luciferase, EGFP, and KRAS(G12D) in a single open reading frame for live imaging of tumor. Sci Rep 2015;5:8053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Urosevic N, Krtolica K, Skaro‐Milic A, Knezevic‐Usaj S, Dujic A. Prevalence of G‐to‐T transversions among K‐ras oncogene mutations in human colorectal tumors in Yugoslavia. Int J Cancer. 1993;54:249‐254. [DOI] [PubMed] [Google Scholar]
- 27. Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials