

Abstract
Embryonic stem cells can self-renew and differentiate, holding great promise for regenerative medicine. They also employ multiple mechanisms to preserve the integrity of their genomes. Nucleotide excision repair, a versatile repair mechanism, removes bulky DNA adducts from the genome. However, the dynamics of the capacity of nucleotide excision repair during stem cell differentiation remain unclear. Here, using immunoslot blot assay, we measured repair rates of UV-induced DNA damage during differentiation of human embryonic carcinoma (NTERA-2) cells into neurons and muscle cells. Our results revealed that the capacity of nucleotide excision repair increases as cell differentiation progresses. We also found that inhibition of the apoptotic signaling pathway has no effect on nucleotide excision repair capacity. Furthermore, RNA-Seq–based transcriptomic analysis indicated that expression levels of four core repair factors, xeroderma pigmentosum (XP) complementation group A (XPA), XPC, XPG, and XPF-ERCC1, are progressively up-regulated during differentiation, but not those of replication protein A (RPA) and transcription factor IIH (TFIIH). Together, our findings reveal that increase of nucleotide excision repair capacity accompanies cell differentiation, supported by the up-regulated transcription of genes encoding DNA repair enzymes during differentiation of two distinct cell lineages.
Keywords: nucleotide excision repair, cancer stem cells, differentiation, neuron, vascular smooth muscle cells, muscle cell, neuron, nucleotide excision repair, stem cell, cell differentiation, genome stability, transcriptomics, NT2 cell, retinoic acid (RA), bone morphogenetic protein-2 (BMP-2)
Introduction
The genome of all living organisms is constantly assaulted by a variety of endogenous and exogenous DNA damaging agents. Cells possess elaborate DNA repair mechanisms to cope with various types of DNA damage. Nucleotide excision repair, one of these repair mechanisms, removes a wide range of bulky and helix-distorting lesions, including UV radiation–induced cis-syn cyclobutane pyrimidine dimers (CPDs)3 and (6–4) pyrimidine-pyrimidone photoproducts ((6–4)PPs), and chemical carcinogens (e.g. benzo[a]pyrene) and cancer chemotherapeutics (e.g. cisplatin)–induced bulky DNA adducts (1–3). The biochemical mechanism of excision repair reaction has been well-characterized in both prokaryotes and eukaryotes, which includes damage recognition, dual incisions bracketing the lesion, release of the excised oligomer, repair synthesis to fill the gap, and ligation (3–6). Nucleotide excision repair occurs in two modes, global repair and transcription-coupled repair, which differ only in the damage recognition step (7, 8). For global repair in humans, six core repair factors, RPA, XPA, XPC, TFIIH (10 subunits including XPB and XPD), XPG, and XPF-ERCC1, are required in the reconstituted in vitro system (9). Specifically, XPF and XPG incise at 19–21 nt 5′ and 5–6 nt 3′ to the lesion, respectively, after damage recognition by cooperative action of XPC, RPA, and XPA and kinetic proofreading by TFIIH, generating predominantly 26- to 27-nt-long excised oligomers (6, 10–13). Then, DNA Pol δ/ϵ resynthesizes the excised fragment and DNA ligase I or XRCC1-ligase III complex seals the 3′ nick (1, 14, 15). For human transcription-coupled repair, CSB translocase recognizes the stalling of elongating RNA polymerase II at a lesion in the transcribed strand and recruits the repair machinery except for XPC to carry out the subsequent excision repair reaction (16–18).
Embryonic stem (ES) cells are derived from the inner mass of embryos at the blastocyst stage of development. Because of their two unique characteristics, self-renewal and pluripotency, ES cells hold great promise for therapeutic purposes for a wide range of human diseases. Maintenance of genome integrity is crucial for ES cells in view of normal embryo development and therapeutic transplantation. In response to DNA damage, ES cells employ multiple strategies: apoptosis, senescence, DNA repair, and translesion DNA synthesis (19). There is compelling evidence to suggest that stem cells have different priorities in the use of various DNA damage counteracting strategies depending on cell type, differentiation stage, and type of DNA damage (20–24). Although the role of nucleotide excision repair in stem cells and terminally differentiated cells has been investigated in various studies (25–29), the main picture emerging from these studies is blurred and often contradictory.
The well-characterized human embryonic carcinoma cell line NTERA-2 (NT2), resembling ES cells closely, can be induced to differentiate into neurons and muscle cells by retinoic acid (RA) and bone morphogenetic protein-2 (BMP-2), respectively (30, 31). In the present study, we utilized NT2 cells to investigate the effects of variable differentiation stages and lineages on nucleotide excision repair. We find that UV resistance and nucleotide excision repair capacity increase along with differentiation of NT2 cells into neurons and muscle cells. We also find that inhibition of the massive apoptosis that has been reported to occur in ES cells has no effect on the repair of UV-induced DNA damage, suggesting the apoptotic signaling pathway does not contribute to the low nucleotide excision repair capacity in the undifferentiated NT2 cells. Furthermore, we show that the expression levels of six core nucleotide excision repair factors, except for RPA and TFIIH, also gradually increase during the differentiation of NT2 cells into the two types of cells.
Results
Differentiation of NT2 cells into neurons and muscle cells
To investigate the effects of distinct differentiation stages and lineages on nucleotide excision repair, we first tested the induction of neurons and muscle cells from NT2 cells. The advantages in utilizing the well-characterized NT2 cells are that nucleotide excision repair in cells at various stages of differentiation can be examined in an identical genetic background, and NT2 cells can differentiate into multiple different types of cells upon treatment with different differentiation agents. To test the induction of neurons from NT2 cells, we treated NT2 cells with RA (10 μm) for 16 days and verified the differentiation toward neuronal lineage and progressive shutdown of stem cell markers, including OCT4, SOX2, NANOG, LIN28A, and KLF4 (Fig. 1A). Of particular interest are that OCT4 and NANOG diminished quickly and neurofilament light (NF-L), unique to neuronal cells, appeared to be detected only after 4 days of RA treatment. For differentiation of NT2 cells into an epithelial lineage, we first used BMP-2, with a concentration of 30 ng/ml, to treat NT2 cells for 21 days and confirmed that the down-regulation of stem cell markers was coupled to up-regulation of α–smooth muscle actin (α-SMA), which is a differentiation marker of smooth muscle cells (Fig. 1B). In contrast, in NT2 cells treated with 0.1% (v/v) DMSO, stem cell markers are constantly expressed and NF-L and α-SMA are barely detectable. Furthermore, expressions of α-SMA in RA treatment group and NF-L in BMP-2 treatment group are negligible. Thus, we validated the induction of both neurons and muscle cells from NT2 cells, which therefore could be used in the subsequent nucleotide excision repair and transcriptional profiling studies.
