

Abstract
Mutations in the GJB2 gene encoding Connexin26 (Cx26) have been linked to skin disorders and genetic deafness. However, the severity and type of the skin disorders caused by Cx26 mutations are heterogeneous. Here we explored the effect of Cx26 KID syndrome associated mutations, G12R, S17F, and D50N on channel function. The Cx26 N14K mutation was also examined which is associated with deafness but has a skin disorder distinct from the KID syndrome mutations. The proteins were all expressed in Xenopus oocytes with levels equal to wild-type Cx26. The G12R, N14K, and D50N mutations resulted in larger hemichannel currents than the wild-type expressing cells, but the S17F mutation resulted in a complete loss of hemichannel activity. Elevated hemichannel activity correlated with an increased cell death. This result could be reversed through the elevation of calcium (Ca2+) in the extracellular media. Functional gap junctions were only produced by paired N14K cells, which had a similar conductance level to wild-type, even though they exhibited a complete loss of voltage sensitivity. This set of data confirms that aberrant hemichannel activity is a common feature of Cx26 mutations associated with KID syndrome, and this may contribute to a loss of cell viability and tissue integrity.
Introduction:
Mutations in connexin genes are associated with diverse hereditary human diseases (White and Paul, 1999;Wei et al., 2004;Anand and Hackam, 2005;Mese et al., 2007). Sensorineural hearing loss (SNHL) is the most common connexin related disease which can be either non-syndromic, or syndromic, when associated with skin disorders and other ectodermal abnormalities (Gerido and White, 2004;Richard, 2005;van Steensel et al., 2004;Lai-Cheong et al., 2007). SNHL is commonly linked to mutations in the Cx26 encoding gene GJB2 (Petit, 2006). Although mutations in Cx26 have been linked to a few rare autosomal dominant cases of non-syndromic deafness (DFNA3), autosomal recessive mutations account for the vast majority of non-syndromic hearing loss associated with GJB2 (DFNB1) (Petit et al., 2001). There is abundant evidence to suggest that loss of function mutations in Cx26 lead to generation of deafness, but not skin disease (Bruzzone et al., 2003;White, 2000;Zhao et al., 2006). In fact, the most common mutation, 35delG, produces a frame shift and early protein truncation in the amino terminus and accounts for up to 85% of all the mutant GJB2 alleles in non-syndromic deaf populations in Europe (Petit et al., 2001).
Syndromic mutations in Cx26 are associated with a variety of skin disorders such as Vohwinkel syndrome, Bart-Pumphrey syndrome, palmoplantar keratoderma, or Keratitis (and hystrix-like) Ichthyosis Deafness syndrome (KID/HID) and always present with autosomal dominant inheritance (Gerido and White, 2004;Richard, 2005;van Steensel et al., 2004;Lai-Cheong et al., 2007) The lack of associated skin disorders in cases of non-syndromic SNHL shows that the function and development of the epidermis is not affected by the simple loss of Cx26 function as in the case of homozygous 35delG patients. Thus, the Cx26 mutations which can cause syndromic deafness associated with skin disease must show some type of alteration of function, but the mechanisms whereby Cx26 mutation leads to pathological changes in the epidermis remain to be elucidated.
Six connexin subunits oligomerize to form a hemichannel (Goodenough and Paul, 2003). A functional gap junction is formed when two plasma membrane hemichannels from adjacent cells align and create a direct communication pathway between their cytoplasms (Harris, 2001). Each connexin subunit is comprised of 4 transmembrane domains connected by two extracellular and one intracellular loop domains, containing cytoplasmic carboxy- and amino-termini. Historically the formation of complete gap junction channels was thought to be the primary function of connexin subunits, although recently non-junctional hemichannels have been speculated to play a role in cellular homeostasis under different physiological conditions (Bennett et al., 2003;Saez et al., 2005).
Since hemichannels may contribute to normal cell function, disease causing connexin mutations could also mediate their effects through alteration of hemichannel activity. Analysis of connexin mutations causing syndromic SNHL associated with skin disease has supported this idea. Examples include the A40V and G45E mutations of Cx26, both of which cause severe forms of KID syndrome, producing neonatal fatality in the case of G45E (Montgomery et al., 2004;Jonard et al., 2008;Janecke et al., 2005). Both of these mutations displayed aberrant hemichannel activity leading to cell death in vitro, and constitutively active hemichannels was suggested to be the cause of the resultant epidermal pathology (Montgomery et al., 2004;Gerido et al., 2007;Stong et al., 2006). These observations suggested that abnormal hemichannel activity may be a general feature of Cx26 mutations associated with KID/HID disorders.
Four additional Cx26 mutations G12R, N14K, S17F, and D50N have also been linked to cases of KID/HID syndrome (Mazereeuw-Hautier et al., 2007;van Geel et al., 2002;van Geel et al., 2002;Richard et al., 2002;van Steensel et al., 2004). All patients exhibited SNHL symptoms, but the accompanying skin disorders varied in both clinical features and severity. KID/HID disorders cover a broad clinical spectrum, although several subtypes can be distinguished based on prevailing clinical features (Mazereeuw-Hautier et al., 2007). However, there is not yet sufficient data to establish clear genotype-phenotype correlations, or to associate specific reported Cx26 mutations with distinct subtypes.
The functional properties of G12R, N14K, S17F, and D50N were characterized using an in vitro expression assay comprised of cRNA injected Xenopus oocytes. All four mutations displayed significantly different membrane currents than wild-type Cx26. Two of the mutations, D50N and G12R, produced large hemichannel currents that increased with cell depolarization and failed to induce any gap junctional conductance between paired cells. N14K also showed abnormal hemichannel activity that was activated at positive voltages, in addition to producing robust junctional conductance in cell pairs. The voltage gating sensitivity of junctions formed by N14K channels was greatly reduced compared to wild-type Cx26. The results for the final mutation tested, S17F, differed from both the other mutations and wild-type Cx26. S17F expression induced neither hemichannel currents nor gap junctional conductance. When cultured in solutions with higher extracellular Ca2+ concentrations all three of the hemichannel forming mutations had reduced levels of activity and increased cell survival. Three of the four KID mutations tested demonstrated significantly increased hemichannel activity compared to the wild-type protein. Taken together with the aforementioned G45E and A40V mutations, increased hemichannel activity appears to be a common feature among GJB2 mutations responsible for KID/HID syndrome.
Results:
Gap Junction Hemichannel Currents in Single Xenopus Oocytes.
The GJB2 mutations G12R, N14K, S17F, and D50N are the result of single amino acid substitutions in the amino-terminus and first extracellular loop Cx26 and are associated with KID/HID syndrome. To assess the functionality of these mutations, wild-type, G12R, N14K, S17F, and D50N-Cx26 proteins were expressed in Xenopus oocytes. Single cells were subjected to depolarizing voltage pulses and membrane currents were recorded (Figure 1). Oocytes injected with H2O showed negligible current flow for voltages from −30 to +60 mV. The hemichannel activity of wild-type Cx26 injected cells was previously reported and was characterized by outward currents that increased with greater depolarization (Gonzalez et al., 2006;Ripps et al., 2004;Gerido et al., 2007). Three of the KID mutants, G12R, N14K, and D50N, all showed a significant increase in this outward current when compared to the either H2O or Cx26 injected cells. This increased membrane current is associated with a reduction in cell membrane resistance and suggests the presence of hemichannels. Conversely, the S17F mutant showed a reduction in membrane current when compared to Cx26 injected cells, and S17F injected cells were similar to H2O injected negative control cells. This suggested that G12R, N14K, and D50N mutants induced aberrant hemichannel activity, while S17F mutants completely eliminated normal hemichannel activity.
Figure 1.
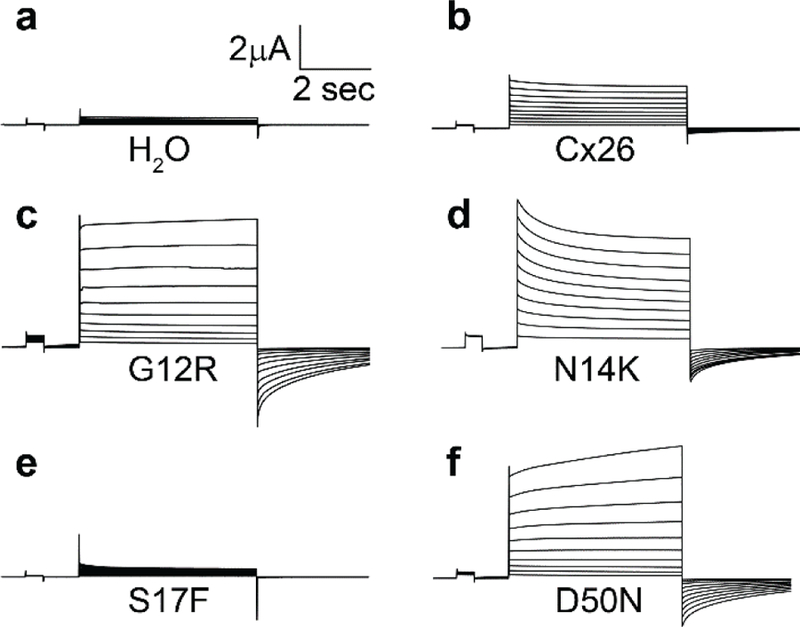
Hemichannel currents recorded from Xenopus oocytes. Cells were held at a potential of −40mV and then membrane currents were recorded at pulses between −30 and +60mV in 10mV steps. (a) H2O injected cells displayed small currents at all potentials. Cells injected with Cx26 (b), G12R (c), N14K (d), and D50N (f) displayed hemichannel currents with different magnitudes. S17F cells (e) had a negligible current at all voltages recorded.
Mean steady state currents were plotted as a function of membrane potential to quantify the hemichannel currents (Figure 2). Control cells injected with H2O showed negligible currents at all tested voltages. Wild-type Cx26 injected cells displayed larger outward currents than H2O injected cells that increased at greater depolarizing voltages. At the highest voltage tested wild-type cells showed a maximum current more than seventeen times greater than the control cells (+60mV, <0.05, students t-test). The G12R, N14K, and D50N expressing cells produced large outward currents at all voltages tested. At +60mV these three mutations exhibited currents that were approximately three times larger than wild-type and more than forty times larger than control cells, differences that were statistically significant (<0.05, one-way ANOVA). This change represents a difference in membrane conductance associated with each mutation that could be attributed to increased hemichannel activity. Conversely, expression of the S17F mutation in cells caused a significant reduction of current compared to wild-type cells and closely mimicked control cells across all voltages. The introduction of this mutation can be attributed to a reduction in hemichannel activity at increased membrane potentials.
Figure 2.

Current-voltage relationship of the wild-type and mutant hemichannels. Cells expressing each mutation were plotted along with H2O (n=10, ▽) and Cx26 (n=8, ◆) injected cells to give a comparison of hemichannel activity. H2O injected cells displayed negligible currents at all voltages tested. Cells injected with (a) G12R (n=7, ○), (b) N14K (n=9, □), and (d) D50N (n=12, △) all had currents that were similar to Cx26 at lower voltages but became dramatically larger than Cx26 at increasingly positive potentials. Oocytes injected with (c) S17F (n=11, ▷) were similar to H2O injected cells and displayed a relative lack of current at all voltageddds tested.
The hemichannel currents recorded from cells expressing the four KID associated mutations were dramatically different from the wild-type currents. This could have been due to changes in relative quantities of the protein that was produced by the cell or due to a change in activity of the hemichannel. To draw a distinction between these two possibilities, we quantified the amount of protein expressed in the Xenopus ooctyes by western blot analysis (Figure 3). The band density for each mutation mimicked that of the wild-type expression, with an intensity normalized to wild-type of close to 1 for all mutations. This showed that the alterations in current in the hemichannels were not the result of a change in total connexin protein levels.
Figure 3.
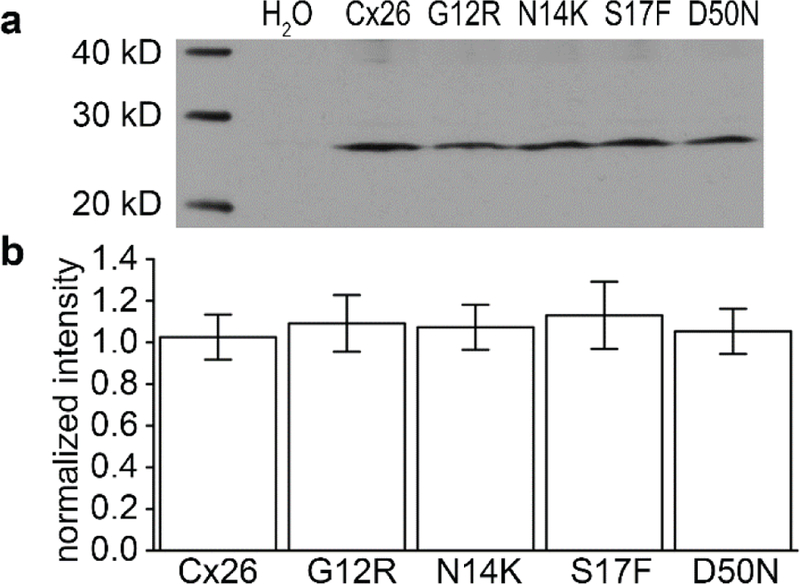
Western blot analysis of protein content from Xenopus oocytes. The expression level of each mutation (G12R, N14K, S17F, and D50N) was similar to Cx26. (a) Membrane extracts were obtained from an equal number of cells and stained with Cx26 antibodies. H2O injected cells were used as a negative control. Cx26, G12R, N14K, S17F, and D50N were seen in their corresponding lanes and in relatively equal amounts. (b) The expression levels were tested via densitometry (n=3) and were found to be equal for all mutations and wild-type (P > 0.05). Data are means ± SE.
Cells Expressing KID Mutations Experienced Ca2+ Dependent Cell Death
Cells expressing the G45E and A40V mutations associated with KID syndrome in the Cx26 gene have been shown to cause an increased rate of cell death (Gerido et al., 2007;Montgomery et al., 2004;Stong et al., 2006). Elevated extracellular Ca2+ levels are known to inhibit connexin hemichannels (Ebihara and Steiner, 1993) and prolonged the lifespan of G45E expressing cells (Stong et al., 2006;Gerido et al., 2007). Cells expressing G12R, N14K, S17F, and D50N mutations were incubated in either Ca2+ free MB solution, or 4mM Ca2+ MB solution immediately after injection with cRNA. Wild-type Cx26 and H2O injected cells were incubated as controls. The effects of Ca2+ on the cell viability depended on the expressed mutation (Figure 4). There was no apparent difference between cells incubated in high Ca2+ or no Ca2+ for conditions with wild-type levels of hemichannel activity or lower (H2O control, Cx26wt, and S17F). These cells were healthy upon visual inspection and showed no signs of cell death after being incubated in media with or without Ca2+. Incubation in media with no Ca2+ had a profound effect for cells expressing G12R, N14K, and D50N. The membranes of these cells exhibited blebbing and ooplasm began leaking from the cells within 8 to 10 hours (Figure 4 top). Typically, G12R, N14K, and D50N expressing cells would undergo lysis and cell death within 24 hours. When G12R, N14K, and D50N were expressed in elevated 4mM Ca2+, the cells were rescued and the membrane integrity was preserved (Figure 4 bottom).
Figure 4.

Hemichannel activity dependent cell death. (a) Cells were injected with either H2O, Cx26, G12R, N14K, S17F, or D50N and incubated in MB-solution overnight. Cells injected with H2O and Cx26 wild-type remained viable and intact. Mutations that caused hemichannel activity (G12R, N14K, and D50N) displayed obvious blebbing and an increased rate of cell death compared to Cx26 cells. The S17F mutation that lacked hemichannel activity did not experience the increased rate of cell death. (b) Injected cells were incubated in 4mM Ca2+ MB solution overnight. The increased rate of cell death and physical deformation of the membrane experienced by mutations with active hemichannels was rescued by elevated Ca2+. Scale bar = 1mm.
To determine if the change in the observed rates of cell death in different Ca2+ concentrations were statistically significant, cell viability was scored by counting the number of cells with visible blebbing every hour between 12 and 24 hours post cRNA injection. Cells expressing wild-type or S17F cRNA showed little variation between 0 and 4 mM Ca2+ media (Figure 5a and b). Oocytes injected with G12R, N14K, and D50N showed very different rates between 0 and 4 mM Ca2+. When incubated in 0 mM Ca2+ these three mutations were statistically different (P < 0.05, students t-test) from wild-type after 24 hours, while S17F injected oocytes were not (P > 0.05, students t-test, Figure 5c). This confirms the observation that cells expressing G12R, N14K, and D50N mutations died at a significantly faster rate than their wild-type counterparts. In contrast, rates of death for S17F injected oocytes were indistinguishable from wild-type. When all cells are incubated in 4 mM Ca2+ instead of 0 mM Ca2+ all of the 4 mutations displayed normal rates of cell death.
Figure 5.
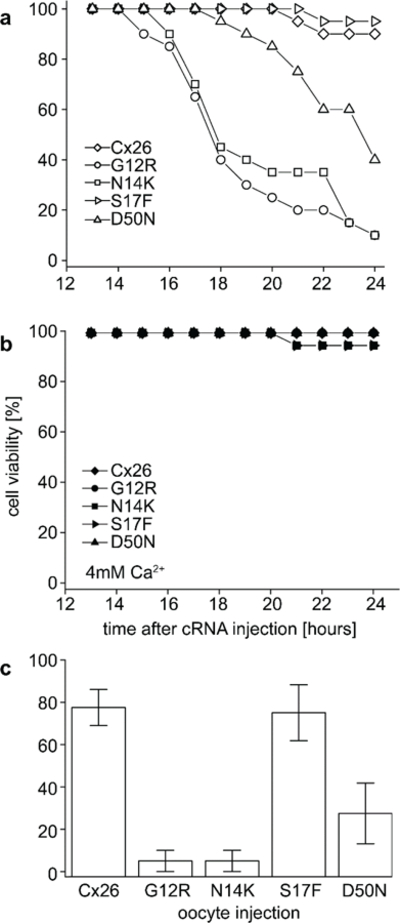
Viability of Cx26 wild-type and mutant expressing cells. (a) Cells in 0 mM Ca2+ media were scored for cell viability based on the appearance of blebbing. Cells were injected with wild-type Cx26, G12R, N14K, S17F, or D50N cRNA and monitored for 24 hours. This data is based on 20 injected cells per experimental condition. (b) Equal numbers of cells expressing the same cRNAs were incubated in 4 mM Ca2+ media overnight and scored in the same fashion. (c) Cell viability at 24 hours post injection for all 5 conditions. The G12R, N14K, and D50N injected cells are statistically different (P < 0.05, students t-test) from wild-type injected cells. Cells injected with S17F were not statistically different from wild-type. Data are the mean ± SE from 4 experiments.
Abnormal Hemichannel Activity was Suppressed by Extracellular Ca2+
Cells expressing KID mutations that resulted in abnormal hemichannel activity also experienced increased cell death in the absence of elevated extracellular Ca2+. This led us to test whether the addition of extracellular Ca2+ that rescued the cells was also blocking the aberrant hemichannel activity of G12R, N14K, and D50N. To assess this, currents were recorded for cells in 0, 1, 2, and 4mM Ca2+ containing MB solutions (Figure 6). Cells containing mutant Cx26 G12R (Figure 6A), N14K (Figure 6B), and D50N (Figure 6C) proteins showed large outward currents in 0mM Ca2+ solutions as documented earlier. The substitution of 1mM Ca2+ led to a significant reduction in membrane currents (p<0.05) but did not display a total reduction in outward current. The aberrant hemichannel currents produced by the mutated proteins in 2 and 4mM Ca2+ continued to decrease. The currents were significantly reduced in a concentration dependent manner, but each mutation showed a different sensitivity to extracellular Ca2+ (Figure 6D). This variation in sensitivity is presumably due to the differential effect of each mutation on channel function. However, the variation does not change the phenotype. The mutations responsible for larger aberrant hemichannels still showed increases in cell survivability and decreases in hemichannel currents in higher levels of extracellular Ca2+. The role of hemichannels in cell death seems to be a common feature of Cx26 mutations associated with KID syndrome.
Figure 6.
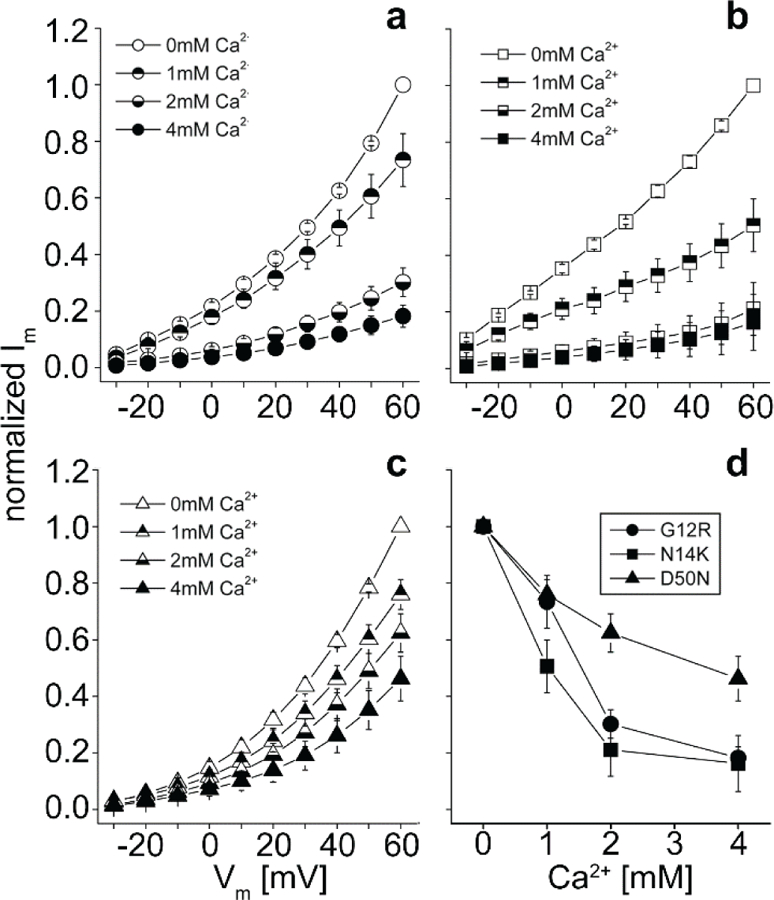
Current-voltage relationships of cells recorded in MB with 0, 1, 2, or 4 mM Ca2+ (open, top closed, bottom closed, and completely closed, respectively). Only cells that expressed hemichannels were recorded. (a-c) Cells were clamped at −40mV and then tested with pulses from −30mV to +60mV via +10mV steps. G12R (○), N14K (□), and D50N (△) mutations caused hemichannel activity that increased at positive potentials. The magnitude of hemichannel current was reduced at all potentials as the extracellular concentration of Ca2+ increased. (d) Currents for each mutation were normalized for steady-state maximal values at each concentration and plotted against the concentration of extracellular Ca2+. There was a distinct difference in the degree and rate of current reduction induced by Ca2+.
