

Abstract
Background
Up to 80% of pancreatic cancer patients suffer from cachexia, a devastating condition that exacerbates underlying disease, reduces quality of life, and increases treatment complications and mortality. Tumour‐induced inflammation is linked to this multifactorial wasting syndrome, but mechanisms and effective treatments remain elusive. Myeloid differentiation factor (MyD88), a key component of the innate immune system, plays a pivotal role in directing the inflammatory response to various insults. In this study, we tested whether MyD88 signalling is essential in the development of pancreatic cancer cachexia using a robust mouse tumour model.
Methods
Sex, age, and body weight‐matched wide type (WT) and MyD88 knockout (MyD88 KO) mice were orthotopically or intraperitoneally implanted with a pancreatic tumour cell line from a syngeneic C57BL/6 KRASG12D/+ P53R172H/+ Pdx‐Cre (KPC) mouse. We observed the effects of MyD88 signalling during pancreatic ductal adenocarcinoma progression and the cachexia development through behavioural, histological, molecular, and survival aspects.
Results
Blocking MyD88 signalling greatly ameliorated pancreatic ductal adenocarcinoma‐associated anorexia and fatigue, attenuated lean mass loss, reduced muscle catabolism and atrophy, diminished systemic and central nervous system inflammation, and ultimately improved survival. Our data demonstrate that MyD88 signalling plays a critical role in mediating pancreatic cancer‐induced inflammation that triggers cachexia development and therefore represents a promising therapeutic target.
Conclusions
MyD88‐dependent inflammation is crucial in the pathophysiology of pancreatic cancer progression and contributes to high mortality. Our findings implicate the importance of innate immune signalling pathways in pancreatic cancer cachexia and a novel therapeutic target.
Keywords: Pancreatic cancer, Cachexia, MyD88, Inflammation, Orthotopic model
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most common fatal malignancies, and less than 8% of patients with PDAC survive 5 years after diagnosis.1 This dismal survival rate is essentially unchanged since the 1960s, despite substantial improvements in the survival rates of other major cancers.2 The high mortality rates are multifactorial, but one important factor is that more than 80% patients with PDAC suffer from cachexia, a devastating wasting syndrome characterized by anorexia, weight loss (including both fat and lean mass), lethargy, fatigue, and increased metabolism.3, 4, 5 Cachexia is strongly associated with poor prognosis, reduced treatment tolerance, worse post‐operative outcome, a marked reduction in quality of life, and significantly decreased survival.6, 7, 8, 9, 10, 11, 12 Unfortunately, there is no effective treatment for this condition.
Despite the tremendous progress in understanding mediators, signalling, and metabolic pathways of cachexia, it is unclear why cachexia is so ubiquitous and severe in PDAC. Although there are likely several factors driving the pathology in this disease, much of the attention has focused on the host inflammatory response to tumour growth. Tumour cells can produce inflammatory cytokines and induce the host's local and systemic inflammatory responses, all of which can trigger many of the metabolic changes associated with cachexia.10, 12, 13, 14, 15 In this way, the response to PDAC is similar to that observed after exposure to bacterial endotoxin, viral RNA, or cytokines, each of which induces robust inflammation that results in anorexia, body weight loss, fatigue, and muscle wasting.16, 17, 18, 19 Over the past decade, studies in both humans and rodents implicate a number of pro‐inflammatory cytokines as key mediators of cachexia in PDAC, including tumour necrosis factor alpha, interleukin‐1 beta, interleukin‐6 (IL‐6), and interferon gamma.12, 14, 20 This in turn led to efforts at developing anti‐cytokine agents (e.g. anti‐tumour necrosis factor alpha and anti‐IL‐6) to treat this syndrome. However, none of these individual cytokine blockers provided clinically relevant attenuation of cachexia.10 It is now apparent that the primary signals driving cachexia depend on tumour type and reflect a complex response to a network of mediators rather than to any single cytokine.21, 22, 23, 24 The complex and redundant nature of signalling molecules involved impedes therapeutic development efforts, highlighting the need to identify targets that represent master regulatory pathways and signals rather than downstream mediators.
MyD88 is a key component of both innate and adaptive immune systems, as it is the universal adaptor protein to all Toll‐like receptors (TLRs) except TLR3, as well as the type‐I interleukin receptor (IL‐1R) family. TLRs recognize specific pathogen‐associated and danger‐associated molecular patterns, which are released by many different kinds of infections and cellular injury. Upon TLR or IL‐1R activation, MyD88 is recruited to initiate and activate downstream signalling cascades leading to production of a wide array of pro‐inflammatory cytokines.25, 26, 27 Therefore, MyD88 is a critical signalling node that directly links a variety of upstream signals to a broad range of downstream signalling mediators.
In the present study, we utilized a murine PDAC model that reliably induces a robust and reproducible cachexia phenotype.28 We tested the hypothesis that MyD88 signalling plays an essential role in mediating PDAC‐induced systemic and central nervous system (CNS) inflammation, thereby serving as a central mechanistic driver of metabolic derangement and cachexia. Using behavioural, histological, and molecular approaches as well as survival evaluation, we demonstrate that blockade of MyD88 signalling ameliorates all features of cachexia and therefore is a logical target for the development of novel therapies to improve the quality of life and survival in patients suffering from PDAC.
Methods
Animals
C57BL/6J WT (Stock# 000664) and MyD88 KO (Stock# 009088) mice were originally purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained in our animal facility. MyD88 KO mice were generated on C57BL/6J background, bred via pairing of homozygous for knockout allele, and genotyped using standard protocols from The Jackson Laboratory. All animals were housed in a room with controlled temperature at 27 °C and 12 h light–dark cycles and provided ad libitum access to water and food (Purina rodent diet 5001; Purina Mills, St. Louis, MO, USA). Sex, age, and body weight‐matched WT and MyD88 KO mice at 7–10 weeks old were used in all experiments. In behavioural and survival experiments, animals were individually housed for acclimation at least 7 days prior to procedures. Pre‐implantation animal selection was randomized but balanced according to initial body weight. Except in survival studies, tumour‐bearing animals were euthanized according to the end points of tumour study policy. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Oregon Health & Science University.
Tumour cell culture and implantation
The KPC tumour cells are derived from a tumour explant of a C57BL/6 mouse modified to express oncogenic KRASG12D and the point mutant TP53R172H under a pancreas‐specific promoter expressed during early development, Pdx1‐Cre, which are kindly provided by Dr Elizabeth Jaffee.29 KPC cells were cultured in RPMI 1640 supplemented with 10% FBS and 1% penicillin–streptomycin at 37°C and 5% CO2. Prior to implantation, cells were harvested, counted, and diluted based on the cell number and volume of implantation. All KPC cells were tested to be negative for mycoplasma contamination. For orthotopic (OT) implantation, animals were anaesthetised with isoflurane, and the abdominal cavity was opened by a small longitudinal incision in the left upper quadrant. After the pancreas was exposed, 40 μL of KPC cell suspension with a total of 3 × 106 cells was directly injected into the pancreas without leakage using a Hamilton syringe. The abdominal wall muscle layer was closed with Polysorb sutures, and the skin incision was closed with surgical clips. For the intraperitoneal (IP) and subcutaneous (SC) models, 0.5 mL of KPC cell suspension with a total of 3 × 106 or 10 × 106 cells was injected intraperitoneally into abdominal cavity or subcutaneously into interscapular space under isoflurane anaesthesia. Mice receiving sham implantation were injected with heat‐killed KPC cells or PBS at an equal volume as that in tumour‐implanted animals.
Food intake and body weight measurement
In behavioural experiments, food intake and body weight were measured manually on a daily basis at a similar time point (3 h after lights on), starting on the day of implantation (Day 0) until the day of sacrifice. Daily screening of food spillage and debris from cage bedding was employed to account for uningested food loss.
Locomotor activity and body temperature measurement
Voluntary home cage locomotor activity (LMA) and body temperature (BT) were measured throughout the entire experimental period using a MiniMitter system (MiniMitter, Bend, OR, USA) as described previously.18 Briefly, under isoflurane anaesthesia, transponders for sensing LMA and BT were implanted subcutaneously into interscapular space, and the animals were returned to their home cage. BT and LMA in x‐axis, y‐axis, and z‐axis were recorded in 5‐min intervals.
Body composition analysis
Body composition (fat and lean mass) was analysed twice on the day of implantation (Day 0) and the day of sacrifice, using magnetic resonance relaxometry (EchoMRI 4‐in‐1 Live Animal Composition Analyser; Echo Medical System, Houston, TX, USA).
Blood and tissue collection
When tumour‐bearing animals reached the predetermined criteria for euthanasia or designed time points, they were deeply anaesthetised with a ketamine‐xylazine‐acepromazine cocktail, and blood was collected via cardiac puncture; 100 μL of blood was placed into an EDTA blood collection tube for haematological analysis, and the rest of blood was used for isolation of serum. Serum was stored in −80°C until analysis. Brain tissue was removed, and hypothalamic and cortical blocks were dissected. Heart, liver, gastrocnemius of both left and right hindlimbs, and tumour were dissected and weighed. For molecular analysis, tissues were snap frozen right after dissection using liquid nitrogen and then stored in −80°C until further analysis. For immunohistochemical analysis of gastrocnemius cyrosections, tissues were embedded with OCT, snap frozen and stored in −80°C until analysis as previously described.17 Ascites and tumour metastasis were grossly noted as present or absent during animal sacrifice and tissue dissection.
Haematology
Whole blood was analysed with a veterinary haematology analyser (HemaVet, 950FS, Drew Scientific, Oxford CT, USA) for haematological parameters, including total white blood cell count, white blood cell differential (neutrophils, lymphocytes, monocytes, eosinophils, and basophils), red blood cells count, haemoglobin concentration, haematocrit, mean corpuscular volume, mean corpuscular haemoglobin, mean corpuscular haemoglobin concentration, and platelet count.
Enzyme‐linked immunosorbent assay
Serum IL‐6 was measured using enzyme‐linked immunosorbent assay kits according to manufacturers' instructions (ThermoFisher Scientific‐Invitrogen).
Real‐time quantitative PCR
Real‐time quantitative PCR was performed as described previously.18 RNA was extracted from tissues using RNeasy kits (Qiagen Inc., Valencia, CA, USA). Reverse transcription and quantitative PCR reagents were obtained from Life Technologies (Carlsbad, CA, USA). Real‐time quantitative PCR reactions were run on an ABI 7300 (Applied Biosystems), using TaqMan universal PCR master mix with the following TaqMan mouse gene expression assays (Life Technologies): Mafbx (Mm00499518_m1), Murf1 (Mm01185221_m1), Foxo1 (Mm 00490672_m1), Il1b (Mm00434228_m1), Il1r1 (Mm00434237_m1), Il6 (Mm01210732_m1), Il10 (Mm99999062_m1), Tnf (Mm00443258_m1), Ifng (Mm01168134_m1), Selp (Mm00441295_m1), Ccl2 (Mm99999056_m1), Crp (Mm00432680_g1), and Myd88 (Mm00440338_m1); 18s (Mm04277571_s1) or Actb (Mm00607939_s1) was used as the endogenous control for brain tissue or muscle and liver tissue, respectively. Relative expression was calculated via the ΔΔCt method and was normalized to sham‐treated group within same genotype of mice or to WT/sham group. Statistical analysis was performed on the ΔCt values.
Quantification of triglycerides in faeces
Fresh faeces were collected from the late stage of PDAC cachexia mice along with sham mice. Positive control samples were collected from mice administered with Orlistat (GlaxoSmithKline) at 60 mg/kg body weight via gavage for 4 consecutive days. For triglyceride extraction, faeces were snap frozen, and 0.02‐g sample was ground and homogenized in 5% NP‐40. Sample mix was heat twice at 80–100°C and then centrifuged. The supernatant was assayed using a triglyceride quantification assay kit according to manufactures' instructions (Abcam, cat # ab178780).
Histopathology
Formalin fixed paraffin‐embedded histologic sections stained with haematoxylin and eosin were evaluated by a pathologist blinded to animal group. Tissue sections of the pancreas, spleen, and liver (minimum of two sections from each of 10 animals per group) were scored as positive or negative for carcinoma, acute inflammation, chronic inflammation, and unusual patterns like extramedullary haematopoiesis. A similar analysis was performed on 10 tumours per group in the tumour bearing animals. Representative fields were photographed, and reproducible patterns within groups were determined by consensus between investigators.
Immunohistochemistry
Cryosections of the gastrocnemius were immunostained and analysed as described previously.17 Briefly, cryosections were cut in section at 20‐μm thickness, fixed with 4% paraformaldehyde for 15 min and blocked in PBS/10% bovine serum albumin (BSA) for 1 h at room temperature. Sections was incubated overnight at room temperature with a rabbit anti‐laminin antibody (Sigma‐Aldrich) diluted 1:250 in PBS/10% BSA, washed with PBS/0.025% Triton X‐100, and incubated in a goat anti‐rabbit Alexa Fluor 488 nm labelled secondary antibody (Invitrogen) diluted 1:500 in PBS/10% BSA. Sections were coverslipped with ProLong Gold Antifade media with DAPI (Invitrogen). Fibre area was measured in images that were acquired on a microscope (DM 4000 B, Leica Microsystems). Images were processed using NIH ImageJ software.
Survival study
Sex, age, and body weight‐matched WT and MyD88 KO mice were implanted on Day 0 via OT or IP injection of KPC tumour cells at 3 × 106 cells (per mouse). All animals were individually housed after implantation and observed twice daily until death. Tumour appearance was confirmed in all animals, and tumours were dissected and weighed by necropsy.
Statistical analysis
Data are expressed as mean ± standard error of the mean for each group. In survival studies, symbols represent data from individual mice. Statistical analyses were performed with GraphPad Prism 5.0 (La Jolla, CA), using the unpaired Student's t‐test, one‐way and two‐way analyses of variance followed by Bonferroni post‐tests, and comparison of survival curves with Log‐rank (Mantel‐Cox) test. P < 0.05 was considered statistically significant.
Results
Blockade of MyD88 signalling attenuates anorexia during development of pancreatic ductal adenocarcinoma cachexia
To elucidate the role of MyD88 signalling in the development of PDAC cachexia, we first characterized a mouse model in which a pancreatic tumour cell line from a syngeneic C57BL/6 KRASG12D/+ P53R172H/+ Pdx‐Cre (KPC) mouse was implanted through three different routes of implantation: OT, IP, and SC. Each route represents a unique advantage. For the OT model, tumours are organ specific and clinically relevant, which is crucial for accurately modelling tumour biology, and therefore, it has emerged as the preferred model for cancer research.30 We primarily utilized the OT model (Supporting Information, Figure S1) in this study (all data presented were obtained from OT model unless otherwise stated). For the IP model, with nonsurgical implantation, there is particularly no trauma‐associated inflammation. We did apply the IP model for a subset of experiments to rule out any unequal effects derived from trauma between WT and MyD88 KO mice. This was also to determine whether the different implantation routes yield dissimilar characteristics or different degrees of the cachexia. Additionally, we found that the SC model produced inconsistent results, and it was not studied further. As loss of appetite and body weight is the most common feature of cachexia,12 we first examined the effect of MyD88 signalling on anorexia and weight loss in PDAC cachexia. Starting at Days 6–8 post‐implantation, WT male and female KPC mice significantly decreased food intake compared to WT/sham groups (Supporting Information, Figure S2A). In contrast, the decrease in food intake in MyD88 KO male and female KPC mice was attenuated compared to MyD88 KO/sham groups (Figure 1A). Because mouse feeding behaviour differs by biological variation and is also highly influenced by environmental changes (Supporting Information, Figure S2A), in order to reduce non‐specific effects, we normalized the daily food intake of tumour groups to same genotype of sham group for a relative comparison (Figure 1A). Accordingly, the decrease in cumulative food intake of the entire experimental period was attenuated in MyD88 KO KPC mice (Figure 1B; Supporting Information, Figure S2b). We did not find evidence of sexual dimorphism in anorexia in either genotype of KPC mice, and a similar feeding response was reproducibly observed in separate experiments with both male and female mice. Because initial body weight was matched within the same genotype and sex of mice, and considering the effect of tumour growth and particularly ascites development, we did not normalize food consumption to body weight. WT KPC mice progressively increased gross body weight compared to WT sham mice due to tumour growth and the development of malignant ascites. The same effect was observed in MyD88KO KPC mice although with delayed kinetics, particularly in the female cohort (Supporting Information, Figure S2C). Tumours were moderately or significantly smaller in male and female MyD88 KO mice compared to WT mice (Supporting Information, Figure S2D). Consistent with the increase in gross body weight, substantial ascites was found in abdominal cavity of both genotypes of KPC mice (Supporting Information, Figure S2E). In addition to cachexia‐specific signalling, local tumour invasion or inflammation could lead to exocrine pancreatic insufficiency as a contributing factor to weight loss. Therefore, we assayed the faeces from sham and KPC mice vs. Orlistat‐treated positive controls and found no evidence of fat malabsorption even in late stage cachexia (Supporting Information, Figure S2F). A similar alteration in food intake, body weight change, and tumour growth was observed in IP KPC animals (Supporting Information, Figure S3A–S3E).
Figure 1.
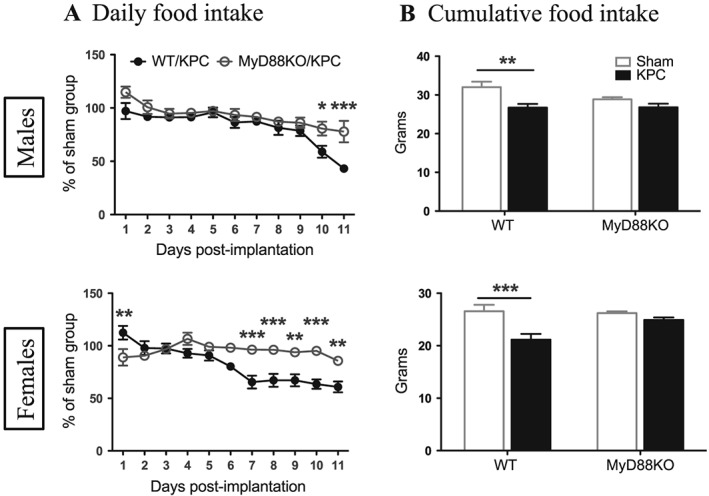
Blockade of MyD88 signalling attenuates anorexia during development of PDAC cachexia. (A) Daily food intake post‐implantation. (B) Cumulative food intake within 11 days post‐implantation. *, P < 0.05; **, P < 0.01; ***, P < 0.001; WT/KPC vs. MyD88KO/KPC (A) or WT/sham vs. WT/KPC (B). Two‐way analysis of variance with post hoc Bonferroni‐corrected t‐test. N = 5–6/group.
Blockade of MyD88 signalling reduces lean mass loss in pancreatic ductal adenocarcinoma cachexia
To determine whether MyD88 signalling is critical for fat mass and lean mass loss during the development of PDAC cachexia, we measured body composition prior to implantation and on the day of sacrifice. WT (both male and female mice) KPC mice lost a pronounced amount of fat mass and lean mass within 11 days of post‐implantation compared to WT sham mice, whereas MyD88 KO KPC mice lost less fat mass and gained lean mass (Figure 2A and 2B; Supporting Information, Figure S4A–S4C). Therefore, the sum loss of fat mass and lean mass was attenuated in MyD88 KO KPC mice (Figure 2C). There was no sexual dimorphism in fat mass or lean mass loss within each genotype. Collectively, these data demonstrate that blockade of MyD88 signalling produces a modest preservation in fat mass and a marked preservation in lean mass in PDAC cachexia. Similar results were obtained with the IP model (Supporting Information, Figure S5A–S5E).
Figure 2.
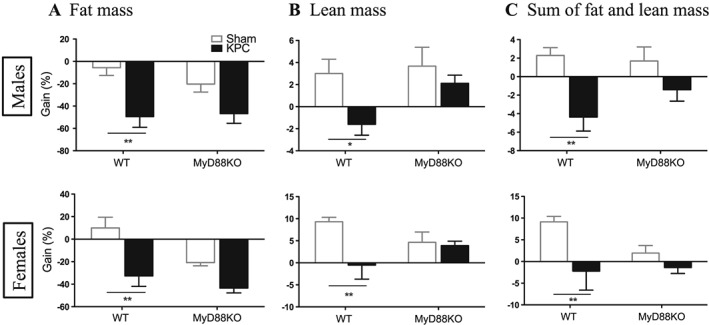
Blockade of MyD88 signalling reduces lean mass loss in pancreatic ductal adenocarcinoma cachexia. Body composition was measured on the day of implantation and the day of sacrifice. Gain% is the net gain normalized to baseline. (A) Fat mass gain. (B) Lean mass gain. (C) Sum of fat mass and lean mass gain. *, P < 0.05; **, P < 0.01; WT/sham vs. WT/KPC. Two‐way analysis of variance with post hoc Bonferroni‐corrected t‐test. N = 5/group.
MyD88 signalling is critical for development of fatigue during pancreatic ductal adenocarcinoma cachexia progression
Fatigue is a very common symptom of cancer at diagnosis and throughout the course of illness.31, 32 The ability to maintain normal daily physical activity is critical not only for wellness and quality of life but may also serve as a tool for early detection of cancer or for monitoring disease progression.33 To examine the effect of MyD88 signalling on physical activity during development of PDAC cachexia, we monitored daily LMA. A similar pattern in the change of daily LMA was observed in separate experiments with male and female mice. Compared to sham groups, both WT male and female KPC mice progressively decreased LMA, particularly during 12‐h dark phase (active phase), starting at Day 6 post‐implantation and remaining in continuous decline over the remainder of experiments (Figure 3A and 3B). In contrast, MyD88 KO male and female KPC mice exhibited a notable (3 days) delay and much smaller decrease in LMA relative to WT (Figure 3A and 3B). Similarly, MyD88 KO IP KPC mice also exhibited a trend toward attenuation in the decrease of LMA, but it was relatively modest compared to MyD88 KO OT KPC mice (Supporting Information, Figure S5F). Again, no sexual dimorphism was observed in either genotype. It is important to note that a decrease in LMA is the first measurable behavioural change in PDAC cachexia and precedes anorexia.28 In addition, BT was simultaneously recorded with the same MiniMitter system. There was no change in BT at the early stage of cachexia, but at the late stage, both WT and MyD88 KO KPC mice gradually developed a similar pattern of hypothermia that consistently coincided with normal physiological BT nadir in the sham groups (middle of active phase) (Supporting Information, Figure S6A and 6B). WT IP KPC mice had deeper hypothermia compared to OT KPC mice (Supporting Information, Figure S6C).
Figure 3.
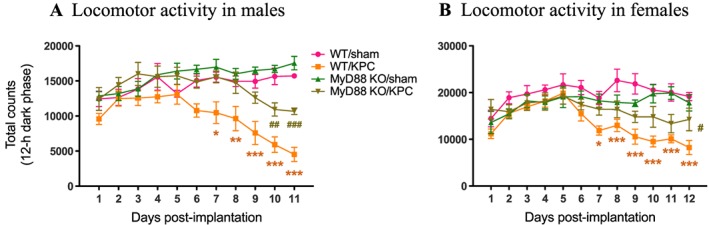
MyD88 signalling is critical for development of fatigue during PDAC cachexia progression. Movement counts were monitored post‐implantation throughout the entire course of experiment. (A) Locomotor activity (LMA) within 12‐h dark phase (active phase) in male mice. (B) LMA within 12‐h dark phase in female mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001; WT/sham vs. WT/KPC. #, P < 0.05; ##, P < 0.01, ###, P < 0.001; MyD88 KO/sham vs. MyD88 KO/KPC. Two‐way analysis of variance with post hoc Bonferroni‐corrected t‐test. N = 5/group.
MyD88 signalling is essential for muscle catabolism and atrophy in pancreatic ductal adenocarcinoma cachexia
To explore the involvement of MyD88 signalling in PDAC cachexia‐related muscle catabolism, we measured muscle weight of gastrocnemius and heart, analysed gene expression related to muscle catabolism and atrophy in both skeletal and cardiac muscle, and quantified gastrocnemius muscle fibre area. Both gastrocnemius and heart weights were dramatically decreased in WT male and female KPC mice, but the decrease was greatly attenuated in MyD88 KO KPC male and female groups (Figure 4A and 4B; Supporting Information, Figure S7A–S7D). Similar attenuation in gastrocnemius and heart muscle loss was also found in MyD88 KO IP KPC mice (Supporting Information, Figure S8A–S8C). When compared to WT KPC mice, MyD88 KO KPC mice demonstrated an attenuation of the gene expression profile seen in catabolic muscle, including muscle atrophy F‐box protein (Mafbx or atrogin‐1), muscle ring finger protein 1 (Murf1), and forkhead box 1 (Foxo1) (Figure 4C; Supporting Information, Figure S9A). Similar results were found in the cardiac muscle (Figure 4D, Supporting Information, Figure S9B).
Figure 4.
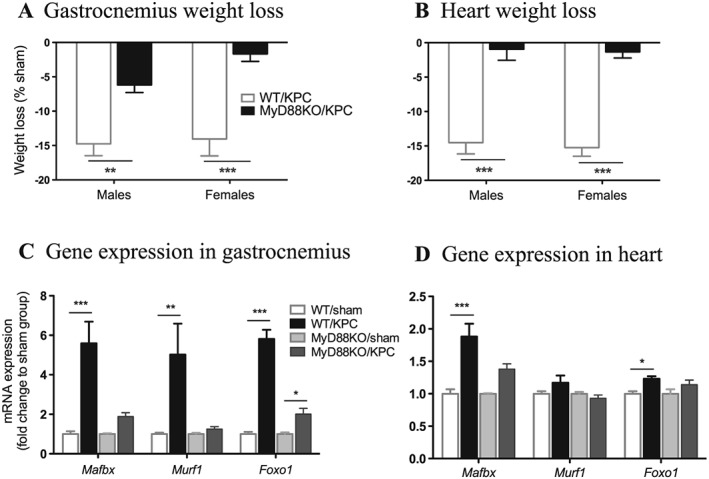
MyD88 signalling is essential for muscle catabolism in pancreatic ductal adenocarcinoma cachexia. Mice were euthanized on Days 11–12 post‐implantation. (A) Gastrocnemius weight (average of left and right gastrocnemii). (B) Heart weight. Muscle weight was normalized to initial body weight and presented as loss (%) to sham mice of the same genotype. (C) Gene expression in gastrocnemius. (D) Gene expression in heart. *, P < 0.05; **, P < 0.01; ***, P < 0.001; WT/KPC vs. MyD88KO/KPC (A, B) or sham vs. KPC (C, D). Two‐way analysis of variance with post hoc Bonferroni‐corrected t‐test. N = 5/group.
To determine how MyD88 signalling affects muscle fibre morphology, we performed anti‐laminin staining in cross sections of gastrocnemius (Figure 5A) and quantified fibre cross‐sectional area (CSA) in more than 300 cells per sample. Fibre CSA was dramatically decreased in WT KPC mice relative to sham animals, but no change was observed in MyD88 KO KPC mice vs. sham mice (Figure 5B). The fibre CSA distribution curve in WT KPC mice was notably shifted to left, indicating an increased portion of smaller fibres, whereas no change was observed in MyD88 KO KPC animals (Figure 5C).
Figure 5.
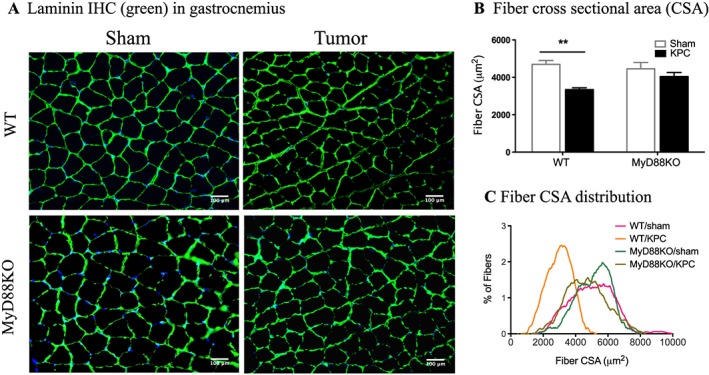
Blockade of MyD88 signalling prevents muscle atrophy in pancreatic ductal adenocarcinoma cachexia. Mice were euthanized on Days 11–12 post‐implantation. (A) Immunohistochemistry (IHC): gastrocnemius cross sections immunostained with antibody against laminin (green). Blue is DAPI for nuclear counterstain. (B) Fibre cross‐sectional area (CSA). (C) Fibre CSA distribution. **, P < 0.01; WT/sham vs. WT/KPC. Two‐way analysis of variance with post hoc Bonferroni‐corrected t‐test. N = 4–5/group. Scale bars: 100 μm.
MyD88 signalling plays a key role in mediating pancreatic ductal adenocarcinoma‐induced systemic and central nervous system inflammation
To demonstrate that PDAC‐induced systemic inflammation is diminished in the absence of MyD88 signalling, we first analysed basic hematologic parameters. Blood leukocyte parameters indicated that total white blood cell count was greatly increased in WT both male and female KPC mice relative to sham animals, which was mainly due to an increase in neutrophils. However, this increase was diminished in MyD88 KO KPC mice (Figure 6A). There was no significant effect on lymphocytes, monocytes, eosinophils, and basophils in KPC animals of both genotypes (Figure 6A; Supporting Information, Tables S1 and S2). Furthermore, blood erythrocyte parameters showed lower haematocrit, red blood cells, and haemoglobin in both genotypes of KPC mice relative to sham mice (Supporting Information, Tables S1 and S2), suggesting that anaemia, a common feature of cachexia, occurred independently of MyD88 signalling. There was a moderate increase in platelets in WT KPC mice but not MyD88 KO KPC mice (Supporting Information, Tables S1 and S2). In the separate experiments with IP male models, we observed similar alterations in haematological profile (Supporting Information, Figure S10A and Table S3).
Figure 6.
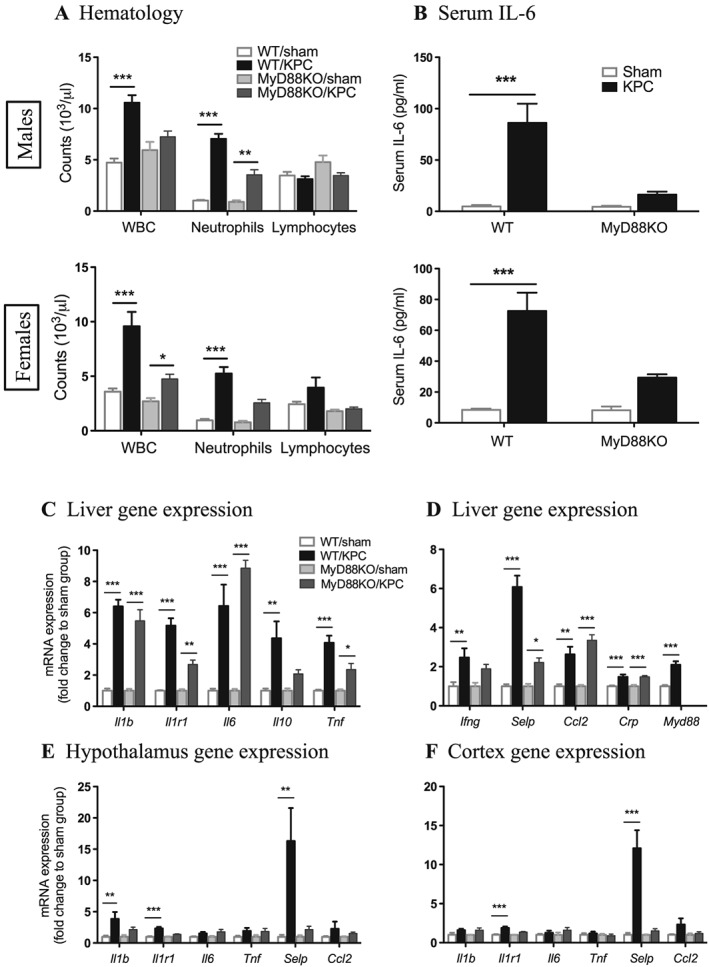
Pancreatic ductal adenocarcinoma‐induced systemic and central nervous system inflammation is diminished by blocking MyD88 signalling. Mice were euthanized on Days 11–12 post‐implantation. (A) White blood cell (WBC) and neutrophil and lymphocyte counts. (B) Serum interleukin‐6 levels. (C, D) Liver inflammatory gene expression. (E) Hypothalamic inflammatory gene expression. (F) Cortex inflammatory gene expression. *, P < 0.05; **, P < 0.01; ***, P < 0.001; sham vs. KPC. Two‐way analysis of variance with post hoc Bonferroni‐corrected t‐test. N = 5/group.
Serum IL‐6 levels were measured by enzyme‐linked immunosorbent assay and found to be elevated in WT KPC mice compared with sham controls. IL‐6 elevation was attenuated in MyD88 KO KPC mice (Figure 6B). Attenuated serum IL‐6 levels were also found in MyD88 KO IP KPC mice (Supporting Information, Figure S10B).
Histopathologic studies of pancreas, liver, and spleen revealed increased organ inflammation associated with PDAC in these animals independent of MyD88 signalling. Sections of the pancreas showed marked acute inflammation associated with tumour cells in WT KPC and MyD88 KO KPC mice that was absent in Sham controls (Supporting Information, Figure S11A). KPC mice had a mixed portal inflammation composed of lymphocytes, neutrophils, and eosinophils (Supporting Information, Figure S11B) that was absent in Sham controls. KPC mice also had enlarged spleens compared with sham controls with enlarged reactive lymphoid follicles, lymphoid infiltration of red pulp, and conspicuous megakaryocytes suggestive of extramedullary haematopoiesis (Supporting Information, Figure S11D). Additionally, spleen weights were significantly increased to a similar degree in both WT KPC and MyD88 KO KPC groups relative to sham controls (Supporting Information, Figure S11E).
The liver tissue was analysed for gene expression of inflammatory mediators that are linked to cancer cachexia.9, 14, 20, 34 Multiple genes, including Il1b, Il1r1, Il6, Il10, Tnf, Ifng, Selp, Ccl2, Crp, and Myd88 were upregulated in WT KPC mice relative to sham mice. These genes were chosen to represent broad classes of inflammatory mediators and pathways, including cytokines (Il1b, Il6, Il10, and Tnf), cytokine receptors and adaptors (Il1r1 and Myd88), interferons (Ifng), and chemokines/acute phase reactants (Ccl2 and Crp). A similar upregulation level of Il1b, Il6, Ccl2, and Crp was found in MyD88 KO mice, and Il1r1, Il10, Tnf, Ifng, and Selp gene expression was modestly or significantly diminished in MyD88 KO KPC mice (Figure 6C and 6D; Supporting Information, Figure S12A and 12B). Overall, we did not observe that inflammatory gene induction in the liver was greatly attenuated in MyD88 KO KPC mice. This observation is consistent with the histopathological examination, indicating that other signalling pathways or mechanisms may facilitate the local inflammation. It was also confirmed that MyD88 KO mice did not have detectable MyD88 gene expression (Figure 6D; Supporting Information, Figure S12B). Next to demonstrate that PDAC‐induced CNS inflammation depends on MyD88 signalling, we analysed key brain regions for inflammatory gene expression. Il1b, Il1r1, and Selp were robustly up‐regulated in the hypothalamus of WT KPC mice relative to sham mice, and this up‐regulation was significantly diminished in MyD88 KO KPC mice (Figure 6E; Supporting Information, Figure S12C). Il1r1 and Selp were also significantly up‐regulated in cortex of WT KPC mice, but no change was observed in MyD88 KO KPC mice (Figure 6F; Supporting Information, Figure S12D). Selp is the gene encoding P‐selectin, an endothelial adhesion molecule that facilitates immune cell transendothelial migration during inflammation. Expression of this transcript is linked to cancer and cancer cachexia.35 The highly upregulated Selp in both peripheral organs and CNS in WT KPC mice was diminished in MyD88 KO KPC mice, suggesting that it could be a key mediator in the network of MyD88 signalling in pancreatic cancer cachexia.
MyD88 signalling contributes to high mortality in pancreatic ductal adenocarcinoma
The improvements noted in inflammation and catabolism implied that blocking MyD88 signalling would also benefit survival during advanced PDAC cachexia. Therefore, survival experiments were performed in sex, age, and initial body weight‐matched WT and MyD88 KO KPC mice. After OT implantation, WT male and female KPC mice survived 14–23 or 12–21 days, respectively, whereas MyD88 KO male and female KPC mice survived 19–35 or 16–28 days (the median survival in WT and MyD88 KO OT male mice was 17.5 vs. 24 days, P = 0.003, and in OT female mice 15.5 vs. 25.5 days, P = 0.0003, Log‐rank (Mantel‐Cox) test; Figure 7A). After IP implantation, WT male and female KPC mice survived 14–18 or 15–22 days, whereas MyD88 KO male and female KPC mice survived 21–26 or 23–32 days (the median survival in WT and MyD88 KO IP male mice was 15.5 vs. 24 days, P < 0.0001, and in IP female mice was 21 vs. 26 days, P = 0.0002) (Supporting Information, Figure S13A). Because body weight approximates the amount of starting ‘reserve’ for animals and is therefore a very important factor in survival, initial body weight between WT and MyD88 KO mice was rigorously matched within each experiment (Figure 7B; Supporting Information, Figure S13B). At necropsy, we found that MyD88 KO KPC mice had larger tumours compared to WT KPC mice (Figure 7C; Supporting Information, Figure S13C), likely due to the longer survival (and tumour growth) time.
Figure 7.
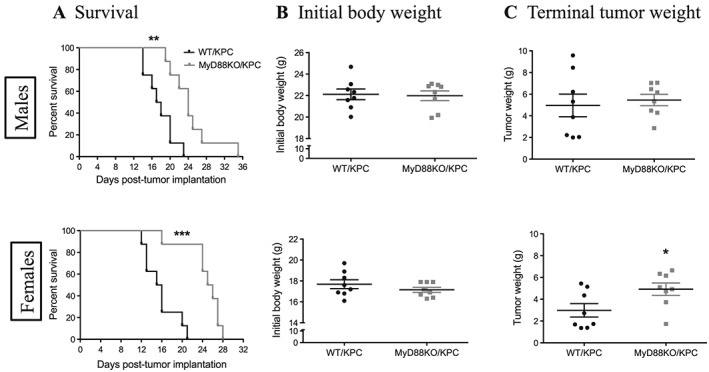
MyD88 signalling contributes to high mortality in PDAC. Sex, age, and body weight‐matched WT and MyD88 KO mice were implanted with 3 × 106 KPC cells through orthotopic route. (A) Survival curves in male and female mice. (B) Initial body weight in male and female mice pre‐tumour implantation. (C) Terminal tumour weight in male and female mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001; WT/KPC vs. MyD88KO/KPC. Comparison of survival curves with Log‐rank (Mantel‐Cox) test. N = 8/group.
Discussion
During cancer progression, tumour‐derived and host‐derived mediators promote excessive inflammation that worsens underling disease and accelerates catabolism, ultimately leading to cachexia.6, 9, 12, 14, 20, 34, 36 This circumstance is common in many cancers but is particularly ubiquitous and severe in pancreatic cancer.9, 12, 34, 37 In the present work, we utilized mouse OT and IP PDAC models and demonstrate the role of MyD88 signalling in mediating development of PDAC cachexia and mortality. Our first focus was on establishing a reliable, consistent, and reproducible experimental model to mimic this complex behavioural and metabolic condition. While there are experimental advantages to SC tumour implantation, it is clear that an OT model is necessary for recapitulating the tumour micro‐environment and the relevant tumour‐host interactions.30, 38, 39, 40 Similar to our PDAC OT allografts, a recent study reported that OT xenografts of human pancreatic cancer induce more profound muscle wasting and systemic inflammation compared to the SC xenografts.41 However, relatively few cachexia studies utilize orthtopic implantation, in part because this approach is technically challenging and induces some degree of surgical trauma and associated post‐operative recovery time.6 In our hands, OT implantation caused a modest acute illness response that was evident for approximately 48 h. At necropsy, 95–100% of tumour tissue was found in the pancreas, with frequent spread into local mesentery but infrequent (<5%) distal metastasis. Therefore, our current model is a good representation of clinical disease, reaching the equivalent of Stage III disease in all cases and Stage IV in a relatively small subset. It would be very interesting to further investigate the interaction and crosstalk between tumour cells, host immune cells, and stroma in this particular OT model. This future work would provide insight into how global MyD88 signalling affects the pancreatic tumour micro‐environment, disease progression, and cachexia development.
Loss of appetite and loss of body weight (both fat and lean mass) are common morbidities that often appear earlier than other symptoms and significantly impact resection rate, treatment tolerance, prognosis, and survival in cancer patients.3, 7, 8, 12, 13 We performed a series of rigorous feeding experiments and directly demonstrated that blockade of MyD88 signalling significantly attenuates anorexia, providing a clear link between MyD88 signalling and PDAC cachexia‐associated anorexia. This result is similar to that found in a murine SC sarcoma model induced by methylcholanthrene,42, 43 suggesting that this feature of cachexia is sensitive to MyD88 signalling blockade in a wide variety of cancer subtypes. However, prior studies of acute and chronic inflammation in animals with MyD88 signalling blockade in various tissues generally did not address alterations in body composition, fatigue, or mortality, so the generality beyond PDAC in these features of cachexia remains unknown.42, 43, 44, 45 We also found that the preservation of fat mass was more variable and less substantial than that observed for lean mass. This may in part reflect the direct impact of MyD88‐dependent signalling in muscle.46, 47 However, our prior work demonstrated that muscle‐specific deletion of MyD88 did not attenuate muscle catabolism in response to acute inflammatory challenges.16 Among multiple central and systemic signalling pathways, the hypothalamic melanocortin system exerts a powerful role in the regulation of feeding behaviour, metabolic activity, energy homeostasis, and body composition and is known to be altered during acute and chronic inflammation and cachexia.18, 48, 49, 50 Indeed, mice and humans with defects in central melanocortin signalling have large increases in lean mass, including skeletal muscle, cardiac muscle, and bone.51, 52 Our results demonstrate that blockade of MyD88 signalling attenuates hypothalamic inflammation in PDAC cachexia, which we hypothesize would decrease hypothalamic melanocortin signalling and thereby lessen the anorexia and excessive loss of lean mass.
Patients with cancer cachexia frequently experience a dramatic decline in physical activity due to underlying disease and cancer treatment, and this is a primary determinant of quality of life.12, 33, 53, 54 Experimentally, fatigue‐related behavioural alteration can be demonstrated in the early stages of cancer cachexia through the use of electronic activity monitors, with the overall decrease in physical activity approaching 40% of baseline.55 With a murine actigraphy system, we demonstrate that decreased home cage movement (fatigue‐related behaviour) is an early and robust symptom of PDAC cachexia, as it was detectable on Day 6 post‐tumour implantation. In the absence of MyD88 signalling, fatigue was greatly delayed in onset and reduced in magnitude. The pathophysiology of fatigue is complex and multifactorial, as it is driven by immune/inflammatory, metabolic, neuroendocrine, and genetic determinants.56 We previously demonstrated that inflammation‐induced and cancer chemotherapy‐induced fatigue is associated with suppressed central orexin neuron activity17, 57. Therefore, it is possible that MyD88 signalling promotes early fatigue behaviour during the evolution of PDAC cachexia by suppressing central orexin signalling. As the disease evolves, it is likely that other mechanisms further exacerbate the loss of physical activity (e.g. muscle weakness and pain). Of course, objective observations in a mouse model do not account for the impact of psychological or social stress frequently experienced by cancer patients and also fail to account for indirect physiological impacts such as gut dysfunction and visceral pain, both of which are also common in pancreatic cancer patients.
Another novel finding of the present work is a marked improvement in survival with PDAC in MyD88 KO mice relative to WT mice. Our OT and IP models demonstrated that MyD88 signalling has a substantial impact on survival during PDAC cachexia, as MyD88 KO PDAC mice consistently had improved survival compared to WT PDAC mice. It is important to note that the improved survival occurred even though tumour size in MyD88 KO mice was larger than that in WT mice at the time of necropsy. The behavioural studies do suggest slower growth kinetics of tumours in MyD88 KO mice, but the survival result clearly demonstrates that MyD88 signalling blockade improves survival independent of effects on tumour burden. Our data regarding tumour growth contrast with the conclusions drawn from a prior study in which a MyD88 inhibitor was delivered systemically in a model of murine pancreatic cancer.58 In this case, the investigators found that peripheral delivery of a MyD88 inhibitor resulted in accelerated carcinogenesis and subsequent tumour growth in p48Cre;KrasG12D with caerulein‐induced pancreatitis. Therefore, it is possible that MyD88 blockade has differential effects on carcinogenesis vs. tumour growth and cancer progression.59 In a more recent study, a small molecule inhibitor of MyD88 prevented colitis‐associated colon cancer, suggesting that the outcomes of this strategy may also be dependent both on the organ (pancreas vs. colon) and on the properties of the MyD88 blocking drug (e.g. kinetics and tissue penetration).58, 60
In conclusion, our study recapitulates the key characteristics of pancreatic cancer cachexia using mouse OT and IP KPC tumour models and demonstrates that MyD88 signalling exacerbates systemic and CNS inflammation, promotes tumour growth, worsens cachexia, and accelerates mortality during pancreatic cancer progression and the cachexia development. MyD88 is a molecular nodal point that connects a wide range of upstream ligand‐receptor complexes (TLRs and IL‐1R family) to a broad array of downstream signalling‐mediators. It has a synergistic role in the integration and modulation of both systemic and CNS inflammation, both of which contribute to mortality in PDAC. Therefore, MyD88 is a logical target for developing novel therapies to ameliorate this condition. Numerous pharmacological agents are available or in development that inhibit MyD88‐dependent signalling pathways, including MyD88 dimerization inhibitors.60, 61 Future studies will be necessary to investigate the promising possibility that MyD88 inhibition during PDAC‐associated cachexia could provide an effective therapy for patients in this crucial area of need.
Funding
This work was supported by the NCI R01 CA184324 to D.L.M and the Brenden‐Colson Center for Pancreatic Care at OHSU.
Author contributions
X.Z. and D.L.M. conceived the study and designed the experiments. X.Z., K.G.B., and K.A.M. performed the animal experiments. X.Z., K.A.M., K.R.P., and T.K.M. assessed the blood, faeces, and tissues. X.Z., T.P.B., B.O., and D.L.M. analysed the data. X.Z. and D.L.M. wrote the manuscript. D.L.M. acquired founding and resources. All authors contributed to the revision and approved the final version of manuscript. The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017.62
Conflict of interest
None declared.
Supporting information
Figure S1 Orthotopic (OT) model of pancreatic cancer cachexia.
Figure. S2 Daily food intake and gross body weight, and terminal tumor mass, ascites and fecal triglyceride.
Figure. S3 IP KPC mice exhibit amelioration in behavioral phenotype when My88 signaling is blocked.
Figure. S4 Blockade of MyD88 signaling reduces lean mass loss in PDAC cachexia.
Figure. S5 IP KPC mice exhibit reduced lean mass loss and less fatigue when MyD88 signaling is absent.
Figure. S6 MyD88 signaling does not affect body temperature during development of PDAC cachexia.
Figure. S7 MyD88 signaling is essential for muscle catabolism in PDAC cachexia (muscle weights).
Figure. S8 MyD88 KO IP KPC mice exhibit reduced muscle catabolism.
Figure. S9 MyD88 signaling is essential for muscle catabolism in PDAC cachexia (gene expression).
Figure. S10 Pancreatic cancer‐induced systemic inflammation is diminished in MyD88 KO IP mice.
Figure. S11 Histopathology in orthotopically implanted WT vs. MyD88KO mice.
Figure. S12 Pancreatic cancer‐induced systemic and CNS inflammatory gene expression.
Figure. S13 MyD88 signaling contributes to high mortality in mice with IP PDAC.
Table S1 Pancreatic cancer induces neutrophil‐dominant leukocytosis and anemia in OT males.
Table S2 Pancreatic cancer induces neutrophil‐dominant leukocytosis and anemia in OT females.
Table S3 Pancreatic cancer induces neutrophil‐dominant leukocytosis and anemia in IP males.
Acknowledgements
We thank Dr Elizabeth Jaffee, of Johns Hopkins University, for kindly providing C57BL/6 KPC epithelial PDAC cells derived in her laboratory. We also thank Mason Norgard and Peter Levasseur for technical support with tissue collection and molecular analysis.
Zhu, X. , Burfeind, K. G. , Michaelis, K. A. , Braun, T. P. , Olson, B. , Pelz, K. R. , Morgan, T. K. , and Marks, D. L. (2019) MyD88 signalling is critical in the development of pancreatic cancer cachexia. Journal of Cachexia, Sarcopenia and Muscle, 10: 378–390. 10.1002/jcsm.12377.
References
- 1. Board, P.D.Q.A.T.E . Pancreatic Cancer Treatment (PDQ(R)): Health Professional Version, in PDQ Cancer Information Summaries. Bethesda (MD): National Cancer Institute (US); 2002. [Google Scholar]
- 2. Ansari D, Tingstedt B, Andersson B, Holmquist F, Sturesson C, Williamsson C, et al. Pancreatic cancer: yesterday, today and tomorrow. Future Oncol 2016;12:1929–1946. [DOI] [PubMed] [Google Scholar]
- 3. Bachmann J, Ketterer K, Marsch C, Fechtner K, Krakowski‐Roosen H, Büchler MW, et al. Pancreatic cancer related cachexia: influence on metabolism and correlation to weight loss and pulmonary function. BMC Cancer 2009;9:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larkin M. Thwarting the dwindling progression of cachexia. Lancet 1998;351:1336. [DOI] [PubMed] [Google Scholar]
- 5. Tisdale MJ. Biology of cachexia. J Natl Cancer Inst 1997;89:1763–1773. [DOI] [PubMed] [Google Scholar]
- 6. Argiles JM, Busquets S, Stemmler B, López‐Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer 2014;14:754–762. [DOI] [PubMed] [Google Scholar]
- 7. Bachmann J, Büchler MW, Friess H, Martignoni ME. Cachexia in patients with chronic pancreatitis and pancreatic cancer: impact on survival and outcome. Nutr Cancer 2013;65:827–833. [DOI] [PubMed] [Google Scholar]
- 8. Bachmann J, Heiligensetzer M, Krakowski‐Roosen H, Büchler MW, Friess H, Martignoni ME. Cachexia worsens prognosis in patients with resectable pancreatic cancer. J Gastrointest Surg 2008;12:1193–1201. [DOI] [PubMed] [Google Scholar]
- 9. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 10. Mueller TC, Burmeister MA, Bachmann J, Martignoni ME. Cachexia and pancreatic cancer: are there treatment options? World J Gastroenterol 2014;20:9361–9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pausch T, Hartwig W, Hinz U, Swolana T, Bundy BD, Hackert T, et al. Cachexia but not obesity worsens the postoperative outcome after pancreatoduodenectomy in pancreatic cancer. Surgery 2012;152:S81–S88. [DOI] [PubMed] [Google Scholar]
- 12. Tan CR, Jamil L, Yaffee P, Tuli R, Nissen N, Lo S, et al. Pancreatic cancer cachexia: a review of mechanisms and therapeutics. Front Physiol 2014;5:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Uomo G, Gallucci F, Rabitti PG. Anorexia‐cachexia syndrome in pancreatic cancer: recent development in research and management. JOP 2006;7:157–162. [PubMed] [Google Scholar]
- 14. Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogene 2016;5:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002;420:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Braun TP, Grossberg AJ, Krasnow SM, Levasseur PR, Szumowski M, Zhu XX, et al. Cancer‐ and endotoxin‐induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J 2013;27:3572–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grossberg AJ, Zhu X, Leinninger GM, Levasseur PR, Braun TP, Myers MG, et al. Inflammation‐induced lethargy is mediated by suppression of orexin neuron activity. J Neurosci 2011;31:11376–11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu X, Levasseur PR, Michaelis KA, Burfeind KG, Marks DL. A distinct brain pathway links viral RNA exposure to sickness behavior. Sci Rep 2016;6:29885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Braun TP, Zhu X, Szumowski M, Scott GD, Grossberg AJ, Levasseur PR, et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic‐pituitary‐adrenal axis. J Exp Med 2011;208:2449–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Matos‐Neto EM, Lima JD, de Pereira WO, Figuerêdo RG, Riccardi DM, Radloff K, et al. Systemic inflammation in cachexia: is tumor cytokine expression profile the culprit? Front Immunol 2015;6:629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zambirinis CP, Miller G. Cancer manipulation of host physiology: lessons from pancreatic cancer. Trends Mol Med 2017;23:465–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Argiles JM, Lopez‐Soriano FJ, Busquets S. Mechanisms and treatment of cancer cachexia. Nutr Metab Cardiovasc Dis 2013;23:S19–S24. [DOI] [PubMed] [Google Scholar]
- 23. Ebadi M, Mazurak VC. Potential biomarkers of fat loss as a feature of cancer cachexia. Mediators Inflamm 2015;2015:820934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tisdale MJ. Molecular pathways leading to cancer cachexia. Physiology (Bethesda) 2005;20:340–348. [DOI] [PubMed] [Google Scholar]
- 25. Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe 2008;3:352–363. [DOI] [PubMed] [Google Scholar]
- 26. Kawai T, Akira S. TLR signaling. Cell Death Differ 2006;13:816–825. [DOI] [PubMed] [Google Scholar]
- 27. Wang JQ, Jeelall YS, Ferguson LL, Horikawa K. Toll‐like receptors and cancer: MYD88 mutation and inflammation. Front Immunol 2014;5:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Michaelis KA, Zhu X, Burfeind KG, Krasnow SM, Levasseur PR, Morgan TK, et al. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle 2017;8:824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Foley K, Rucki AA, Xiao Q, Zhou D, Leubner A, Mo G, et al. Semaphorin 3D autocrine signaling mediates the metastatic role of annexin A2 in pancreatic cancer. Sci Signal 2015;8:ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Herreros‐Villanueva M, Hijona E, Cosme A, Bujanda L. Mouse models of pancreatic cancer. World J Gastroenterol 2012;18:1286–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grossberg AJ, Vichaya EG, Christian DL, Molkentine JM, Vermeer DW, Gross PS, et al. Tumor‐associated fatigue in cancer patients develops independently of IL1 signaling. Cancer Res 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ryan JL, Carroll JK, Ryan EP, Mustian KM, Fiscella K, Morrow GR. Mechanisms of cancer‐related fatigue. Oncologist 2007;12:22–34. [DOI] [PubMed] [Google Scholar]
