

Abstract
Background
Recent studies indicate important roles for long noncoding RNAs (lncRNAs) in the regulation of gene expression by acting as competing endogenous RNAs (ceRNAs). However, the specific role of lncRNAs in skeletal muscle atrophy is still unclear. Our study aimed to identify the function of lncRNAs that control skeletal muscle myogenesis and atrophy.
Methods
RNA sequencing was performed to identify the skeletal muscle transcriptome (lncRNA and messenger RNA) between hypertrophic broilers and leaner broilers. To study the ‘sponge’ function of lncRNA, we constructed a lncRNA‐microRNA (miRNA)‐gene interaction network by integrated our previous submitted skeletal muscle miRNA sequencing data. The primary myoblast cells and animal model were used to assess the biological function of the lncIRS1 in vitro or in vivo.
Results
We constructed a myogenesis‐associated lncRNA‐miRNA‐gene network and identified a novel ceRNA lncRNA named lncIRS1 that is specifically enriched in skeletal muscle. LncIRS1 could regulate myoblast proliferation and differentiation in vitro, and muscle mass and mean muscle fibre in vivo. LncIRS1 increases gradually during myogenic differentiation. Mechanistically, lncIRS1 acts as a ceRNA for miR‐15a, miR‐15b‐5p, and miR‐15c‐5p to regulate IRS1 expression, which is the downstream of the IGF1 receptor. Overexpression of lncIRS1 not only increased the protein abundance of IRS1 but also promoted phosphorylation level of AKT (p‐AKT) a central component of insulin‐like growth factor‐1 pathway. Furthermore, lncIRS1 regulates the expression of atrophy‐related genes and can rescue muscle atrophy.
Conclusions
The newly identified lncIRS1 acts as a sponge for miR‐15 family to regulate IRS1 expression, resulting in promoting skeletal muscle myogenesis and controlling atrophy.
Keywords: Atrophy, ceRNA, IRS1, Myogenesis, lncRNA‐miRNA‐gene network
Abbreviations
- miRNA
microRNA
- lncRNAs
long noncoding RNAs
- CCK‐8
cell counting kit‐8
- NC
negative control
- ceRNA
competing endogenous RNA
- MREs
miRNA response elements
- IPA
ingenuity pathway analysis
- EdU
5‐ethynyl‐2′‐deoxyuridine
Introduction
A decrease in adult muscle mass and fibre size is called ‘atrophy’ and is characterized by enhanced protein degradation.1, 2, 3 An increase in muscle mass, called ‘hypertrophy’, is associated with increased protein synthesis.3 Therefore, the maintenance of muscle mass is controlled through a balance between protein synthesis and protein degradation pathways.1 Skeletal muscle atrophy occurs in a variety of conditions, such as cancer cachexia, prolonged periods of muscle inactivity, and aging itself. Unfortunately, there are no effective pharmacological treatments for this devastating disease.
Skeletal muscle myogenesis and hypertrophy is a multistep process that comprises four major stages: in the initial stage, muscle precursor cells differentiate from the somite; in the second stage, myogenic precursor proliferation and differentiation gives rise to myoblasts; in the third stage, myoblast differentiation gives rise to myotubes; and finally, myofibers are formed from the myotubes.4 These complex cellular and development processes depend on the precise spatiotemporal expression of regulatory factors. Insulin receptor substrate 1 (IRS1) is a signalling adapter protein, essential for skeletal muscle growth and protein homeostasis. Mice lacking IRS1 have reduced growth rate by 40–70% compared to controls.5, 6 IRS1 plays a key role in transmitting signals from the insulin and insulin‐like growth factor‐1 (IGF‐1) receptors to intracellular signalling pathway PI3K/AKT. IGF1‐PI3K/AKT pathway is a key intracellular signalling mechanism controlling muscle hypertrophy.7 In addition, evidence suggests that in complex organism development, RNA contains a hidden layer of regulatory information. It not only functions as a messenger between DNA and the protein but also plays a role in the modulation of gene expression.8 Long noncoding RNAs (lncRNAs), a novel class of regulatory RNAs, commonly defined as transcribed RNAs with sizes ranging from 200 bp to >100 kb, are involved in numerous important biological processes.9, 10 For example, SRA, a lncRNA, was demonstrated to facilitate myoblast differentiation by controlling the transcriptional activity of MyoD.11 Salmena12 previously presented a competing endogenous RNA (ceRNA) hypothesis that was supported by many evidences.13, 14 LncRNAs can function as ceRNAs protect messenger RNAs (mRNAs) by acting as molecular sponges for microRNAs (miRNAs) that specifically inhibit the target mRNAs (Figure S1). For example, Linc‐MD1 acts as a ceRNA by sponging miR‐133 and miR‐135 to regulate the expression of MAML1 and MEF2C, which are transcription factors that activate late‐differentiation muscle genes.15 In addition, lncRNA H19, which is highly expressed in the developing embryo and in adult muscle, functions as a molecular sponge for the let‐7 family and thereby regulates muscle differentiation.16 Although functions of these lncRNAs have been partially characterized, most of their roles for myogenesis and anti‐atrophy are still poorly understand. Therefore, the aim of our study is to explore the role of lncRNAs in regulating of skeletal muscle myogenesis and anti‐atrophy.
In this study, we first focused on skeletal muscle myogenesis associated‐lncRNAs and constructed a ceRNA network (lncRNA‐miRNA‐gene network) in silico. We investigated the function of a novel lncRNA named lncIRS1 in skeletal muscle myogenesis. LncIRS1 was highly expressed in skeletal muscle and promoted proliferation and differentiation of myoblast. Mechanistic investigations showed that lncIRS1 functioned as a ceRNA by sponging miR‐15a, miR‐15b‐5p, and miR‐15c‐5p, which then activated IRS1 and regulated the IGF‐1 signalling pathway, a critical pathway of skeletal muscle myogenesis. Moreover, the effect of lncIRS1 overexpression and knockdown on skeletal muscle atrophy induced by dexamethasone was investigated. Our study provides some clues regarding lncIRS1 mechanism of regulation in skeletal muscle myogenesis and atrophy.
Material and methods
Animals
Seven‐week‐old of hypertrophic (WRR) and leaner broilers (XH) were used. All human and animal studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
RNA sequencing (RNA‐Seq)
Breast muscle tissues from WRR and XH broilers were used for RNA‐seq. Total RNA was isolated using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. RNA quantity and quality were evaluated on an Agilent 2100 Bioanalyzer (Agilent technologies, Waldbronn, Germany), and RNA integrity was further examined using agarose gel electrophoresis. Ribosomal RNA (rRNA) was removed from the total RNA using Epicentre Ribo‐ZeroTM rRNA Removal Kit (Epicentre, Madison, Wisconsin, USA) following the manufacturer's instructions. Subsequently, the RNA from four broilers within each group was mixed in equal amounts to construct a pooled sample for each group. High‐throughput RNA‐seq was performed on the Illumina Hiseq 2000 platform (Illumina, San Diego, CA, USA). The raw Illumina sequencing reads were cleaned by removing empty reads, adapter sequences, reads with over 10% N sequence, and low‐quality reads in which the number of bases with a quality value Q ≤ 10 was greater than 50%. In addition, rRNA reads were identified by blasting against the rRNA database (http://www.arb‐silva.de/) using SOAP software and removed from the dataset. The filtered reads were mapped to the chicken reference genome (ftp://ftp.ensembl.org/pub/release‐73/fasta/gallus_gallus/dna/) using Tophat2. The mapped reads were assembled and transcripts were constructed using Cufflinks 2.0.2. The Ensembl, NCBI RefGene, and UCSC databases were chosen as annotation references for lncRNA analyses. RNA length ≥ 200 nt, CPC score ≤ 0, CPAT probability ≤ 0.364, and phyloCSF score ≤ −20 were used to evaluate the coding potential of transcripts. The RefSeq and Ensembl databases were used for gene analysis. The expression levels of the transcripts are expressed as fragments per kilobase of transcript per million mapped reads values. Differentially expressed transcripts for were identified using Cuffdiff, using q‐value <0.001 and |(fold change)| ≥ 2 as the cut‐off. The sequencing data obtained from the RNA‐Seq were released to the GEO database under accession number GSE58755.
Construction of lncRNA‐miRNA‐gene interaction network
Previous study proposed a ceRNA hypothesis that protein‐coding RNAs and lncRNAs containing the common one or more miRNA response elements (MREs) can compete for binding to miRNAs and regulate each other's expression.12 To study the ‘sponge’ function of lncRNA, we constructed a lncRNA‐miRNA‐gene interaction network by integrated our previous submitted skeletal muscle miRNA sequencing data (GSE62971). The lncRNA‐miRNA‐gene crosstalk network construction involved the following three components: (i) interactions between miRNAs and genes, (ii) interactions between lncRNAs and miRNAs, and (iii) interaction between genes and genes. Firstly, the potential target genes of differentially expressed miRNAs were predicted with TargetScan; miRNAs repress their target genes expression at post‐transcriptional level. Therefore, the predicted target genes that have an opposition expression patterns of their corresponding miRNAs were selected as candidate targets for differentially expressed miRNAs. Secondly, RNAhybrid, a tool for finding the minimum free energy hybridization of a long and a short RNA, was used to predict the target lncRNA of differentially expressed miRNA. As miRNA can repress the expression of mRNA, it also can inhibit the expression of its target lncRNAs. Therefore, the predicted target lncRNAs that have an opposition expression patterns of their corresponding miRNAs were selected as candidate target lncRNAs for differentially expressed miRNAs. Thirdly, ingenuity pathway analysis (IPA), a database of known and predicted protein interactions, was used to construct gene–gene interactions. Finally, lncRNA‐miRNA‐mRNA interaction network was constructed using Cytoscape. The Molecule Annotation System 3.0 (http://bioinfo.capitalbio.com/mas3) was used to identify GO terms and pathways enriched in genes belonging to lncRNA‐miRNA‐gene interaction network.
In situ hybridization
Whole‐mount in situ hybridization of chick embryos was performed according to a standard in situ hybridization protocol.17 Digoxigenin‐labelled probes were synthesized to detect IRS1 and lncIRS1. IRS1: Forward: ACCTGGACTTGGTGAAGGATTGC; Reverse: AATTAACCCTCACTAAAGGGA GACCCATTTACAGAGGAAGAGGAGGAG. LncIRS1: Forward: GTATTGGTCA CTCTGGGTCAG; Reverse: AATTAACCCTCACTAAAGGGAGATCTTCACTTT CTCCTTGGGTA. The whole embryos at stage HH10 were fixed with 4% paraformaldehyde overnight at 4°C, dehydrated in a graded series of methanol, and stored at −20°C (overnight). Next, the embryos were hybridized with IRS1 or lncIRS1 digoxigenin‐labelled probe overnight at 65°C. After hybridization, the bound RNA probe was visualized by incubation with alkaline phosphatase‐conjugated anti‐digoxigenin antibodies, and the colour was developed in NBT/BCIP (Roche, Basel, Switzerland). The whole‐mount stained embryos were photographed under a stereomicroscope (Olympus MVX10).
Cell culture, treatment, and transfection
Chicken embryonic fibroblast cell line (DF‐1 cells) were cultivated with Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY, USA) supplemented with 10% foetal bovine serum (Gibco) and 0.2% penicillin/streptomycin (Gibco) in a humidified atmosphere with 5% (v/v) CO2 at 37°C.
Chicken primary myoblasts were isolated from E11 chicken leg muscles as previously described.18 Chicken primary myoblasts were cultured with growth medium consisting of RPMI‐1640 medium (Gibco), 20% foetal bovine serum, and 0.2% penicillin/streptomycin. After myoblasts achieving 100% cell confluence, the growth medium was then removed and replaced with differentiation medium (RPMI‐1640 medium, 2% horse serum and 0.2% penicillin/streptomycin). For cell treatments, myotubes were treated with vehicle and 10 μM dexamethasone (Sigma‐Aldrich, St. Louis, Mo, USA) in RPMI‐1640 for 24 h. All transient transfections were performed with Lipofectamine 3000 reagent (Invitrogen, USA) according to manufacturer's direction.
RNA extraction, complementary DNA (cDNA) synthesis, and quantitative real‐time PCR (qRT‐PCR)
Total RNA was extracted from tissues or cells using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) as recommended by the supplier. A PARIS Kit (Ambion, Life Technologies, USA) was used to harvest the cytoplasmic and nuclear cell lysates of myoblast, following the manufacturer's protocol; cDNA synthesis for RNA (mRNA and lncRNA) was carried out using the PrimeScript RT Reagent Kit with gDNA Eraser (Perfect Real Time) (TaKaRa, Otsu, Japan). For miRNA, bulge‐Loop™ miRNA qRT‐PCR primers specific for miR‐15 family and U6 were designed by RiboBio (RiboBio, Guangzhou, China), and ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan) was used to synthesize cDNA. Real‐time quantitative PCR (qPCR) reactions were performed on a Bio‐Rad CFX96 Real‐Time Detection System using iTaq Universal SYBR Green Supermix Kit (Bio‐Rad Laboratories Inc., USA). Data analyses were performed using the 2−△△Ct method as described previously.19 Chicken β‐actin and U6 were used as internal controls.
RNA oligonucleotides and plasmids construction
The gga‐miR‐15 family mimics (miR‐15a, miR‐15b‐5p, miR‐15c‐5p, miR‐16‐5p, and miR‐16c‐5p), mimic negative control, siRNA target against the IRS1 gene (si‐IRS1), siRNA target against the lncIRS1 gene (si‐lncIRS1), and siRNA nonspecific control were designed and synthesized by RiboBio (Guangzhou, China).
The IRS1 overexpression construct was generated by amplifying the IRS1 coding sequence, which subsequently integrated into the HindIII/KpnI restriction sites of the pcDNA3.1 overexpression plasmid (named pcDNA3.1‐IRS1). The full‐length sequence of lncIRS1 was subcloned into the BamHI/XhoI restriction sites of the pcDNA3.1 overexpression plasmid (named pcDNA3.1‐lncIRS1). The empty pcDNA3.1 vector was used as control plasmid.
MiR‐15 family binding sites in IRS1 3'UTR or lncIRS1 (three binding sites: Site 1 at position 2651–2657 of lncIRS1; Site 2 at position 3438–3444 of lncIRS1; and Site 3 at position 3775–3781 of lncIRS1) were amplified by PCR using a cDNA template synthesized from total RNA. Then, the PCR products were subcloned into XhoI/XbaI restriction sites in the pmirGLO dual‐luciferase reporter vector to generate the pmirGLO‐IRS1 reporter, pmirGLO‐site 1 of lncIRS1 reporter, pmirGLO‐site 2 of lncIRS1 reporter, and pmirGLO‐site 3 of lncIRS1 reporter.
For identification of the coding ability of lncIRS1, seven possible Open Reading Frames (ORFs) of lncIRS1 were amplified and cloned into pSDS‐20218 vector (SiDanSai, Shanghai, China). Chicken β‐actin gene was subcloned into pSDS‐20218 vector as positive control.
5′ and 3′ rapid amplification of cDNA ends (RACE)
A SMARTer RACE cDNA Amplification Kit (Clontech, Osaka, Japan) was used to obtain the full‐length sequences of lncIRS1 and IRS1, following the manufacturer's instructions. Nested‐PCR reactions were performed. The products of the RACE PCR were cloned into the pJET 1.2/blunt cloning vector (CloneJET PCR Cloning Kit; Fermentas, Glen Burnie, MD, USA) and sequenced by Sangon Biotech (Shanghai, China).
Dual‐luciferase reporter assay and RNA immunoprecipitation
DF‐1 cells were seeded in 96‐well plates 1 day before transfection and then co‐transfected with (I) pmirGLO‐IRS1 reporter, (II) pmirGLO‐site 1 of lncIRS1 reporter, (III) pmirGLO‐site 2 of lncIRS1 reporter, and (IV) pmirGLO‐site 3 of lncIRS1 reporter, and miRNA mimics or mimic NC using Lipofectamine 3000 reagent. After 48 h, luciferase activity analysis was performed using a Fluorescence/Multi‐Detection Microplate Reader (BioTek, Winooski, VT, USA) and a Dual‐GLO Luciferase Assay System Kit (Promega, USA). The firefly luciferase activities were normalized to Renilla luminescence in each cell‐well.
RNA immunoprecipitation used the Magna RIP RNA‐Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA, USA) and the anti‐Argonaute2 (AGO2) antibody (Abcam, Cambridge, UK) according to the manufacture's protocol. After the antibody was recovered by protein A + G beads, qRT‐PCR was performed to detect IRS1, lncIRS1, and miR‐15 family in the precipitates.
Western blotting
Myoblast cells were seeded in six‐well plates and transfected with overexpression plasmid, siRNA, or miRNA mimics for 48 h. Cells and muscle tissue were harvested, washed with 1× phosphate buffered saline (PBS) and lysed in RIPA lysis buffer. Immunoblotting was performed using standard procedures and various antibodies. The primary antibodies used were anti‐IRS1 (1:1000; catalogue no. ab5603, Abcam, Cambridge, UK), anti‐phospho‐AKT (1:1000; catalogue no. #4040, Cell Signaling Technology, Danvers, MA, USA), anti‐AKT (1:1000; catalogue no. #9272, Cell Signaling Technology), anti‐MyHC (1:50; catalogue no. B103, Developmental Studies Hybridoma Bank, Iowa City, Iowa, USA), anti‐MyoG (1:1000; catalogue no. orb6492, Biorbyt, Cambridge, UK), anti‐Foxo1 (1:1000; catalogue no. 82358, Thermo Fisher Scientific, Meridian Road, IL, USA), anti‐phospho‐Foxo1 and anti‐phospho‐Foxo4 (1:1000; catalogue no. #9461, Cell Signaling Technology), anti‐Foxo3 (1:1000; catalogue no. NBP2‐24579, Novus Biologicals, Littleton, CO, USA), anti‐phospho‐Foxo3 (1:1000; catalogue no. bs‐3140R, Bioss, China), anti‐Foxo4 (1:1000; catalogue no. bs‐2766R, Bioss), anti‐Fbx32/Atrogin‐1 (1:1000; catalogue no. ab74023, Abcam), and anti‐GAPDH (1:10 000; catalogue no. MB001H, Bioworld, St Louis Park, MN, USA). The goat anti‐mouse IgG (H & L)‐HRP (1:10 000; catalogue no. BS12478, Bioworld Technology, Inc.), and goat anti‐rabbit IgG (H & L)‐HRP (1:10 000; catalogue no. BS13278, Bioworld Technology, Inc.) were used as a secondary antibody.
Flow cytometric analysis
For the flow cytometry analysis of the cell cycle, myoblast cells were seeded in 12‐well plates. When the cells grew to a density of 50% confluence, they were transfected with overexpression plasmid, siRNA, or miRNA mimics. After transfection for 48 h, the cells were collected and fixed overnight in 70% ethanol at 4°C. Subsequently, the fixed cells were stained with 50 μg/mL propidium iodide solution (Sigma Life Science, St. Louis, MO, USA) containing 10 μg/mL RNase A (Takara, Japan) and 0.2% (v/v) Triton X‐100 (Sigma Life Science, St. Louis, MO, USA) and then incubated at 37°C in the dark for 30 min. Flow cytometry analysis was performed on a BD Accuri C6 flow cytometer (BD Biosciences, USA), and data were processed using FlowJo7.6 software.
5‐Ethynyl‐2′‐deoxyuridine (EdU) assay
Myoblast cells were seeded in 12‐well plates. When the cells grew to a density of 50% confluence, they were transfected with overexpression plasmid, siRNA, or miRNA mimics. After transfection for 48 h, myoblasts were exposed to 50 μM EdU (RiboBio, China) for 2 h at 37°C. Subsequently, the cells were fixed in 4% paraformaldehyde for 30 min, neutralized using 2 mg/mL glycine solution, and then permeabilized by adding 0.5% Triton X‐100. A solution containing EdU (Apollo Reaction Cocktail; RiboBio, China) was added and the cells were incubated at room temperature for 30 min. The nuclear stain Hoechst 33342 was then added, and incubation was continued for another 30 min. A fluorescence microscope (DMi8; Leica, German) was used to capture three randomly selected fields to visualize the number of EdU‐stained cells.
Cell counting kit‐8 (CCK‐8) assay
Primary myoblasts were seeded in 96‐well plates and cultured in growth medium. After being transfected, cell proliferation was monitored using a TransDetect CCK (TransGen Biotech, Beijing, China) according to the manufacturer's protocol. Absorbance was measured using a Model 680 Microplate Reader (Bio‐Rad, Hercules, California, USA) by optical density at a wavelength of 450 nm.
Lentiviral vector construction, production, and infection
LncIRS1 knockdown
Lentiviral vectors were constructed to produce lentiviruses expressing short hairpin RNA (shRNA) against lncIRS1. Three shRNA sequences were designed by Shanghai Hanbio Biotechnology Co., Ltd (shRNA‐1: 5′‐GGTCTCATGTACAACGTAT‐3′, shRNA‐2: 5′‐GCAACTTAAAGGAAGGCAT‐3′, shRNA‐3: 5′‐GCACAACCCTAACAGAAAT‐3′). These shRNA sequences were subcloned into the pLKO.1 vector between Agel/EcoRI restriction enzyme sites, and high titre lentiviruses were produced. Briefly, the expression vectors were co‐transfected with packaging plasmid psPAX2 (Addgene, USA) and envelope plasmid pMD2.G (Addgene, USA) into 293T cells. Infectious particles were harvested at 48 and 72 h after transfection, filtered through 0.45‐m‐pore cellulose acetate filters, concentrated by ultracentrifugation, redissolved in sterile HBSS, aliquoted, and stored at −80°C.
LncIRS1 overexpression
For the construction of lncIRS1‐overexpression lentiviral vector, the full‐length of lncIRS1 produced from breast muscle cDNA using PCR was subcloned into the BamHI/EcoRI restriction enzyme sites of the lentiviral expression vector (pWPXL). Lentivirus was produced as described earlier.
Lentiviral intramuscular injections
Injections were performed as previously described by Luo20 with some modifications. Briefly, 1‐day‐old chicks were received three intramuscular doses (single dose injection started at Days 1, 3, and 5) of lentivirus into the breast muscle at dosage of 1 × 108 IU/mL.
Sampling and in vivo validation of lentiviral vectors
Breast muscle samples were taken from pLKO.1‐lncIRS1 (n = 15), pLKO.1‐control (n = 15), pWPXL‐lncIRS1 (n = 15), and pWPXL control (n = 15) group injected chickens 9 days after the initial injection. After removing the breast muscle, 4‐mm slices were taken based on designated anatomical markers using standard razor blades. Protein and RNA extraction were performed for Western blot and qPCR analysis.
Histology image analysis
Breast muscle tissues of chicken were obtained from shRNA: pLKO.1‐lncIRS1, pLKO‐control; and overexpression: pWPXL‐lncIRS1, pWPXL‐control infected muscle (n = 15/case). On Day 9 after initial dose, the tissues were quickly harvested from all groups and fixed in 10% formalin (10% formalin: 90% PBS). Fixed tissues were paraffin embedded, sectioned and stained with haematoxylin and eosin, and then subjected to image analysis.
Immunofluorescence analysis
The immunofluorescence was performed in myoblast cells cultured in 24‐well plates. After transfection for 48 h, cells were fixed in 4% formaldehyde for 20 min and then washed three times with PBS (5 min each). Subsequently, the cells were permeabilized with 0.1% Triton X‐100 for 15 min and blocked with goat serum for 1 h. After overnight incubation at 4°C with anti‐MyHC antibody (1:50; B103, Developmental Studies Hybridoma Bank, Iowa City, Iowa, USA), the cells were treated with Dylight 594‐conjugated AffiniPure Goat Anti‐Mouse IgG (H + L) (1:100; BS10027, Bioworld, USA) and incubated in dark for 1 h. The cell nuclei were visualized using DAPI staining (Beyotime, Jiangsu, China). Images were obtained with a fluorescence microscope (DMi8; Leica, German). The area of cells labelled with anti‐MyHC was measured by using ImageJ software (National Institutes of Health), and the total myotube area was calculated as a percentage of the total image area covered by myotubes.
Statistical analysis
Each experiment was performed in triplicate. The data are presented as the mean ± standard error of the mean of each set of three independent experiments. Where applicable, the statistical significance of the data was tested using one‐sample or paired t‐tests. The types of tests and the P‐values, when applicable, are indicated in the figure legends.
Results
LncRNA‐miRNA‐gene interaction network
There are 239 differentially expressed lncRNAs (DE‐lncRNAs) (Figure 1A, Table S1) and 763 differentially expressed genes (Figure 1B, Table S2) were identified with a Benjamini q‐value <0.001 and |(fold change)| ≥ 2 as the cut‐off. Our previous study has presented the miRNA expression profile between hypertrophic and leaner broilers,21 and 101 differentially expressed miRNAs were identified with a Benjamini q‐value of 0.001 as the cut‐off (Figure 1C, Table S3). Protein‐coding RNAs (genes) and lncRNAs, which share the common MREs, can both compete for binding to miRNAs and regulate each other. In this study, we constructed a putative lncRNA‐miRNA‐gene crosstalk network involved in skeletal muscle myogenesis. The lncRNA‐miRNA‐gene crosstalk network construction involved the following three components: interactions between miRNAs and genes, interactions between lncRNAs and miRNAs, and interaction between genes and genes. Firstly, miRNA‐gene interaction network contains 378 negatively correlated pairs of miRNA‐gene, involving 79 miRNAs and 92 genes (Figure S2, Table S4). Secondly, the target lncRNAs of the 79 differentially expressed miRNAs from the miRNA‐gene network were predicted by RNAhybrid. The lncRNA‐miRNA interaction network contains 483 negatively correlated pairs of lncRNA‐miRNA, involving 173 lncRNAs and 68 miRNAs (Figure S3, Table S5). Thirdly, to narrow the field of the key genes involved in skeletal muscle myogenesis, the genes of miRNA‐gene network were used to construct gene–gene interactions by IPA online software. In IPA analysis, a total of four gene networks were identified (Figure S4). Importantly, one of the four gene–gene networks was involved in cellular compromise, organismal injury and abnormalities, and skeletal and muscular disorders (Figure S4c; Table S6), and 16 differentially expressed genes from this network were used for lncRNA‐miRNA‐gene network construction. Finally, miRNA‐gene interactions, lncRNA‐miRNA interactions, and gene–gene interactions were integrated to construct possible lncRNA‐miRNA‐gene network by Cytoscape. The lncRNA‐miRNA‐gene interaction network contains 733 pairs of lncRNA‐miRNA‐gene, involving 130 lncRNAs, 31 miRNAs, and 15 genes (Figure 1D). MiR‐15 family, including miR‐15a, miR‐15b‐5p, miR‐15c‐5p, miR‐16‐5p, and miR‐16c‐5p are the core component of this interaction network with a large number of genes and lncRNAs connected (Figure 1D). GO and KEGG pathway analyses of the identified genes from the lncRNA‐miRNA‐gene network revealed that these genes were enriched in transcription regulation‐related processes, such as transcription factor activity and regulation of transcription, DNA dependent (Figure S5a). Notably, according to the KEGG analysis, IRS1 gene from lncRNA‐miRNA‐gene network was enriched in skeletal muscle myogenesis related pathway, such as growth hormone signalling pathway, insulin signalling pathway, and IGF‐1 signalling pathway (Figures S5b and S5c). IGF‐1 signalling pathway plays a major role in the regulation of skeletal muscle myogenesis (Figure S5d). IRS1 gene is thought to be down‐stream of the IGF1 receptor (Figure S5d), and IRS1 null mice have much reduced growth (30–60% of control).22, 23 Interestingly, IRS1 is the predicted target gene of miR‐15 family which is the core component miRNAs in the lncRNA‐miRNA‐mRNA network.
Figure 1.
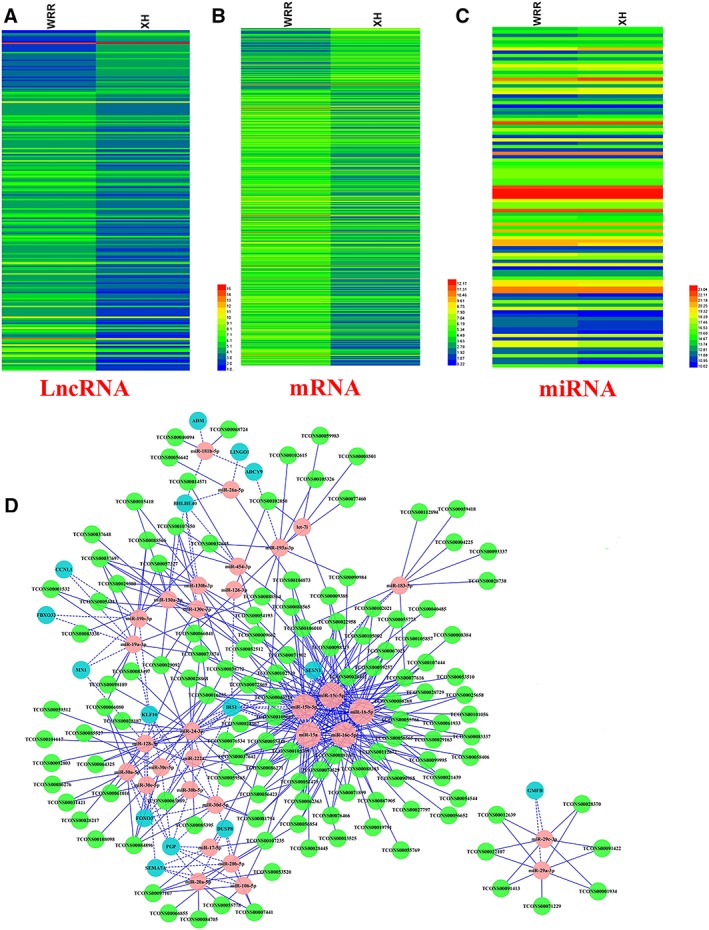
MiR‐15 family is the core of lncRNA‐miRNA‐gene interaction network. (A) Heat map of 239 differentially expressed lncRNAs (DE‐lncRNAs) between hypertrophic and leaner broilers, with rows representing lncRNAs and columns representing breast muscle tissues from WRR and XH (B) Heat map of 763 differentially expressed genes (DEGs) between hypertrophic and leaner broilers, with rows representing mRNAs and columns representing breast muscle tissues from WRR and XH. (C) Heat map of 101 differentially expressed miRNAs (DE‐miRNAs) between hypertrophic and leaner broilers, with rows representing miRNAs and columns representing breast muscle tissues from WRR and XH. (D) LncRNA‐miRNA‐gene interaction network consists of 130 lncRNAs (green circles), 31 miRNAs (pink circles), and 15 genes (blue circles). MiR‐15 family, including miR‐15a, miR‐15b‐5p, miR‐15c‐5p, miR‐16‐5p, and miR‐16c‐5p are the core component of this interaction network with a large number of genes and lncRNAs connected. Dashed lines represent the interactions between differentially expressed miRNAs and theirs corresponding target genes. Solid lines represent the interactions between differentially expressed miRNAs and theirs corresponding target lncRNAs. lncRNA, long noncoding RNA; miRNA, microRNA; mRNA, messenger RNA.
LncIRS1 is up‐regulated in hypertrophic broilers
RNA‐seq revealed that 52 lncRNAs were down‐regulated and 187 were up‐regulated (Table S1) in hypertrophic and leaner broilers. We identified an lncRNA (TCONS_00086268, named lncIRS1) which is upregulated in hypertrophic broiler (Figure 2A and 2B). The 5′ and 3′ ends of lncIRS1 were determined by RACE system (Figure 2C and 2D, Table S7). Predictions of the coding identify of lncIRS1 by the Coding Potential Calculator suggested a low coding potential and low evolutionary conservation consistent with a non‐coding RNA (Figure 2E).24 In order to verify this prediction, we analysed the coding ability of seven potential ORFs of lncIRS1. Western blot analysis suggestive that lncIRS1 is an lncRNA with no protein‐encoding potential (Figure 2F). The NCBI BLAST indicated that the lncIRS1 was 4623 bp long, located on Chromosome 5 and spanned from 8 080 605 to 8 085 228. We further investigated the subcellular localization of lncIRS1, and the RT‐PCR result confirmed that it is an RNA molecule present in the cytoplasm and nucleus (Figure 2G). Moreover, we identified the 5′ and 3′ ends of IRS1 by RACE (Figure 2H and 2I, Table S7).
Figure 2.
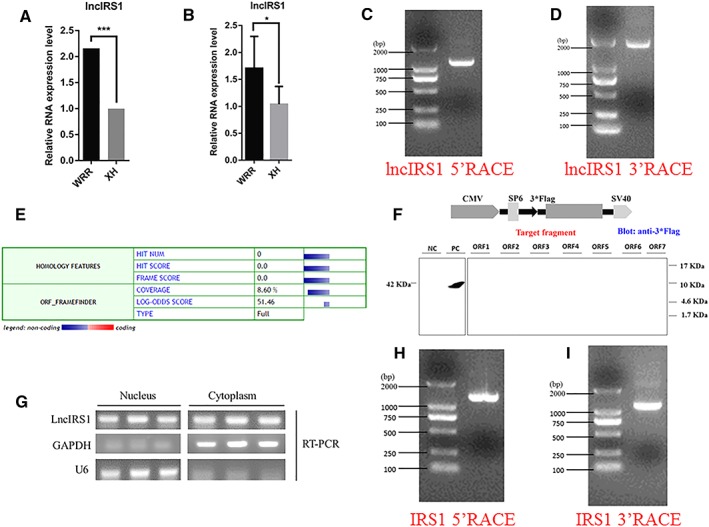
Identification of lncIRS1. (A) RNA sequencing found that lncIRS1 expression was up‐regulated in hypertrophic broilers compared with leaner broilers (q value=0.000652) and this expression pattern was confirmed by qPCR. The data are shown as the mean ± SEM with *P < 0.05 (B). (C) Results of lncIRS1 5'RACE. 5'RACE product, 1169 bp. (D) Result of lncIRS1 3'RACE. 3'RACE product, 1909 bp. (E) The coding ability predication of lncIRS1. Analysis was obtained from the coding potential calculator (http://cpc.cbi.pku.edu.cn/) based on evolutionary conservation and ORF attributes. (F) Western blot analysis of the coding ability of lncIRS1. The possible seven ORFs of lncIRS1 were cloned into the eukaryotic expression verctor pSDS‐20218 with a 3*Flag tag. Untransfected DF‐1 cells were used as negative control (NC) and DF‐1 cells transfected with pSDS‐20218‐β‐actin were used as a positive control (PC). The upper panel shows the model target fragment in the pSDS vector. The lower left panel is the Western blot result of the NC and PC, and the lower right panel is the Western blot result of ORFs 1–7; all samples were probed with Flag antibody (G) LncIRS1 is localized in the cytoplasm and nucleus. RNA was isolated from nucleus and cytoplasm fraction of myoblasts was used to analyse the expression level of lncIRS1 by RT‐PCR. GAPDH and U6 serve as cytoplasmic and nuclear localization controls, respectively. (H) Results of IRS1 5'RACE. 5'RACE product, 1184 bp. (I) Result of IRS1 3'RACE. 3'RACE product, 1075 bp. LncRNA, long noncoding RNA; RACE, rapid amplification of cDNA ends; RT‐PCR, real‐time PCR.
