

Abstract
The P-type ATPase protein family includes, in addition to ion pumps such as Ca2+-ATPase and Na+,K+-ATPase, also phospholipid flippases that transfer phospholipids between membrane leaflets. P-type ATPase ion pumps translocate their substrates occluded between helices in the center of the transmembrane part of the protein. The large size of the lipid substrate has stimulated speculation that flippases use a different transport mechanism. Information on the functional importance of the most centrally located helices M5 and M6 in the transmembrane domain of flippases has, however, been sparse. Using mutagenesis, we examined the entire M5-M6 region of the mammalian flippase ATP8A2 to elucidate its possible function in the lipid transport mechanism. This mutational screen yielded an informative map assigning important roles in the interaction with the lipid substrate to only a few M5-M6 residues. The M6 asparagine Asn-905 stood out as being essential for the lipid substrate–induced dephosphorylation. The mutants N905A/D/E/H/L/Q/R all displayed very low activities and a dramatic insensitivity to the lipid substrate. Strikingly, Asn-905 aligns with key ion-binding residues of P-type ATPase ion pumps, and N905D was recently identified as one of the mutations causing the neurological disorder cerebellar ataxia, mental retardation, and disequilibrium (CAMRQ) syndrome. Moreover, the effects of substitutions to the adjacent residue Val-906 (i.e. V906A/E/F/L/Q/S) suggest that the lipid substrate approaches Val-906 during the translocation. These results favor a flippase mechanism with strong resemblance to the ion pumps, despite a location of the translocation pathway in the periphery of the transmembrane part of the flippase protein.
Keywords: ATPase, membrane lipid, membrane transport, lipid binding protein, lipid transport, lipid-protein interaction, mutant, molecular modeling, membrane enzyme, transporter, CAMRQ syndrome, enzyme mechanism, mutagenesis, P4-ATPase, phosphatidylserine transport
Introduction
ATP8A2 is a membrane-bound enzyme, a “flippase,” that utilizes the energy liberated by ATP hydrolysis to translocate (flip) the amino phospholipids phosphatidylserine (PS)2 and phosphatidylethanolamine (PE), but not phosphatidylcholine (PC), from the exoplasmic leaflet to the cytoplasmic leaflet of cellular membrane lipid bilayers, thereby creating an essential asymmetric lipid distribution between the membrane leaflets (1). Flippases, also known as P4-ATPases, belong to the P-type ATPase family. Compared with the ion pump members of this enzyme family, flippases are poorly understood, although as many as 14 of the 36 P-type ATPases encoded by the human genome are flippases (1, 2), and certain mutations in flippases, including ATP8A2, are associated with severe disorders (1, 3–9).
P4-ATPases are made up of three cytoplasmic domains, N (nucleotide-binding), P (phosphorylation), and A (“actuator”), linked to 10 transmembrane helices (M1–M10), like the ion-pumping P-type ATPases Ca2+-ATPase, Na+,K+-ATPase, H+,K+-ATPase, and H+-ATPase (P2- and P3-ATPases). In addition, most P4-ATPases are composed of an accessory subunit comparable in many respects to the β-subunit of Na+,K+- and H+,K+-ATPases. During the ATP hydrolysis cycle, ATP8A2, like other P-type ATPases, is phosphorylated at a conserved aspartate in the P-domain. Dephosphorylation depends on catalysis by a conserved glutamate in the A-domain and is stimulated by the binding of the lipid substrate from the exoplasmic bilayer leaflet, just like dephosphorylation of the Na+,K+-ATPase is stimulated by the binding of extracellular K+. These features argue for a catalytic cycle of flippases that involves four major conformational states, E1, E1P, E2P, and E2 (“P” indicating phosphorylation; see. Fig. 1) similar to those known from the P-type ATPase ion pumps (10–12), but the translocation pathway for the lipid substrate and the signal-transducing event associating binding and translocation of the lipid substrate with the dephosphorylation occurring in the cytoplasmic part of the flippase are unknown. The fact that the lipid substrate is about 10-fold larger (in linear size; much more if volume is considered) than the ions handled by the Ca2+-ATPase and the Na+-K+-ATPase underscores the central enigma in understanding lipid flipping, the so-called “giant substrate problem” (13): how and where in the flippase protein can such a large substrate be selected, bound, and translocated? Does the large size of the lipid substrate imply that the transport mechanism differs fundamentally from that of the ion pumps? In the latter, the transported ions are bound at residues in transmembrane helices M4, M5, M6, and M8, which are located centrally in the transmembrane domain, shielded from the lipid phase by other transmembrane helices. In connection with the transport process, the ions become occluded between these central helices (14–16). Because of the large size of the lipid substrate and the need for the lipid to reorient itself during the flipping process, a different transport mechanism has been proposed for flippases: the so-called “credit card model.” Here, the translocation of the phospholipid is thought to occur at the periphery of the flippase protein facing the lipid bilayer phase, and only the phospholipid headgroup interacts with the protein, whereas the hydrocarbon tail is dragged along, jutting out into the membrane lipid bilayer (17–20). Mutagenesis studies of various flippases of mammals, yeast, and plants have pinpointed residues in transmembrane helices M1, M2, M3, and M4 as important for the recognition and propagation of the phospholipid headgroup (18–21). Of these helices, M1–M3 are peripherally located relative to the central core of the protein, thus facing the lipid bilayer. Mutational sensitivity of the isoleucine of the conserved PISL motif in M4 (Ile-364 of bovine ATP8A2) in combination with structural modeling has implicated Ile-364 as part of a “hydrophobic gate” moving the lipid headgroup along a peripheral pathway in accordance with the credit card model (18). However, the importance of certain other M4 residues has been considered evidence in favor of a more centrally located binding site for the lipid headgroup (21). So far, there is only sparse information on the functional importance of residues in the central core helices M5 and M6 of flippases, although in the ion pumps, M5 and M6 contain most of the key residues involved in ion occlusion and translocation (14–16, 22, 23). Here, we examined the M5-M6 region of ATP8A2 to elucidate the possible function of this region in the lipid transport mechanism. Our mutational screen of the entire region yielded an informative map assigning important roles to only a few M5-M6 residues in the interaction with the lipid substrate. Significantly, the asparagine Asn-905 of M6 stood out as being essential for the lipid substrate–induced dephosphorylation. Strikingly, Asn-905 aligns with key ion-binding residues of the ion pumps, thus providing evidence that the mechanism of flippases resembles that of P-type ATPase ion pumps much more than previously realized, and the N905D mutation was recently identified as one of the mutations causing the neurological disorder cerebellar ataxia, mental retardation, and disequilibrium (CAMRQ) syndrome (9).
Figure 1.
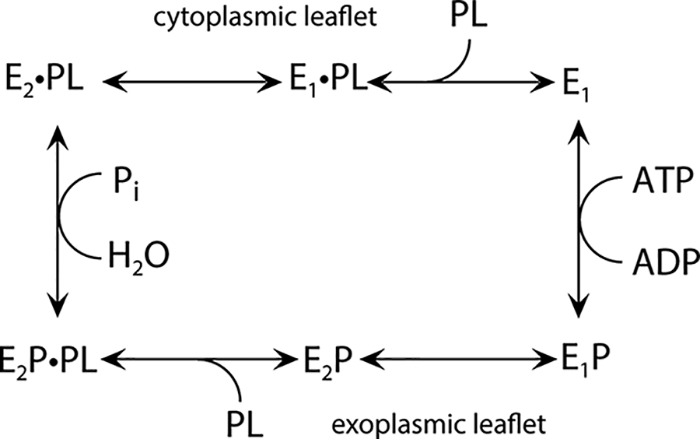
Proposed reaction cycle of ATP8A2. This scheme emphasizes similarities to the Na+,K+-ATPase (1). E1 is the state allowing phosphorylation (P) of ATP8A2 in the presence of ATP, forming E1P. The subsequent conformational change from E1P to E2P makes the enzyme ready to bind the phospholipid (PL) substrate (i.e. PS or PE) from the exoplasmic leaflet. The binding of PL to E2P activates the dephosphorylation, similarly to K+ binding from the extracellular side to the Na+,K+-ATPase, and Pi is released, resulting in E2·PL (12). The dot indicates noncovalent bond(s). PL is released to the cytoplasmic leaflet following a conformational change to E1·PL (18), and a new reaction cycle can begin. There are also differences from the Na+,K+-ATPase and other P2-ATPases. Hence, the flippase seems to lack a substrate transported toward the exoplasmic side and is, thus, phosphorylated spontaneously from ATP (1). In the P2-ATPases, the binding of such a substrate to the E1 form is mandatory for activation of the phosphoryl transfer from ATP.
Results
Asn-905 is the functionally most critical residue of the present screen
Alignment of flippases and P-type ion pumps of known structure defines the borders of M5 and M6 of the flippases and locates nine M5-M6 residue positions in flippases that correspond to ion-binding residues of the ion pumps Ca2+-ATPase (SERCA), Na+,K+-ATPase, and H+,K+-ATPase (Fig. 2). The corresponding residues in the bovine flippase ATP8A2 studied here are in the following text, Fig. 3, and Table 1 marked by an asterisk: *Tyr-869, *Cys-870, *Tyr-872, *Lys-873, *Asn-874, and *Leu-877 of M5 and and *Asn-905, *Phe-908, and *Thr-909 of M6. We examined the functional importance of the M5 and M6 residues of ATP8A2 in a total alanine-scanning mutagenesis analysis of the ATPase activity summarized in Fig. 3 and Table 1 (asparagine was used as substituent when the WT residue was an alanine). At certain residues, including some of those aligning with ion-binding residues in the ion pumps, the alanine scan was supplemented with additional substitutions, varying size, polarity, and charge of the substituent. In total, the analysis of 61 new mutants is reported in Fig. 3 and Table 1. For completeness of this M5-M6 survey, alanine scanning data for four residues of M5 and M6 (Lys-865, Lys-873, Asn-874, and Val-906) that have previously been published (12, 18) are included.
Figure 2.

Alignment of amino acid sequences corresponding to the M5-M6 region of flippases and ion pumps. The identification of the transmembrane helices M5 and M6 of ATP8A2 (residues in cyan boxes) is based on the crystal structure of Ca2+-ATPase (14). Lys-873 (K873) and Asn-905 (N905) of the bovine ATP8A2 studied here are indicated. For the ion pumps, residues involved in ion binding are shown in gray boxes.
Figure 3.

Vmax and apparent affinity (K0.5) for PS or PE of M5 and M6 mutants. Residues at positions corresponding to ion-binding residues in the ion pumps are indicated by an asterisk. The ATPase activity per mg of purified flippase protein was determined and analyzed in the presence of varying concentrations of PS or PE as described under “Experimental procedures.” Examples of the determined concentration dependences and the fitted lines are shown in Fig. 4. The apparent lipid affinities are expressed as the concentration giving half-maximum activation (K0.5). Vmax generally refers to the plateau value obtained at the highest lipid concentration (1000 μm PS or 2000 μm PE). For a few mutants showing inhibition at high lipid concentration, Vmax was taken as the peak value. The Vmax is shown in percentage of the WT Vmax, which was 111 ± 43 μmol/min/mg for PS and 42 ± 16 μmol/min/mg for PE (average ± S.D.). All numbers on which this figure is based are found in Table 1. All the individual data points collected are shown as circles, and the columns show the average values. The error bars indicate the S.D. For further information on statistics, see Table 1. E indicates that the expression level of the mutant was too low for reliable determination of the ATPase activity. L indicates that although the mutant was well expressed, the ATPase activity was too low for reliable determination of substrate affinity.
Table 1.
ATPase activity parameters of M5-M6 mutants
The data set for ATPase activities underlying the column diagrams in Fig. 3 is shown here. The Vmax of the mutants is here given in % of the WT Vmax, which was 111 ± 43 μmol/min/mg for PS and 42 ± 16 μmol/min/mg for PE (average ± S.D.). The K0.5(PS) and K0.5(PE) values were extracted as described in “Experimental Procedures”. Calculated Vmax(PE)/Vmax(PS) and K0.5(PE)/K0.5(PS) ratios (all based on the values in % WT) are also shown to indicate changes to the lipid substrate selectivity. The four mutants for which the data were obtained in previous studies are indicated by superscripts corresponding to the references (12) and (18). The horizontal line in the middle separates M5 and M6 residues. Mutations to residues aligning with ion binding residues in the ion pumps are marked by *. The superscript inh indicates that inhibition was observed at the highest concentrations of the lipid substrate (cf. the legend to Fig. 4). S.D. is indicated by ±. The number of independent experiments contributing to the determination of each parameter is indicated by n. Numbers in parentheses and the arrow indicate fold change relative to WT, ↑ for increase and ↓ for decrease (an increase of K0.5 corresponds to reduced apparent affinity). “E” indicates that the expression level of the mutant was too low for reliable determination of the specific ATPase activity. “L” indicates that although the mutant was well expressed, the ATPase activity was too low for reliable determination of the parameter.

The ATP8A2 wildtype (WT) and mutants were expressed in mammalian cells (HEK293T) together with the accessory CDC50A protein, and the detergent-solubilized protein complexes were immunoaffinity-purified followed by reconstitution in PC vesicles. Only the mutants V876R, *T909D, and *T909N showed an expression level too low for reliable functional analysis (<10% WT in repeated expression analysis; indicated by “E” in Fig. 3 and Table 1). The ATPase activity per mg of the purified protein was determined with either of the two substrates of ATP8A2, PS and PE, within the concentration ranges 0–1000 μm PS and 0–2000 μm PE. For this assay, the reconstituted vesicles containing purified ATP8A2/CDC50A complexes together with PC were resolubilized by addition of CHAPS detergent, thus ensuring access of the added PS or PE lipid substrate to the flippase. The concentration of PC was varied inversely with that of PS or PE to maintain a constant total lipid concentration. Neither the WT ATP8A2 nor the mutants showed any dependence of the ATPase activity on the PC concentration. These measurements provided the maximal ATPase activity per mg protein with either PS or PE as substrate (in the following denoted as Vmax(PS) and Vmax(PE), respectively), as well as the apparent affinity for activation with either lipid substrate, extracted from the data in the form of the concentration giving half-maximum activation, K0.5(PS) and K0.5(PE) (Fig. 3 and Table 1, with examples of the lipid concentration dependences shown in Fig. 4). For WT ATP8A2, PS is the primary substrate: the Vmax(PS) was 2–3-fold higher than the Vmax(PE), and the K0.5(PS) was about one-tenth of the K0.5(PE), corresponding to ∼10-fold higher apparent affinity for PS (legend to Fig. 3 and Table 1).
Figure 4.
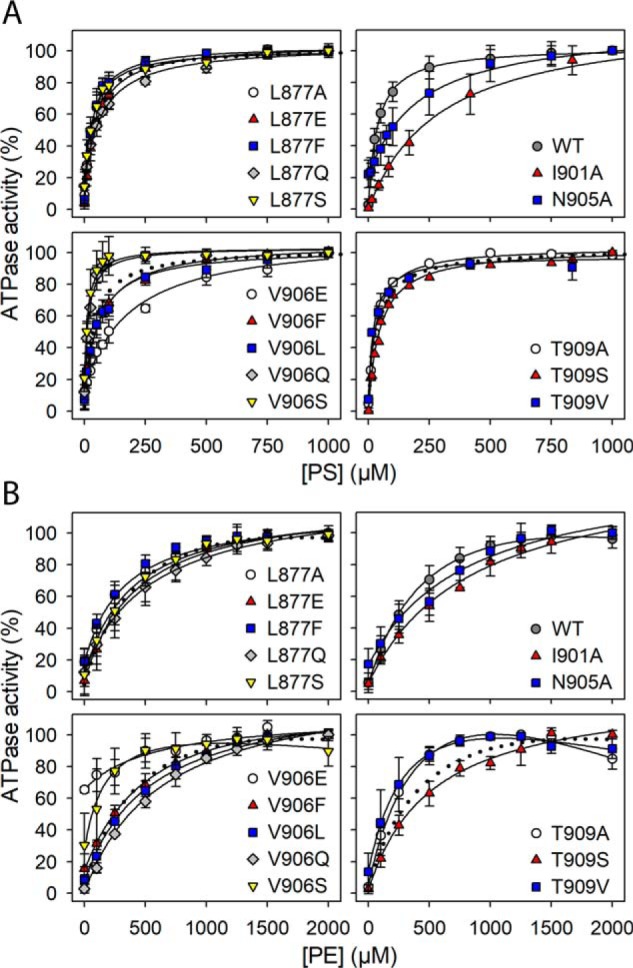
Examples of the PS and PE concentration dependences of ATPase activity summarized as K0.5 values in Fig. 3. The ATPase activity was determined in the presence of CHAPS detergent and the indicated concentrations of PS (A) or PE (B), keeping a constant total lipid concentration by varying also the concentration of PC (inert with respect to ATPase activity). The maximum ATPase activity per mg of flippase protein was taken as 100% (see legend to Fig. 3 for absolute value). The lipid concentration giving half-maximum activation (K0.5) was determined as described under “Experimental procedures” with the lines showing the best fits. Inhibition was in some cases observed at the highest concentrations of the lipid substrate and is most likely due to displacement of the E2–E1 equilibrium in favor of E2 (see Fig. 1 and Ref. 18). Error bars (shown only when larger than the size of the symbols) represent S.D.
The alanine scan showed that only a relatively small fraction of the residues in M5 and M6 are really critical to function. Mutations giving rise to a ≥10-fold change to Vmax and/or a ≥5-fold change to the apparent affinity for one or both of the lipid substrates are mentioned below. *Asn-905 of M6 was identified as the functionally most critical residue of the present screen and was further examined by a series of substitutions: *N905/D/E/H/L/Q/R. All the *Asn-905 mutations, including *N905A, reduced the ATPase activity per mg of protein to a very low level, ≤2% of WT, both with PS and PE as substrate. The expression level of the *Asn-905 mutants was WT-like except for N905R, which expressed 20–50% of WT. *Lys-873 of M5 is in the same functionally very critical category as *Asn-905, with the *K873A mutant showing a Vmax of only 2–3% of WT with PS and even less with PE (0.5% of WT). For *N874A, the Vmax(PS) was reduced less markedly (to 18% of WT), but the Vmax(PE) was only ∼2% of WT. Likewise, the Vmax(PS) of Y904A was reduced to 31% of WT, but the Vmax(PE) was reduced more, to ∼3% of WT, and the Vmax(PS) of V906E was 22% of WT with the Vmax(PE) ∼3% of WT. Hence, there are examples of the maximal activities with PS and PE as substrates being differently affected, as also seen for the apparent affinities in certain cases (see the Vmax(PE)/Vmax(PS) and K0.5(PE)/K0.5(PS) ratios in Table 1). I901A may also be mentioned here, with Vmax(PS) 69% of WT and Vmax(PE) only 11% of WT. However, for G902L, the Vmax(PS) and the Vmax(PE) were both reduced to ∼10% of WT. G902A was WT-like, suggesting that the main reason for the disturbance seen for G902L is the bulkiness of the side chain, giving steric problems.
Of the *Asn-905 mutants, only *N905A showed sufficient ATPase activity for a reliable determination of the PS and PE concentration dependences. The apparent PS affinity of *N905A was 8-fold reduced (K0.5 increased) relative to WT, whereas the apparent PE affinity was only ∼2-fold reduced. Among the other M5 and M6 mutants, *K873A showed the largest reduction of the apparent PS affinity (14-fold relative to WT). The apparent PE affinity of *K873A could not be determined due to the very low Vmax(PE). I901A and V906E showed ≥5-fold reduction of the apparent PS affinity with much less reduction of the apparent PE affinity.
