

Abstract
Cachexia is a complex metabolic process that is associated with several end‐stage organ diseases. It is known to be also associated with advanced dementia, although the pathophysiologic mechanisms are still largely unknown. The present narrative review is aimed at presenting recent insights concerning the pathophysiology of weight loss and wasting syndrome in dementia, the putative mechanisms involved in the dysregulation of energy balance, and the interplay among the chronic clinical conditions of sarcopenia, malnutrition, and frailty in the elderly. We discuss the clinical implications of these new insights, with particular attention to the challenging question of nutritional needs in advanced dementia and the utility of tube feeding in order to optimize the management of end‐stage dementia.
Keywords: Advanced dementia, Cachexia, Failure to thrive, Frailty, Sarcopenia, Supportive care
Introduction
Cachexia is a complex metabolic process associated with underlying terminal illnesses including end‐stage renal disease, cancer, advanced heart and lung failure, and others. It is mainly characterized by anorexia and loss of fat and muscle mass.1 Approximately 10–40% of patients with chronic diseases, including heart failure, chronic obstructive pulmonary disease, cancer, human immunodeficiency virus, and renal and hepatic failure become cachexic. However, reliable estimates for the incidence of cachexia in the elderly are not available. The interplay between sarcopenia, malnutrition, and inactivity, which show increasing prevalence with ageing, makes this group particularly vulnerable to cachexia.2
Cachexia usually presents as severe wasting, and it is frequently associated with insulin resistance (IR), myolysis, and systemic inflammation. The Special Interest Group on cachexia–anorexia in chronic wasting diseases of the European Society for Clinical Nutrition and Metabolism recently proposed a consensus definition to differentiate between cachexia and sarcopenia.3 Accordingly, a diagnosis of cachexia is based on a set of core criteria,3 namely, the presence of an underlying chronic disease, an unintentional weight loss of >5% of the usual body weight during the last 6 months, and anorexia or anorexia‐related symptoms.
Cachexia is viewed as a multifactorial syndrome that is characterized by an acute loss of body weight (fat and muscle mass) and increased protein catabolism in the setting of an underlying disease. Cachexia increases morbidity and mortality, and as such, it is a highly relevant clinical condition. Major contributors to cachexia include systemic inflammation, increased muscle proteolysis, and impaired carbohydrate, protein, and lipid metabolism.3
Taking this as the current scientific background, the present narrative review is aimed at assessing the impact of cachexia in advanced dementia. A literature search in PubMed, Medline, and the Cochrane databases of all articles published with the medical subject heading keywords ‘aging’, ‘dementia’, ‘Alzheimer's disease’, ‘cachexia’, ‘sarcopenia’, ‘older adults’, ‘frailty’, ‘nutrition’, and ‘anorexia of ageing’ was carried out. The keywords were matched in all of the potential combinations, and all types of studies, from bench and animal models to the clinical field, were included. The review provides an overview of the anorexia of ageing, sarcopenia, frailty, and cachexia in older adults and their clinical interplay.
In particular, the evidence from bench to bedside on the cachexia of advanced dementia is provided, with a focus on the pathophysiology and underlying molecular mechanisms. The review concludes with a discussion of the nutritional needs of elderly patients with advanced dementia, including implications and challenges for future research.
Anorexia of ageing
One of the core characteristics of cachexia is the presence of anorexia. The anorexia of ageing, namely, the loss of appetite and decreased food intake later in life, was first recognized as a physiological syndrome more than 30 years ago, framing a key paradigm for geriatric syndromes. From this, the major cause of the anorexia of ageing lies on an alteration in fundal compliance, with an increase in antral stretch and enhanced cholecystokinin activity, leading to early satiety. It is further hypothesized that the anorexia of ageing arises from inflammation‐driven losses of appropriate hypothalamic responses to orexigenic and anorexigenic signals. The mechanisms that are involved in age‐related changes in the specific activities of brain areas such as the hypothalamus in response to peripheral stimuli including nutrients, hormones, and adipokines are complex.4 Reduced appetite and a decreased total energy expenditure are common in older individuals, but the frail elderly and those with chronic co‐morbidities often show an increased basal metabolism. Reduced total energy expenditure, along with biological and physiological changes (reduced lean body mass, changes in hormonal profiles, fluid–electrolyte dysregulation, delayed gastric emptying, and diminished sense of smell and taste), lead to the anorexia of ageing.5 In addition, multiple morbidities, polypharmacy, and social and psychological vulnerabilities all play a role in the complex aetiology of anorexia and in the subsequent onset of malnutrition in the elderly. The physiological anorexia of ageing places the older person at increased risk of weight loss and wasting syndrome when an acute illness intervenes. Indeed, the pro‐inflammatory cytokine burst usually accompanies the onset of an acute illness, and it has been associated with the risk of a sudden worsening of age‐related anorexia.5 In turn, the physiologic anorexia of ageing that increases the risk of sudden weight loss and malnutrition in the presence of a physical or psychological illness may also precipitate sarcopenia.6 The development of the anorexia of ageing is influenced by co‐morbidity and directly interferes with nutritional status and favours malnourishment.5, 6, 7, 8, 9 Functional impairment, social and environmental factors, and polypharmacy represent only some of the risk factors associated with age‐related weight loss.8
Notwithstanding the evidence so far, the interplay among anorexia, sarcopenia, and cachexia remains only partly understood, especially in the highly vulnerable elderly population,5 representing an ongoing research challenge.
Sarcopenia
It is noteworthy that cachexic older adults are co‐morbid for sarcopenia, but those who are sarcopenic are rarely co‐morbid for cachexia. Sarcopenia is a core characteristic of the proposed definition for cachexia,10 and sarcopenia and cachexia are two major markers of malnutrition in older adults. Sarcopenia has been reported to affect 5–13% of persons aged 60 to 70 years and up to 50% of those aged over 80 years.11, 12 Reduced muscle mass, tone, and strength in the elderly are independently associated with increased risk of functional impairment, falls, disability, decreased physical performance, poorer quality of life, and mortality.13 In the year 2000, costs attributed directly to sarcopenia accounted for 1.5% of the total healthcare expenditure, and it is estimated that a 10% reduction in the prevalence of sarcopenia would save 1.1 billion dollars in health‐related costs.14
Frailty and cachexia
Frailty in the geriatric patient is a syndrome resulting from cumulative age‐related declines across multiple physiologic systems, with impaired homeostatic reserve and a diminished ability to withstand environmental stressors. This syndrome is associated with greater vulnerability to adverse health outcomes such as falls, hospitalizations, institutionalization, and mortality.
There are three main paradigms of frailty. Fried et al.15 emphasizes the physiologic view of frailty, defining it as a physiological syndrome characterized by the reduction of functional reserves and a diminished resistance to stressors due to the cumulative decline of multiple physiological systems, which causes vulnerability and adverse consequences. According to Fried's definition, the onset of frailty is heralded by at least three of the following readily identifiable changes: unintended weight loss, exhaustion, weakness, slow gait speed, and reduced physical activity. There is a significant overlap between frailty and sarcopenia. In fact, most of the frail elderly population is sarcopenic, which suggests a common pathogenic mechanism.
The second theory of frailty supports the biopsychosocial model proposed by Gobbens et al.16 which views frailty as a dynamic state characterized by losses in any functional domain (physical, psychic, or social) that are ‘caused by the influence of multiple variables that increase the risk of adverse health outcomes’.
Finally, Rockwood et al.17 has proposed an alternative definition of frailty, which is based on counting the number of clinical deficits accumulated over time. This theory also views frailty as a dynamic condition that is caused by a loss of physiological systems complexity, reflected by the functional state, diseases, cognitive deficits, psychosocial risk factors, and geriatric syndromes, including malnutrition and sarcopenia According to the Rockwood frailty index, the frailty trajectory is a continuum that ends with a failure to thrive that mainly resembles cachexia.
Again, the conceptual framework of frailty in the elderly views sarcopenia and malnutrition as the main drivers for the acceleration of frailty, with cachexia as the end‐stage status (Figure 1 ).
Figure 1.
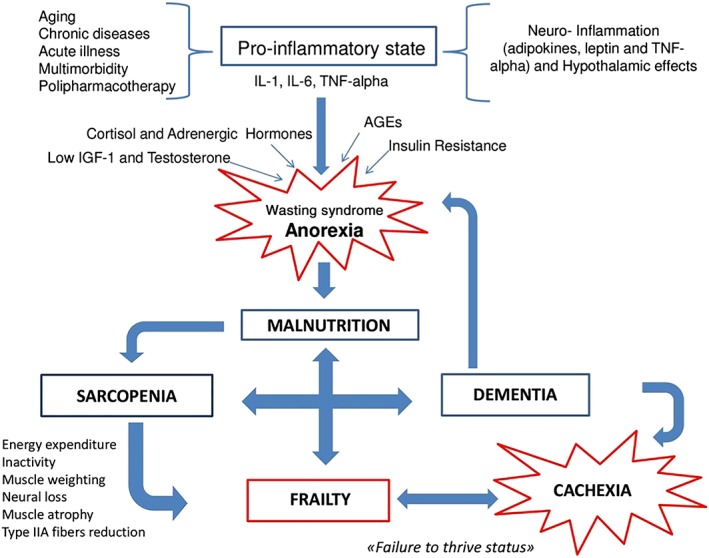
The interplay between anorexia/malnutrition, sarcopenia, cachexia, and dementia. AGEs, advanced glycation end products; IL, interleukin; TNF‐alpha, tumour necrosis factor alpha.
In line with these assumptions, the general concept of frailty goes beyond physical factors to encompass psychological and social dimensions as well. This may include cognitive decline and dementia as core features of frailty, which significantly shape its clinical trajectory over time.
The main core features of these intertwined clinical entities are illustrated in Table 1.
Table 1.
Definition and core features of cachexia, sarcopenia, anorexia of ageing, and frailty syndrome
| Definition | Anorexia | Comorbidity | Functional limitation | Energy intake | Resting energy expenditure | |
|---|---|---|---|---|---|---|
| Cachexia | Unintentional weight loss of >5% of the usual body weight during the last 6 months | +++ | +++ | +++ |
|
|
| Sarcopenia | Low muscle mass | + | +/− | +++ |
|
|
| Low muscle strength | ||||||
| Low physical performance | ||||||
| Anorexia of ageing | Loss of appetite and decreased food intake later in life | +++ | +/− | +/− |
|
|
| Frailty syndrome | Multisystem syndrome of low physiological reserves, with a diminished capacity to respond to stressors | ++ | ++ | ++ |
Cachexia and dementia
It is noteworthy that scant data are available on the role of sarcopenia in dementia and that even fewer studies are focused on the link between cachexia and dementia, especially in its advanced stages.18 This putative association becomes particularly relevant in light of the anticipated global increase in the incidence of dementia, with up to nine million new cases of Alzheimer's disease (AD) projected by the year 2040. The estimated prevalence of severe dementia among individuals older than 85 (the oldest old) is much higher, ranging from 25 to 45%.19 Alzheimer's dementia is a progressive degenerative disorder that leads to an eventual loss of independence, and it is one of the leading causes of death in the elderly.20 The natural history of dementia spans over 10 years, and the later stages of the disease are marked by substantial unintentional weight loss, malnutrition, sarcopenia, anorexia, lethargy, altered immune function, and cachexia.21
It is well known that patients with dementia develop nutritional disorders early and over the course of their disease. The National Institute of Neurological and Communicative Disorders and Strokes Task Force on AD includes weight loss among the clinical features that are consistent with a diagnosis of AD.22 This weight loss is a multifactorial event, related not only to the cognitive impairment that is associated with loss of appetite and reduced food intake but also to AD‐linked alterations of energy consumption due to hypothalamic feeding dysregulation, olfactory changes and psycho‐behavioural disturbances, and the dysphagia (apraxia of swallowing) that is a common feature in the later stages of the disease, and which may also worsen malnutrition.23
Loss of body weight in AD is also typically associated with sarcopenia, which leads to further functional decline, greater disability, and increased clinical vulnerability, and perpetuates the cycle of altered food consumption and decreased energy intake.
While it is still argued that the weight loss associated with AD may be entirely prevented by counteracting all of these predisposing causes, there is also strong evidence that weight loss is probably a true manifestation of the disease itself.
Although epidemiological evidence suggests that middle‐aged individuals with obesity are at a higher risk of developing dementia, older adults seem to display a more likely association between weight loss and dementia. The physiological weight loss that accompanies ageing is exaggerated in older patients with AD, even years before the cognitive decline sets in.24 Moreover, weight loss seems to progress according to the natural history of the disease, leading to failure to thrive in the advanced stages with common features that resemble cachexia.25
Unintentional weight loss may in fact be a harbinger of AD, appearing before the detectable clinical onset of dementia. Although the association between early weight loss and dementia is still far from being understood, it is known that patients with AD sometimes exhibit a paradoxical pattern of overeating concomitant with weight loss.26 This pattern may suggest a hypermetabolic state,27, 28 although it is unknown whether metabolic abnormalities occur in patients with AD, because no significant changes in basal metabolism have yet been observed in association with the disease.29, 30
From the clinical point of view, there are several behaviours that are commonly associated with Alzheimer's dementia that could contribute to daily metabolic disturbances, including wandering, increased physical activity, and sundowning with circadian disturbances, all of which involve increased energy expenditure.31, 32, 33, 34
The unexplained weight loss and low body mass index that have been associated with AD may also be linked to an increase in the resting metabolic rate.35 This finding supports the theory that central regulators of body composition and energy balance, such as hypothalamic control of adipose metabolism, are altered in AD.
Further research suggests that body composition may influence AD‐associated energy demands. To test this hypothesis, the resting metabolic state that counts for a large part of the daily energy expenditure for the maintenance of homeostasis has been investigated.30
The debate continues over whether the wasting syndrome is a prodromal symptom of dementia or whether it appears during the intermediate stages of the disease, driving sarcopenia to cachexia in the late stages of an irreversible failure to thrive.
In line with that, whether cachexia may represent the first clinical symptom of dementia or a mere side symptom along with its natural history is still far from being understood. So far, further studies with regard to these questions are warranted to implement this conceptual framework.
However, so far, in the advanced stages of dementia, the acceleration of weight loss seems to share identifiable features with cachexia.
Pathophysiology
Metabolic homeostasis
It has been suggested that neuroinflammation contributes to the wasting syndrome in dementia, leading to cachexia.36, 37 Appetite‐controlling metabolites and adipokines have been implicated in this pathophysiology. Alterations in plasma levels of leptin and tumour necrosis factor alpha (TNF‐α) are prominent anorexic signals in patients with AD, as supported by the finding of a female gender dimorphism in the levels of circulating anorectic adipokines in AD patients.38 Pro‐inflammatory cytokines are known to mimic appetite‐regulating peptides and to suppress glucose‐sensitive neurons. Leptin and TNF‐α have been mostly implicated in this immunological hypothesis, linking AD and anorexia–cachexia. Nonetheless, the role of these adipokines and their pathophysiological contribution to cachexia remains only partially elucidated.
AD has also been defined as an eating disorder, and the role of several appetite‐controlling metabolites has been investigated. A few in vivo models, particularly regarding the cachexia of dementia, have provided evidence of the metabolic consequences of β‐amyloid‐induced dementia.
Namely, experiments in rat models have demonstrated that the injection of β‐amyloid into the hippocampus induces metabolic disturbances and involuntary weight loss, both of which are early potential indicators of AD.39
Several models of amyloid deposition in AD have described reduced body weight and increasing feeding behaviour.27, 28, 40 Specifically, Tg 2576 amyloid precursor protein mice have exhibited weight reduction and increased energy expenditure, without any perturbation of feeding behaviour.41, 42
In addition, abnormalities in circadian rhythms were observed in the same murine model, combined with decreased body weight, hyperactivity, and increased agitation.43, 44, 45, 46
In keeping with that, the previous results39 expanded this research. In fact, β‐amyloid injection into the hippocampus induced immediate morphological alterations and weight loss after 3 weeks in both diabetic and non‐diabetic rats, and, interestingly, visceral fat was preserved at the expense of lean mass. These reports are consistent with the hypothalamic‐mediated cachexia that causes loss of appetite, anorexia, involuntary weight loss, and lethargy in patients with advanced dementia.
The mouse model of tau protein accumulation in AD has also advanced the understanding of cachexia in advanced dementia. Hyperactivity, increased agitation, and decreased body weight have all been observed in animal models of tau deposition, including THY‐22 (P301S mutation under a Thy1.2 promotor) and Tg4510 mice.47, 48
Further evidence shows that transgenic mice overexpressing the tau protein seemed to eat more yet weighed less than their non‐transgenic littermates.
In addition, this recent in vivo model measured the time course of changes in the metabolic state over the lifespan of the tau‐depositing Tg4510 mice. Interestingly, the mice weighed less at older ages, and this was paralleled by the pathologic accumulation of tau and by dramatic increases in physical activity.
A wasting phase began at 12 months, near the end of the Tg4510 mouse lifespan, with a considerable decrease in the resting metabolic rate, although hyperactivity and food intake were maintained. Hyperphosphorylated tau was detected in the hypothalamus at both 7 and 10 months, which may have contributed to the variation in energy expenditure, food efficiency, and body weight.
Furthermore, the volume of adipose tissue and the plasma leptin levels were significantly decreased in aged mice, indicating a dysregulation of the hypothalamic adipostat control mechanism and energy storage. This last finding is in line with the results of Ishii et al.,42 who reported that amyloid precursor protein Tg2576 mice showed hypothalamic leptin signalling dysfunction, leading to body weight deficits. The observations seem to correlate to the dysfunctional orexigenic effects of neuropeptide Y in the hypothalamus.
Originally, in the THY‐Tau mouse model, a reduction of serotoninergic neurons was observed,22 possibly being involved in the reduction of serotonin (5HT) as an anorexic neurotransmitter.48
It is noteworthy that the Tg4510 model of tauopathy depicted a biphasic energy metabolism response. In particular, the phenotype observed in those mice aged 12 months bears some resemblance to the symptoms of cachexia.
These recent findings support a key relevance of tau in weight loss and metabolic dysregulation, especially in light of the hypothesis that hypermetabolism is responsible for weight loss in AD.
The in vivo tauopathy model of AD showed that a progressively hyperactive phenotype with an increased metabolic rate was not adequately compensated by increased food intake in 7‐month‐old mice. As these mice reached the end of their lifespan (average longevity is 11.5 months), the hyperactivity persisted, but it was ultimately compensated by a reduced resting metabolic rate, despite lower energy intake, at 12 months of age.
These animal models of cachexia‐related metabolic disturbances and dysregulated metabolic homeostasis could add knowledge to the understanding of both cognitive deficit and muscle wasting syndrome in dementia, until its final stages.