LncIRS1 promotes proliferation and differentiation of myoblast
LncIRS1 was predominantly expressed in breast muscle and heart and followed by liver and leg muscle (Figure 3A). During skeletal muscle development, muscle precursor cells differentiate from the somite.25 Whole‐mount in situ hybridization shows that lncIRS1 is expressed in somite (Figure 3B). This may imply that lncIRS1 plays an important role in myogenesis. Overexpression of lncIRS1 reduced the number of cells that progressed to G0/G1 phase and increased the number of cells that progressed to S phase (Figure 3C) and significantly promoted myoblast proliferation (Figure 3D). Conversely, lncIRS1 knockdown increased cell cycle arrest in the G0/G1 phase (Figure 3C) and significantly reduced myoblast proliferation (Figure 3E). EdU staining also demonstrated that the proliferation rate of lncIRS1 overexpression cells was significantly increased compared with that of the control cells (Figure 3F and 3G). Conversely, proliferation was significantly inhibited after lncIRS1 knockdown (Figure 3F and 3G). After immunofluorescence staining, we found lncIRS1 overexpression promoted myoblast differentiation and significantly increased the total areas of myotubes (Figure 3H and 3J), while knockdown of lncIRS1 reduced myoblast differentiation (Figure 3I and 3K). The expression of myoblast differentiation marker genes (MyoD, MyHC, MyoG, and Myomarker) and IGF‐1 pathway related genes (IGF1, IGF1R, and IGF2R) were down‐regulated in lncIRS1 knockdown cells compared to control cells (Figure 3L and 3M). Together, the data suggest that lncIRS1 enriched in muscle and can promote myoblast proliferation and differentiation.
Figure 3.
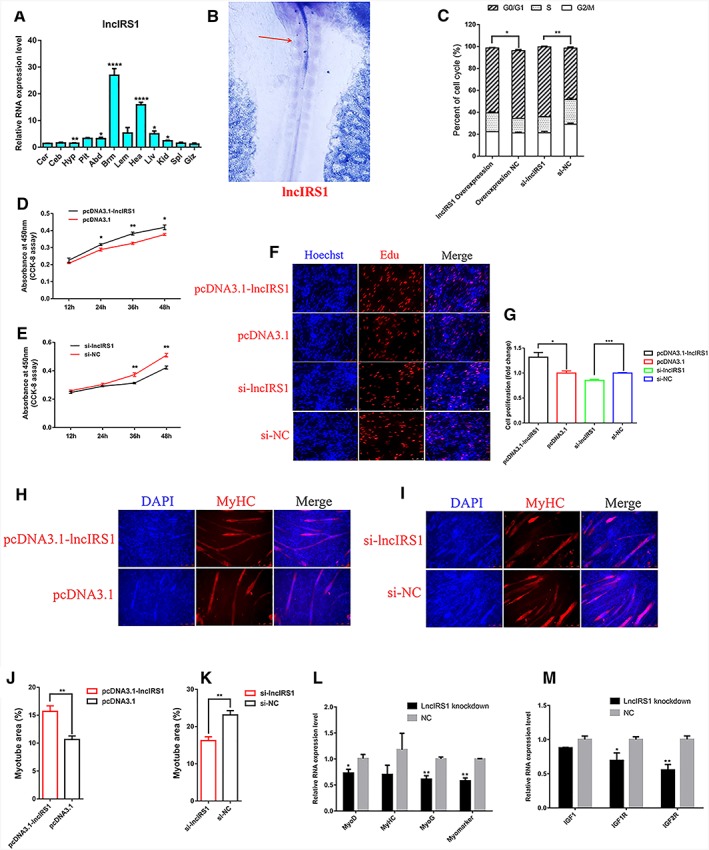
Skeletal muscle‐enriched lncIRS1 promotes myoblast proliferation and differentiation. (A) The RNA expression level of lncIRS1 in 12 different tissues in leaner broilers. Cer, cerebrum; Ceb, cerebellum; Hyp, hypothalamus; Pit, pituitary; Abd, abdominal fat; Brm, breast muscle; Lem, leg muscle; Hea, heart; Liv, liver; Kid, kidney; Spl, spleen; Giz, gizzard. The relative RNA expression of lncIRS1 in different tissues was normalized to Giz. (B) Whole‐mount in situ hybridization shows that lncIRS1 is expressed in the forming somites in HH10 chick embryo. Red arrow indicates lncIRS1 expression location (C) Cell cycle analysis of myoblasts at 48 h after transfection of pcDNA3.1‐lncIRS1 and pcDNA3.1 empty plasmid, or si‐lncIRS1 and si‐NC. (D) CCK‐8 assay was performed to assess the effect of lncIRS1 overexpression on myoblast proliferation. (E) CCK‐8 assay was performed to assess the effect of lncIRS1 knockdown on myoblast proliferation. (F) EdU and Hoechst (nuclei) staining analysis after transfection of pcDNA3.1‐lncIRS1 and pcDNA3.1 empty plasmid, or si‐lncIRS1 and si‐NC in proliferating myoblast, scale bars are 50 μm. (G) The proliferation rate of myoblast cells transfected with pcDNA3.1‐lncIRS1 and pcDNA3.1 empty plasmid, or si‐lncIRS1 and si‐NC. (H, I) Immunofluorescence Myoblast cells transfected with pcDNA3.1‐lncIRS1 and pcDNA3.1 empty plasmid, or si‐lncIRS1 and si‐NC were induced to differentiate for 72 hr then stained with MyHC antibody and DAPI (nuclei). Scale bars are 100 μm. (J, K) Myotube area (%) at 72 h after transfection of pcDNA3.1‐lncIRS1 or si‐lncIRS1. (L) Knockdown of lncIRS1 decreased the RNA expression level of myoblast cell differentiation associated genes, including MyoD, MyoG, MyHC, and MyoMarker. (M) Knockdown of lncIRS1 decreased the RNA expression level of IGF‐1 pathway associated genes, including IGF1, IGF1R, and IGF2R. Results are shown as the mean ± SEM of three independent experiments. Independent sample t‐test was used to analysis the statistical differences between groups. * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
LncIRS1 acts as a molecular sponge for miR‐15a, miR‐15b‐5p, and miR‐15c‐p to regulate IRS1 expression
LncIRS1 locates in both cytoplasm and nucleus (Figure 2G), and it is significantly upregulated in hypertrophic broiler. We speculated that lncIRS1 regulate myogenesis by function as a ceRNA. In lncRNA‐miRNA‐gene network, miR‐15 family is the core component of this network, and IRS1 is a target gene of these miRNAs. Interestingly, bioinformatics analysis reveals three or one predicted miRNA binding site in the lncIRS1 (Figure 4A) or IRS1 (Figure 4B), respectively. During myoblast proliferation and differentiation, IRS1 (Figure 4C) and lncIRS1 (Figure 4D) RNA expression level were upregulated, while the opposite expression pattern was presented in miR‐15a (Figure 4E), miR‐15b‐5p (Figure 4F), and miR‐15c‐5p (Figure 4G) but not miR‐16‐5p (Figure 4H) or miR‐16c‐5p (Figure 4I). Using dual‐luciferase reporter gene assays, we validated that miR‐15a, miR‐15b‐5p, and miR‐15c‐5p can directly interact with the three predicted binding sites of lncIRS1 (Figure 4J–4L), and these miRNAs can also directly interact with IRS1 (Figure 4M). To determine whether lncIRS1 could regulate IRS1 expression by sequester miRNAs, we co‐transfected pmirGLO‐IRS1, miRNAs, and lncIRS1 overexpression plasmid into DF‐1 cells. The luciferase activity increased in response to lncIRS1 in a dose‐dependent manner, suggesting that ectopically expressed lncIRS1 specifically sequestered miR‐15a (Figure 4N), miR‐15b‐5p (Figure 4O), and miR‐15c‐5p (Figure 4P), thereby preventing theirs from inhibiting luciferase activity. Overexpression of these miRNAs could simultaneously reduce the expression of lncIRS1 (Figure 4Q, Figure S6a) and IRS1 (Figure 4R, Figure S6b). We further performed RNA immunoprecipitation using antibody against AGO2 which is a key component of the RNA‐induced silencing complex. As expected, lncIRS1 (Figure 4S), IRS1 (Figure 4T), miR‐15a, miR‐15b‐5p, and miR‐15c‐5p (Figure 4U) were significantly enriched in AGO2 pellet (AGO2 antibody versus IgG). Western blot confirmed that the AGO2 antibody precipitated the AGO2 protein from our cellular extract (Figure 4V). Overexpression of miR‐15a, miR‐15b‐5p, and miR‐15c‐5p can significantly reduce the protein level of IRS1 (Figure 4W). Together, these data suggest that lncIRS1 can regulate the expression of IRS1 by sequester miR‐15a, miR‐15b‐5p, and miR‐15c‐5p.
Figure 4.
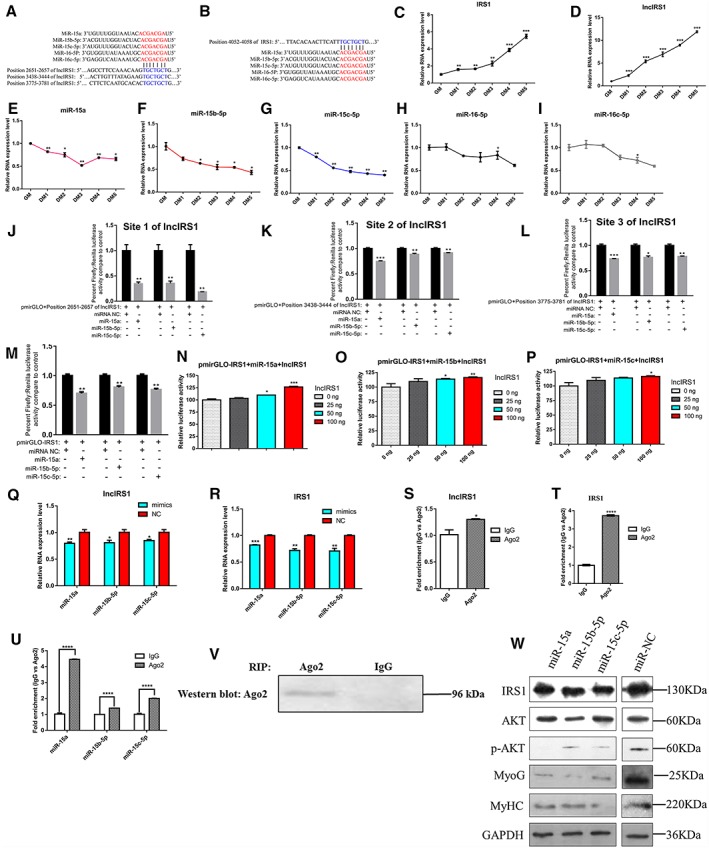
LncIRS1 functions as a ceRNA for miR‐15a, miR‐15b‐5p, and miR‐15c‐5p. (A) Three miR‐15 family binding sites in lncIRS1 were identified by RNAhybrid. (B) The miR‐15 family binding site in IRS1 was identified by TargetScan. Expression analysis of IRS1 (C), lncIRS1 (D), miR‐15 a (E), miR‐15b‐5p (F), miR‐15c‐5p (G), miR‐16‐5p (H), and miR‐16c‐5p (I) during myoblasts proliferation (GM) and differentiation (DM), using qRT‐PCR. The dual‐luciferase reporter assay verified that Site 1 (position 2651–2657 of lncIRS1) (J), Site 2 (position 3438–3444 of lncIRS1) (K) and Site 3 (position 3775–3781 of lncIRS1) (L) of lncIRS1 could be bound by miR‐15a, miR‐15b‐5p, and miR‐15c‐5p. (M) The dual‐luciferase reporter assay verified that IRS1 could be bound by miR‐15a, miR‐15b‐5p, and miR‐15c‐5p; pmirGLO‐IRS1 or miR‐15a (N), miR‐15b‐5p (O), and miR‐15c‐5p (P) were transfected into DF‐1 cells, together with 0, 25, 50, or 100 ng of sponge plasmid of lncIRS1. Overexpression of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p decreased the RNA expression level of lncIRS1 (Q) and IRS1 (R). Association of lncIRS1, IRS1, and miR‐15a, miR‐15b‐5p, and miR‐15c‐5p with AGO2. Cellular lysates from chicken myoblast cells were used for RNA immunoprecipitation with AGO2 antibody. Detection of lncIRS1 (S); IRS1 (T); miR‐15a, miR‐15b‐5p, and miR‐15c‐5p (U) using qRT‐PCR; and detection of AGO2 using IP‐Western analysis (V). (W) Overexpression of miR‐15a, miR‐15b‐5p, and miR‐15c‐5p decreased the protein expression level of IRS1, p‐AKT, MyoG, and MyHC. Results are shown as the mean ± SEM of three independent experiments. Independent sample t‐test was used to analysis the statistical differences between groups. * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001. RIP, RNA immunoprecipitation.