The differential effects of the various Val-906 substituents on the apparent lipid substrate affinities are noteworthy. Hence, the V906A, V906Q, and V906S mutants exhibited increased apparent affinity for PS (2–3-fold reduced K0.5(PS)) relative to WT as well as increased (V906A and V906S) or WT-like (V906Q) apparent affinity for PE. The V906F and V906L mutants exhibited slightly reduced (∼2-fold) and WT-like apparent affinity, respectively, for both lipid substrates. By contrast, the apparent PS affinity of V906E was more markedly reduced (∼5-fold), whereas the apparent PE affinity of V906E was WT-like. This dependence on the substituent side chain and the lipid substrate could be a consequence of physical nearness during the translocation (see the Discussion).
The overall result of the M5-M6 survey is that *Asn-905 of M6 and the previously studied residues *Lys-873 and *Asn-874 of M5 (12) are highly mutation-sensitive hot spots. Marked functional effects were moreover seen for mutations to residues flanking *Asn-905, including Ile-901, Gly-902 (only for substituents larger than alanine), Tyr-904, and Val-906 (only for substitution with the negatively charged glutamate).
Dephosphorylation induced by the lipid substrate is blocked in the *Asn-905 mutants
The drastic lowering of the PS- and PE-stimulated ATPase activity in the *Asn-905 mutants led us to study the partial reactions of the enzyme cycle. For WT ATP8A2, the phosphorylation of the E1 form appears to occur spontaneously in the presence of ATP. The lipid substrate enters the reaction cycle later and activates the dephosphorylation step (Fig. 1 and Ref. 12; see also the Discussion). Fig. 5A shows that the *Asn-905 mutants were fully competent with respect to phosphorylation from [γ-32P]ATP. The steady-state phosphorylation level was in some of these mutants higher than that of the WT. Fig. 5, B–D, depict the results of studying the PS dependence of dephosphorylation in the WT and the *Asn-905 mutants as well as V906E and V906F. The enzyme was phosphorylated in the presence of [γ-32P]ATP, and then PS was added at various concentrations followed by acid quenching 5 s later. CHAPS detergent was present to allow the PS to access the flippase protein. For the WT and V906F, the dephosphorylation initiated by PS binding was very rapid, resulting in almost complete dephosphorylation within the 5 s at PS concentrations above 250 μm. The PS concentration giving half-maximum dephosphorylation of V906F was ∼2-fold increased, compared with WT, in agreement with the reduction of the apparent PS affinity seen in the ATPase activity measurement (Table 1). V906E likewise showed PS-dependent dephosphorylation, and the PS concentration dependence corresponded to the ∼5-fold reduced apparent PS affinity seen in the ATPase activity measurement. In addition, V906E exhibited a high plateau level, corresponding to the highest PS concentrations, of as much as ∼68% of the phosphorylation level seen in the absence of PS. As described previously (18), such a plateau level indicates a reduced maximal dephosphorylation rate (for V906E only ∼32% of the phosphoenzyme disappeared within the 5 s at the highest PS concentrations studied), consistent with the reduced Vmax(PS) of V906E seen in the ATPase activity measurement (Table 1).
Figure 5.
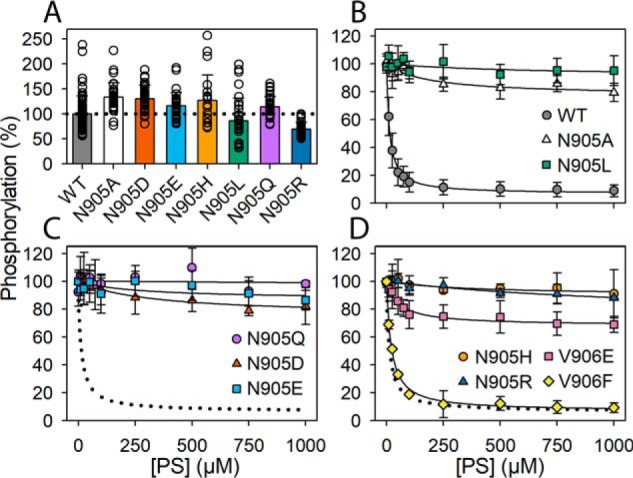
Phosphorylation and PS concentration dependence of dephosphorylation. A, phosphorylation in the presence of 2 μm [γ-32P]ATP was carried out for 30 s at 0 °C in the presence of CHAPS detergent as described under “Experimental procedures.” The level of phosphorylation per mg of purified flippase protein is shown in percentage of WT. All the individual data points collected are shown as circles, and the columns show the average values. B–D, to initiate dephosphorylation, PS dissolved in the presence of CHAPS detergent was added at the final concentrations indicated on the abscissa. After 5 s, the reaction was terminated by acid quenching. The phosphorylation level with PC added instead of PS was taken as 100%. For direct comparison in each panel, the dotted lines reproduce the WT from B. Error bars (shown only when larger than the size of the symbols) represent S.D.
Importantly, Fig. 5, B–D shows that the *Asn-905 mutants exhibited a dramatic insensitivity to PS, with no or very little dephosphorylation occurring within the 5 s (plateau values of ≥75%). Only *N905A and *N905D showed significant PS-dependent dephosphorylation, and the amplitude (∼25%) was too small to allow an accurate determination of the K0.5(PS) for activation of dephosphorylation. Roughly, from Fig. 5, B and C, the K0.5(PS) values for *N905A and *N905D appear ∼10-fold higher than that corresponding to WT. In additional experiments the dephosphorylation time course was followed up to 30 s, confirming that the *Asn-905 mutants exhibited a drastic slowing of the dephosphorylation even at a PS concentration of 1000 μm, i.e. about 100-fold higher than the K0.5 for PS activation of the dephosphorylation of WT, and that the effect was a little less pronounced for *N905A and *N905D than for the other *Asn-905 mutants (Fig. 6).
Figure 6.
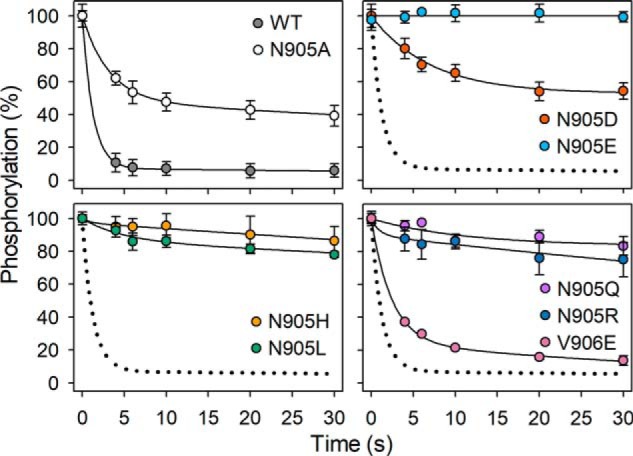
Time course of dephosphorylation induced by PS. Phosphorylation in the presence of 2 μm [γ-32P]ATP was carried out for 30 s at 0 °C in the presence of CHAPS detergent as described under “Experimental procedures.” Thereafter (at time 0), 100 μm unlabeled ATP (to terminate phosphorylation from [γ-32P]ATP) with PS (final concentration 1000 μm) and PC to maintain a total lipid concentration of 1 mg/ml was added, and the time course of dephosphorylation was followed by acid quenching at the indicated time intervals. For direct comparison in each panel, the dotted lines reproduce the WT from the upper left panel. Error bars (shown only when larger than the size of the symbols) represent S.D.