Figure 1.
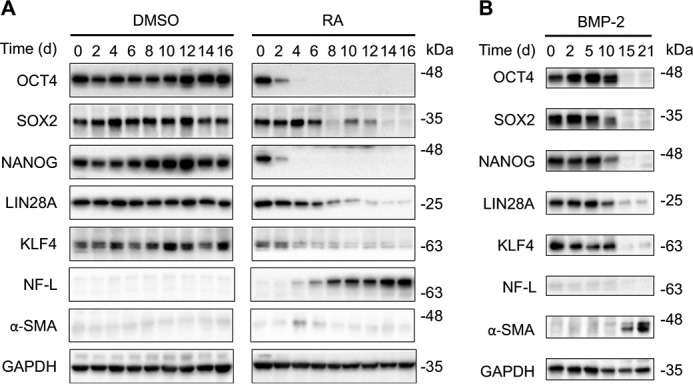
Differentiation of NT2 cells into neurons and muscle cells. A, NT2 cells were treated with 10 μm RA or 0.1% DMSO for up to 16 days. At the indicated days, whole cell extracts were prepared and analyzed by Western blotting with antibodies against markers of stem cell, neuron, muscle cell, and loading control GAPDH. B, differentiation induction was analyzed as in A except that NT2 cells were treated with 30 ng/ml BMP-2 for up to 21 days.
Up-regulation of XPC protein and enhancement of UV resistance upon differentiation of NT2 cells
XPC protein is indispensable for global repair by being essential for DNA damage recognition and recruiting the other excision repair factors (12, 32, 33). It has been shown that XPC physically interacts with RAD23B and CETN2 to form a tight complex, which was reported to function as an OCT4/SOX2 coactivator to enhance expression of pluripotency genes such as NANOG in ES cells (34–36). However, a follow-up study has shown that inducible knockout of the XPC C-terminal region, which contains the interaction sites with RAD23B and CETN2, has no impairment on OCT3/4 transcriptional activity and the maintenance of pluripotency (37), casting doubt on the coactivator role of XPC. The intriguing roles of XPC in nucleotide excision repair and potentially in transcriptional regulation prompted us to examine the level of XPC protein in multi-lineage differentiation of NT2 cells. First, we used XP-C mutant cells and NT2 cells to confirm the specificity of the XPC antibody and verified that XPC protein can be detected in NT2 cells but not in XP-C mutant cells (Fig. 2A). Surprisingly, we found that the level of XPC protein gradually increases during the treatment with RA or BMP-2 for up to 21 days (Fig. 2B). The up-regulation of XPC protein level is more striking during the differentiation of NT2 cells into muscle cells.
Figure 2.

Up-regulation of XPC protein and enhancement of UV resistance during differentiation. A, verification of anti-XPC antibody. Whole cell extracts were prepared from XP-C and NT2 cells, and analyzed by Western blotting with antibodies against XPC and GAPDH. B, Western blots showing up-regulation of XPC protein level during differentiation of NT2 cells into neurons and muscle cells. NT2 cells were treated with 10 μm RA or 40 ng/ml BMP-2 for up to 21 days, and whole cell extracts were prepared at indicated time points and analyzed by Western blotting with anti-XPC antibody. Stain-free total protein staining was used as loading control. C, enhancement of UV resistance in the neural-lineage differentiation. NT2 cells were treated with 10 μm RA for up to 21 days and resazurin cell viability assays were performed within 24 h following 2.5, 5, 10 J/m2 UV irradiation at day 0, 5, 10, and 21.
ES cells have been reported to be hypersensitive to UV exposure and undergo apoptosis rapidly after UV irradiation to eliminate the damaged cells (27, 38). To investigate the effect of differentiation stage on UV sensitivity, we treated NT2 cells at different stages of neural lineage differentiation with various UV doses and determined their UV sensitivity by resazurin cell viability assay. As expected, undifferentiated NT2 cells have lower cell viability within 24 h after even 2.5 J/m2 of UV irradiation, whereas the differentiated NT2 cells became more and more UV resistant with increasing differentiation stage (Fig. 2C). To conclude, we observed up-regulation of XPC protein in multi-lineage differentiation of NT2 cells and found that UV resistance progressively increases during differentiation.
Increase of nucleotide excision repair capacity accompanies multi-lineage differentiation
To determine the effect of RA-induced differentiation on nucleotide excision repair capacity, we measured the repair of (6–4)PPs and CPDs induced by various doses of UV in NT2 cells treated with RA for up to 21 days. Specifically, we irradiated NT2 cells after 0, 5, 10, and 21 days of RA treatment with 2.5, 5, and 10 J/m2 of UV and used immunoslot blot assay to quantify the repair rates of (6–4)PPs and CPDs within 24 h. We found that nucleotide excision repair capacity increases progressively during the RA-induced differentiation (Fig. 3). As expected, (6–4)PPs repair, which is almost complete within 4 h, is much faster than the repair of CPDs. The removed fraction of (6–4)PPs in undifferentiated NT2 cells gradually decreases as UV dose increases from 2.5 to 10 J/m2, and the rate of removal is significantly slower than differentiated NT2 cells treated with RA for 10 and 21 days (Fig. 3, A–F). For CPD repair, undifferentiated NT2 cells also have lower repair capacity, and the difference between undifferentiated and differentiated NT2 cells after 21 days' RA treatment reaches statistical significance following UV irradiation at 5 and 10 J/m2 doses (Fig. 3, G–L).
Figure 3.

Nucleotide excision repair capacity increases during differentiation of NT2 cells into neurons. A–F, immunoslot blot assays showing repair rates of (6–4)PPs in NT2 cells at various differentiation stages following UV treatment. NT2 cells were irradiated with 2.5, 5, 10 J/m2 UV following 0, 5, 10, and 21 days of 10 μm RA treatment and repair rates of (6–4)PPs within 4 h following UV irradiation were analyzed by immunoslot blot with anti-(6–4)PP antibody. SYBR gold staining was used to measure total loading DNA, and (6–4)PP signals were normalized to total DNA and plotted as a function of time. Results shown are the mean ± S.E. from three biological replicates, where error bars denote S.E. Statistical differences were tested by two-way ANOVA, followed by pairwise multiple comparisons. Significance from the post hoc Bonferroni multiple comparison test is indicated (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001). Statistical significances are denoted by asterisk in red, pink, and orange colors for differences between 21 days and 0 days, 10 days and 0 days, and 5 days and 0 days, respectively. G–L, repair rates of CPDs in NT2 cells at various differentiation stages within 24 h following UV treatment were analyzed and presented as in A–F except that anti-CPD antibody was used.
As different cellular differentiation lineage will eventually produce different types of terminally differentiated cells with various nucleotide excision repair capacity, the enhancement of nucleotide excision repair observed in the above studies may be unique in the neural lineage differentiation. We therefore determined the dynamics of nucleotide excision repair capacity over differentiation of NT2 cells into muscle cells. Briefly, we treated NT2 cells with 40 ng/ml BMP-2 for 0, 5, 10, and 21 days and measured the removal rate of (6–4)PPs and CPDs after 2.5, 5, and 10 J/m2 of UV irradiation by immunoslot blot assay. We found a gradual increase of nucleotide excision repair capacity with increasing differentiation stage (Fig. 4). Compared with undifferentiated NT2 cells, differentiated NT2 cells at day 21 of BMP-2 treatment have statistically significantly higher repair rates of (6–4)PPs and CPDs. In contrast, after different doses of UV irradiation (2.5, 5, and 10 J/m2) repair rates for (6–4)PPs and CPDs in differentiated NT2 cells at day 5 and day 10 of BMP-2 treatment are not statistically significantly different with those in undifferentiated NT2 cells, with the exception of differentiated NT2 cells at day 10 of BMP-2 treatment after 5 J/m2 of UV irradiation (Fig. 4, A–L).
Figure 4.

Nucleotide excision repair capacity increases during differentiation of NT2 cells into muscle cells. A–F, repair rates of (6–4)PPs in NT2 cells at various differentiation stages following UV treatment were analyzed and presented as in Fig. 3, A–F except that 40 ng/ml BMP-2 was used to induce differentiation. G–L, repair rates of CPDs in NT2 cells at various differentiation stages following UV treatment were analyzed and presented as in Fig. 3, G–L except that 40 ng/ml BMP-2 was used to induce differentiation.
Taken together, our results indicate that increase of nucleotide excision repair capacity accompanies differentiation of NT2 cells into neurons and muscle cells. These results suggest that the increase of nucleotide excision repair capacity after differentiation, which is not likely to be confined to the two specific lineages of NT2 cell differentiation, may commonly occur in all lineages of ES cell differentiation.
Inhibition of apoptosis has no effect on nucleotide excision repair capacity
ES cells are highly UV sensitive and undergo massive apoptosis after UV irradiation (39). In response to UV-induced DNA damage, the tumor suppressor p53 and the histone variant H2AX are phosphorylated by ATM and/or ATR kinases (40, 41). In the induction of apoptosis, a variety of cellular proteins are cleaved including the executioner caspase 3 and the DNA repair protein poly (ADP-ribose) polymerase (PARP). Therefore, it is reasonable to assume that this large-scale apoptosis dismantles cellular architecture and thus interferes with all essential cellular functions for survival including nucleotide excision repair (27). To test for this potential explanation for low repair activity, we used Z-VAD-FMK (42), an irreversible pan-caspase inhibitor, to validate the inhibition of apoptosis following UV treatment in undifferentiated and differentiated NT2 cells at day 21 of RA treatment, and then we measured repair rates for (6–4)PPs and CPDs, caused by 10 J/m2 of UV irradiation, within 24 h in undifferentiated and differentiated NT2 cells with or without inhibition of apoptosis. Compared with terminally differentiated cells, ES cells are more vulnerable to apoptosis. As expected, phosphorylation of tumor suppressor p53 on Ser-15 and histone variant H2AX on Ser-139 and cleavage of caspase 3 and PARP in undifferentiated NT2 cells were higher than those in differentiated NT2 cells after UV treatment (Fig. 5A). In contrast, as apoptotic signaling pathway was blocked by Z-VAD-FMK inhibitor, cleaved caspase 3 and PARP were barely detectable (Fig. 5B). Because Z-VAD-FMK has no effect on phosphorylation of p53 and H2AX, the dynamics of phosphorylation of p53 on Ser-15 and H2AX on Ser-139 were similar in the presence and absence of apoptosis inhibitor (Fig. 5B). Importantly, we found that inhibition of apoptosis does not significantly change the repair rates for (6–4)PPs and CPDs in undifferentiated and differentiated NT2 cells (Fig. 5, C–F).
Figure 5.