Gap Junctions in Paired Xenopus Oocytes
The rescue of cells expressing G12R, N14K, and D50N proteins by elevated Ca2+ allowed us to test whether any of our mutations created fully functional intercellular channels using dual whole-cell voltage clamp in paired oocytes (Figure 7). Cells injected with the cRNAs for G12R, N14K, S17F, D50N, or wild-type Cx26 were incubated and paired in 4mM Ca2+. Water injected cells were used as a negative control. Cell pairs expressing wild-type Cx26 proteins showed junctional conductances that were 12-fold higher than those for control pairs. Oocyte pairs for G12R, S17F, and D50N showed conductances that were significantly lower than wild-type pairs (p<0.05) and were tantamount to control pairs. This suggested the lack of properly functioning gap junctions for these mutations. It has previously been shown by dye transfer assay that S17F showed no gap junctional coupling (Richard et al., 2002). Curiously, pairs of N14K expressing cells showed a junctional conductance that was significantly higher than negative controls (p<0.05) and indistinguishable from wild-type values.
Figure 7.
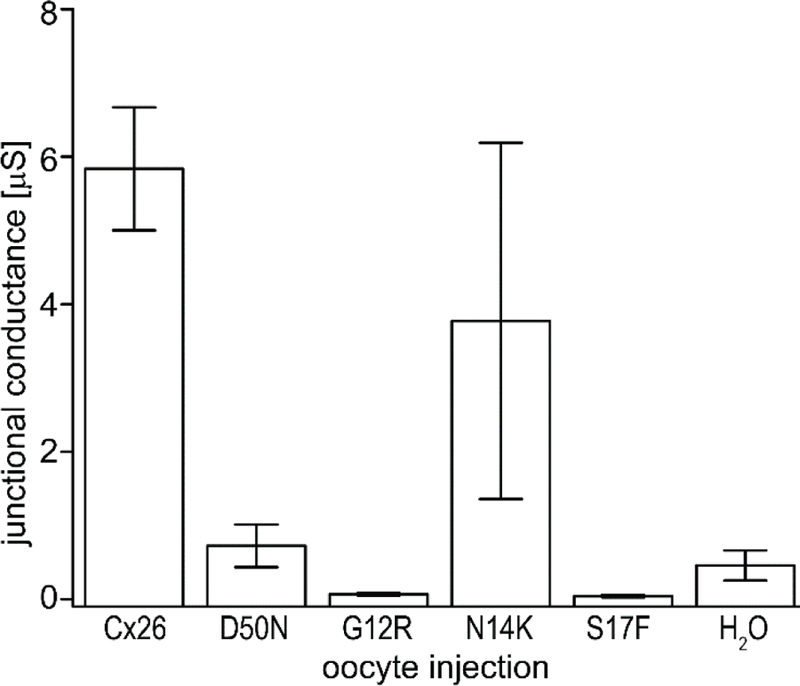
Comparison of junctional conductances recorded from paired Xenopus oocytes via dual whole cell voltage clamp. Pairs of cells expressing Cx26 (n=43), D50N (n=17), G12R (n=12), N14K (n=19), S17F (n=22), and H2O (n=41) were clamped and held at - 40mV. One cell of the pair was subjected to a voltage pulse while the other cell was held constant. The junctional current between cells was recorded and divided by the voltage step to calculate junctional conductance. N14K was the only expressed mutation with a conductance greater than that of H2O cells. Oocytes with Cx26 and N14K produced a similar junctional conductance. Data are means ± SE.
The formation of gap junction channels by N14K allowed for analysis of the voltage gating properties. Oocyte pairs expressing N14K showed an apparent loss of the voltage gating seen in their wild-type counterparts. A common feature of wild-type Cx26 traces was the asymmetric nature of decay at higher voltages that was not seen in N14K cell pairs (Figure 8A-B). The steady state conductance values were normalized, plotted against transjunctional voltage, and fit to a Boltzmann equation (Figure 8C). The complete loss of voltage gating can clearly be seen by the linearity of the N14K data that has a value of unity at all voltages tested and a slope near zero. This apparent difference can be attributed to the N14K mutation and may be part of observed variation in KID phenotype for individuals with this mutation.
Figure 8.

Properties of Cx26 and N14K intercellular junctions. (a) Cx26 gap junctions displayed an asymmetric decay in junctional current at transjunctional potentials ≥ ± 100mV. (b) Gap junctions from pairs of N14K expressing cells showed a complete loss of voltage sensitivity. (c) When normalized junctional conductance was plotted against transjunctional potential, the slope of the line produced by N14K pairs was zero. The Cx26 gap junctional data could be fit to a Boltzmann equation.
Discussion:
The appearance of KID syndrome is marked by syndromic deafness and associated skin diseases. Mutations causing nonsyndromic deafness are found throughout the GJB2 protein, but KID syndrome variants are restricted to the amino-terminus and first extracellular loop. It was previously shown that the KID mutations G45E and A40V resulted in increased hemichannel activity (Montgomery et al., 2004;Gerido et al., 2007). The increased hemichannel currents caused cell death which could be blocked through the addition of extracellular Ca2+. In this study we examined the behavior of four additional GJB2 mutations associated with SNHL and skin disorders. The G12R, N14K, and D50N mutations led to increased hemichannel activity and cell death. The S17F mutation produced a distinct loss of hemichannel activity altogether, with no cellular lethality. The cell death phenotype of G12R, N14K, and D50N could be rescued through the addition of extracellular Ca2+. These findings are consistent with previous studies describing hemichannel activity associated with Cx26 and suggest a common characteristic among the KID associated mutations (Gerido et al., 2007;Stong et al., 2006;Montgomery et al., 2004).