Insulin and glucose homeostasis
The pathophysiology of the wasting syndrome in dementia is even more poorly understood. It is known that AD is associated with impaired glucose signalling, which is a key risk factor for diabetes, and some have proposed that AD should be considered type 3 diabetes. The progressive onset of IR that occurs with ageing is a result of the progressive metabolic remodelling that characterizes the ageing process. It affects anthropometric, endocrine, and metabolic parameters, and it is manifested by the aberrant regulation of glucose and protein metabolism.49, 50
However, the evidence consistently supports the hypothesis that the IR of ageing is not simply a routine metabolic finding. Rather, it may also be a major risk factor for many age‐related diseases. Age‐related IR is widely known to be associated with altered lipid metabolism, impaired endothelial function, pro‐thrombotic status, and increased inflammatory response, and it is also involved in the regulatory functions of the brain.
Reductions in glucose metabolism have been found in the hippocampus of AD patients, and intravenous insulin infusion to maintain normal plasma glucose levels can improve cognitive function in both AD patients and in cognitively healthy volunteers. This finding reliably indicates that normal glucose metabolism is required for optimal cognitive performance, and the achievement and maintenance of good glycaemic control may be the most important way to prevent the onset and progression of cognitive decline in diabetes.
The pathophysiologic impact of diabetes in cognitive impairment has not been completely elucidated, and the impact of diabetes on the brain, particularly in relation to cognitive decline, is still under investigation. IR, hyperinsulinaemia, hyperglycaemia, hypoglycaemia, neuroinflammation, and vascular disease certainly play a role. Oxidative stress, advanced glycation products, and impaired insulin signalling in the brain also underlie the association between diabetes and cognitive dysfunction.
Insulin is able to cross the blood–brain barrier and exerts its effects by binding to a specific receptor, which is distributed throughout the brain (mainly in the hippocampus and the cortex), and although its role in the brain is not fully understood,51, 52, 53 insulin is known to be not only a regulator of energy homoeostasis and food intake but also a modulator of brain activities such as learning and memory, mainly through its influence upon the release and reuptake of other neurotransmitters. Accordingly, patients with AD were found to have lower cerebrospinal fluid insulin levels.54, 55
Type 2 diabetes is also associated with cognitive impairment, but as mentioned previously, it is characterized by hyperinsulinaemia and IR. Insulin is neurotrophic at moderate concentrations. However, high levels of insulin in the brain are associated with reduced β‐amyloid clearance.55, 56 This is because insulin‐degrading enzyme (IDE) is required for both insulin and β‐amyloid degradation in microglia and neurons.56 However, IDE is more selective for insulin than for β‐amyloid. Therefore, hyperinsulinaemia in type 2 diabetes essentially deprives the brain of its main β‐amyloid clearance mechanism, leading to the inevitable accumulation of β‐amyloid in the brain and promoting many of the pathological changes associated with AD.57
The accumulation of adiposity in the body that is characteristic of clinical obesity is also linked to insulin dysregulation, and an estimated 80% of obese patients will exhibit IR. Insulin inhibits the action of lipase in adipocytes, which leads to a decrease in the release of free fatty acids (FFAs) from the adipose tissue. This process is compromised by IR, leading to chronic elevations in plasma FFA levels, which promote neuroinflammation, induce the tissue accumulation of β‐amyloid, and inhibit β‐amyloid clearance, all of which, as described previously, are involved in the pathogenesis of cognitive decline.57
Elevated concentrations of plasma FFA strongly and specifically inhibit IDE, which plays a pivotal role in the modulation of insulin signalling and β‐amyloid clearance. Moreover, FFAs also appear to be involved in the formation and accumulation of amyloid and tau filaments in the brain tissue. FFAs directly and indirectly activate the inflammatory response though interactions with TNF‐α and other pro‐inflammatory cytokines that are also involved in the pathogenesis of AD and, in turn, are correlated with IR and hyperinsulinaemia.
There is also a direct correlation between adiposity and blood leptin levels, which may be affected in conditions associated with cognitive decline. Furthermore, a growing body of evidence shows that leptin also has various effects on brain health, cognition, and ageing. Accordingly, the discovery of leptin receptors in the hippocampus, hypothalamus, amygdala, and cerebellum strongly indicates the potential existence of regulatory mechanisms.57, 58
Notwithstanding all of the molecular pathways involved in the pathogenesis of AD, and the key findings concerning the dual role of insulin in cognition and in the regulation of metabolism, it is still largely unknown whether these same mechanisms can also be implicated in the development of cachexia associated with the later stages of neurodegeneration.
Mitochondrial dysfunction and oxidative stress
Age‐related changes in redox homeostasis have been proposed to play a role in sarcopenia and cachexia. However, to date, the evidence for this comes mainly from the investigation of cancer‐related muscle wasting syndromes.59, 60
In the broader conceptual framework, oxidative stress causing increased levels of reactive oxygen species is among the commonest mechanisms of cachexia. In particular, soluble atrophic factors that are produced by various chronic diseases induce an oxidative imbalance characterized by increasing levels of oxidant species, such as O2−, H2O2, and OH, and decreasing levels of antioxidant species, such as catalase, glutathione peroxidase, and superoxide dismutase. This oxidative burst is mediated by the mitochondria as well as by xanthine oxidase and NADPH oxidase complex. Oxidative stress, in turn, increases oxidation‐dependent protein modification, autophagy deregulation, myonuclear apoptosis, mitochondrial dysfunction, and ubiquitin proteasome and calpain activity.61
Recent data obtained from experiments in knockout and transgenic rodents seem to support a role for an unbalanced mitochondrial redox environment in age‐related mitochondrial dysfunction and impaired mitophagy, and it is now thought that mitochondrial dysfunction also plays a role in the loss of muscle in age‐ and cancer‐related cachexia.62 Namely, mitochondrial quality control derangements targeting mitochondrial dynamics, mitochondrial tagging for disposal, and mitophagy signalling are considered key checkpoints for the development of cancer cachexia. Moreover, dysfunctional mitochondrial quality control and neuroinflammation have been considered to be at the crossroads of ageing and muscle wasting disorders. In particular, mitochondria‐derived damage‐associated molecular patterns have been specifically implicated in experimental sarcopenia and cachexia models.63
However, our comprehensive literature search found that the current body of evidence mainly concerns cachexia associated with cancer, end‐stage pulmonary disease, heart failure, and renal failure. Therefore, there is a knowledge gap in regarding cachexia and dementia, and our understanding of the pathophysiological background is currently based on mere extrapolation from these other findings.
In summary, the study of the relationship between cachexia and advanced dementia is in its infancy, and there are persuasive arguments to support further investigation of these unexplored signalling pathways.
From bench to bedside
Nutritional needs in advanced dementia
Cachexia is a hypercatabolic state, and, theoretically, nutritional interventions containing 1.5 g/kg/day of protein should be recommended to counteract catabolism.64
However, in line with both European Society for Clinical Nutrition and Metabolism65 and National Institute for Health and Care Excellence guidelines,66 there is not sufficient evidence to support the use of dietary supplements to maintain the nutritional status of patients with dementia. Enteral nutrition may be useful in patients with mild to moderate dementia and reversible malnutrition, but neither guideline recommends the use enteral nutrition in the terminal phase of dementia, although the physician's decision must ultimately rest on each patient's general prognosis and preferences.67
Conversely, the Japan Gastroenterological Endoscopy Society has recommended percutaneous endoscopic gastrostomy (PEG) placement in patients with malnutrition due to cerebrovascular disease or dementia, with the assumption that early PEG placement is associated with longer survival.68 The impact of tube feeding on mortality was equivalent for patients with dementia, without dementia, or in those diagnosed with other neurological conditions, while patients with dementia had decreased mortality compared with those with strokes and increased mortality compared with those with tumours. Trials of caloric supplementation or targeted protein supplements, such as branched‐chain amino acids or creatine, have not shown consistent clinical benefit for cachexia prevention.69
As dementia progresses, it becomes increasingly difficult to maintain body weight through conventional feeding.70 Some researchers have reported the benefit of high‐calorie supplementation in more moderate stages of dementia, with clinical improvement of nutritional indices including body mass index, arm circumference, arm muscle circumference, and total lymphocyte count.71
However, nutritional interventions for patients with severe dementia are inconsistent, and a series of biases such as co‐morbidity hamper the generalization of existing results.65, 72
Although cachexia represents the final stage of frailty and dementia, it is systematically disregarded among cachexia‐related clinical conditions such as cancer or end‐stage organ failure.73
From a pathophysiological perspective, sarcopenia, frailty, and dementia are largely mediated by a hypercatabolic and inflammatory state, with deleterious effects on muscle mass and brain, and while nutritional interventions in sarcopenia resulted in fundamental therapeutic advantages, especially in the earlier stages, nutritional interventions have demonstrated minimal effect on cachexia in general, and there is no evidence specific to cachexia associated with dementia. Thus, cachexia might be considered a refractory symptom in the clinical trajectory of dementia, culminating in the failure to thrive, where the current definition of “refractory symptom” is based on the palliative concept of resistance to change‐oriented stimuli.74 That is, it is a symptom that may be not adequately controlled in spite of adequate and maximal therapy.