MiR‐15a, miR‐15b‐5p, and miR‐15c‐5p suppress myoblast proliferation and differentiation
We identified miR‐15a, miR‐15b‐5p, and miR‐15c‐5p are down‐regulated in hypertrophic broiler (Figure 5A and 5B). To observe the effects of these miRNAs on myoblast proliferation, we transfected myoblasts with miRNA mimics or miRNA negative control (miR‐NC) and then monitored the proliferation status of cells using flow cytometry analysis, CCK‐8 assay, and EdU staining. Flow cytometry analysis of the cell cycle revealed that myoblasts transfected with the miR‐15a, miR‐15b‐5p, or miR‐15c‐5p mimic could arrest myoblasts in the G0/G1 phase (Figure 5C). CCK‐8 and EdU staining also demonstrated that the proliferation rate of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p transfected myoblasts was significantly reduced compared with that of the miR‐NC transfected cells (Figure 5D–5F). After immunofluorescence staining, we found miR‐15a, miR‐15b‐5p, or miR‐15c‐5p overexpression inhibited myoblast differentiation and significantly reduced the total areas of myotubes (Figure 5G and 5H). The expression of IGF‐1 pathway related genes (IGF1, IGF1R, and IGF2R) and myoblast differentiation marker genes (MyoD, MyHC, MyoG, and Myomarker) were down‐regulated in miR‐15a, miR‐15b‐5p, or miR‐15c‐5p overexpression myoblasts compared to miR‐NC transfected myoblasts (Figure 5I–5O). Overexpression of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p reduced the protein expression level of MyoG and MyHC. The kinase AKT is a central component in IGF‐1 signalling pathway which plays a major role in the regulation of skeleteal muscle growth. As shown in Figure 4W, overexpression of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p reduced the phosphorylation level of AKT and had no obvious change in total AKT. Together, the data suggest that miR‐15a, miR‐15b‐5p, and miR‐15c‐5p inhibit myoblast proliferation and differentiation via IGF‐1 pathway.
Figure 5.
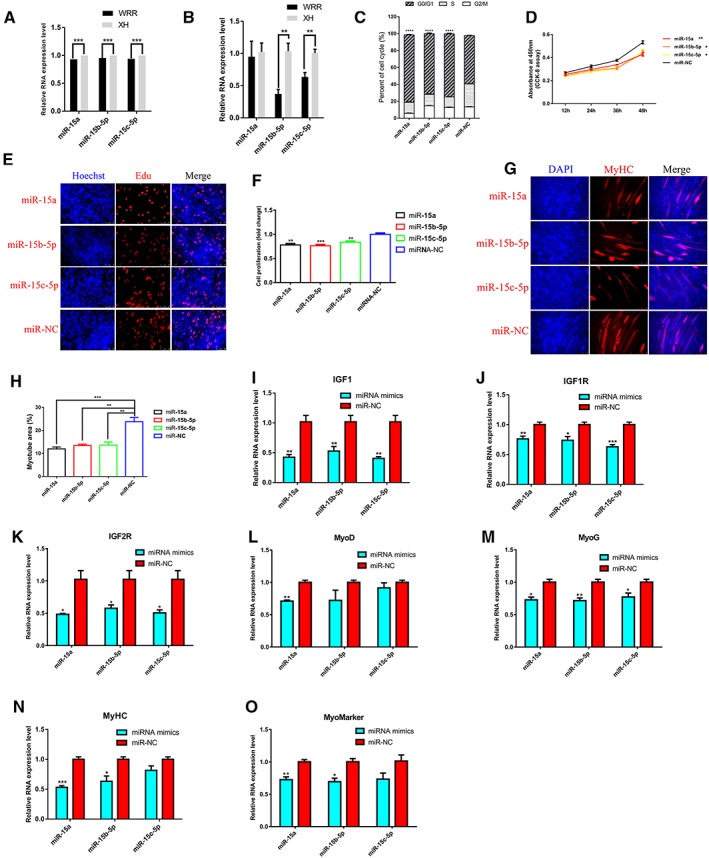
MiR‐15a, miR‐15b‐5p, and miR‐15c‐5p inhibit myoblast proliferation and differentiation. (A) RNA‐seq analysis found that miR‐15a, miR‐15b‐5p, and miR‐15b‐5p were down‐regulated in hypertrophic broilers compared with leaner broilers, and this expression pattern was validated by qRT‐PCR (B). (C) Cell cycle analysis of myoblasts at 48 h after transfection of miR‐15a, miR‐15b‐5p, miR‐15c‐5p, and miR‐NC. (D) CCK‐8 assay was performed to assess the effect of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p overexpression on myoblast proliferation. (E) EdU and Hoechst (nuclei) staining analysis after transfection of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p in proliferating myoblast, scale bars are 50 μm. (F) The proliferation rate of myoblast cells transfected with miR‐15a, miR‐15b‐5p, or miR‐15c‐5p. (G) Myoblast cells transfected with miR‐15a, miR‐15b‐5p, or miR‐15c‐5p were induced to differentiate for 72 hr then stained with MyHC antibody and DAPI (nuclei). Scale bars are 100 μm. (H) Myotube area (%) at 72 h after transfection of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p. Overexpression of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p decreased the RNA expression level of IGF‐1 pathway associated genes, including IGF1 (I), IGF1R (J), and IGF2R (K). Overexpression of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p decreased the RNA expression level of myoblast cell differentiation associated genes, including MyoD (L), MyoG (M), MyHC (N), and MyoMarker (O). Results are shown as the mean ± SEM of three independent experiments. Independent sample t‐test was used to analysis the statistical differences between groups. * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
IRS1 is involved in proliferation and differentiation of myoblast via IGF‐1 signalling pathway
IRS1 is thought to be downstream of the IGF‐1 receptor and associated with mice body weight.7 Here, we identified IRS1 is upregulated in hypertrophic broiler (Figure 6A and 6B), and predominantly expressed in skeletal muscle (Figure 6C). Whole‐mount in situ hybridization shows that IRS1 is expressed in somite (Figure 6D), which is consistent with lncIRS1 (Figure 3B). Overexpression of IRS1 reduced the number of cells that progressed to G0/G1 phase, increased the number of cells that progressed to S phase (Figure 6E), and significantly promoted myoblast proliferation (Figure 6F). Conversely, IRS1 knockdown increased cell cycle arrest in the G0/G1 phase (Figure 6E) and significantly reduced myoblast proliferation (Figure 6G). EdU staining also demonstrated that the proliferation rate of IRS1 overexpression cells was significantly increased compared with that of the control cells (Figure 6H and 6I). Conversely, proliferation was significantly inhibited after IRS1 knockdown (Figure 6H and 6I). After immunofluorescence staining, we found IRS1 overexpression promoted myoblast differentiation and significantly increased the total areas of myotubes (Figure 6J and 6K), while knockdown of IRS1 reduced myoblast differentiation (Figure 6L and 6M). The expression of myoblast differentiation marker genes (MyoD, MyHC, MyoG, and Myomarker) and IGF‐1 pathway related genes (IGF1, IGF1R, and IGF2R) were down‐regulated in IRS1 knockdown cells compared to control cells (Figure 6N and 6O). Together, these data suggest that IRS1 promotes myoblast proliferation and differentiation via IGF‐1 pathway.
Figure 6.
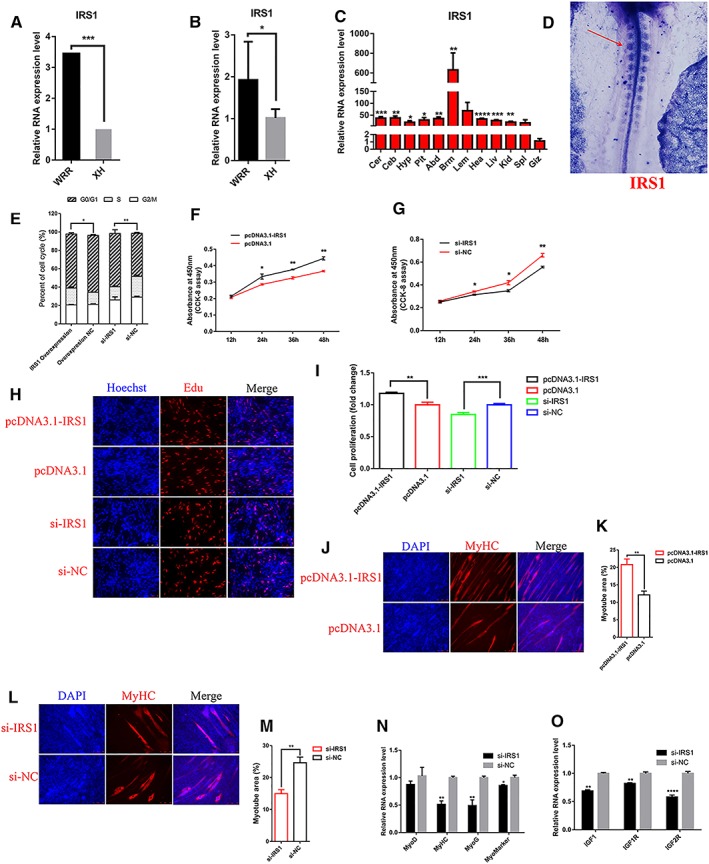
IRS1 promotes myoblast proliferation and differentiation. (A) RNA‐seq analysis found that IRS1 was up‐regulated in hypertrophic broilers, and this expression pattern was validated by qRT‐PCR (B). (C) The RNA expression level of IRS1 in 12 different tissues in leaner broilers and expression was normalized to Giz. (D) Whole‐mount in situ hybridization showed that IRS1 are expressed in the forming somites in HH10 chick embryo Red arrow indicates IRS1 expression location. (E) Cell cycle analysis of myoblasts at 48 h after transfection of pcDNA3.1‐IRS1 and pcDNA3.1 empty plasmid, or si‐IRS1 and si‐NC. (F) CCK‐8 assay was performed to assess the effect of IRS1 overexpression on myoblast proliferation. (G) CCK‐8 assay was performed to assess the effect of IRS1 knockdown on myoblast proliferation. (H) EdU and Hoechst (nuclei) staining analysis after transfection of pcDNA3.1‐IRS1 and pcDNA3.1 empty plasmid, or si‐IRS1 and si‐NC in proliferating myoblast, scale bars are 50 μm. (I) The proliferation rate of myoblast cells transfected with pcDNA3.1‐IRS1 and pcDNA3.1 empty plasmid, or si‐IRS1 and si‐NC. (J, L) Myoblast cells transfected with pCDNA3.1‐IRS1, or si‐IRS1 were induced to differentiate for 72 hr then stained with MyHC antibody and DAPI (nuclei). Scale bars are 100 μm. (K, M) Myotube area (%) was determined at 72 h after transfection of pcDNA3.1‐IRS1 or si‐IRS1. (N) Knockdown of IRS1 decreased the RNA expression level of myoblast cell differentiation associated genes, including MyoD, MyoG, MyHC, and MyoMarker. (O) Knockdown of IRS1 decreased the RNA expression level of IGF‐1 pathway associated genes, including IGF1, IGF1R, and IGF2R. Results are shown as the mean ± SEM of three independent experiments. Independent sample t‐test was used to analysis the statistical differences between groups. * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
LncIRS1 modulated IRS1 expression and promoted myoblast proliferation and differentiation by sponging miR‐15a, miR‐15b‐5p, and miR‐15c‐p
To determine whether lncIRS1 functions as a molecular sponge for miR‐15a, miR‐15b‐5p, and miR‐15c‐p, we performed qPCR analysis of lncIRS1 and IRS1 level in lncIRS1 or IRS1 knockdown myoblast, respectively. Knockdown of lncIRS1 reduce the expression level of lncIRS1 and simultaneously inhibit IRS1 expression (Figure 7A). Similarly, knockdown of IRS1 reduce the expression level of IRS1 and lncIRS1 (Figure 7B). Furthermore, the rescue assay shown that lncIRS1 overexpression can promote the expression of IRS1 (Figure 7C, Figure S6c). Overexpression of lncIRS1 (OV‐lncIRS1) increased the protein level of IRS1 (Figure 7D), as the same effect of IRS1 transfected (OV‐IRS1). Overexpression of lncIRS1 increased the phosphorylation level of AKT, which confirmed that lncIRS1 activates the IGF‐1 pathway via IRS1 protein. Overexpression of lncIRS1 promotes the myoblast differentiation marker genes (MyoG and MyHC) expression (Figure 7D). The opposite results were observed by knockdown of lncIRS1 or IRS1 (Figure 7E). These data demonstrated that lncIRS1 modulates IRS1 to activate the IGF‐1 signalling pathway, affecting myoblast proliferation and differentiation by sponge miR‐15a, miR‐15b‐5p, and miR‐15c‐p.