Inhibition of apoptosis pathway has no effect on nucleotide excision repair capacity. A, NT2 cells were treated with 10 μm RA or 0.1% DMSO for 21 days. At the indicated time points following UV (10 J/m2) treatment, whole cell extracts were prepared and analyzed by Western blotting with antibodies against the indicated proteins. Stain-free total protein staining was used as loading control. B, Western blotting assays were performed as in A except that apoptosis inhibitor (Z-VAD-FMK) was added 30 min prior to UV treatment. The final Z-VAD-FMK concentration was 20 μm. C and D, immunoslot blot assays showing repair rates of (6–4)PPs in NT2 cells at various differentiation stages after RA treatment. Repair rates of (6–4)PPs within 4 h after UV (10 J/m2) irradiation were analyzed as in Fig. 3, A–F except that apoptosis inhibitor (AI) was added 30 min prior to UV treatment. All experiments were repeated three times and representative results are shown. The results are presented as mean ± S.D. Statistical differences were tested by two-way ANOVA, followed by Sidak's multiple comparisons. No statistically significant difference was found. AI denotes group with apoptosis inhibitor treatment. No AI denotes control group. E and F, immunoslot blot assays showing repair rates of CPDs in NT2 cells at various differentiation stages after RA treatment. Repair rates within 24 h after UV (10 J/m2) irradiation were analyzed and presented as in C and D except that anti-CPD antibody was used. No statistically significant difference was found. AI denotes group with apoptosis inhibitor treatment. No AI denotes control group.
Our results, together with cell viability assays, confirmed that after UV irradiation undifferentiated NT2 cells undergo massive apoptosis, whereas differentiated cells have little or no induction of apoptosis. Furthermore, our findings indicated that inhibition of the apoptosis signaling pathway has no effect on nucleotide excision repair capacity.
Transcriptional regulation of core nucleotide excision repair factors in multi-lineage differentiation
Although we have found that the XPC protein level increases as differentiation progresses and that the inhibition of apoptosis does not affect nucleotide excision repair capacity, the molecular mechanisms underlying the gradual increase of nucleotide excision repair capacity during multi-lineage differentiation of NT2 cells remain poorly understood. Regulation of one or more repair factors, which play a rate-limiting role in nucleotide excision repair (e.g. XPA), may contribute to the changes of repair capacity during multi-lineage differentiation. However, it is impractical to measure levels of all factors related to excision repair by Western blotting, because of lack of reliable antibodies for all excision repair proteins.
We therefore performed transcriptomic analysis by RNA-Seq and profiled transcriptional dynamics along the differentiation trajectory of the genes associated with nucleotide excision repair during multi-lineage differentiation of NT2 cells (Fig. 6). Consistent with our differentiation induction results shown in Fig. 1, we observed gradual shutdown of stem cell marker genes (OCT4, SOX2, NANOG, LIN28A, and KLF4) and up-regulation of marker genes specific to the neural and epithelial lineage (NEFL and ACTA2) (Fig. S1). Interestingly, genes coding for XPA, XPC, XPG and XPF-ERCC1 were up-regulated progressively during the multi-lineage differentiation (Fig. 6A). In contrast, RPA1, RPA2, and RPA3, the three subunits of RPA complex, were down-regulated in the multi-lineage differentiation process (Fig. 6A). We also observed that genes coding for the 10 subunits of TFIIH complex including XPB and XPD were down-regulated although there were no statistically significant differences for some genes of the TFIIH complex (Fig. 6A). In addition, CSB and DDB2 were up-regulated but CSA and DDB1 were down-regulated (Fig. 6B).
Figure 6.
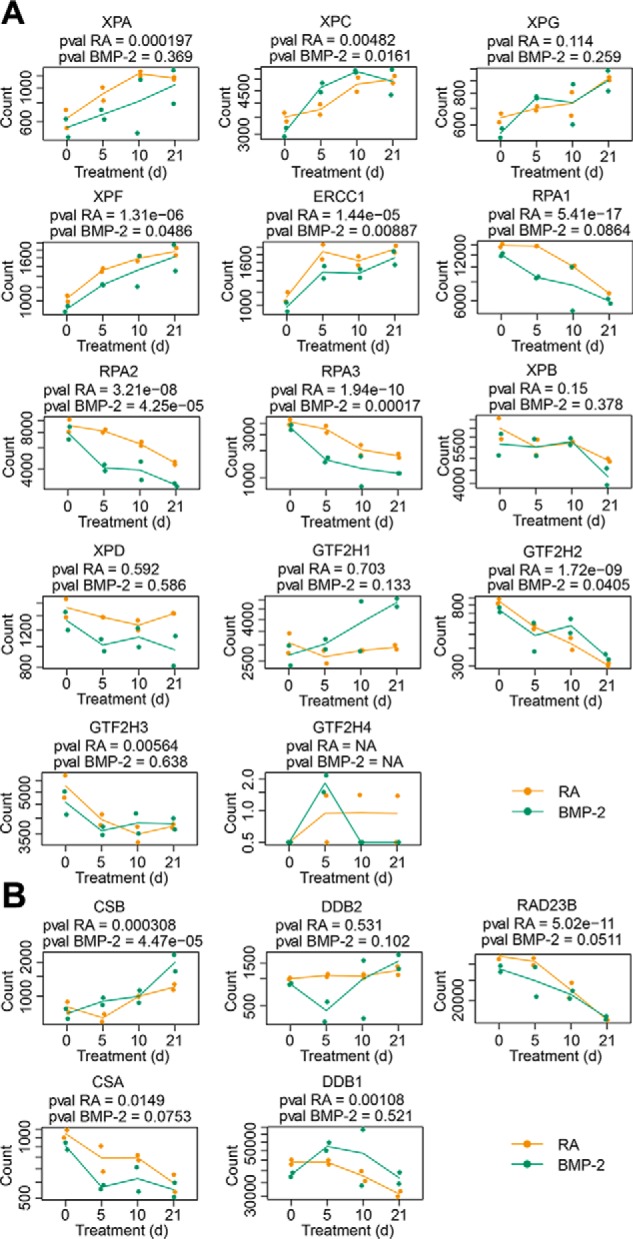
Transcriptional regulation of core nucleotide excision repair factors in multi-lineage differentiation. A, up-regulation of XPA, XPC, XPG, and XPF-ERCC1 and down-regulation of RPA and TFIIH during differentiation of NT2 cells into neurons or muscle cells. B, up-regulation of CSB and DDB2, and down-regulation of CSA and DDB1 during multi-lineage differentiation. p values from likelihood-ratio tests between the neuronal and epithelial lineages after multiple hypothesis testing correction are included. The dots indicate the count of RNA-Seq reads for RA (shown in orange color) and BMP-2 (shown in green color) treated groups. Each group has two biological replicates. The solid lines denote the mean value of counts from the two biological replicates.