In line with these assumptions, Callahan75 has recommended that in late‐stage dementia, therapy and care should be shifted towards end‐of‐life and palliative issues, in order to maximize dignity and quality of life.76 Patients with advanced dementia commonly experience burdensome symptoms such as dyspnoea, pain, and agitation, similarly to patients who are dying with cancer,77 with fewer tools for systematic assessment.78
Artificial nutrition and advanced dementia
Dysphagia is a major risk factor for poor clinical outcomes, including pneumonia, malnutrition, and cachexia. Dysphagia secondary to advanced dementia is a progressive, irreversible, and incurable condition with a multifactorial pathogenesis that involves apraxia, cognitive fluctuation, impulsivity, reduced physical mobility, poor dentition, and dependence for feeding and medications. Tube feeding [artificial nutrition and hydration (ANH)] has been proposed as a means of protein and calorie supplementation for patients in the final stages of dementia to maintain skin integrity, prevent aspiration pneumonia and other infectious complications, improve the functional status, and extend survival. ANH has been popularized as a caring and nurturing intervention, while forgoing such measures has been equated with neglect and abandonment.79 As such, the implementation of ANH in the advanced stages of dementia has become a controversial and emotional issue.
PEG tubes are routinely used for tube feeding in advanced dementia patients with neurogenic dysphagia and cachexia, and as many as 30% of all PEG tubes are placed in patients with dementia.80 The current data on feeding tube placement in patients with severe dementia (Table 1) are mainly retrospective in nature, with extrapolation from mixed populations, and to date, feeding tubes have shown little clinical benefit for the amelioration of malnutrition and the prevention of pressure sores and aspiration pneumonia. Similarly, no clinical benefit has been observed for the improvement of functional status, quality of care, and overall survival.
To the contrary, PEG placement is often burdensome, with high rates of tube‐related complications, including pain, suture breakage, abdominal wall cellulitis and abscess, stoma inflammation, bleeding, stenosis of the stoma, and haematoma, as well as mechanical complications, including erosions, tube leakage or blockage, tube migration and loose fixation plate, and tube malfunction due to a kinked or fractured tube, which are frequently overlooked. Major complications, such as pleuro‐pneumonitis, ileus, reflux, bowel obstruction, anorexia, fever, sepsis, and fluid overload and metabolic disturbances, are also reported, and agitation, self‐extubation,81 abuse of physical restraints, and loss of special aspects of food contribute to diminished well‐being and quality of life. The direct mortality from the placement of a PEG tube is generally low, ranging from 0 to 2%, but the complication rates may range from 15 to 70%.
Henderson et al. have found that that weight loss and severe depletion of lean and fat body mass persisted in tube‐fed patients with advanced dementia even after a standard enteral formula was provided daily for 1 year.82
Similarly, other studies have shown that nutritional markers such as haemoglobin, haematocrit, albumin, and serum cholesterol levels do not improve after a feeding tube is placed.83
It has also been demonstrated that weight loss progressively worsened in parallel with the duration of the tube feeding.84 This last finding seems to highlight the irreversible progression of cachexia alongside the limitations and potentially detrimental effects of ANH.
It seems that the scientific background turns out to be a key determinant, supporting the need for future research aimed at establishing the best clinical interventions according to an updated pathophysiological conceptual framework.
Moreover, it has been reported that the incidence of decubitus ulcers did not significantly differ between patients with dementia who had feeding tubes and those without ANH.85, 86, 87 Similarly, Teno et al. observed a higher incidence of new pressure ulcers, with poorer healing of existing ulcers, in patients with advanced dementia who were tube fed, compared with those without feeding tubes.88 These previous studies also showed that feeding tubes did not reduce the frequency of aspiration pneumonia in patients with advanced dementia.85 In particular, a metanalysis compared the incidence of aspiration between jejunostomy tubes and traditional PEG tubes with no additional clinical benefits between the two types.89
In addition, a retrospective study and several cohort nursing home studies have not shown any role for PEG feeding in improving mental status, functional status, mobility, or overall survival over a period of 18 months,84, 90 and the initiation of tube feeding during hospitalization was also not associated with increased survival.91, 92, 93
In contrast, few studies have reported no harm outcome from tube feeding placement in patients with dementia, even if retrospective in nature.94, 95 Namely, Takenoshita et al. has demonstrated that tube feeding decreased pneumonia and antibiotic use in patients with advanced dementia, extending survival rate as well.94 Similarly, enteral nutrition for patients with dementia was reported to prolong survival. Additionally, PEG tube feeding was observed to be safer than nasogastric tube feeding among patients in psychiatric hospitals.95
These last findings do not necessarily indicate that tube feeding should be administered in patients with severe dementia. However, the scientific debate on which ethical decision should be formulated, when facing the nutritional issue in advanced dementia and cachexia, should carefully consider patient's quality of life, before deciding the use or disuse of tube feeding.
A 2009 Cochrane review of observational studies concluded that there was insufficient evidence to support the benefits of tube feeding in patients with advanced dementia in terms of survival, quality of life, nutrition, functional status, prevention of aspiration, or prevention and healing of pressure ulcers.96
Thus, feeding tubes do not appear to be a useful palliative measure, and the use of ANH in end‐stage dementia should be generally discouraged, as it will only prolong the process of dying and may also increase discomfort and suffering.
Although the line between cachexia and advanced dementia is established along with the refractory nature of this clinical entity, no specific research has been conducted on the relationship between cachexia itself and any meaningful clinical interventions (Table 2).
Table 2.
Evidence for the efficacy of percutaneous endoscopic gastrostomy tube feeding in the improvement of overall mortality
| Authors | Study design | Benefita | Subjects | Advanced dementia | Follow‐up months | Size of series, n | Type of feeding | Mean age, years | Mortality |
|---|---|---|---|---|---|---|---|---|---|
| Alvarez‐Fernandez et al.97 | Prospective observational cohort | No | C | DSM‐IV FAST > 7A | 30 | 67 | 14 NG | 82.2 | 37.3% at 2 years |
| Cintra et al.98 | Prospective observational cohort | No | H, C | FAST >7A | 6 | 67 | 31 EN (28 NG) | 84.79 |
At 3 months, 41.9% EN; At 6 months, 58.1% EN |
| Non‐randomized | |||||||||
| 36 CG | |||||||||
| Not blinded | |||||||||
| Peck et al.85 | Prospective cohort | Yes | NH | MMSE | 6 | 104 | 52 EN | 87 | NA |
| 52 CG | |||||||||
| Meier et al.92 | Prospective observational cohort | No | H | FAST >6D | 60 | 99 | EN | 84 | At 6 months, 50% |
| Mitchell et al.99 |
Prospective observational cohort MDS |
No | NH | CPS > 4 | 24 | 1386 | 135 EN | NA | NA |
| Kuo et al.100 |
Prospective observational cohort MDS |
No | NH | CPS > 4 | 12 | 97 111 | 3337 (53.6/1000) EN | 84.8 | At 1‐year, 64.1% |
| Teno et al.91 | Cohort national data set (MDS) | No | NH | CPS > 6 | 6 | 36 492 | 1.957 (5.4%) EN (PEG) | 84.9 | NA |
| Teno et al.88 | Cohort national data set (MDS) | No | NH | CPS > 6 | 6 | 18 021 | 1.124 EN (PEG) CG | 82.5 |
30‐day mortality rate, 2.0% PEG; 180‐day mortality rate, 24% PEG |
| Nair et al.101 | Prospective cohort | No | C, H | NA | 6 | 88 | 55 EN (PEG) 33 CG | 83.3 | 44% EN vs. 6% CG |
| Murphy and Lipman 102 | Retrospective observational cohort | No | C | NA | 24 | 41 | 23 EN (PEG) | NA | NA |
| Kaw and Sekas84 | Retrospective observational cohort | No | NH | NA | 18 | 46 | EN (PEG) | 73.6 |
At 12‐months, 50%; At 18‐months, 60% |
| Ticinesi et al.103 | Prospective observational non‐randomized un‐blinded | no | H, C | FAST ≥ 5 CDR ≥ 1 | 24 | 184 | 54 EN (PEG) | 82.2 | 70% PEG vs. 40% EN |
| Henderson et al.82 | Prospective cohort | No | NH | NA | 12 | 40 | EN | NA | NA |
| Ciocon et al.83 | Prospective cohort | No | C | NA | 11 | 70 | 15 PEG 55 NG | NA | 40% |
| Callahanet al.90 | Prospective cohort | No | C | FAST | 14 | 70 | 15 PEG 55 NG | 78.9 |
At 30 days, 22%; At 1 year, 50% |
| Arinzon et al.104 | Prospective cohort | No | H | MMSE | 167 | 57 EN (42 NG, 15 PEG)110 CG | 80.17 | EN 42% vs. CG 27% | |
| Jaul et al.105 | Prospective cohort | Yes | H | MMSE | 17 | 95 | 69 EN (62 NG, 7 PEG) 26 CG | 79 | NA |
| Rudberg et al.106 | Prospective cohort (MDS) | Yes | NH | CPS > 6 | 12 | 1545 | 353 EN (mixed tube type) 1192 CG | 84.63 | At 1 year, 60% CG vs. 50% EN |
| Takenoshita et al.94 | Retrospective observational cohort | Yes | H | CDR‐SoB FAST | 24 | 58 | 46 EN 12 CG | 79.6 | NA |
| Takayama et al.95 | Retrospective observational cohort | Yes | H | FAST | 24 | 185 | 150 EN (60 PEG 90 NG) 35 CG | 76.6 | NA |
C, community dwelling patients; CDR, clinical dementia rating; CDR‐SoB, clinical dementia rating sum of boxes; CG, control group in oral nutrition; CPS, Cognitive Performance Score; DSM‐IV, Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; EN, enteral nutrition by a feeding tube; FAST, Functional Assessment Staging; H, hospitalized patients; MDS, minimum data set; NA, not available; NG, nasogastric tube; NH, nursing home; PEG, percutaneous endoscopic gastrostomy.