Figure 7.
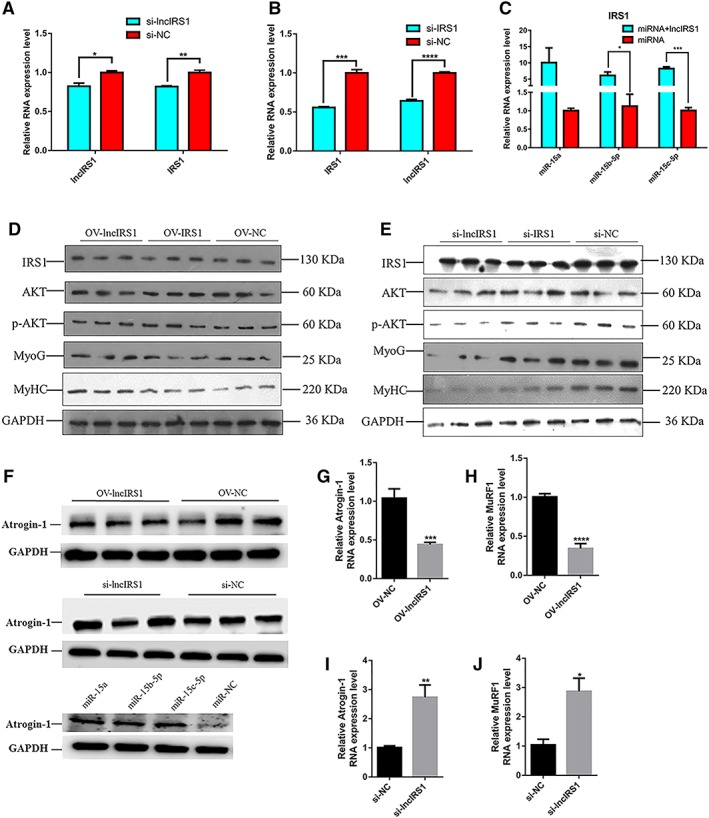
LncIRS1 promotes myoblast differentiation via IGF‐1 signalling pathway. (A) mRNA expression of lncIRS1 and IRS1 after lncIRS1 knockdown in myoblasts. (B) mRNA expression of lncIRS1 and IRS1 after IRS1 knockdown in myoblasts. (C) Co‐expression of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p with lncIRS1 up‐regulated the expression level of IRS1. (D) Overexpression of lncIRS1 or IRS1 increased the protein expression level of IRS1, p‐AKT, MyoG, and MyHC. (E) Knockdown of lncIRS1 or IRS1 reduced the protein expression level of IRS1, p‐AKT, MyoG, and MyHC. (F) Effect of lncIRS1 overexpression, lncIRS1 knockdown, and miR‐15 family on Atrogin‐1 protein expression. (G) Effect of lncIRS1 overexpression on Atrogin‐1 RNA expression. (H) Effect of lncIRS1 overexpression on MuRF1 RNA expression. (I) Effect of lncIRS1 knockdown on Atrogin‐1 RNA expression. (J) Effect of lncIRS1 knockdown on MuRF1 RNA expression in all panels, Myoblast cells transfected with above‐indicated chemicals (plasmid DNA, siRNAs, or miRNAs) were induced to differentiate for 48 hr then detected with qRT‐PCR or Western blot analysis. Results are shown as the mean ± SEM of three independent experiments. Independent sample t‐test was used to analysis the statistical differences between groups. * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
LncIRS1 could rescue skeletal muscle atrophy
We first investigated the impact of lncIRS1 and miR‐15 family on atrophy‐related gene expressions in primary myotubes. LncIRS1 overexpression decreased the expression of two atrophy‐related genes (atrogenes), Atrogin‐1 and MuRF1, at either mRNA levels or protein level of Atrogin‐1 compared to the control group (Figure 7F–7H). In contrast, myotubes transfected with lncIRS1 knockdown increased the expression of Atrogin‐1 and MuRF1 at either mRNA levels or protein level of Atrogin‐1 compared to those transfected with the small interfering RNA negative control (Figure 7F, 7I, and 7J). In addition, overexpression of miR‐15a, miR‐15b‐5p, or miR‐15c‐5p promotes the protein expression of Atrogin‐1 (Figure 7F).
The dexamethasone (synthetic glucocorticoid) has been known to promote protein breakdown and to induce Atrogin‐1 and MuRF1 expression in myotube cultures and adult muscles.26, 27 IGF downstream signalling (AKT‐FOXO signalling) has been reported to play a major role in eliciting the myofiber atrophy.28 We next asked if lncIRS1 rescue muscle atrophy, we performed lncIRS1 overexpression and knockdown during dexamethasone‐induced myotube atrophy in vitro. We then assessed AKT and FOXO signalling by determining the levels of Foxo1, phosphorylated Foxo1 (p‐Foxo1), Foxo3, phosphorylated Foxo3 (p‐Foxo3), Foxo4, phosphorylated Foxo4 (p‐Foxo4), AKT, phosphorylated AKT (p‐AKT), and Atrogin‐1 by Western blot analysis. Result showed that protein expression of p‐Foxo1, p‐Foxo3, p‐Foxo4, and p‐AKT in dexamethasone‐treated myotubes was increased after transfection with lncIRS1 overexpression plasmid (OV‐lncIRS1) compared with control (OV‐NC) (Figure 8A–8E). The protein expression level of Atrogin‐1 was decreased in lncIRS1 overexpression group (Figure 8F). In contrast, the protein expression (p‐Foxo1, p‐Foxo3, p‐Foxo4, and p‐AKT) was decreased after interference with si‐lncIRS1 (Figure 8G–8K). The protein expression level of Atrogin‐1 was increased in si‐lncIRS1 group (Figure 8L). Taken together, our data demonstrated that lncIRS1 regulates the expression of atrophy‐related genes and further can rescue dexamethasone‐induced atrophy in cultured myotubes.
Figure 8.
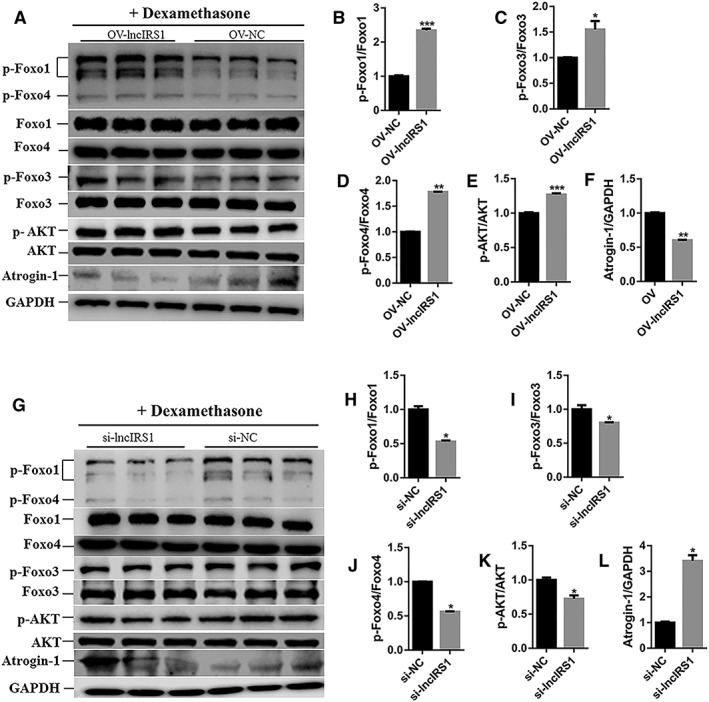
LncIRS1 could rescue skeletal muscle atrophy. (A) Effect of lncIRS1 overexpression on AKT‐FOXO signalling pathway. Chicken primary myotubes isolated from leg muscle of E11 were first treated with dexamethasone for 24 hr to induce atrophy and then transfected with lncIRS1 overexpression plasmid for 48 hr, followed by Western blot analysis. (B–E) Effect of lncIRS1 overexpression on protein phosphorylation level of AKT‐FOXO signalling pathway components, including p‐foxo1 (B), p‐foxo3 (C), p‐foxo4 (D), and p‐AKT (E). (F) Effect of lncIRS1 overexpression on protein expression level of Atrogin‐1 during dexamethasone treated myotubes. (G) Effect of lncIRS1 knockdown on AKT‐FOXO signalling pathway. Chicken primary myotubes isolated from leg muscle of E11 were first treated with dexamethasone for 24 hr to induce atrophy and then transfected with si‐lncIRS1 for 48 hr, followed by Western blot analysis. (H–K) Effect of lncIRS1 knockdown on protein phosphorylation level of AKT‐FOXO pathway, contains p‐foxo1 (H), p‐foxo3 (I), p‐foxo4 (J), and p‐AKT (K). (L) Effect of lncIRS1 knockdown on Atrogin‐1 protein expression level during dexamethasone treated myotubes. In all panels, error bars indicate the standard error of the mean. Independent sample t‐test was used to analysis the statistical differences between groups. * P < 0.05, ** P < 0.01, and *** P < 0.001.
LncIRS1 controls muscle mass and muscle fibre cross‐sectional area
To test whether lncIRS1 regulates muscle growth in vivo, 1‐day‐old chicks were injected with lentiviral‐mediated lncIRS1 overexpression (pWPXL‐lncIRS1) and lentiviral‐mediated lncIRS1 knockdown (pLKO.1‐lncIRS1). Compared with control group (pWPXL‐control), the expression level of lncIRS1 or IRS1 gene was significantly higher in pWPXL‐lncIRS1 (Figure 9A and 9B). Breast muscle mass (breast muscle weight) and muscle fibre cross‐sectional area were both increased in the lncIRS1 overexpression groups compared with those in control groups (Figure 9C–9E), while they were decreased in lncIRS1 knockdown groups (Figure 9F–9J). In addition, we performed Western blot analysis for AKT‐FOXO signalling in lentiviral‐lncIRS1 overexpression and knockdown. The results showed that protein expression of p‐Foxo1, p‐Foxo3, p‐Foxo4, and p‐AKT in lncIRS1 overexpression group (pWPXL‐lncIRS1) had higher level than did control group (pWPXL‐control), while lncIRS1 knockdown group (pLKO.1‐lncIRS1) had lower expression level than did the controls (pLKO.1‐control) (Figure 9K). Moreover, the protein levels of myogenic markers (MyoG and MyHC) showed similar changes with the parameters of breast muscle mass (Figure 9K). We conclude that lncIRS1 is associated with muscle mass and muscle fibre in vivo.