Discussion
The present data make it clear that the central helices, M5 and M6, of the transmembrane domain contain residues that are crucial players in the mechanism of ATP8A2. Mutation-sensitive “hot spots” in M5 and M6 seem to be critical for both the maximal PS- or PE-stimulated activity and the apparent affinities for PS and PE. It is significant that the hot spot residues are located at positions corresponding to ion-binding residues in the ion pumps and that alanine substitution of most of the other M5-M6 residues examined resulted in no or relatively small effects. The M6 asparagine *Asn-905 pinpointed here as a hot spot residue is essential for the PS-induced dephosphorylation. The *Asn-905 mutants exhibited no or very little dephosphorylation, even at high PS concentration, which explains their low ATPase activity. This asparagine is highly conserved among P4-ATPases. The corresponding residues in the ion pumps Ca2+-ATPase (Asn-796 in rabbit SERCA1), Na+,K+-ATPase (Asp-804 in pig α1), and H+,K+-ATPase (Glu-820 in pig α1) are known to participate directly in the interaction with the ions being transported (14–16, 22) (see Fig. 2). Alanine mutation of Asn-796 of the Ca2+-ATPase disrupts Ca2+ occlusion and blocks the dephosphorylation of E2P, with the latter effect being similar to the present observation for mutations to *Asn-905 of ATP8A2. The dephosphorylation block of the Ca2+-ATPase seems to be associated with a role of Asn-796 in proton countertransport, i.e. transport in the same direction across the membrane as the PS transport by ATP8A2. The binding of protons to be countertransported is thought to be part of the signal transduction activating the dephosphorylation (23, 24). The equivalent residues in Na+,K+-ATPase (Asp-804) and H+,K+-ATPase (Glu-820) are essential for the binding of K+ to E2P (22, 25). In H+,K+-ATPase, charge neutralization of Glu-820 by substitution with glutamine led to an anomalous, K+-independent dephosphorylation and resulting constitutive activation of ATP hydrolysis (22, 26), thus clearly indicating a role in the signal transduction involved in dephosphorylation.
The ATP8A2 M5 residues *Lys-873 and *Asn-874, which were previously studied (12) and have been included here in Fig. 3 and Table 1, appear almost as critical for function as *Asn-905. They are located in positions corresponding to a serine and an asparagine that contribute to bind and occlude the ions being transported in Ca2+-ATPase (Asn-768 in rabbit SERCA1) and Na+,K+-ATPase (Ser-775 and Asn-776 in pig α1). The H+,K+-ATPase has a lysine at the same position as *Lys-873 of ATP8A2. During the proton translocation by the H+,K+-ATPase, this lysine seems to assist the dissociation of the proton on the luminal side by forming a salt bridge with the proton-binding glutamate, Glu-820, aligning with *Asn-905 of the flippase.
Further along this line, the critical ATP8A2 M4 isoleucine, Ile-364, previously implicated as part of a hydrophobic gate moving the lipid headgroup (see the Introduction), aligns with a most essential glutamate involved in ion binding and occlusion in the ion pumps (Glu-309 in rabbit SERCA1, Glu-327 in pig α1 Na+,K+-ATPase, and Glu-343 in pig α1 H+,K+-ATPase) (18). Hence, adding together all this evidence for conservation between flippases and P-type ion pumps of key functional elements in the transmembrane domain, it is evident that they must share more mechanistic similarities than previously realized. Thus, there are not only similarities in the catalysis of ATP hydrolysis by conserved residues in the cytoplasmic P- and A-domains but certainly also similarities relating to the transport event and energy coupling taking place in the transmembrane domain, including the signal transduction responsible for activation of the dephosphorylation induced by the lipid substrate.
Besides the similarities between flippases and P-type ion pumps, there are also marked differences, including the apparent lack in flippases of a substrate being transported from the cytoplasmic side toward the exoplasmic side (see Fig. 1). Such a cis-transported substrate (cis with respect to ATP) is a hallmark of the P-type ion pumps (Ca2+, Na+, and H+ for Ca2+-ATPase, Na+,K+-ATPase, H+,K+-ATPase, and H+-ATPase), and its binding to the E1 form is mandatory for activation of the phosphoryl transfer from ATP, whereas no ion (H+, SO42−, acetate, Cl−, Ca2+, Na+, and K+ are just some of those examined) or other substrate has been identified as a trigger of the phosphorylation of a flippase (1).
Another significant difference between flippases and P-type ion pumps is the large size of the substrate of flippases, which has led to the proposal of transport pathways that are more peripherally located in the transmembrane domain (credit card model) than the narrow space between the central transmembrane helices, M4, M5, M6, and M8, where the ions translocated by the ion pumps become occluded during their translocation, as described in the Introduction. Two different, but partially overlapping, peripheral transport pathways in the flippases have been proposed: the two-gate mechanism (20) and the hydrophobic gate mechanism (18), which involve transmembrane helices M1, M2, M3, and M4 or M1, M2, M4, and M6, respectively (see Fig. 7, red star on E2 model). The data set we now have for the centrally placed M5 and M6 helices emphasizes the need for explaining the roles of the crucial M5 and M6 residues in the translocation process, in particular the key role of *Asn-905 in the substrate-induced dephosphorylation. Fig. 7 shows a cross-sectional view of the transmembrane domain seen from the cytoplasmic side in the E2 and E2P structural models of ATP8A2 obtained by homology modeling based on the Ca2+-ATPase crystal structures and refinement by molecular dynamics simulation (18). In these models, *Lys-873 and *Asn-874 of M5 together with *Asn-905 and *Thr-909 of M6 are located in the center of the transmembrane domain, close together and presumably interacting through hydrogen bonds. The present findings show that *Asn-905 is of such crucial importance that not even glutamine could substitute to allow PS-induced dephosphorylation and, thus, ATPase activity. The polar, oxygen- and nitrogen-containing, but uncharged, glutamine side chain has chemical properties similar to asparagine, differing only by the additional length corresponding to a single carbon atom. Hence, its inability to replace the asparagine functionally indicates that a precise steric fit is crucial. For each of the two modeled conformational states in Fig. 7, the central region between M4, M5, M6, and M8 was the most stable part of the transmembrane domain during the molecular dynamics refinement (Fig. 8). The hydrogen bonding network between the critical residues of M5 and M6 in the center of the transmembrane domain appears stable for each conformational state and could aid the correct placement of critical residues in other segments such as M4, which in the hydrophobic gate mechanism (18) interacts with the lipid during its translocation. The implication is that *Asn-905, despite its functional importance, is only indirectly involved in interaction with the lipid. According to the modeling, *Asn-905 is close to the proline kink of M4 near the hydrophobic gate residue *Ile-364 (PISL motif). In the E2 model, *Asn-905 is within hydrogen bonding distance of the M4 residue Asn-360, one helix turn from *Ile-364, and juxtaposed to Asn-359, previously shown to be critical to the affinity for the lipid substrate (18). Moreover, in addition to the hydrogen bond between *Lys-873 and *Asn-905, a new hydrogen bond appears to be established between *Lys-873 and Ser-365 in connection with the transition from E2P to E2 (Fig. 8). This serine is located in M4 right beside *Ile-364, so this event might be involved in the movement of *Ile-364 that pushes the lipid along. With such a scenario, each of the mutations to *Asn-905 analyzed in the present study would be expected to largely disturb the hydrogen bonding network and, thereby, indirectly the binding of the lipid substrate and/or the signal transduction leading to dephosphorylation, which likely involves M4 with its direct connection to the P-domain. Mutation of *Asn-905 would also affect the position of its hydrogen bonding partner, *Lys-873, which like *Asn-905 is important for the dephosphorylation (12) and is connected via M5 to the P-domain and therefore may as well be part of the mechanism of signal transduction to the phosphorylation site.