For more details on the effectiveness/complications of enteral nutrition, see Table A1.
In summary, in line with these assumptions, no approved treatment or recommendation for reversing cachexia and advanced dementia can be formulated at present.
End‐of‐life issues, advanced dementia, and palliative care
Patients who survive to the final phase of dementia are more likely to die from cachexia, and the development of cachexia is a common final background that needs inclusion in a palliative conceptual framework.67 The elaboration of grief, surrogate decision‐making, and lack of effectiveness of nutrition might magnify the caregiver's burden,2 and the final phase of dementia unleashes an onslaught of emotions. Sorrow, grief, fear, and anger may pervade the final stages of this terminal illness. However, our understanding of end‐of‐life issues thus far is extrapolated from terminal illnesses other than dementia.
The clinical course of advanced dementia has been described in the Choices, Attitudes and Strategies for Care of Advanced Dementia at the End of life (CASCADE) study, which prospectively enrolled nursing home residents and showed a median survival of 1.3 years, with the most common clinical complications being eating problems (86%), febrile episodes (53%), and pneumonia (41%).107, 108
Similarly, the Study of Pathogen Resistance and Exposure to Antimicrobials in Dementia (SPREAD) investigated nursing home residents with advanced dementia, indicating that urinary and respiratory tract infections were the main causes for 1‐year mortality.109
Approximately half of the patients with advanced dementia receive a diagnosis of pneumonia in the last 2 weeks of life, but palliative care needs may be overlooked in favour of inappropriate use of antibiotics, representing disproportionate therapeutic aggressiveness.110
In a cohort study of nursing home residents with advanced dementia, hospice care was provided for only 22.3% and 29.9% of those facing imminent death.111 The factors associated with greater likelihood of hospice referral were the presence of an eating problem and the perception by family members that the resident had fewer than 6 months to live. Those who received hospice services had fewer unmet needs during the last week of life.112
These findings emphasize the need to improve end‐of‐life care for elders with advanced dementia.
The application of palliative care principles to the natural history of AD, and especially to its advanced stages, should guide communication about treatment goals and family education in order to avoid potentially futile and onerous clinical interventions. The rate of hospice placement for end‐stage dementia has improved dramatically over the past two decades,113 but end‐of‐life care in these patients remains a challenging issue.113
Lack of prognostic tools in advanced dementia may be a key barrier to delivering hospice services and excellent end‐of‐life care for these patients.114 Indeed, better prognostic awareness among family members has been associated with decreased use of burdensome interventions during the last 90 days of life among nursing home residents with advanced dementia.115
The best practices for tailoring interventions and hospice care expertise to the individual needs of patients with advanced dementia are far from being fully understood, but up‐to‐date knowledge, forethought, and compassion allow hospice providers and geriatric palliative care providers to ensure that patients with dementia will die with peace and dignity.76
In terms of the strong need to reconcile the pan‐culturally symbolic act of feeding with dysphagia or refusal to eat and cachexia in end‐stage dementia, clinical observations have confirmed the benefit of minimal interventions including swabs, sips of water, ice chips, lubrication of the lips, and oral comfort feeding.116 Oral comfort feeding (hand feeding) meets the symbolic significance of food, and re‐establishes the pleasure of sharing food and of the social interaction with the caregiver.117 It is also effective for eliminating feelings of hunger or thirst,118 and it may represent a valuable alternative to ANH.72 Comfort feeding offers a clear, goal‐oriented alternative to tube feeding and eliminates the troublesome care/no care dichotomy imposed by current recommendations against ANH.
Conclusions and future directions
In the 50 years since Bernard Isaacs defined the ‘geriatric giants’ of immobility, instability, incontinence, and impaired intellect/memory, the understanding of those giants has evolved to include four additional syndromes: frailty, sarcopenia, the anorexia of ageing, and dementia.119 In the current review, we hypothesized that cachexia represents the final common pathway of the four modern‐day giants. Cachexia and the failure to thrive commonly occur near the end of life, and the failure to thrive is characterized by the patient's inability to tolerate aggressive and inappropriate treatments.
To date, investigations on cachexia and dementia are relatively rare, and caution is advised in the interpretation of results that are merely extrapolated from cachexia associated with other end‐stage diseases.
Therefore, if cachexia represents a true symptom of dementia or a side symptom is still an unsolved question.
It is conceivable to draw three different pathways that might underpin the role of cachexia in dementia. First, it could be hypothesized that cachexia represents the progression of brain neurodegeneration; the more amyloid burden affects hypothalamus and the feeding regulation system, the more cachexia develops as a side clinical effect.
Moreover, it could be considered a multifactorial origin for cachexia in dementia, including the metabolic deregulation (basal metabolism and energy expenditure) and the cognitive and behavioural correlates as key determinant factors for the onset and progression of the wasting syndrome, especially in the later stages of dementia.
Not least, cachexia could be attributed as being a metabolic disorder. Namely, a systemic increase of pro‐inflammatory cytokines might promote IR as well as secreting wasting tissue factors, ultimately responsible for the dysfunction in specific organs and the metabolic driven progression of the wasting syndrome to cachexia, according to the natural history of dementia.
To date, clinical interventions aimed at reversing cachexia in advanced dementia have proven mostly ineffective, even if largely unexplored.
Few efforts have been made to understand the pathophysiology of cachexia in dementia, and the current evidence to this regard is insufficient to fill the knowledge gap.
Future research should aim to synthesize evidence from the bench and the bedside to provide a useful pathophysiological framework that supports a clinical decision‐making process towards the most precisely tailored interventions for this vulnerable patient population.
Furthermore, prospective studies are needed to better depict the trajectory of cachexia in dementia. Greater understanding of patient distress, prognoses, decision‐making, and family burden will allow the caregiving community to identify the most effective strategies to improve end‐of‐life care in patients with advanced dementia.
Conflict of interest
All authors declare that they have no conflict of interest.
Acknowledgements
The authors would like to thank the BioMed Proofreading® LLC for the English editing service. The manuscript does not contain clinical studies or patient data. The authors certify that they comply with the ‘Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017.120
Appendix 1.
Table A1.
Effectiveness/complications of enteral nutrition
| Authors | Complication rate (%) | Effectiveness | Survival | Factors influencing survival |
|---|---|---|---|---|
| Alvarez‐Fernandez et al.97 | 77.6% during 1 year.Pressure sores: 43.3%.UTI: 32.8%.Pneumonia: 29.9%.Dehydration: 29.9%. | NA | NA |
Pneumonia Permanent NG Albumin < 3.4 g/dL |
| Cintra, 201498 | Pressure ulcers: 1.31 ± 1.55 CGvs. 2.74 ± 3.31 ENAspiration pneumonia: 58.1% vs. 25.0% | NA | NA |
Feeding route Dementia Number of pressure ulcers |
| Peck et al.85 | Aspiration pneumonia: 58% EN vs. 17% CGDecubitus ulcers: 21% EN vs. 14% CGRestraints: 71% EN vs. 56% CG | Increased weight 48% EN vs. 17% CG | NA | NA |
| Meier et al.92 | NA | NA | Median 175 days.No increased survival | NA |
| Mitchell et al.99 | NA | NA | No increased survival | NA |
| Kuo et al.100 | Tube replacement: 20% Hospitalization rate: 1.01% | NA | Median 56 days | NA |
| Teno et al.91 | NA | NA | No increased survival | Timing of PEG tube insertion not associated with improved survival |
| Teno et al.88 | Duplicate risk of new pressure ulcer | No improvement in healing of existing pressure ulcers | NA | NA |
| Nair et al.101 | Cellulitis 7%, GI bleeding > 5%, fever 21% within 72 h of PEG placement (common cause: aspiration pneumonia14%) | No improvement in PS by Karnofsky.No difference in weight, BMI, triceps skinfold thickness, midarm muscle circumference, serum cholesterol, or total lymphocyte count | NA |
Albumin > 2.8 g/dL Predictor of survival >6 months |
| Murphy and Lipman102 | 4.3% | NA | No increased survival (median 59 vs. 60 days) | NA |
| Kaw and Sekas84 | 34.7% | No improvement in functional and nutritional status | NA | Albumin ≥ 3.5 g/dL associated with improved survival |
| Ticinesi et al.103 | NA | NA | No increased survival | EN |
| Henderson et al.82 | NA | No improvement in nutritional status | NA | NA |
| Ciocon et al.83 | Aspiration pneumonia 43% in NG, 56% in PEG.Agitation and self‐extubation 67% in NG, 44% in PEG. | NA | NA | NA |
| Callahan et al.90 | NA | No improvement in functional, nutritional, or subjective health status | NA | NA |
| Arinzon et al.104 | 61% EN vs. 34% CG higher Norton scale in EN | Improvements in laboratory data (haemoglobin, lymphocyte count, serum proteins, renal function). No improvement in cognitive and functional status. | NA | NA |
| Jaul et al.105 | NA | No improvement in nutritional status or on healing pre‐existing pressure ulcers | Significantly higher in EN group | NA |
| Rudberg et al.106 | NA | NA | Survival at 1 year: 39% CG vs. 50% EN | Feeding tubes associated with a reduced risk of death |
| Takenoshita et al.94 | Decreased pneumonia and antibiotic use | NA | EN group significantly longer survival (23 vs. 2 months) | Feeding tubes associated with a reduced risk of death |
| Takayama et al.95 | NA | NA | EN group significantly longer survival (711 vs. 61 days) | Feeding tube and female gender associated with a reduced risk of death |
BMI, body mass index; CG, control group in oral nutrition; EN, enteral nutrition by a feeding tube; GI, gastrointestinal bleeding; NA: not available; NG, nasogastric tube; PEG, percutaneous endoscopic gastrostomy; PS, performance status; UTI, urinary tract infection.