Figure 9.
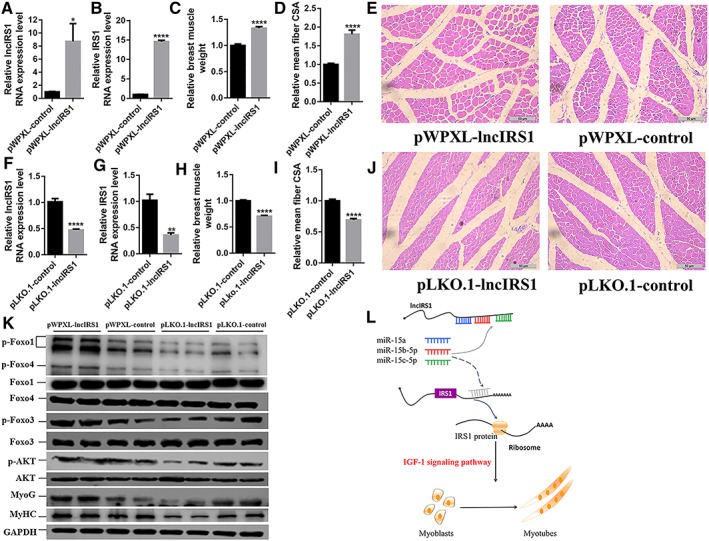
LncIRS1 regulates muscle mass and mean muscle fibre in vivo. (A) qRT‐PCR of lncIRS1 after lncIRS1 overexpression prepared with lentivirus and intramuscularly injected into the breast muscle. (B) qRT‐PCR of IRS1 after lncIRS1 overexpression prepared with lentivirus and intramuscularly injected into the breast muscle. (C–E) Breast muscle weight (C), mean muscle fibre cross‐section area (CSA) (D), and H‐E staining (E) of breast muscle fibre cross section infected with pWPXL‐lncIRS1 or pWPXL‐control scale bars are 50 μm. (F) qRT‐PCR analysis of lncIRS1 expression in pLKO.1‐lncIRS1 overexpression group or pLKO.1‐control group prepared with lentivirus and intramuscularly injected into the chicken breast muscle. (G) qRT‐PCR analysis of IRS1 expression in pLKO.1‐lncIRS1 overexpression group or pLKO.1‐control group prepared with lentivirus and intramuscularly injected into the chicken breast muscle. (H–J) Breast muscle weight (H), mean muscle fibre cross‐section area (CSA) (I), and H‐E staining (J) of breast muscle fibre cross section infected with pLKO.1‐lncIRS1 or pLKO.1‐control scale bars are 50 μm. (K) The protein expression level of AKT‐FOXO signalling components and myogenic‐related markers (MyoG and MyHC) during chicken infection with lentivirus containing pWPXL‐lncIRS1 and pWPXL‐control, or pLKO.1‐lncIRS1 and pLKO.1‐control, as analysed by Western blot. (L) Molecular mechanism of lncIRS1 promotes myoblast proliferation, differentiation, and muscle fibre. LncIRS1 controls IRS1 protein level and further activates IGF‐1 signalling pathway by functioning as ceRNA to sponge miR‐15a, miR‐15b‐5p, and miR‐15c‐5p. In all panels, error bars indicate the standard error of the mean. Independent sample t‐test was used to analysis the statistical differences between groups. * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
Discussion
In recent years, studies revealed that lncRNAs containing MREs act as molecular sponges to effectively inhibit miRNA function.29, 30 Lnc‐mg promotes myogenesis by protecting IGF2 transcripts from miR‐125b‐mediated suppression.31 Another, lncRNA, FER1L4, play important roles in post‐transcriptional regulation in gastric cancer by competing for shared miR‐106a‐5p.32 In addition, a circular RNA that contains about 70 MREs with miR‐7 was identified.33 The circular RNA function as efficient miRNA sponge to sequesters miR‐7 away from its targets and suppresses its function.
Many studies suggest the important role of lncRNAs in skeletal muscle myogenesis while highlighting the necessity to systematically identify lncRNAs altered in skeletal muscle development. In this study, we constructed the ceRNA network jointed by lncRNAs, miRNAs, and mRNAs. MiR‐15a, miR‐15b‐5p, and miR‐15b‐5p are the core of this network, and they commonly target with IRS1 which is the key signal transduction gene of IGF‐1 signalling pathway. In addition, we identify a skeletal muscle‐enriched lncIRS1 that promotes myogenesis in vitro. In the initial stage of skeletal muscle development, muscle precursor cells differentiate from the somite.4 In this study, we identified that IRS1 and lncIRS1 were abundantly expressed in somite. During myoblasts proliferation and differentiation, the expression level of IRS1 and lncIRS1 are gradually increased, while the opposite expression pattern of miR‐15a, miR‐15b‐5p, and miR‐15c‐5p were observed. Overexpression of those miRNAs simultaneously decreased the expression level of IRS1 and lncIRS1. By functioning as a ceRNA, lncIRS1 bocks miR‐15a, miR‐15b‐5p, and miR‐15c‐5p to control the abundance of IRS1 protein and phosphorylation level of AKT. IRS1 is downstream of the IGF‐1 receptor and is well known to be principal substrate for IGF‐1, which mediated the IGF1‐PI3K/AKT signalling pathway.7 In mice, knockout of IRS1 down‐regulated the expression of IGF‐1 and led to 40–70% weight loss.5, 6
Muscle atrophy is due to a decrease in muscle mass and fibre size. White muscle fibre atrophy occurs under conditions of tough nutritional restriction.34 In mammals, inactivity and starvation lead to skeletal muscle atrophy.35 Muscle atrophy also results from a co‐morbidity of some common diseases, such as AIDS, congestive heart failure, cancer, chronic obstructive pulmonary disease, renal failure, and severe burns. Atrogin‐1 and MuRF1 expressions correlated with loss of muscle protein.36 In our study, lncIRS1 controls the expression of Atrogin‐1 and MuRF1 genes. It has been reported that dexamethasone can increase atrophy‐related gene expressions in myotubes, while IGF‐I inhibits their expression and blocks dexamethasone effects.26 The converse of atrophy is hypertrophy, which is induced by an increase in protein synthesis and is characterized by an increase in muscle fibre size. In contrast to atrophy, however, high‐rigour marker genes of the hypertrophy condition have not been well validated. Skeletal muscle hypertrophy is accompanied by the increased expression of IGF‐1.37 AKT is a central component in IGF1‐PI3K/AKT pathway; it controls not only protein synthesis via the kinases mammalian target of rapamycin and glycogen synthase kinase 3β but also protein degradation via the FOXO family transcription factor. In this study, we used myoblast models of hypertrophy for studying the underlying mechanisms that regulate skeletal muscle hypertrophy. Recent studies have shown that the hypertrophy‐inducing IGF‐1‐PI3K/AKT pathway could dominantly block the atrophy‐inducing effects of dexamethasone, via AKT mediated phosphorylation and subsequent inhibition of the FOXO family of transcription factors.38, 39 In this study, we identified that lncIRS1 can activate IGF1‐PI3K/AKT pathway by acting as a sponge for miR‐15 family to relieve IRS1 expression (Figure 9L). Overexpression of lncIRS1 promotes the expression of IGF‐1, activates AKT phosphorylation, and increases skeletal muscle proliferation and total myotube numbers. From a clinical perspective, lncIRS1 can promote the expression of IGF‐1 and modulate key atrophy induced genes, such as Atrogin‐1 and MuRF1 via the PI3K/AKT pathway, which may be used as a therapeutic target for muscle atrophy.
It has been reported that lncRNA MAR1 played a positive role in skeletal muscle differentiation and growth in vitro and in vivo.40 To test the function of the lncIRS1 in vivo, we performed lncIRS1 overexpression or knockdown by direct injection of lentiviral particles in breast muscle. In our study, direct injection of overexpression of lncIRS1 lentiviral particles into the breast muscle weakened the muscle atrophy, evidenced by the enhanced breast muscle weight, higher muscle fibre fusion index and higher myogenic marker (MyoG and MyHC) protein expression levels than did the negative control group. However, in the lentiviral‐lncIRS1 knockdown experiment, the opposite was true. These results show the therapeutic potential of lncIRS1 in muscle atrophy.
In conclusion, we identified a novel lncIRS1 acts as a sponge for miR‐15 family to activate IGF1‐PI3K/AKT signalling, resulting in promoting muscle proliferation, differentiation, and muscle mass, which may be used as a potential therapeutic agent for treating muscle atrophy.
Conflict of interest
Z.L., B.C., B.A.A., X.Z., M.Z., P.H., Q.N., and X.Z. declare that they have no conflict of interest.
Supporting information
Table S1. Differential expression analysis of lncRNAs in breast muscle between hypertrophic and leaner broilers.
Table S2. Differential expression analysis of genes in breast muscle between hypertrophic and leaner broilers.
Table S3. Differential expression analysis of miRNAs in breast muscle between hypertrophic and leaner broilers.
Table S4. Differentially expressed miRNAs and theirs corresponding target genes with differential expression in hypertrophic and leaner broilers.
Table S5. Differentially expressed miRNAs and theirs corresponding target lncRNAs with differential expression in hypertrophic and leaner broilers.
Table S6. Summary of the represented networks generated by IPA analysis.
Table S7. The full length sequences of lncIRS1 and IRS1
Table S8. Information of Primers.
Fig S1 A competing endogenous RNA (ceRNA) hypothesis.
LncRNAs and mRNAs, which share the common miRNA response elements (MREs), could influence each other's levels by competing for a limited pool of miRNA. (a) Upregulation of lncRNA increases cellular concentrations of particular MREs and can result in the derepression of mRNA that contains the same MREs. (b) While downregulation of lncRNA would lead to decreased levels of particular MREs, and, consequently inhibited the expression of mRNA.
Fig S2 miRNA‐gene interaction network
Hypertrophic broiler vs. leaner broiler, differentially expressed miRNAs and their corresponding differentially expressed target genes with opposition expression pattern were used to construct a miRNA‐gene interaction network. In this network, miRNAs are displayed as pink circles, and miRNA target genes are displayed as blue circles. Dashed lines represent the interactions between differentially expressed miRNAs and theirs corresponding target genes.
Fig S3 miRNA‐lncRNA interaction network
Hypertrophic broiler vs. leaner broiler, differentially expressed miRNAs and their corresponding differentially expressed target lncRNAs with opposition expression pattern were used to construct a miRNA‐lncRNA interaction network. In this network, miRNAs are displayed as pink circles, and miRNA target lncRNAs are displayed as green circles. Solid lines represent the interactions between differentially expressed miRNAs and theirs corresponding target lncRNAs.
Fig S4 Ingenuity Pathway Analysis (IPA) identification of gene networks
IPA online software was used to identify the gene‐gene interaction networks, using the genes in the miRNA‐gene network as input. (a‐d) A total of four major gene networks were identified. Nodes shaded in blue represent the genes from miRNA‐gene network are also involved in the IPA interaction network. White nodes represent transcription factors (as opposed to gene from miRNA‐gene network) that are associated with the regulation of some of these genes, based on the evidence stored in the IPA knowledgebase. Edges and node are annotated with labels that illustrate the nature of the relationship between the genes and their functions.
Fig S5 GO and pathway analysis of the gene from lncRNA‐miRNA‐gene network
(a) The top 10 enriched terms for three GO categories (biological process, cellular component and molecular function) for the genes belonging to lncRNA‐miRNA‐gene network. (b) The top 10 enriched pathway for the genes belonging to lncRNA‐miRNA‐gene network. Pink bars represent the pathway associated with muscle growth. (c) The network between genes and theirs associated pathway. Yellow oval represent the genes that from lncRNA‐miRNA‐gene network, while blur bar represent these pathway that they enrich in. (d) Pathway analysis indicates that the genes (IRS1) from lncRNA‐miRNA‐gene are enriched in the IGF‐1 signaling pathway. IRS1 (nodes shaded) function in the IGF‐1 signaling pathway.