Figure 7.

Structural relations in ATP8A2 E2P and E2 models of relevance to the present data. These models were obtained by homology modeling based on Ca2+-ATPase crystal structures with refinement by molecular dynamics simulation corresponding to 100 ns (18). The transmembrane segments are here viewed from the cytoplasmic side of the membrane and are shown in cartoon with the most important residues discussed in the text in stick representation (carbon, yellow; oxygen, red; and nitrogen, blue). The color code for transmembrane segments is as follows: M1, orange; M2, purple; M3, blue; M4, wheat; M5, brown; M6, green; and M7–M10, gray. The red * on the E2 model indicates the peripheral (hydrophobic gate) pathway discussed.
Figure 8.

Asn-905 and Lys-873 are part of a “stability cluster.” The upper panels show in top view from the cytoplasmic side of the membrane domains of ATP8A2 the E2P and E2 models (see Fig. 7), aligned with M7–M10 helix backbones as reference. In the E2P model, the respective side-chain oxygen atoms of Thr-909 and Asn-905 are only 2.8 and 3.4 Å from the side-chain nitrogen atom of Lys-873, suggesting stable interactions among these residues. In the E2 model, the side-chain oxygen of Ser-365 is only 3.1 Å from the side-chain nitrogen atom of Lys-873. These interactions may stabilize the position of M4 with Ile-364, which together with other hydrophobic residues shown, including Ile-362 and Leu-367 of M4, Phe-88 of M1, Ile-115 of M2, and Val-906 of M6, make up a hydrophobic gate (18). The lower panels demonstrate the stability of the core region in the molecular dynamics simulation (18). The root mean square fluctuation of the 20-ns molecular dynamics simulation compared with the original homology models was computed and assigned to cartoon figures with blue signifying low and red signifying high divergence from the original models. Lys-873 is shown in yellow licorice as above.
The analysis of Val-906 mutants showed that replacement with alanine, serine, or glutamine led to increased apparent affinity for PS, whereas the negatively charged glutamate, despite being isosteric with glutamine, led to a marked, 5-fold reduction of the apparent affinity for PS but had little effect on the apparent affinity for PE, similar to glutamine. Because the headgroup of PS carries net negative charge, whereas that of PE is electrically neutral, the low PS affinity of the V906E mutant may be related to electric repulsion between the negative glutamate side chain and the PS headgroup during passage of the lipid substrate relatively close to the position of Val-906. In the structural models, the orientations of the side chains of *Asn-905 and Val-906 differ critically, the former pointing into the center between M4, M5, M6, and M8 as discussed above, whereas the latter points into the proposed hydrophobic gate peripheral pathway between M1, M2, M4, and M6 (indicated by the red star on the E2 model in Fig. 7). The data are therefore in accordance with the lipid headgroup moving along the latter pathway, and it is conceivable that a conformational change elicited by the presence of the lipid headgroup here can activate the dephosphorylation via *Asn-905.
As an alternative to the interpretation of the data in terms of the hydrophobic gate peripheral transport pathway, a central pathway between M4, M5, M6 and M8, similar to that of the ion pumps, might be considered (the “canonical” pathway (20)). A widening of the central space between *Lys-873, *Asn-874, *Asn-905, and *Thr-909 to accommodate the lipid substrate here would disrupt the modeled tight interactions between these residues shown in Fig. 7, implying that they would interact with the lipid substrate instead of with each other. Against this scenario as a possible reason for the functional importance of Lys-873, *Asn-874, and *Asn-905 stands the present finding that most of the other M5-M6 residues were mutation-insensitive, which would be rather surprising if the “giant” lipid substrate were bound between these helices. Evidence against a direct involvement of *Asn-905 in the binding of the lipid headgroup is furthermore provided by the finding that all the *Asn-905 substituents examined in this study caused a drastic effect on the dephosphorylation, irrespective of polarity and charge. Larger differences between the effects of the various *Asn-905 substitutions, similar to those seen for the Val-906 substitutions, would be expected if the negatively charged headgroup of PS interacted with *Asn-905. In particular, the negative charge of the aspartate substituent should repel PS. The functional effect was, on the contrary, a little less pronounced for *N905A and *N905D than for the other *Asn-905 mutants, which is best explained by the size of their side chains being more similar to that of the genuine asparagine.
Concluding remarks
*Asn-905 is here shown to be a key residue in the mechanism determining the sensitivity of the ATP8A2 phosphoenzyme to the lipid substrate. This finding in combination with the alignment showing that *Asn-905 is located in a position corresponding to key ion-binding asparagine/aspartate/glutamate residues of Ca2+-ATPase, Na+-K+-ATPase, and H+,K+-ATPase indicates that the signal transduction mechanism of the flippase is fundamentally very similar to that of the ion pumps. Our analysis shows that the new information can be reconciled with a model in which the translocation pathway is peripherally located (e.g. the hydrophobic gate mechanism (18)), although a centrally located transport pathway is not excluded.