Minaglia, C. , Giannotti, C. , Boccardi, V. , Mecocci, P. , Serafini, G. , Odetti, P. , and Monacelli, F. (2019) Cachexia and advanced dementia. Journal of Cachexia, Sarcopenia and Muscle, 10: 263–277. 10.1002/jcsm.12380.
References
- 1. Morley JE. Undernutrition: a major problem in nursing homes. J Am Med Dir Assoc 2011;12:243–246. [DOI] [PubMed] [Google Scholar]
- 2. Ali S, Garcia JM. Sarcopenia, cachexia and aging: diagnosis, mechanisms and therapeutic options – a mini‐review. Gerontology 2014;60:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muscaritoli M, Anker SD, Argilés J, Aversa Z, Bauer JM, Biolo G, et al. Consensus definition of sarcopenia, cachexia and pre‐cachexia: joint document elaborated by Special Interest Groups (SIG) “cachexia–anorexia in chronic wasting diseases” and “nutrition in geriatrics”. Clin Nutr 2010. Apr;29:154–159. [DOI] [PubMed] [Google Scholar]
- 4. Conte C, Cascino A, Bartali B, Donini L, Rossi‐Fanelli F, Laviano A. Anorexia of aging. Curr Nutr Food Sci 2009;5:9–12. [Google Scholar]
- 5. Morley JE. Anorexia of aging: a key component in the pathogenesis of both sarcopenia and cachexia. J Cachexia Sarcopenia Muscle 2017. Aug;8:523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morley JE. Pathophysiology of the anorexia of aging. Curr Opin Clin Nutr Metab Care 2013;1:27–32. [DOI] [PubMed] [Google Scholar]
- 7. Landi F, Russo A, Liperoti R, Tosato M, Barillaro C, Pahor M, et al. Anorexia, physical function, and incident disability among the frail elderly population: results from ilSIRENTE study. J Am Med Dir Assoc 2010. May;11:268–274. [DOI] [PubMed] [Google Scholar]
- 8. Landi F, Lattanzio F, Dell'Aquila G, Eusebi P, Gasperini B, Liperoti R, et al. Prevalence and potentially reversible factors associated with anorexia among older nursing home residents: result from the ULISSE project. J Am Med Dir Assoc 2013;14:119–124. [DOI] [PubMed] [Google Scholar]
- 9. Loreck E, Chimakurthi R, Steinle NI. Nutritional assessment of the geriatric patient: a comprehensive approach toward evaluating and managing nutrition. Clin Ger 2012;20:20–26. [Google Scholar]
- 10. Evans WJ, Morley JE, Argilés J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr 2008;27:793–799. [DOI] [PubMed] [Google Scholar]
- 11. Morley JE. Sarcopenia: diagnosis and treatment. J Nutr Health Aging 2008;12:452–456. [DOI] [PubMed] [Google Scholar]
- 12. Bauer JM, Kaiser MJ, Sieber CC. Sarcopenia in nursing home residents. J Am Med Dir Assoc 2008. Oct;9:545–551. [DOI] [PubMed] [Google Scholar]
- 13. Janssen I, Heymsfield SB, Ross R. Low relative skeletal muscle mass (sarcopenia) in older persons is associated with functional impairment and physical disability. J Am Ger Soc 2002;50:889–896. [DOI] [PubMed] [Google Scholar]
- 14. Cruz‐Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. Sarcopenia: European consensus on definition and diagnosis: report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010. Jul;39:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fried LP, Ferrucci L, Darer J, Williamson JD, Anderson G. Untangling the concepts of disability, frailty, and comorbidity: implications for improved targeting and care. J Gerontol A Biol Sci Med Sci 2004. Mar;59:255–263. [DOI] [PubMed] [Google Scholar]
- 16. Gobbens RJ, Luijkx KG, Wijnen‐Sponselee MT, Schols JM. In search of an integral conceptual definition of frailty: opinions of experts. J Am Med Dir Assoc 2010. Jun;11:338–343. [DOI] [PubMed] [Google Scholar]
- 17. Rockwood K, Andrew M, Mitnitski A. A comparison of two approaches to measuring frailty in elderly people. J Gerontol A Biol Sci Med Sci 2007. Jul;62:738–743. [DOI] [PubMed] [Google Scholar]
- 18. Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001. Mar;56:M146–M156. [DOI] [PubMed] [Google Scholar]
- 19. Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013. May 7;80:1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Katzman R. Editorial: the prevalence and malignancy of Alzheimer disease. A major killer. Arch Neurol 1976;33:217–218. [DOI] [PubMed] [Google Scholar]
- 21. Folstein MF, Whitehouse PJ. Cognitive impairment of Alzheimer disease. Neurobehav Toxicol Teratol 1983. Nov‐Dec;5:631–634. [PubMed] [Google Scholar]
- 22. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984. Jul;34:939–944. [DOI] [PubMed] [Google Scholar]
- 23. Morley JE. Anorexia of aging: physiologic and pathologic. Am J Clin Nutr 1997;66:760–773. [DOI] [PubMed] [Google Scholar]
- 24. Johnson DK, Wilkins CH, Morris JC. Accelerated weight loss may precede diagnosis in Alzheimer disease. Arch Neurol 2006. Sep;63:1312–1317. [DOI] [PubMed] [Google Scholar]
- 25. Emmerzaal TL, Kiliaan AJ, Gustafson DR. 2003–2013: a decade of body mass index, Alzheimer's disease, and dementia. J Alzheimers Dis 2015;43:739–755. [DOI] [PubMed] [Google Scholar]
- 26. Wolf‐Klein GP, Silverstone FA, Levy AP. Nutritional patterns and weight change in Alzheimer patients. Int Psychogeriatr 1992. Summer;4:103–118. [DOI] [PubMed] [Google Scholar]
- 27. Vloeberghs E, Van Dam D, Franck F, Serroyen J, Geert M, Staufenbiel M, et al. Altered ingestive behavior, weight changes, and intact olfactory sense in an APP overexpression model. Behav Neurosci 2008. Jun;122:491–497. [DOI] [PubMed] [Google Scholar]
- 28. Morgan D, Gordon MN. Amyloid, hyperactivity, and metabolism: theoretical comment on Vloeberghs et al. (2008). Behav Neurosci 2008. Jun;122:730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Donaldson K. E, Carpenter W. H, Toth M. J, Goran M.I., Newhouse P, Poehlman E.T. No evidence for a higher resting metabolic rate in noninstitutionalized Alzheimer's disease patients. J Am Geriatr Soc 1996. Oct;44(10):1232–1234. [DOI] [PubMed] [Google Scholar]
- 30. Niskanen L, Piirainen M, Koljonen M, Uusitupa M. Resting energy expenditure in relation to energy intake in patients with Alzheimer's disease, multi‐infarct dementia and in control women. Age Ageing 1993. Mar;22:132–137. [DOI] [PubMed] [Google Scholar]
- 31. Devanand DP, Jacobs DM, Tang MX, Del Castillo‐Castaneda C, Sano M, Marder K, et al. The course of psychopathologic features in mild to moderate Alzheimer disease. Arch Gen Psychiatry 1997. Mar;54:257–263. [DOI] [PubMed] [Google Scholar]
- 32. Devanand DP, Miller L, Richards M, Marder K, Bell K, Mayeux R, et al. The Columbia University Scale for Psychopathology in Alzheimer's disease. Arch Neurol. 1992. Apr;49:371–376. [DOI] [PubMed] [Google Scholar]
- 33. Lopez OL, Wisniewski SR, Becker JT, Boller F, De Kosky ST. Psychiatric medication and abnormal behavior as predictors of progression in probable Alzheimer disease. Arch Neurol 1999. Oct;56:1266–1272. [DOI] [PubMed] [Google Scholar]
- 34. Scarmeas N, Brandt J, Blacker D, Albert M, Hadjigeorgiou G, Dubois B, et al. Disruptive behavior as a predictor in Alzheimer disease. Arch Neurol 2007. Dec;64:1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reyes‐Ortega G, Guyonnet S, Ousset PJ, Nourhashemi F, Vellas B, Albarède JL, et al. Weight loss in Alzheimer's disease and resting energy expenditure (REE), a preliminary report. J Am Geriatr Soc 1997. Nov;45:1414–1415. [DOI] [PubMed] [Google Scholar]
- 36. Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement. 2016. Jun;12:719–732. [DOI] [PubMed] [Google Scholar]
- 37. Burfeind KG, Michaelis KA, Marks DL. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin Cell Dev Biol 2016. Jun;54:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Intebi AD, Garau L, Brusco I, Pagano M, Gaillard RC, Spinedi E. Alzheimer's disease patients display gender dimorphism in circulating anorectic adipokines. Neuroimmunomodulation 2002. –2003;10:351–358. [DOI] [PubMed] [Google Scholar]
- 39. James D, Kang S, Park S. Injection of β‐amyloid into hippocampus induces metabolic disturbances and involuntary weight loss which may be early indicators of Alzheimer's disease. Aging Clin Exp Res 2014. Feb;26:93–98. [DOI] [PubMed] [Google Scholar]
- 40. Pugh PL, Richardson JC, Bate ST, Upton N, Sunter D. Non‐cognitive behaviours in an APP/PS1 transgenic model of Alzheimer's disease. Behav Brain Res 2007. Mar 12;178:18–28. [DOI] [PubMed] [Google Scholar]