Fig S6 LncIRS1 acts as a sponge of miR‐15a, miR‐15b‐5p and miR‐15c‐5p to regulate the expression of IRS1
MiR‐15a, miR‐15b‐5p and miR‐15c‐5p inhibit the RNA expression level of lncIRS1 (a), and simultaneously depress the expression of IRS1 (b). (c) Co‐expression of miR‐15a, miR‐15b‐5p or miR‐15c‐5p, and lncIRS1 upregulate the RNA expression level of IRS1.
Acknowledgements
This work was supported by the Ten‐Thousand Talents Program (W03020593), Natural Scientific Foundation of China (31761143014), the Chinese Postdoctoral Science Foundation (2017M622715), Natural Science Foundation of Guangdong Province (2018A030310209), the China Agriculture Research System (CARS‐41‐G03), and Open Fund of Guangdong Provincial Key Laboratory of Agro‐animal Genomics and Molecular Breeding, South China Agricultural University.
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.41
Li, Z. , Cai, B. , Abdalla, B. A. , Zhu, X. , Zheng, M. , Han, P. , Nie, Q. , and Zhang, X. (2019) LncIRS1 controls muscle atrophy via sponging miR‐15 family to activate IGF1‐PI3K/AKT pathway. Journal of Cachexia, Sarcopenia and Muscle, 10: 391–410. 10.1002/jcsm.12374.
References
- 1. Jagoe RT, Goldberg AL. What do we really know about the ubiquitin‐proteasome pathway in muscle atrophy? Curr Opin Clin Nutr Metab Care 2001;4:183–190. [DOI] [PubMed] [Google Scholar]
- 2. Hasselgren PO. Glucocorticoids and muscle catabolism. Curr Opin Clin Nutr Metab Care 1999;2:201–205. [DOI] [PubMed] [Google Scholar]
- 3. Goldspink DF, Garlick PJ, McNurlan MA. Protein turnover measured in vivo and in vitro in muscles undergoing compensatory growth and subsequent denervation atrophy. Biochem J 1983;210:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Z, Ouyang H, Zheng M, Cai B, Han P, Abdalla BA, et al. Integrated analysis of long non‐coding RNAs (LncRNAs) and mRNA expression profiles reveals the potential role of LncRNAs in skeletal muscle development of the chicken. Front Physiol 2016;7:687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dong X, Park S, Lin X, Copps K, Yi X, White MF. Irs1 and Irs2 signaling is essential for hepatic glucose homeostasis and systemic growth. J Clin Invest 2006;116:101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate‐1. Nature 1994;372:182–186. [DOI] [PubMed] [Google Scholar]
- 7. Schiaffino S, Mammucari C. Regulation of skeletal muscle growth by the IGF1‐Akt/PKB pathway: insights from genetic models. Skelet Muscle 2011;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet 2014;15:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guttman M, Rinn JL. Modular regulatory principles of large non‐coding RNAs. Nature 2012;482:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fatica A, Bozzoni I. Long non‐coding RNAs: new players in cell differentiation and development. Nat Rev Genet 2014;15:7–21. [DOI] [PubMed] [Google Scholar]
- 11. Caretti G, Schiltz RL, Dilworth FJ, Di Padova M, Zhao P, Ogryzko V, et al. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev Cell 2006;11:547–560. [DOI] [PubMed] [Google Scholar]
- 12. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 2011;146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 2007;4:721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding‐independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010;465:1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011;147:358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L, et al. The imprinted H19 lncRNA antagonizes let‐7 microRNAs. Mol Cell 2013;52:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henrique D, Adam J, Myat A, Chitnis A, Lewis J, Ish‐Horowicz D. Expression of a Delta homologue in prospective neurons in the chick. Nature 1995;375:787–790. [DOI] [PubMed] [Google Scholar]
- 18. Luo W, Wu H, Ye Y, Li Z, Hao S, Kong L, et al. The transient expression of miR‐203 and its inhibiting effects on skeletal muscle cell proliferation and differentiation. Cell Death Dis 2014;5:e1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 20. Luo W, Chen J, Li L, Ren X, Cheng T, Lu S, et al. c‐Myc inhibits myoblast differentiation and promotes myoblast proliferation and muscle fibre hypertrophy by regulating the expression of its target genes, miRNAs and lincRNAs. Cell Death Differ 2018; 10.1038/s41418-018-0129-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ouyang H, He X, Li G, Xu H, Jia X, Nie Q, et al. Deep sequencing analysis of miRNA expression in breast muscle of fast‐growing and slow‐growing broilers. Int J Mol Sci 2015;16:16242–16262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin‐like growth factors in embryonic and postnatal growth. Cell 1993;75:73–82. [PubMed] [Google Scholar]
- 23. Araki E, Lipes MA, Patti ME, Bruning JC, Haag BR, Johnson RS, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS‐1 gene. Nature 1994;372:186–190. [DOI] [PubMed] [Google Scholar]
- 24. Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, et al. CPC: assess the protein‐coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 2007;35:W345–W349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luo W, Nie Q, Zhang X. MicroRNAs involved in skeletal muscle differentiation. J Genet Genomics 2013;40:107–116. [DOI] [PubMed] [Google Scholar]
- 26. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 27. Wang L, Luo GJ, Wang JJ, Hasselgren PO. Dexamethasone stimulates proteasome‐ and calcium‐dependent proteolysis in cultured L6 myotubes. Shock 1998;10:298–306. [DOI] [PubMed] [Google Scholar]
- 28. Levine S, Biswas C, Dierov J, Barsotti R, Shrager JB, Nguyen T, et al. (2011) Increased proteolysis, myosin depletion, and atrophic AKT‐FOXO signaling in human diaphragm disuse. Am J Respir Crit Care Med 2011;183:483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ebert MS, Sharp PA. Emerging roles for natural microRNA sponges. Curr Biol 2010;20:R858–R861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karreth FA, Pandolfi PP. ceRNA cross‐talk in cancer: when ce‐bling rivalries go awry. Cancer Discov 2013;3:1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu M, Liu J, Xiao J, Yang L, Cai M, Shen H, et al. Lnc‐mg is a long non‐coding RNA that promotes myogenesis. Nat Commun 2017;8:14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xia T, Liao Q, Jiang X, Shao Y, Xiao B, Xi Y, et al. Long noncoding RNA associated‐competing endogenous RNAs in gastric cancer. Sci Rep 2014;4:6088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013;495:333–338. [DOI] [PubMed] [Google Scholar]
- 34. Maddock DM, Burton MP. Some effects of starvation on the lipid and skeletal muscle layers of the winter flounder, Pleuronectes americanus . Can J Zool 1994;72:1672–1679. [Google Scholar]
- 35. Fuster G, Busquets S, Almendro V, López‐Soriano FJ, Argilés JM. Antiproteolytic effects of plasma from hibernating bears: a new approach for muscle wasting therapy? Clin Nutr 2007;26:658–661. [DOI] [PubMed] [Google Scholar]
- 36. Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL. IGF‐I stimulates muscle growth by suppressing protein breakdown and expression of atrophy‐related ubiquitin ligases, Atrogin‐1 and MuRF1. Am J Physiol Endocrinol Metab 2004;287:E591–E601. [DOI] [PubMed] [Google Scholar]
- 37. DeVol DL, Rotwein P, Sadow JL, Novakofski J, Bechtel PJ. Activation of insulin‐like growth factor gene expression during work‐induced skeletal muscle growth. Am J Physiol 1990;259:E89–E95. [DOI] [PubMed] [Google Scholar]
- 38. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase Atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The IGF‐1/PI3K/Akt pathway prevents expression of muscle atrophy‐induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 2004;14:395–403. [DOI] [PubMed] [Google Scholar]
- 40. Zhang ZK, Li J, Guan D, Liang C, Zhuo Z, Liu J, et al. A newly identified lncRNA MAR1 acts as a miR‐487b sponge to promote skeletal muscle differentiation and regeneration. J Cachexia Sarcopenia Muscle 2018;9:613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differential expression analysis of lncRNAs in breast muscle between hypertrophic and leaner broilers.
Table S2. Differential expression analysis of genes in breast muscle between hypertrophic and leaner broilers.
Table S3. Differential expression analysis of miRNAs in breast muscle between hypertrophic and leaner broilers.
Table S4. Differentially expressed miRNAs and theirs corresponding target genes with differential expression in hypertrophic and leaner broilers.
Table S5. Differentially expressed miRNAs and theirs corresponding target lncRNAs with differential expression in hypertrophic and leaner broilers.
Table S6. Summary of the represented networks generated by IPA analysis.
Table S7. The full length sequences of lncIRS1 and IRS1
Table S8. Information of Primers.
Fig S1 A competing endogenous RNA (ceRNA) hypothesis.
LncRNAs and mRNAs, which share the common miRNA response elements (MREs), could influence each other's levels by competing for a limited pool of miRNA. (a) Upregulation of lncRNA increases cellular concentrations of particular MREs and can result in the derepression of mRNA that contains the same MREs. (b) While downregulation of lncRNA would lead to decreased levels of particular MREs, and, consequently inhibited the expression of mRNA.
Fig S2 miRNA‐gene interaction network
Hypertrophic broiler vs. leaner broiler, differentially expressed miRNAs and their corresponding differentially expressed target genes with opposition expression pattern were used to construct a miRNA‐gene interaction network. In this network, miRNAs are displayed as pink circles, and miRNA target genes are displayed as blue circles. Dashed lines represent the interactions between differentially expressed miRNAs and theirs corresponding target genes.
Fig S3 miRNA‐lncRNA interaction network
Hypertrophic broiler vs. leaner broiler, differentially expressed miRNAs and their corresponding differentially expressed target lncRNAs with opposition expression pattern were used to construct a miRNA‐lncRNA interaction network. In this network, miRNAs are displayed as pink circles, and miRNA target lncRNAs are displayed as green circles. Solid lines represent the interactions between differentially expressed miRNAs and theirs corresponding target lncRNAs.
Fig S4 Ingenuity Pathway Analysis (IPA) identification of gene networks
IPA online software was used to identify the gene‐gene interaction networks, using the genes in the miRNA‐gene network as input. (a‐d) A total of four major gene networks were identified. Nodes shaded in blue represent the genes from miRNA‐gene network are also involved in the IPA interaction network. White nodes represent transcription factors (as opposed to gene from miRNA‐gene network) that are associated with the regulation of some of these genes, based on the evidence stored in the IPA knowledgebase. Edges and node are annotated with labels that illustrate the nature of the relationship between the genes and their functions.
Fig S5 GO and pathway analysis of the gene from lncRNA‐miRNA‐gene network
(a) The top 10 enriched terms for three GO categories (biological process, cellular component and molecular function) for the genes belonging to lncRNA‐miRNA‐gene network. (b) The top 10 enriched pathway for the genes belonging to lncRNA‐miRNA‐gene network. Pink bars represent the pathway associated with muscle growth. (c) The network between genes and theirs associated pathway. Yellow oval represent the genes that from lncRNA‐miRNA‐gene network, while blur bar represent these pathway that they enrich in. (d) Pathway analysis indicates that the genes (IRS1) from lncRNA‐miRNA‐gene are enriched in the IGF‐1 signaling pathway. IRS1 (nodes shaded) function in the IGF‐1 signaling pathway.
Fig S6 LncIRS1 acts as a sponge of miR‐15a, miR‐15b‐5p and miR‐15c‐5p to regulate the expression of IRS1
MiR‐15a, miR‐15b‐5p and miR‐15c‐5p inhibit the RNA expression level of lncIRS1 (a), and simultaneously depress the expression of IRS1 (b). (c) Co‐expression of miR‐15a, miR‐15b‐5p or miR‐15c‐5p, and lncIRS1 upregulate the RNA expression level of IRS1.