- 33. Franc M, Michalski B, Kuczerawy I, Szuta J, Skrzypulec‐Plinta V. Cancer related fatigue syndrome in neoplastic diseases. Prz Menopauzalny 2014;13:352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsoli M, Robertson G. Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol Metab 2013;24:174–183. [DOI] [PubMed] [Google Scholar]
- 35. Tan BH, Fladvad T, Braun TP, Vigano A, Strasser F, Deans DC, et al. P‐selectin genotype is associated with the development of cancer cachexia. EMBO Mol Med 2012;4:462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Onesti JK, Guttridge DC. Inflammation based regulation of cancer cachexia. Biomed Res Int 2014;2014:168407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fearon KC, Baracos VE. Cachexia in pancreatic cancer: new treatment options and measures of success. HPB (Oxford) 2010;12:323–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang YJ, Lee CL, Wang Q, Zhou ZW, Yang F, Jin C, et al. Establishment of an orthotopic pancreatic cancer mouse model: cells suspended and injected in Matrigel. World J Gastroenterol 2014;20:9476–9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Qiu W, Su GH. Development of orthotopic pancreatic tumor mouse models. Methods Mol Biol 2013;980:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loi M, Di Paolo D, Becherini P, Zorzoli A, Perri P, Carosio R, et al. The use of the orthotopic model to validate antivascular therapies for cancer. Int J Dev Biol 2011;55:547–555. [DOI] [PubMed] [Google Scholar]
- 41. Delitto D, Judge SM, Delitto AE, Nosacka RL, Rocha FG, DiVita BB, et al. Human pancreatic cancer xenografts recapitulate key aspects of cancer cachexia. Oncotarget 2017;8:1177–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ruud J, Bäckhed F, Engblom D, Blomqvist A. Deletion of the gene encoding MyD88 protects from anorexia in a mouse tumor model. Brain Behav Immun 2010;24:554–557. [DOI] [PubMed] [Google Scholar]
- 43. Ruud J, Wilhelms DB, Nilsson A, Eskilsson A, Tang YJ, Ströhle P, et al. Inflammation‐ and tumor‐induced anorexia and weight loss require MyD88 in hematopoietic/myeloid cells but not in brain endothelial or neural cells. FASEB J 2013;27:1973–1980. [DOI] [PubMed] [Google Scholar]
- 44. Ogimoto K, Harris MK Jr, Wisse BE. MyD88 is a key mediator of anorexia, but not weight loss, induced by lipopolysaccharide and interleukin‐1 beta. Endocrinology 2006;147:4445–4453. [DOI] [PubMed] [Google Scholar]
- 45. Wisse BE, Ogimoto K, Tang J, Harris MK Jr, Raines EW, Schwartz MW. Evidence that lipopolysaccharide‐induced anorexia depends upon central, rather than peripheral, inflammatory signals. Endocrinology 2007;148:5230–5237. [DOI] [PubMed] [Google Scholar]
- 46. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas‐Ahner J, et al. NF‐kappaB‐mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 2013;123:4821–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hindi SM, Shin J, Gallot YS, Straughn AR, Simionescu‐Bankston A, Hindi L, et al. MyD88 promotes myoblast fusion in a cell‐autonomous manner. Nat Commun 2017;8:1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci 2005;8:571–578. [DOI] [PubMed] [Google Scholar]
- 49. Marks DL, Ling N, Cone RD. Role of the central melanocortin system in cachexia. Cancer Res 2001;61:1432–1438. [PubMed] [Google Scholar]
- 50. Scarlett JM, Zhu X, Enriori PJ, Bowe DD, Batra AK, Levasseur PR, et al. Regulation of agouti‐related protein messenger ribonucleic acid transcription and peptide secretion by acute and chronic inflammation. Endocrinology 2008;149:4837–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 2003;348:1085–1095. [DOI] [PubMed] [Google Scholar]
- 52. Braun TP, Orwoll B, Zhu X, Levasseur PR, Szumowski M, Nguyen ML, et al. Regulation of lean mass, bone mass, and exercise tolerance by the central melanocortin system. PLoS One 2012;7:e42183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brown DJ, McMillan DC, Milroy R. The correlation between fatigue, physical function, the systemic inflammatory response, and psychological distress in patients with advanced lung cancer. Cancer 2005;103:377–382. [DOI] [PubMed] [Google Scholar]
- 54. Brown JC, Harhay MO, Harhay MN. Self‐reported major mobility disability and mortality among cancer survivors. J Geriatr Oncol 2018;9:459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dahele M, Skipworth RJ, Wall L, Voss A, Preston T, Fearon KC. Objective physical activity and self‐reported quality of life in patients receiving palliative chemotherapy. J Pain Symptom Manage 2007;33:676–685. [DOI] [PubMed] [Google Scholar]
- 56. Saligan LN, Olson K, Filler K, Larkin D, Cramp F, Sriram Y, et al. The biology of cancer‐related fatigue: a review of the literature. Support Care Cancer 2015;23:2461–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Weymann KB, Wood LJ, Zhu X, Marks DL. A role for orexin in cytotoxic chemotherapy‐induced fatigue. Brain Behav Immun 2014;37:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med 2012;209:1671–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zambirinis CP, Miller G. Signaling via MYD88 in the pancreatic tumor microenvironment: a double‐edged sword. Oncoimmunology 2013;2:e22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xie L, Jiang FC, Zhang LM, He WT, Liu JH, Li MQ, et al. Targeting of MyD88 homodimerization by novel synthetic inhibitor TJ‐M2010‐5 in preventing colitis‐associated colorectal cancer. J Natl Cancer Inst 2016;108:djv364. [DOI] [PubMed] [Google Scholar]
- 61. Olson MA, Lee MS, Kissner TL, Alam S, Waugh DS, Saikh KU. Discovery of small molecule inhibitors of MyD88‐dependent signaling pathways using a computational screen. Sci Rep 2015;5:14246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Orthotopic (OT) model of pancreatic cancer cachexia.
Figure. S2 Daily food intake and gross body weight, and terminal tumor mass, ascites and fecal triglyceride.
Figure. S3 IP KPC mice exhibit amelioration in behavioral phenotype when My88 signaling is blocked.
Figure. S4 Blockade of MyD88 signaling reduces lean mass loss in PDAC cachexia.
Figure. S5 IP KPC mice exhibit reduced lean mass loss and less fatigue when MyD88 signaling is absent.
Figure. S6 MyD88 signaling does not affect body temperature during development of PDAC cachexia.
Figure. S7 MyD88 signaling is essential for muscle catabolism in PDAC cachexia (muscle weights).
Figure. S8 MyD88 KO IP KPC mice exhibit reduced muscle catabolism.
Figure. S9 MyD88 signaling is essential for muscle catabolism in PDAC cachexia (gene expression).
Figure. S10 Pancreatic cancer‐induced systemic inflammation is diminished in MyD88 KO IP mice.
Figure. S11 Histopathology in orthotopically implanted WT vs. MyD88KO mice.
Figure. S12 Pancreatic cancer‐induced systemic and CNS inflammatory gene expression.
Figure. S13 MyD88 signaling contributes to high mortality in mice with IP PDAC.
Table S1 Pancreatic cancer induces neutrophil‐dominant leukocytosis and anemia in OT males.
Table S2 Pancreatic cancer induces neutrophil‐dominant leukocytosis and anemia in OT females.
Table S3 Pancreatic cancer induces neutrophil‐dominant leukocytosis and anemia in IP males.