Taken together, our transcriptomic analysis showed that expression levels of XPA, XPC, XPG, and XPF-ERCC1 increased, but those of RPA and TFIIH decreased during the multi-lineage differentiation process. As terminally differentiated neurons and muscle cells are nondividing, the down-regulation of both RPA and TFIIH during differentiation is likely because of the corresponding decrease in DNA replication and transcription. Therefore, our results suggest that the up-regulation of XPA, XPC, XPG, and XPF-ERCC1, together with some other nucleotide excision repair-related genes such as CSB and DDB2, may contribute to the gradual increase of nucleotide excision repair capacity during multi-lineage differentiation of NT2 cells.
Discussion
ES cells, which are rapidly dividing and self-renewing, are essential for embryo development. They are generally thought to have better capability to cope with attacks on the genome compared with nondividing, terminally differentiated cells (43). In this study, we monitored the dynamic changes of nucleotide excision repair capacity at various stages of differentiation of human embryonic carcinoma cells (NT2) into neurons and muscle cells. Our results showed that nucleotide excision repair capacity gradually increases as differentiation progresses. We found that XPC protein level increases along with differentiation toward the two different lineages as well. We confirmed that undifferentiated stem cells are more UV sensitive than differentiated cells and undergo apoptosis rapidly after UV irradiation. Our RNA-Seq data analysis indicated that, of all six core nucleotide excision repair factors, XPA, XPC, XPG, and XPF-ERCC1 were up-regulated, but RPA and TFIIH were down-regulated during multi-lineage differentiation. The accompanied increase of nucleotide excision repair capacity over the differentiation course may be explained by the up-regulation of expression levels for the four core repair factors. ES cells utilize multiple strategies, including apoptosis, senescence, DNA repair and translesion DNA synthesis, to preserve the genomic integrity upon DNA damage (19). Our findings suggest that stem cells might primarily use apoptotic pathway to eliminate cells containing massively damaged DNA, whereas postmitotic differentiated cells, which have lost self-renewal capacity, mainly employ nucleotide excision repair to cope with DNA damage.
Unlike somatic cells, ES cells have unique cell cycle features: a short G1 phase and deficiency in G1/S checkpoint (44). Upon UV irradiation, these unique features will allow ES cells containing damaged DNA enter the S phase, which will ultimately cause apoptosis. This may explain the massive apoptosis occurring after UV irradiation in stem cells. Moreover, the strong apoptotic response in ES cells was speculated to impair the nucleotide excision repair activity (27, 39). However, in our study we used an irreversible pan-caspase inhibitor to block the apoptosis pathway and found there was essentially no significant difference in repair capacities between apoptosis inhibitor–treated and control groups. As this apoptosis inhibitor used in our study only targets caspases, we cannot exclude the possibility that very early apoptotic signals may interfere with nucleotide excision repair activity instead.
A variety of studies (22, 26–29) comparing nucleotide excision repair capacity between stem cells and differentiated cells showed discrepant results. Those discrepancies may originate from the different extents of differentiation caused by different differentiation induction procedures and repair rate measurement techniques. In our studies, we monitored the repair rates of UV damage at various stages of differentiation and in two distinct differentiation lineages. With the advancement of high-throughput sequencing-based methods for mapping of DNA damage and repair (45–50), the nucleotide-resolution genome-wide DNA damage formation and repair profiles in stem cells and differentiated cells can be achieved and analyzed in future studies to better understand the molecular mechanisms underlying the dynamics of nucleotide excision repair capacity over differentiation process.
Experimental procedures
Cell lines, culture conditions, and differentiation induction
The NTERA-2 cl.D1 (NT2/D1) cell line was purchased from American Type Culture Collection (ATCC). XP-C (XP4PA-SV-EB, GM15983) cells, a human skin fibroblast cell line, were purchased from the National Institute of General Medical Sciences Human Genetic Cell Repository (Coriell Institute). All cell lines were cultured in DMEM (Gibco) supplemented with 10% FBS (Sigma-Aldrich) at 37 °C in a 5% CO2 humidified incubator.
For induction of NT2 cells into neurons, 10 mm all-trans RA (Sigma), dissolved in DMSO, was added into the culture medium at a final concentration of 10 μm. BMP-2 (GenScript) at a final concentration of 30 ng/ml was firstly used to test the muscle cell induction, and then 40 ng/ml BMP-2 was used in later experiments. As a negative control, DMSO was added into the culture medium for a final concentration of 0.1%. NT2 cells were treated with differentiation induction agents or DMSO for up to 21 days, and the culture medium was changed every 2 days.
Antibodies and reagents
The iPS Cell Reprogramming Antibody Kit (9092s), containing antibodies against stem cell markers (OCT4, SOX2, NANOG, LIN28A, and KLF4), and antibodies against NF-L (2837), cleaved caspase-3 (9661), PARP (9542), phosphor-p53 (Ser-15; 9284), and phosphor-H2AX (Ser-139; 9718) were purchased from Cell Signaling Technology. The other primary antibodies included antibodies against XPC (sc-74410) from Santa Cruz Biotechnology; α-SMA (A2547) from Sigma; GAPDH (GTX627408) from GeneTex; CPDs (NMDND001) and (6–4)PPs (NMDND002) from Cosmo Bio. The secondary antibodies including peroxidase-linked anti-mouse IgG (NA931V) and anti-rabbit IgG (NA934V) were purchased from GE Healthcare. The pan-caspase inhibitor Z-VAD-FMK (tlrl-vad) was obtained from InvivoGen.
UV irradiation
After treatment with different agents, culture medium was removed and cells were then exposed to 254-nm UV light (1 J/m2/sec) from a GE germicidal lamp for 2.5, 5, and 10 s. Then, culture medium was added and cells were further incubated in the chamber. At different time points as indicated, cells were washed twice with ice-cold PBS, scraped from the Petri dish, and collected by centrifugation.