The most commonly occurring KID mutation analyzed in this paper is D50N. Reported cases display profound SNHL, photophobia, keratitis, and associated skin disorders (van Steensel et al., 2002;Mazereeuw-Hautier et al., 2007;Richard et al., 2002). The recorded properties of D50N and G12R agreed with the theory that KID mutations displayed aberrant hemichannel activity and both mutations failed to induce electrical coupling between paired cells. The cellular death observed in these cases could be rescued by introduction of Ca2+ to the extracellular media during incubation. However, the main difference between the two mutations was an apparent change in their sensitivity to the concentration of Ca2+. Hemichannel activity in D50N was less responsive to changes in the extracellular concentration. The response of G12R was quicker and more complete. Both cell populations could be rescued from cell death, but they showed a different reaction to extracellular Ca2+ levels. The reduction of D50N currents was smaller than G12R, however D50N also had a lower magnitude of hemichannel activity in 0 mM Ca2+. Elevation of external calcium reduces the amplitude of hemichannel currents by shifting the voltage activation curve to more positive potentials (Ebihara and Steiner, 1993), such that the mutations with larger hemichannel activity (G12R and N14K) may reflect different activation voltages and experience a larger reduction in current. The underlying cause remains unknown, but may be attributed to the location of the mutations relative to the membrane. The G12R and N14K mutations are bathed in the intracellular solution and D50N is found on the first extracellular loop.
The reported case of N14K was found to have SNHL similar to all of the other mutations reported (van Steensel et al., 2004). However, the patient bearing this mutation was described as having a phenotype more similar to Clouston syndrome (caused by mutations in GJB6) than KID syndrome due to the lack of keratitis and overall mild associated skin conditions. These observations were consistent with our functional data. The mutant channels led to cell death and elevated hemichannel activity similar to other KID associated mutations in addition to the formation of complete gap junctions with a conductance similar to wild-type channels. The difference between N14K channels and Cx26 wild-type gap junctions was the complete loss of voltage dependent gating. The amino terminus has been shown to be crucial for voltage gating properties of channels (Oh et al., 1999;Barrio et al., 1991). It was also reported that the amino terminus of Cx26 has a region of flexibility around the first 12–14 amino acid residues which is important to cellular function (Arita et al., 2006). The loss of voltage gating seen for N14K also suggests that this residue plays a key role in voltage gating. Although several KID mutations have shown no electrical coupling between cells, G45E cells were coupled and the resulting channels demonstrated an increase in voltage sensitivity (Gerido et al., 2007). This suggests that KID mutations that form gap junction channels have reduced conductance at modest transjunctional potentials. The relative loss of conductance compared to wild-type could be another property shared by KID mutations. The lack of voltage sensitivity seen by N14K cells may be responsible for causing a phenotype closer to Clouston Syndrome by producing conductances across all transjunctional voltages.
One of the most severe phenotypes observed in patients was due to S17F mutation. It has been reported that this condition lead to SNHL, visual impairment, and in one case lethal carcinoma of the tongue (Mazereeuw-Hautier et al., 2007;Richard et al., 2002). Oddly enough, this mutation also had a different phenotype in our results, displaying a complete lack of both gap junctional coupling and hemichannel activity. It was similar to negative controls in both experimental recordings. It was also reported that S17F showed a complete lack of coupling in HeLa cells via a dye transfer assay with caboxyfluorescein (Richard et al., 2002). That was consistent with our observations which demonstrated the inability of S17F mutant channels to couple cells. These data suggest that S17F may operate by an alternative method of function previously not classified.
After classification of the functional activity of the four mutations analyzed in this paper, we concluded that aberrant hemichannel activity is a common feature among KID associated mutations. It has been shown that Cx26 plays an important role in keratinocyte growth and differentiation (Kelsell et al., 2001). There seems to be a distinct pathology for syndromic hearing loss and skin associated disorders. As was displayed with the N14K mutation, a combination of effects can be caused by the dual activity of Cx26 as both an intercellular channel and a hemichannel interacting with the extracellular solution. With the data from G12R, N14K, S17F, A40V, G45E, and D50N mutations of Cx26 one can conclude that the altered hemichannel activity of these mutants plays an important role in cell signaling (Gerido et al., 2007). The mutation of G11R in Cx30 also was reported to produce hemichannel activity and lead to cell death (Essenfelder et al., 2004). This group also showed the association of A88V with induced cell death in Xenopus oocytes. Aberrant hemichannel activity and cell death are common features of Cx26 and Cx30 mutations associated with KID syndrome and other epidermal disorders, however, the precise mechanism whereby this may contribute to a loss of cell viability and tissue integrity remains to be elucidated.
Methods:
Molecular cloning
Human wild-type Cx26 was cloned into the BamHI restriction site of the pCS2+ expression vector for functional studies in Xenopus laevis oocytes (Rouan et al., 2001). DNA primers (Table S1) with BamHI restriction sites (Integrated DNA Technologies, Inc. Coralville, IA) were designed to generate the G12R, N14K, S17F, and D50N mutations by standard PCR mutagenesis (Mese et al., 2004;Gerido et al., 2007). The G12R, N14K and S17F specific sense primers were paired with a Cx26 wild-type antisense primer and amplified by polymerase chain reaction (PCR) with conditions of 95°C for 4 minutes, followed by 25 cycles of 94°C for 30s, 60°C for 30s, 72°C for 45s, and 72°C for 2 minutes. The D50N mutation was created by the overlap extension method (Horton et al., 1990) using D50N specific primers in conjunction with Cx26 sense and antisense primers. PCR products were gel purified using the QIAquik gel extraction kit (QIAGEN, Valencia, CA), digested with BamHI and cloned into pBlueScript (Stratagene, La Jolla, CA) and sequenced on both strands (GeneWiz North Brunswick, NJ). Mutants with the correct sequence were subcloned into the pCS2+ vector (Turner and Weintraub, 1994).