Since 2013 it has been known that the I364M mutation of the hydrophobic gate residue (I376M in human) causes the neurological disorder CAMRQ syndrome (7, 18). Remarkably, the *N905D mutation studied here (N917D in human) was very recently identified as the cause of CAMRQ syndrome in a different family (9). Our results showing that the N905D variant lacks activity is consistent with the loss in function of ATP8A2 as the mechanism responsible for this disorder.
Experimental procedures
Mutagenesis of the pcDNA3 vector encoding the bovine ATP8A2 protein, coexpression with bovine CDC50A in HEK293T cells, and purification by immunoaffinity chromatography, taking advantage of a C-terminal 1D4 tag on ATP8A2, followed by reconstitution into PC vesicles and quantification of the protein content by Coomassie Blue staining of SDS-polyacrylamide gels with the purified ATP8A2/CDC50A preparations have been described previously in detail (12, 18, 27). The functional analysis was carried out on the reconstituted vesicles resolubilized in CHAPS detergent to allow addition of the lipid substrates PS and PE. The specific ATPase activity (μmol of Pi/min/mg of purified protein) was determined by following the liberation of Pi at 37 °C for 15 min in the presence of 7.5 mm ATP, 46 mm Hepes-Tris (pH 7.5), 150 mm NaCl, 12 mm MgCl2, 1 mm DTT, 9 mm CHAPS, and 0–1000 μm PS or 0–2000 μm PE with various concentrations of PC added to maintain a total lipid concentration of 2.5 mg/ml (approximately 3000 μm). The reaction was terminated by addition of an equal volume of 12% (w/v) SDS, and the Pi liberation was measured colorimetrically (27).
Phosphorylation with [γ-32P]ATP was carried out at 0 °C for 30 s in the presence of 2 μm [γ-32P]ATP, 50 mm Hepes-Tris (pH 7.5), 150 mm NaCl, 1 mm MgCl2, 1 mm DTT, and 10 mm CHAPS. The dephosphorylation was initiated by addition of various concentrations of PS (up to a maximum of 1000 μm) and PC (the latter to maintain a constant total lipid concentration of 1 mg/ml) dissolved in 50 mm Hepes-Tris (pH 7.5), 150 mm NaCl, 1 mm MgCl2, 1 mm DTT, and 10 mm CHAPS. The reaction was terminated after 5 s with 7% TCA and 1 mm H3PO4 or 25% TCA and 100 mm H3PO4 followed by precipitation on ice and SDS-PAGE under acidic conditions, taking advantage of the acid stability of the acylphosphate intermediate. The gels were fixed in 10% acetic acid and dried. Quantification of the radioactive phosphorylated bands on the gel was carried out with the Packard CycloneTM Storage Phosphor System.
All data analyses were carried out with SigmaPlot software (SPSS, Inc.). Error bars in figures (shown only when larger than the size of the symbols) represent S.D. To determine the K0.5(PS) and K0.5(PE) (lipid concentrations giving half-maximum activation), the one-site binding equation (Equation 1) was fitted to the data points corresponding to the concentration dependence of the ATPase activity.
| (Eq. 1) |
L is the concentration of PS or PE, and y0 is the minute ATPase activity determined in the absence of PS or PE (only PC present).
In cases where the highest PS or PE concentrations resulted in inhibition, only the data points corresponding to the rising part were fitted using Equation 1, to extract K0.5, and to draw the lines shown the following equation was used (Equation 2).
| (Eq. 2) |
Imax is the maximal inhibition, and IC50 is the lipid concentration giving half-maximum inhibition.
Author contributions
S. A. M., B. V., R. S. M., A. L. V., and J. P. A. conceptualization; S. A. M., L. S. M., A. L. V., and J. P. A. data curation; S. A. M., L. S. M., B. V., A. L. V., and J. P. A. formal analysis; S. A. M., L. S. M., and J. P. A. validation; S. A. M., L. S. M., A. L. V., and J. P. A. investigation; S. A. M., A. L. V., and J. P. A. visualization; S. A. M., L. S. M., A. L. V., and J. P. A. methodology; S. A. M. and J. P. A. writing-original draft; S. A. M., L. S. M., B. V., R. S. M., A. L. V., and J. P. A. writing-review and editing; B. V., R. S. M., A. L. V., and J. P. A. supervision; R. S. M. and J. P. A. resources; R. S. M. and J. P. A. funding acquisition; A. L. V. and J. P. A. software; J. P. A. project administration.
This work was supported in part by Danish Council for Independent Research Grant DFF 6110-00271 (to J. P. A.), Novo Nordisk Foundation Grant NNF16OC0022510 (to J. P. A.), Canadian Institutes of Health Research Grant PJT-148649 (to R. S. M.), and National Institutes of Health Grant EY002422 (to R. S. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PS
- phosphatidylserine
- PE
- phosphatidylethanolamine
- PC
- phosphatidylcholine
- CAMRQ
- cerebellar ataxia, mental retardation, and disequilibrium
- N
- nucleotide-binding
- P
- phosphorylation
- A
- actuator
- M1–M10
- the ten transmembrane helices numbered from the N-terminal end of the protein
- SERCA
- sarco/endoplasmic reticulum Ca2+-ATPase
- PL
- phospholipid.