Western blotting
Cells were lysed for 10 min on ice in RIPA buffer (10 mm Tris-Cl, pH 8.0, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mm NaCl, 1 mm PMSF) containing protease inhibitors (Roche Applied Science). Then, cell lysates were sonicated for 30 s, and equal amounts of cell lysates were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and then probed by the indicated antibodies. 10% Stain-Free Precast Gels (Bio-Rad) were used and gel loading control was done by stain-free detection following the manufacturer's instructions. Prestained Protein Ladder (Fermentas) was used to determine molecular weight. Chemiluminescence signals were visualized with ECL Prime Western Blotting Detection Reagent (GE Amersham Bioscience) by using a ChemiDoc Imaging System (Bio-Rad). All experiments were repeated three times and representative results are shown.
Cell viability assay
To assess cell viability, cells were seeded in 96-well plates and subjected to 0, 2.5, 5, or 10 J/m2 dose of UV exposure after 24 h incubation. Cells were then incubated at 37 °C in an incubator for up to 24 h. During the incubation, resazurin (Sigma) was added into the culture medium to a final concentration of 0.02%, and cells were incubated for 4 h prior to the measurement of resazurin reduction by SpectraMax M3 microplate reader (Molecular Devices). After subtraction of background signal, the cell viability was expressed as the percentage of signal intensity relative to cells without UV treatment. All experiments have three biological replicates and each sample was assayed in triplicates. Mean values ± S.D. were plotted (Fig. 2C).
Immunoslot blot assay
Repair of UV-induced (6–4)PPs and CPDs was measured as described previously (51). Briefly, cells after treatment were harvested at the indicated time points and genomic DNA was extracted with a QIAamp DNA Mini Kit (Qiagen). An equal amount of genomic DNA (250 ng for (6–4)PPs and 100 ng for CPDs) was immobilized onto a nitrocellulose membrane by using a Bio-Dot SF apparatus (Bio-Rad) and probed with anti-(6–4)PP or anti-CPD antibody. The peroxidase-linked anti-mouse IgG was used as a secondary antibody and chemiluminescence signals were detected with ECL Prime Western Blotting Detection Reagent (GE Amersham Bioscience) by using a ChemiDoc Imaging System (Bio-Rad). SYBR gold staining was then applied to the membrane as gel loading control. All signal intensities were quantified by Image Lab Software (Bio-Rad). All experiments were repeated three times and representative results are shown. Estimated mean ± S.E. for each group, as well as results from two-way ANOVA analysis, followed by post hoc pairwise multiple comparisons, were plotted using Prism 6 (GraphPad Software) and shown in Figs. 3 and 4.
RNA-Seq and transcriptomic analysis
NT2 cells were harvested at different time points (day 0, 5, 10, and 21) after treatment with RA or BMP-2. Total RNA was then isolated using RNA Reagent (Invitrogen) and PureLink RNA Mini Kit (Invitrogen) according to the manufacturer's instructions. Each sample has two biological replicates with a total of 14 samples sequenced (Table S1). Purification of mRNA, library preparation and paired-end sequencing (2 × 150 bp) on a HiSeq 4000 platform (Illumina) were performed by Novogene.
Reads from RNA-Seq were aligned using STAR (52), followed by a filtering step to remove (i) reads with mapping quality less than 20, (ii) read pairs with unexpected orientations and greater than 500-Kb mapping distance between the read pair, and (iii) reads that were mapped to greater than 10 positions in the genome. Read counts for each gene were obtained using FeatureCounts (53). After removing genes with zero read counts across all samples and genes without corresponding annotations by Ensembl, we obtained raw read counts of 25,952 genes across 14 samples. Read counts were further transformed into reads per kilobase million (RPKM) for visualization (Fig. 6; Fig. S1). Pairwise Spearman correlation between samples was calculated using the RPKM of 2000 genes with highest variance and the heat map is shown in Fig. S2. Likelihood ratio test for differential expression analysis across all time points was carried out using DESeq2 (54) for the neuronal and epithelial lineages, respectively, with p values adjusted by the Benjamini-Hochberg procedure.
Data and code availability
All RNA-Seq data in this study have been deposited in Gene Expression Omnibus (GEO) with accession number GSE125370. All code used in this study is available at https://github.com/yuchaojiang/damage_repair/tree/master/NT2.4
Author contributions
W. Li and A. S. conceptualization; W. Li and W. Liu data curation; W. Li, W. Liu, R. W., O. A., Y. J., and A. S. formal analysis; W. Li, W. Liu, and A. K. investigation; W. Li, O. A., and Y. J. methodology; W. Li writing-original draft; W. Li, O. A., Y. J., and A. S. writing-review and editing; O. A. and Y. J. software; Y. J. resources; A. S. supervision; A. S. funding acquisition; A. S. project administration.
Supplementary Material
This work was supported by National Institutes of Health Grants GM118102 and ES027255 (to A. S.) and CA142538 (to Y. J.) and UNC Lineberger Comprehensive Cancer Center developmental award 2017T109 (to Y. J.). This work was also supported by the Financial Funds of China Scholarship Council (201506315077) and the Fundamental Research Funds of Xiamen University (to W. Liu) and by EMBO Installation Grant Award funded by the Scientific and Technological Research Council of Turkey (TUBITAK) (to O. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2 and Table S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- CPD
- cyclobutane pyrimidine dimer
- (6–4)PP
- (6–4) pyrimidine-pyrimidone photoproduct
- TFIIH
- transcription factor IIH
- ES
- embryonic stem
- NT2
- NTERA-2
- RA
- retinoic acid
- BMP-2
- bone morphogenetic protein-2
- RPA
- replication protein A
- XP
- xeroderma pigmentosum
- NF-L
- neurofilament light
- α-SMA
- α–smooth muscle actin
- PARP
- poly (ADP-ribose) polymerase
- ANOVA
- analysis of variance.