In vitro transcription, oocyte microinjection, and pairing
Frog use was approved by the Institutional Animal Care and Use Committee. Human Cx26, G12R, N14K, S17F, and D50N were linearized using the NotI restriction site of pCS2+, and transcribed using the SP6 mMessage mMachine (Ambion, Austin, TX). Adult Xenopus females were anesthetized with ethyl 3-aminobenzoate methanesulfonate and ovarian lobes were surgically removed and digested for 1.5 hours in a solution containing 50mg/ml collagenase B, and 50mg/ml hyaluronidase in modified Barth’s medium (MB) without Ca2+. Stage V-VI oocytes were collected and injected first with 10ng of antisense Xenopus Cx38 oligonucleotide to eliminate endogenous connexins (Barrio et al., 1991;Bruzzone et al., 1993). Antisense oligonucleotide treated oocytes were then injected with wild-type Cx26, G12R, N14K, S17F, and D50N cRNA transcripts (5ng/cell), or H2O as a negative control. cRNA injected oocytes were then cultured in Ca2+ free MB, or MB with elevated Ca2+ (4 mM CaCl2) and cultured until ready for electrophysiological recording or image capture. For measurements of gap junctional conductance, the vitelline envelopes were removed in a hypertonic solution (200mM aspartic acid, 10mM HEPES, 1mM MgCl2, 10mM EGTA, and 20mM KCL at pH 7.4), and the oocytes were manually paired with the vegetal poles apposed in MB with elevated Ca2+.
Electrophysiological hemichannel current recordings
8–12 hours after cRNA injection, macroscopic recordings of hemichannel currents were recorded from single Xenopus oocytes using a GeneClamp 500 amplifier controlled by a PC-compatible computer through a Digidata 1320 interface (Axon Instruments, Foster City, CA). pClamp 8.0 software (Axon Instruments) was used to program stimulus and data collection paradigms. To obtain hemichannel I-V curves, cells were initially clamped at −40mV and subjected to 5 second depolarizing voltage steps ranging from-30 to +60mV in 10mV increments. To test the effect of extracellular Ca2+ on hemichannel currents, oocytes were switched between MB media without Ca2+, or MB supplemented with elevated Ca2+ (1, 2, and 4mM CaCl2) via a perfusion system that washed 25ml of solution through the 35mm dish prior to recording. Cells were allowed to rest in the new solution for 2–5 minutes prior to recording.
Dual whole-cell voltage clamp
Gap junctional coupling between oocyte pairs was measured using the dual whole-cell voltage clamp technique (Spray et al., 1981). Current and voltage electrodes (1.2mm diameter, omega dot; Glass Company of America, Millville, NJ) were pulled to a resistance of 1–2MΩ with a horizontal puller (Narishige, Tokyo, Japan) and filled with solution containing 3M KCl, 10mM EGTA, and 10mM HEPES, pH 7.4. Dual voltage clamp experiments were performed using the same amplifier, data acquisition interface, computer, and software used for hemichannel current recordings. For measurements of junctional conductance, both cells in a pair were initially clamped at −40mV to eliminate any transjunctional potential. One cell was then subjected to alternating pulses of ± 20mV, while the current produced by the change in voltage was recorded in the second cell. The current delivered to the second cell was equal in magnitude to the junctional current, and the junctional conductance was calculated by dividing the measured current by the voltage difference, Gj = Ij/(V1-V2).
To determine voltage gating properties, transjunctional potentials (Vj) of opposite polarity were generated by hyperpolarizing or depolarizing one cell in 20mV steps (range = ± 120mV) while clamping the second cell at −40mV. Currents were measured at the end of the voltage pulse, at which time they approached steady state (Ijss). Macroscopic conductance (Gjss) was calculated by dividing Ijss by Vj, normalized to the values determined at ±20mV, and plotted against Vj. Data describing the relationship of Gjss as a function of Vj were analyzed using Origin 6.1 (Microcal Software, Northampton, MA) and fit to a Boltzmann relation of the form: Gjss = (Gjmax-Gjmin)/1 + exp[A(Vj-V0)] = Gjmin, where Gjss is the steady state junctional conductance, Gjmax (normalized to unity) is the maximum conductance, Gjmin is the residual conductance at large values of Vj, and V0 is the transjunctional voltage at which Gjss = (Gjmax-Gjmin)/2. The constant A = nq/kT represents the voltage sensitivity in terms of gating charge as the equivalent number (n) of electron charges (q) moving through the membrane, k is the Boltzmann constant, and T is the absolute temperature.
Preparation of oocyte samples for Western blot analysis and quantification
Oocytes used for electrophysiological recording were collected in 2ml tubes and frozen at −80 C. Oocytes were homogenized in 1 ml of buffer containing 5 mM Tris pH 8.0, 5 mM EDTA and protease inhibitors using a series of mechanical passages through needles of diminishing caliber (20, 22, 26 Ga). Extracts were centrifuged at 1000 g at 4°C for 5 min. The supernatant was then centrifuged at 100,000 g at 4°C for 30 min. Membrane pellets were resuspended in SDS sample buffer (1μl per oocyte), samples were separated on 15% SDS gels and transferred to nitrocellulose membranes. Blots were blocked with 5% BSA in 1x PBS with 0.02% NaN3 for 1 hour and probed with a polyclonal Cx26 antibody, at a 1:500 dilution (Zymed laboratories, San Francisco, CA) followed by incubation with alkaline-phosphatase conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). Band intensities were quantified using Kodak 1D Image Analysis software (Eastman Kodak, Rochester, NY). Values from three independent experiments were normalized to the mean value of band intensity of the wild-type Cx26 sample.
Supplementary Material
Acknowledgements:
This work was supported by NIH grants EY13163 and DC06652 (T.W.W.). We thank Dr. Gülistan Meşe for critically reading the manuscript.
Footnotes
Conflict of Interest:
The authors state no conflict of interest.