- 41. Brownlow ML, Benner L, D' Agostino D, Gordon MN, Morgan D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer's pathology. PLoS One 2013. Sep 12;8:e75713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ishii M, Wang G, Racchumi G, Dyke JP. Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J Neurosci 2014. Jul 2;34:9096–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brownlow ML, Joly‐Amado A, Azam S, Elza M, Selenica ML, Pappas C, et al. Partial rescue of memory deficits induced by calorie restriction in a mouse model of tau deposition. Behav Brain Res 2014. Sep 1;271:79–88. [DOI] [PubMed] [Google Scholar]
- 44. Leboucher A, Laurent C, Fernandez‐Gomez F. J, Burnouf S, Troquier L, Eddarkaoui S et al. Detrimental effects of diet‐induced obesity on tau pathology are independent of insulin resistance in tau transgenic mice. Diabetes 2013. May;62(5):1681–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dickey C, Kraft C, Jinwal U, Koren J, Johnson A, Anderson L, et al. Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am J Pathol 2009. Jan;174:228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005. Jul 15;309:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jul P, Volbracht C, de Jong IE, Helboe L, Elvang AB, Pedersen JT. Hyperactivity with agitative‐like behavior in a mouse tauopathy model. J Alzheimers Dis 2016;49:783–795. [DOI] [PubMed] [Google Scholar]
- 48. Van der Jeugd A, Blum D, Raison S, Eddarkaoui S, Buee L, D'Hooge R. Observations in THY‐Tau22 mice that resemble behavioural and psychological signs and symptoms of dementia. Behav Brain Res 2013. Apr 1;242:34–39. [DOI] [PubMed] [Google Scholar]
- 49. Barbieri M, Boccardi V, Papa M, Paolisso G. Metabolic journey to healthy longevity. Horm Res 2009. Jan;71:24–27. [DOI] [PubMed] [Google Scholar]
- 50. Barbieri M, Gambardella A, Paolisso G, Varricchio M. Metabolic aspects of the extreme longevity. Exp Gerontol 2008. Feb;43:74–78. [DOI] [PubMed] [Google Scholar]
- 51. Zhao WQ, Alkon DL. Role of insulin and insulin receptor in learning and memory. Mol Cell Endocrinol 2001;177:125–134. [DOI] [PubMed] [Google Scholar]
- 52. Garrido G, Furuie S, Buchpiguel C, Bottino C, Almeida O, Cid C, et al. Relation between medial temporal atrophy and functional brain activity during memory processing in Alzheimer's disease: a combined MRI and SPECT study. J Neurol Neurosurg Psychiatry 2002;73:508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Watson GS, Craft S. Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer's disease. Eur J Pharmacol 2004;490:97–113. [DOI] [PubMed] [Google Scholar]
- 54. Geijselaers SLC, Aalten P, Ramakers IHGB, De Deyn PP, Heijboer AC, Koek HL, et al. Association of cerebrospinal fluid (CSF) insulin with cognitive performance and CSF biomarkers of Alzheimer's disease. J Alzheimers Dis 2018;61:309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shiiki T, Ohtsuki S, Kurihara A, Naganuma H, Nishimura K, Tachikawa M, et al. Brain insulin impairs amyloid‐beta (1‐40) clearance from the brain. J Neurosci 2004. Oct 27;24:9632–9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin‐degrading enzyme regulates the levels of insulin, amyloid beta‐protein, and the beta‐amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 2003. Apr 1;100:4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Luchsinger JA, Gustafson DR. Adiposity, type 2 diabetes, and Alzheimer's disease. J Alzheimers Dis 2009;16:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sims‐Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol 2010;6:551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Argilés JM, López‐Soriano FJ, Busquets S. Muscle wasting in cancer: the role of mitochondria. Curr Opin Clin Nutr Metab Care 2015. May;18:221–225. [DOI] [PubMed] [Google Scholar]
- 60. Argilés JM, Busquets S, Stemmler B, López‐Soriano FJ. Cachexia and sarcopenia: mechanisms and potential targets for intervention. Curr Opin Pharmacol 2015. Jun;22:100–106. [DOI] [PubMed] [Google Scholar]
- 61. Abrigo J, Elorza AA, Riedel CA, et al. Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxid Med cell Longevity 2018;2018:2063179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marzetti E, Lorenzi M, Landi F, Picca A, Rosa F, Tanganelli F, et al. Altered mitochondrial quality control signaling in muscle of old gastric cancer patients with cachexia. Exp Gerontol 2017. Jan;87:92–99. [DOI] [PubMed] [Google Scholar]
- 63. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Bossola M, et al. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res 2018. Jan;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Op den Kamp CM, Langen RC, Haegens A, Schols AM. Muscle atrophy in cachexia: can dietary protein tip the balance? Curr Opin Clin Nutr Metab Care 2009. Nov;12:611–616. [DOI] [PubMed] [Google Scholar]
- 65. Volkert D, Chourdakis M, Faxen‐Irving G, Frühwald T, Landi F, Suominen MH, et al. ESPEN guidelines on nutrition in dementia. Clin Nutr. 2015. Dec;34:1052–1073. [DOI] [PubMed] [Google Scholar]
- 66. National Institute for Health and Care Excellence . Dementia: support people with dementia and their carers in health and social care. 2006. Available online: http://www.nice.org.uk/guidance/cg42/resources/guidance‐dementia‐pdf (accessed on 1 July 2014).
- 67. Brooke J, Ojo O. Enteral nutrition in dementia: a systematic review. Nutrients 2015. Apr 3;7:2456–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kumagai R, Kubokura M, Sano A, Shinomiya M, Ohta S, Ishibiki Y, et al. Clinical evaluation of percutaneous endoscopic gastrostomy tube feeding in Japanese patients with dementia. Psychiatry Clin Neurosci 2012. Aug;66:418–422. [DOI] [PubMed] [Google Scholar]
- 69. Baldwin C, Spiro A, Ahern R, Emery PW. Oral nutritional interventions in malnourished patients with cancer: a systematic review and meta‐analysis. J Natl Cancer Inst 2012. Mar 7;104:371–385. [DOI] [PubMed] [Google Scholar]
- 70. Pivi GAK, de Andrade Vieira NM, da Ponte JB, de Moraes DSC, Bertolucci PHF. Nutritional management for Alzheimer's disease in all stages: mild, moderate, and severe. Nutrire 2017;42. [Google Scholar]
- 71. Pivi GAK, Silva RV, Juliano Y. A prospective study of nutrition education and oral nutritional supplementation in patients with Alzheimer's disease. Nutr J 2011;10:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Arcand Marcel. End‐of‐life Issues in Advanced Dementia. Part 2: Management of Poor Nutritional Intake, Dehydration, and Pneumonia. Vol 61: April 2015, Canadian Family Physician; [PMC free article] [PubMed] [Google Scholar]
- 73. Huijberts S, Buurman BM, De Rooij SE. End‐of‐life care during and after an acute hospitalization in older patients with cancer, end‐stage organ failure, or frailty: a sub‐analysis of a prospective cohort study. Palliat Med 2016. Jan;30:75–82. [DOI] [PubMed] [Google Scholar]
- 74. Cherny NI, Portenoy RK. Sedation in the management of refractory symptoms: guidelines for evaluation and treatment. J Palliat Care 1994. Summer;10:31–38. [PubMed] [Google Scholar]
- 75. Callahan D. Terminating life‐sustaining treatment of the demented. Hastings Cent Rep 1995;25:25–31. [PubMed] [Google Scholar]
- 76. Stewart JT, Schultz SK. Palliative care for dementia. Psychiatr Clin North Am 2018. Mar;41:141–151. [DOI] [PubMed] [Google Scholar]
- 77. Soares LGL, Japiassu AM, Gomes LC, Pereira R, Peçanha C, Goldgaber T. Prevalence and intensity of dyspnea, pain, and agitation among people dying with late stage dementia compared with people dying with advanced cancer: a single‐center preliminary study in Brazil. Ann Palliat Med 2018. May 26. pii: apm.2018;05. [DOI] [PubMed] [Google Scholar]
- 78. Monacelli F, Signori A, Roffredo L, Pace K, Nencioni A, Pickering G, et al. Algoplus® scale in older patients with dementia: a reliable real‐world pain assessment tool. J Alzheimers Dis 2017;56:519–527. [DOI] [PubMed] [Google Scholar]
- 79. McCann R. Lack of evidence about tube feeding–food for thought. JAMA 1999. Oct 13;282:1380–1381. [DOI] [PubMed] [Google Scholar]
- 80. Rabeneck L, Wray NP, Petersen NJ. Long‐term outcomes of patients receiving percutaneous endoscopic gastrostomy tubes. J Gen Intern Med 1996. May;11:287–293. [DOI] [PubMed] [Google Scholar]
- 81. Gillick MR. Rethinking the role of tube feeding in patients with advanced dementia. N Engl J Med 2000;342:206–210. [DOI] [PubMed] [Google Scholar]
- 82. Henderson CT, Trumbore LS, Mobarhan S, Benya R, Miles TP. Prolonged tube feeding in long‐term care: nutritional status and clinical outcomes. J Am Coll Nutr 1992. Jun;11:309–325. [DOI] [PubMed] [Google Scholar]