References
- 1. Wood R. D. (1997) Nucleotide excision repair in mammalian cells. J. Biol. Chem. 272, 23465–23468 10.1074/jbc.272.38.23465 [DOI] [PubMed] [Google Scholar]
- 2. Sancar A. (2016) Mechanisms of DNA repair by photolyase and excision nuclease (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 55, 8502–8527 10.1002/anie.201601524 [DOI] [PubMed] [Google Scholar]
- 3. Hu J., Selby C. P., Adar S., Adebali O., and Sancar A. (2017) Molecular mechanisms and genomic maps of DNA excision repair in Escherichia coli and humans. J. Biol. Chem. 292, 15588–15597 10.1074/jbc.R117.807453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adebali O., Chiou Y. Y., Hu J., Sancar A., and Selby C. P. (2017) Genome-wide transcription-coupled repair in Escherichia coli is mediated by the Mfd translocase. Proc. Natl. Acad. Sci. U.S.A. 114, E2116–E2125 10.1073/pnas.1700230114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Adebali O., Sancar A., and Selby C. P. (2017) Mfd translocase is necessary and sufficient for transcription-coupled repair in Escherichia coli. J. Biol. Chem. 292, 18386–18391 10.1074/jbc.C117.818807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li W., Adebali O., Yang Y., Selby C. P., and Sancar A. (2018) Single-nucleotide resolution dynamic repair maps of UV damage in Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. U.S.A. 115, E3408–E3415 10.1073/pnas.1801687115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mellon I., Spivak G., and Hanawalt P. C. (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 51, 241–249 10.1016/0092-8674(87)90151-6 [DOI] [PubMed] [Google Scholar]
- 8. Hanawalt P. C., and Spivak G. (2008) Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 10.1038/nrm2549 [DOI] [PubMed] [Google Scholar]
- 9. Mu D., Park C. H., Matsunaga T., Hsu D. S., Reardon J. T., and Sancar A. (1995) Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem. 270, 2415–2418 10.1074/jbc.270.6.2415 [DOI] [PubMed] [Google Scholar]
- 10. Huang J. C., Svoboda D. L., Reardon J. T., and Sancar A. (1992) Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc. Natl. Acad. Sci. U.S.A. 89, 3664–3668 10.1073/pnas.89.8.3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Svoboda D. L., Taylor J. S., Hearst J. E., and Sancar A. (1993) DNA repair by eukaryotic nucleotide excision nuclease. Removal of thymine dimer and psoralen monoadduct by HeLa cell-free extract and of thymine dimer by Xenopus laevis oocytes. J. Biol. Chem. 268, 1931–1936 [PubMed] [Google Scholar]
- 12. Reardon J. T., and Sancar A. (2003) Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 17, 2539–2551 10.1101/gad.1131003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu J., Choi J. H., Gaddameedhi S., Kemp M. G., Reardon J. T., and Sancar A. (2013) Nucleotide excision repair in human cells: Fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem. 288, 20918–20926 10.1074/jbc.M113.482257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem. 65, 43–81 10.1146/annurev.bi.65.070196.000355 [DOI] [PubMed] [Google Scholar]
- 15. Moser J., Kool H., Giakzidis I., Caldecott K., Mullenders L. H., and Fousteri M. I. (2007) Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase IIIα in a cell-cycle-specific manner. Mol. Cell 27, 311–323 10.1016/j.molcel.2007.06.014 [DOI] [PubMed] [Google Scholar]
- 16. Selby C. P., and Sancar A. (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem. 272, 1885–1890 10.1074/jbc.272.3.1885 [DOI] [PubMed] [Google Scholar]
- 17. Selby C. P., and Sancar A. (1997) Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 94, 11205–11209 10.1073/pnas.94.21.11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fousteri M., Vermeulen W., van Zeeland A. A., and Mullenders L. H. (2006) Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell 23, 471–482 10.1016/j.molcel.2006.06.029 [DOI] [PubMed] [Google Scholar]
- 19. Nouspikel T. (2013) Genetic instability in human embryonic stem cells: Prospects and caveats. Future Oncol. 9, 867–877 10.2217/fon.13.22 [DOI] [PubMed] [Google Scholar]
- 20. Heyer B. S., MacAuley A., Behrendtsen O., and Werb Z. (2000) Hypersensitivity to DNA damage leads to increased apoptosis during early mouse development. Genes Dev. 14, 2072–2084 [PMC free article] [PubMed] [Google Scholar]
- 21. Potten C. S., Wilson J. W., and Booth C. (1997) Regulation and significance of apoptosis in the stem cells of the gastrointestinal epithelium. Stem Cells (Dayton) 15, 82–93 10.1002/stem.150082 [DOI] [PubMed] [Google Scholar]
- 22. de Waard H., Sonneveld E., de Wit J., Esveldt-van Lange R., Hoeijmakers J. H., Vrieling H., and van der Horst G. T. (2008) Cell-type-specific consequences of nucleotide excision repair deficiencies: Embryonic stem cells versus fibroblasts. DNA Repair 7, 1659–1669 10.1016/j.dnarep.2008.06.009 [DOI] [PubMed] [Google Scholar]
- 23. Tichy E. D., and Stambrook P. J. (2008) DNA repair in murine embryonic stem cells and differentiated cells. Exp. Cell Res. 314, 1929–1936 10.1016/j.yexcr.2008.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rocha C. R., Lerner L. K., Okamoto O. K., Marchetto M. C., and Menck C. F. (2013) The role of DNA repair in the pluripotency and differentiation of human stem cells. Mutat. Res. 752, 25–35 10.1016/j.mrrev.2012.09.001 [DOI] [PubMed] [Google Scholar]
- 25. Pedersen R. A., and Cleaver J. E. (1975) Repair of UV damage to DNA of implantation-stage mouse embryos in vitro. Exp. Cell Res. 95, 247–253 10.1016/0014-4827(75)90548-0 [DOI] [PubMed] [Google Scholar]
- 26. Rasko I., Georgieva M., Farkas G., Santha M., Coates J., Burg K., Mitchell D. L., and Johnson R. T. (1993) New patterns of bulk DNA repair in ultraviolet irradiated mouse embryo carcinoma cells following differentiation. Somat. Cell Mol. Genet. 19, 245–255 10.1007/BF01233072 [DOI] [PubMed] [Google Scholar]