References
- 1. Andersen J. P., Vestergaard A. L., Mikkelsen S. A., Mogensen L. S., Chalat M., and Molday R. S. (2016) P4-ATPases as phospholipid flippases—structure, function, and enigmas. Front. Physiol. 7, 275 10.3389/fphys.2016.00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Halleck M. S., Pradhan D., Blackman C., Berkes C., Williamson P., and Schlegel R. A. (1998) Multiple members of a third subfamily of P-type ATPases identified by genomic sequences and ESTs. Genome Res. 8, 354–361 10.1101/gr.8.4.354 [DOI] [PubMed] [Google Scholar]
- 3. Bull L. N., van Eijk M. J., Pawlikowska L., DeYoung J. A., Juijn J. A., Liao M., Klomp L. W., Lomri N., Berger R., Scharschmidt B. F., Knisely A. S., Houwen R. H., and Freimer N. B. (1998) A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet. 18, 219–224 10.1038/ng0398-219 [DOI] [PubMed] [Google Scholar]
- 4. Stapelbroek J. M., Peters T. A., van Beurden D. H., Curfs J. H., Joosten A., Beynon A. J., van Leeuwen B. M., van der Velden L. M., Bull L., Oude Elferink R. P., van Zanten B. A., Klomp L. W., and Houwen R. H. (2009) ATP8B1 is essential for maintaining normal hearing. Proc. Natl. Acad. Sci. U.S.A. 106, 9709–9714 10.1073/pnas.0807919106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Folmer D. E., Elferink R. P., and Paulusma C. C. (2009) P4 ATPases—lipid flippases and their role in disease. Biochim. Biophys. Acta 1791, 628–635 10.1016/j.bbalip.2009.02.008 [DOI] [PubMed] [Google Scholar]
- 6. Meguro M., Kashiwagi A., Mitsuya K., Nakao M., Kondo I., Saitoh S., and Oshimura M. (2001) A novel maternally expressed gene, ATP10C, encodes a putative aminophospholipid translocase associated with Angelman syndrome. Nat. Genet. 28, 19–20 [DOI] [PubMed] [Google Scholar]
- 7. Onat O. E., Gulsuner S., Bilguvar K., Nazli Basak A., Topaloglu H., Tan M., Tan U., Gunel M., and Ozcelik T. (2013) Missense mutation in the ATPase, aminophospholipid transporter protein ATP8A2 is associated with cerebellar atrophy and quadrupedal locomotion. Eur. J. Hum. Genet. 21, 281–285 10.1038/ejhg.2012.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martín-Hernández E., Rodríguez-García M. E., Camacho A., Matilla-Dueñas A., García-Silva M. T., Quijada-Fraile P., Corral-Juan M., Tejada-Palacios P., de Las Heras R. S., Arenas J., Martín M. A., and Martínez-Azorín F. (2016) New ATP8A2 gene mutations associated with a novel syndrome: encephalopathy, intellectual disability, severe hypotonia, chorea and optic atrophy. Neurogenetics 17, 259–263 10.1007/s10048-016-0496-y [DOI] [PubMed] [Google Scholar]
- 9. Alsahli S., Alrifai M. T., Tala S. A., Mutairi F. A., and Alfadhel M. (2018) Further delineation of the clinical phenotype of cerebellar ataxia, mental retardation, and disequilibrium syndrome type 4. J. Cent. Nerv. Syst. Dis. 10, 1179573518759682 10.1177/1179573518759682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toyoshima C. (2008) Structural aspects of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Arch. Biochem. Biophys. 476, 3–11 10.1016/j.abb.2008.04.017 [DOI] [PubMed] [Google Scholar]
- 11. Kaplan J. H. (2002) Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 71, 511–535 10.1146/annurev.biochem.71.102201.141218 [DOI] [PubMed] [Google Scholar]
- 12. Coleman J. A., Vestergaard A. L., Molday R. S., Vilsen B., and Andersen J. P. (2012) Critical role of a transmembrane lysine in aminophospholipid transport by mammalian photoreceptor P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. U.S.A. 109, 1449–1454 10.1073/pnas.1108862109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Puts C. F., and Holthuis J. C. (2009) Mechanism and significance of P4 ATPase-catalyzed lipid transport: lessons from a Na+/K+-pump. Biochim. Biophys. Acta 1791, 603–611 10.1016/j.bbalip.2009.02.005 [DOI] [PubMed] [Google Scholar]
- 14. Toyoshima C., Nakasako M., Nomura H., and Ogawa H. (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405, 647–655 10.1038/35015017 [DOI] [PubMed] [Google Scholar]
- 15. Kanai R., Ogawa H., Vilsen B., Cornelius F., and Toyoshima C. (2013) Crystal structure of a Na+-bound Na+,K+-ATPase preceding the E1P state. Nature 502, 201–206 10.1038/nature12578 [DOI] [PubMed] [Google Scholar]
- 16. Morth J. P., Pedersen B. P., Toustrup-Jensen M. S., Sørensen T. L., Petersen J., Andersen J. P., Vilsen B., and Nissen P. (2007) Crystal structure of the sodium-potassium pump. Nature 450, 1043–1049 10.1038/nature06419 [DOI] [PubMed] [Google Scholar]
- 17. Stone A., and Williamson P. (2012) Outside of the box: recent news about phospholipid translocation by P4 ATPases. J. Chem. Biol. 5, 131–136 10.1007/s12154-012-0078-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vestergaard A. L., Coleman J. A., Lemmin T., Mikkelsen S. A., Molday L. L., Vilsen B., Molday R. S., Dal Peraro M., and Andersen J. P. (2014) Critical roles of isoleucine-364 and adjacent residues in a hydrophobic gate control of phospholipid transport by the mammalian P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. U.S.A. 111, E1334–E1343 10.1073/pnas.1321165111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baldridge R. D., and Graham T. R. (2012) Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc. Natl. Acad. Sci. U.S.A. 109, E290–E298 10.1073/pnas.1115725109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baldridge R. D., and Graham T. R. (2013) Two-gate mechanism for phospholipid selection and transport by type IV P-type ATPases. Proc. Natl. Acad. Sci. U.S.A. 110, E358–E367 10.1073/pnas.1216948110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jensen M. S., Costa S. R., Duelli A. S., Andersen P. A., Poulsen L. R., Stanchev L. D., Gourdon P., Palmgren M., Günther Pomorski T., and López-Marqués R. L. (2017) Phospholipid flipping involves a central cavity in P4 ATPases. Sci. Rep. 7, 17621 10.1038/s41598-017-17742-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abe K., Irie K., Nakanishi H., Suzuki H., and Fujiyoshi Y. (2018) Crystal structures of the gastric proton pump. Nature 556, 214–218 10.1038/s41586-018-0003-8 [DOI] [PubMed] [Google Scholar]
- 23. Andersen J. P., and Vilsen B. (1994) Amino acids Asn796 and Thr799 of the Ca2+-ATPase of sarcoplasmic reticulum bind Ca2+ at different sites. J. Biol. Chem. 269, 15931–15936 [PubMed] [Google Scholar]
- 24. Obara K., Miyashita N., Xu C., Toyoshima I., Sugita Y., Inesi G., and Toyoshima C. (2005) Structural role of countertransport revealed in Ca2+ pump crystal structure in the absence of Ca2+. Proc. Natl. Acad. Sci. U.S.A. 102, 14489–14496 10.1073/pnas.0506222102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nielsen J. M., Pedersen P. A., Karlish S. J., and Jorgensen P. L. (1998) Importance of intramembrane carboxylic acids for occlusion of K+ ions at equilibrium in renal Na,K-ATPase. Biochemistry 37, 1961–1968 10.1021/bi972524q [DOI] [PubMed] [Google Scholar]
- 26. Swarts H. G., Koenderink J. B., Willems P. H., Krieger E., and De Pont J. J. (2005) Asn792 participates in the hydrogen bond network around the K+-binding pocket of gastric H,K-ATPase. J. Biol. Chem. 280, 11488–11494 10.1074/jbc.M412321200 [DOI] [PubMed] [Google Scholar]
- 27. Coleman J. A., Kwok M. C., and Molday R. S. (2009) Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J. Biol. Chem. 284, 32670–32679 10.1074/jbc.M109.047415 [DOI] [PMC free article] [PubMed] [Google Scholar]