Reference List
- 1.Anand RJ, Hackam DJ: The role of gap junctions in health and disease. Crit Care Med 33:S535–S538 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Arita K, Akiyama M, Aizawa T, Umetsu Y, Segawa I, Goto M, Sawamura D, Demura M, Kawano K, Shimizu H: A novel N14Y mutation in Connexin26 in keratitis-ichthyosis-deafness syndrome: analyses of altered gap junctional communication and molecular structure of N terminus of mutated Connexin26. Am J Pathol 169:416–423 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrio LC, Suchyna T, Bargiello T, Xu LX, Roginski RS, Bennett MV, Nicholson BJ: Gap junctions formed by connexins 26 and 32 alone and in combination are differently affected by applied voltage. Proc Natl Acad Sci U S A 88:8410–8414 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett MV, Contreras JE, Bukauskas FF, Saez JC: New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci 26:610–617 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruzzone R, Haefliger JA, Gimlich RL, Paul DL: Connexin40, a component of gap junctions in vascular endothelium, is restricted in its ability to interact with other connexins. Mol Biol Cell 4:7–20 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, Petit C, D’Andrea P, White TW: Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett 533:79–88 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Ebihara L, Steiner E: Properties of a nonjunctional current expressed from a rat connexin46 cDNA in Xenopus oocytes. J Gen Physiol 102:59–74 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Essenfelder GM, Bruzzone R, Lamartine J, Charollais A, Blanchet-Bardon C, Barbe MT, Meda P, Waksman G: Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum Mol Genet 13:1703–1714 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Gerido DA, DeRosa AM, Richard G, White TW: Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am J Physiol Cell Physiol (2007). [DOI] [PubMed]
- 10.Gerido DA, White TW: Connexin disorders of the ear, skin, and lens. Biochim Biophys Acta 1662:159–170 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez D, Gomez-Hernandez JM, Barrio LC: Species specificity of mammalian connexin-26 to form open voltage-gated hemichannels. FASEB J 20:2329–2338 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Goodenough DA, Paul DL: Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol 4:285–294 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Harris AL: Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys 34:325–472 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Horton RM, Cai ZL, Ho SN, Pease LR: Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535 (1990). [PubMed] [Google Scholar]
- 15.Janecke AR, Hennies HC, Gunther B, Gansl G, Smolle J, Messmer EM, Utermann G, Rittinger O: GJB2 mutations in keratitis-ichthyosis-deafness syndrome including its fatal form. Am J Med Genet A 133:128–131 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Jonard L, Feldmann D, Parsy C, Freitag S, Sinico M, Koval C, Grati M, Couderc R, Denoyelle F, Bodemer C, Marlin S, Hadj-Rabia S: A familial case of Keratitis-Ichthyosis-Deafness (KID) syndrome with the GJB2 mutation G45E. Eur J Med Genet 51:35–43 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Kelsell DP, Dunlop J, Hodgins MB: Human diseases: clues to cracking the connexin code? Trends Cell Biol 11:2–6 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Lai-Cheong JE, Arita K, McGrath JA: Genetic diseases of junctions. J Invest Dermatol 127:2713–2725 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, Man SY, Bodemer C, Prins C, Antille C, Saurat JH, Atherton D, Harper JI, Kelsell DP, Hovnanian A: Keratitis-ichthyosis-deafness syndrome: disease expression and spectrum of connexin 26 (GJB2) mutations in 14 patients. Br J Dermatol 156:1015–1019 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Mese G, Londin E, Mui R, Brink PR, White TW: Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss. Hum Genet 115:191–199 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Mese G, Richard G, White TW: Gap junctions: basic structure and function. J Invest Dermatol 127:2516–2524 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Montgomery JR, White TW, Martin BL, Turner ML, Holland SM: A novel connexin 26 gene mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J Am Acad Dermatol 51:377–382 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Oh S, Rubin JB, Bennett MV, Verselis VK, Bargiello TA: Molecular determinants of electrical rectification of single channel conductance in gap junctions formed by connexins 26 and 32. J Gen Physiol 114:339–364 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petit C: From deafness genes to hearing mechanisms: harmony and counterpoint. Trends Mol Med 12:57–64 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Petit C, Levilliers J, Hardelin JP: Molecular genetics of hearing loss. Annu Rev Genet 35:589–646 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Richard G: Connexin disorders of the skin. Clin Dermatol 23:23–32 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynanen M, Jabs EW, Bale SJ, DiGiovanna JJ, Uitto J, Russell L: Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet 70:1341–1348 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ripps H, Qian H, Zakevicius J: Properties of connexin26 hemichannels expressed in Xenopus oocytes. Cell Mol Neurobiol 24:647–665 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rouan F, White TW, Brown N, Taylor AM, Lucke TW, Paul DL, Munro CS, Uitto J, Hodgins MB, Richard G: trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci 114:2105–2113 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Saez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV: Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta 1711:215–224 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spray DC, Harris AL, Bennett MV: Equilibrium properties of a voltage-dependent junctional conductance. J Gen Physiol 77:77–93 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stong BC, Chang Q, Ahmad S, Lin X: A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope 116:2205–2210 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Turner DL, Weintraub H: Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev 8:1434–1447 (1994). [DOI] [PubMed] [Google Scholar]
- 34.van Geel M, van Steensel MA, Kuster W, Hennies HC, Happle R, Steijlen PM, Konig A: HID and KID syndromes are associated with the same connexin 26 mutation. Br J Dermatol 146:938–942 (2002). [DOI] [PubMed] [Google Scholar]
- 35.van Steensel MA, Steijlen PM, Bladergroen RS, Hoefsloot EH, Ravenswaaij-Arts CM, van Geel M: A phenotype resembling the Clouston syndrome with deafness is associated with a novel missense GJB2 mutation. J Invest Dermatol 123:291–293 (2004). [DOI] [PubMed] [Google Scholar]
- 36.van Steensel MA, van Geel M, Nahuys M, Smitt JH, Steijlen PM: A novel connexin 26 mutation in a patient diagnosed with keratitis-ichthyosis-deafness syndrome. J Invest Dermatol 118:724–727 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Wei CJ, Xu X, Lo CW: Connexins and cell signaling in development and disease. Annu Rev Cell Dev Biol 20:811–838 (2004). [DOI] [PubMed] [Google Scholar]
- 38.White TW: Functional analysis of human Cx26 mutations associated with deafness. Brain Res Brain Res Rev 32:181–183 (2000). [DOI] [PubMed] [Google Scholar]
- 39.White TW, Paul DL: Genetic diseases and gene knockouts reveal diverse connexin functions. Annu Rev Physiol 61:283–310 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Zhao HB, Kikuchi T, Ngezahayo A, White TW: Gap junctions and cochlear homeostasis. J Membr Biol 209:177–186 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.