- 83. Ciocon JO, Silverstone FA, Graver LM, Foley CJ. Tube feedings in elderly patients. Indications, benefits, and complications. Arch Intern Med 1988;148:429–433. [PubMed] [Google Scholar]
- 84. Kaw M, Sekas G. Long‐term follow‐up of consequences of percutaneous endoscopic gastrostomy (PEG) tubes in nursing home patients. Dig Dis Sci 1994;39:738–743. [DOI] [PubMed] [Google Scholar]
- 85. Peck A, Cohen CE, Mulvihill MN. Long‐term enteral feeding of aged demented nursing home patients. J Am Geriatr Soc 1990. Nov;38:1195–1198. [DOI] [PubMed] [Google Scholar]
- 86. Finucane TE. Malnutrition, tube feeding and pressure sores: data are incomplete. J Am Geriatr Soc 1995. Apr;43:447–451. [DOI] [PubMed] [Google Scholar]
- 87. Finucane TE, Christmas C, Travis K. Tube feeding in patients with advanced dementia: a review of the evidence. JAMA 1999. Oct 13;282:1365–1370. [DOI] [PubMed] [Google Scholar]
- 88. Teno JM, Gozalo P, Mitchell SL, Kuo S, Fulton AT, Mor V. Feeding tubes and the prevention or healing of pressure ulcers. Arch Intern Med 2012;172:697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lazarus BA, Murphy JB, Culpepper L. Aspiration associated with long‐term gastric versus jejunal feeding: a critical analysis of the literature. Arch Phys Med Rehabil 1990. Jan;71:46–53. [PubMed] [Google Scholar]
- 90. Callahan CM, Haag KM, Weinberger M, Tierney WM, Buchanan NN, Stump TE, et al. Outcomes of percutaneous endoscopic gastrostomy among older adults in a community setting. J Am Geriatr Soc 2000. Sep;48:1048–1054. [DOI] [PubMed] [Google Scholar]
- 91. Teno JM, Gozalo PL, Mitchell SL, Kuo S, Rhodes RL, Bynum JPW, et al. Does feeding tube insertion and its timing improve survival? J Am Geriatr Soc 2012. October;60:1918–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Meier DE, Ahronheim JC, Morris J, Baskin‐Lyons S, Morrison RS. High short‐term mortality in hospitalized patients with advanced dementia: lack of benefit of tube feeding. Arch Intern Med 2001;161:594–559. [DOI] [PubMed] [Google Scholar]
- 93. Mitchell SL, Tetroe JM. Survival after percutaneous endoscopic gastrostomy placement in older persons. J Gerontol A Biol Sci Med Sci 2000. Dec;55:M735–M739. [DOI] [PubMed] [Google Scholar]
- 94. Takenoshita S, Kondo K, Okazaki K, Hirao A, Takayama K, Hirayama K, et al. Tube feeding decreases pneumonia rate in patients with severe dementia: comparison between pre‐ and post‐intervention. BMC Geriatr 2017. Nov 21;17:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Takayama K, Hirayama K, Hirao A, Kondo K, Hayashi H, Kadota K, et al. Survival times with and without tube feeding in patients with dementia or psychiatric diseases in Japan. Psychogeriatrics 2017. Nov;17:453–459. [DOI] [PubMed] [Google Scholar]
- 96. Candy B, Sampson EL, Jones L. Enteral tube feeding in older people with advanced dementia: findings from a Cochrane systematic review. Int J Palliat Nurs 2009. Aug;15:396–404. [DOI] [PubMed] [Google Scholar]
- 97. Alvarez‐Fernández B, García‐Ordoñez MA, Martínez‐ Manzanares C, Gómez‐Huelgas R. Survival of a cohort of elderly patients with advanced dementia: nasogastric tube feeding as a risk factor for mortality. Int J Geriatr Psychiatry 2005. Apr;20:363–370. [DOI] [PubMed] [Google Scholar]
- 98. Cintra MT, de Rezende NA, de Moraes EN, Cunha LC, da Gama Torres HO. A comparison of survival, pneumonia, and hospitalization in patients with advanced dementia and dysphagia receiving either oral or enteral nutrition. J Nutr Health Aging. 2014. Dec;18:894–899. [DOI] [PubMed] [Google Scholar]
- 99. Mitchell SL, Kiely DK, Lipsitz LA. The risk factors and impact on survival of feeding tube placement in nursing home residents with severe cognitive impairment. Arch Intern Med 1997. Feb 10;157:327–332. [PubMed] [Google Scholar]
- 100. Kuo S, Rhodes RL, Mitchell SL, Mor V, Teno JM. Natural history of feeding tube use in nursing home residents with advanced dementia. J Am Med Dir Assoc 2009. May;10:264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nair S, Hertan H, Pitchumoni CS. Hypoalbuminemia is a poor predictor of survival after percutaneous endoscopic gastrostomy in elderly patients with dementia. American Journal of Gastroenterology 2000;95:133–136. [DOI] [PubMed] [Google Scholar]
- 102. Murphy LM, Lipman TO. Percutaneous endoscopic gastrostomy does not prolong survival in patients with dementia. Arch Intern Med 2003;163:1351–1353. [DOI] [PubMed] [Google Scholar]
- 103. Ticinesi A, Nouvenne A, Lauretani F, Prati B, Cerundolo N, Maggio M, et al. Survival in older adults with dementia and eating problems: to PEG or not to PEG? Clin Nutr 2016;35:1512–1516. [DOI] [PubMed] [Google Scholar]
- 104. Arinzon Z, Peisakh A, Berner YN. Evaluation of the benefits of enteral nutrition in long‐term care elderly patients. J Am Med Dir Assoc 2008;9:657–662. [DOI] [PubMed] [Google Scholar]
- 105. Jaul E, Singer P, Calderon‐Margalit R. Tube feeding in the demented elderly with severe disabilities. Israel Medical Association Journal 2006;8:870–874. [PubMed] [Google Scholar]
- 106. Rudberg MA, Egleston BL, Grant MD, Brody JA. Effectiveness of feeding tubes in nursing home residents with swallowing disorders. J Parenter Enteral Nutr 2000;24:97–102. [DOI] [PubMed] [Google Scholar]
- 107. Mitchell SL, Teno JM, Kiely DK, Shaffer ML, Jones RN, Prigerson HG, et al. The clinical course of advanced dementia. N Engl J Med 2009. Oct 15;361:1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mitchell SL, Kiely DK, Jones RN, Prigerson H, Volicer L, Teno JM. Advanced dementia research in the nursing home: the CASCADE study. Alzheimer Dis Assoc Disord 2006. Jul‐Sep;20:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mitchell SL, Shaffer ML, Loeb MB, Givens JL, Habtemariam D, Kiely DK, et al. Infection management and multidrug‐resistant organisms in nursing home residents with advanced dementia. JAMA Intern Med 2014. Oct;174:1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Campos‐Calderón C, Montoya‐Juárez R, Hueso‐Montoro C, Hernández‐López E, Ojeda‐Virto F, García‐Caro MP. Interventions and decision‐making at the end of life: the effect of establishing the terminal illness situation. BMC Palliat Care 2016. Nov 7;15:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kiely DK, Givens JL, Shaffer ML, Teno JM, Mitchell SL. Hospice use and outcomes in nursing home residents with advanced dementia. J Am Geriatr Soc 2010. Dec;58:2284–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Di Giulio P, Toscani F, Villani D, Brunelli C, Gentile S, Spadin P. Dying with advanced dementia in long‐term care geriatric institutions: a retrospective study. J Palliat Med 2008;1:1023–1028. [DOI] [PubMed] [Google Scholar]
- 113. Mitchell SL, Black BS, Ersek M, Hanson LC, Miller SC, Sachs GA, et al. Advanced dementia: state of the art and priorities for the next decade. Ann Intern Med 2012. Jan 3;156:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Mitchell SL, Miller SC, Teno JM, Kiely DK, Davis RB, Shaffer ML. Prediction of 6‐month survival of nursing home residents with advanced dementia using ADEPT vs hospice eligibility guidelines. JAMA 2010. Nov 3;304:1929–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mitchell SL, Kiely DK, Hamel MB, et al. Estimating prognosis for nursing home residents with advanced dementia. JAMA 2004;291:2734–2740. [DOI] [PubMed] [Google Scholar]
- 116. McCann RM, Hall WJ, Groth‐Juncker A. Comfort care for terminally ill patients. The appropriate use of nutrition and hydration. JAMA 1994. Oct 26;272:1263–1266. [DOI] [PubMed] [Google Scholar]
- 117. Palecek EJ, Teno JM, Casarett DJ, Hanson LC, Rhodes RL, Mitchell SL. Comfort feeding only: a proposal to bring clarity to decision‐making regarding difficulty with eating for persons with advanced dementia. J Am Geriatr Soc 2010;58:580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mathew R, Davies N, Manthorpe J, Iliffe S. Making decisions at the end of life when caring for a person with dementia: a literature review to explore the potential use of heuristics in difficult decision‐making. BMJ Open 2016;6:e010416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Morley JE. The new geriatric giants. Clin Geriatr Med 2017. Aug;33:xi–xii. [DOI] [PubMed] [Google Scholar]
- 120. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]