- 27. Van Sloun P. P., Jansen J. G., Weeda G., Mullenders L. H., van Zeeland A. A., Lohman P. H., and Vrieling H. (1999) The role of nucleotide excision repair in protecting embryonic stem cells from genotoxic effects of UV-induced DNA damage. Nucleic Acids Res. 27, 3276–3282 10.1093/nar/27.16.3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nouspikel T., and Hanawalt P. C. (2000) Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Mol. Cell Biol. 20, 1562–1570 10.1128/MCB.20.5.1562-1570.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsu P. H., Hanawalt P. C., and Nouspikel T. (2007) Nucleotide excision repair phenotype of human acute myeloid leukemia cell lines at various stages of differentiation. Mutat. Res. 614, 3–15 10.1016/j.mrfmmm.2006.06.008 [DOI] [PubMed] [Google Scholar]
- 30. Lee V. M., and Andrews P. W. (1986) Differentiation of NTERA-2 clonal human embryonal carcinoma cells into neurons involves the induction of all three neurofilament proteins. J. Neurosci. 6, 514–521 10.1523/JNEUROSCI.06-02-00514.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chadalavada R. S., Houldsworth J., Olshen A. B., Bosl G. J., Studer L., and Chaganti R. S. (2005) Transcriptional program of bone morphogenetic protein-2-induced epithelial and smooth muscle differentiation of pluripotent human embryonal carcinoma cells. Funct. Integr. Genomics 5, 59–69 10.1007/s10142-005-0132-7 [DOI] [PubMed] [Google Scholar]
- 32. Sugasawa K., Ng J. M., Masutani C., Iwai S., van der Spek P. J., Eker A. P., Hanaoka F., Bootsma D., and Hoeijmakers J. H. (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell 2, 223–232 10.1016/S1097-2765(00)80132-X [DOI] [PubMed] [Google Scholar]
- 33. Wakasugi M., and Sancar A. (1998) Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc. Natl. Acad. Sci. U.S.A. 95, 6669–6674 10.1073/pnas.95.12.6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fong Y. W., Inouye C., Yamaguchi T., Cattoglio C., Grubisic I., and Tjian R. (2011) A DNA repair complex functions as an Oct4/Sox2 coactivator in embryonic stem cells. Cell 147, 120–131 10.1016/j.cell.2011.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cattoglio C., Zhang E. T., Grubisic I., Chiba K., Fong Y. W., and Tjian R. (2015) Functional and mechanistic studies of XPC DNA-repair complex as transcriptional coactivator in embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 112, E2317–E2326 10.1073/pnas.1505569112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang E. T., He Y., Grob P., Fong Y. W., Nogales E., and Tjian R. (2015) Architecture of the human XPC DNA repair and stem cell coactivator complex. Proc. Natl. Acad. Sci. U.S.A. 112, 14817–14822 10.1073/pnas.1520104112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ito S., Yamane M., Ohtsuka S., and Niwa H. (2014) The C-terminal region of Xpc is dispensable for the transcriptional activity of Oct3/4 in mouse embryonic stem cells. FEBS Lett. 588, 1128–1135 10.1016/j.febslet.2014.02.033 [DOI] [PubMed] [Google Scholar]
- 38. Sabapathy K., Klemm M., Jaenisch R., and Wagner E. F. (1997) Regulation of ES cell differentiation by functional and conformational modulation of p53. EMBO J. 16, 6217–6229 10.1093/emboj/16.20.6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vreeswijk M. P., Westland B. E., Hess M. T., Naegeli H., Vrieling H., van Zeeland A. A., and Mullenders L. H. (1998) Impairment of nucleotide excision repair by apoptosis in UV-irradiated mouse cells. Cancer Res. 58, 1978–1985 [PubMed] [Google Scholar]
- 40. Tibbetts R. S., Brumbaugh K. M., Williams J. M., Sarkaria J. N., Cliby W. A., Shieh S. Y., Taya Y., Prives C., and Abraham R. T. (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13, 152–157 10.1101/gad.13.2.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kitagawa R., and Kastan M. B. (2005) The ATM-dependent DNA damage signaling pathway. Cold Spring Harbor Symp. Quant. Biol. 70, 99–109 10.1101/sqb.2005.70.002 [DOI] [PubMed] [Google Scholar]
- 42. Garcia-Calvo M., Peterson E. P., Leiting B., Ruel R., Nicholson D. W., and Thornberry N. A. (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 273, 32608–32613 10.1074/jbc.273.49.32608 [DOI] [PubMed] [Google Scholar]
- 43. Nouspikel T., and Hanawalt P. C. (2002) DNA repair in terminally differentiated cells. DNA Repair 1, 59–75 [DOI] [PubMed] [Google Scholar]
- 44. Becker K. A., Ghule P. N., Therrien J. A., Lian J. B., Stein J. L., van Wijnen A. J., and Stein G. S. (2006) Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J. Cell Physiol. 209, 883–893 10.1002/jcp.20776 [DOI] [PubMed] [Google Scholar]
- 45. Hu J., Adar S., Selby C. P., Lieb J. D., and Sancar A. (2015) Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 29, 948–960 10.1101/gad.261271.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu J., Lieb J. D., Sancar A., and Adar S. (2016) Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 113, 11507–11512 10.1073/pnas.1614430113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mao P., Smerdon M. J., Roberts S. A., and Wyrick J. J. (2016) Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 113, 9057–9062 10.1073/pnas.1606667113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li W., Hu J., Adebali O., Adar S., Yang Y., Chiou Y. Y., and Sancar A. (2017) Human genome-wide repair map of DNA damage caused by the cigarette smoke carcinogen benzo[a]pyrene. Proc. Natl. Acad. Sci. U.S.A. 114, 6752–6757 10.1073/pnas.1706021114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hu J., Li W., Adebali O., Yang Y., Oztas O., Selby C. P., and Sancar A. (2019) Genome-wide mapping of nucleotide excision repair with XR-seq. Nat. Protoc. 14, 248–282 10.1038/s41596-018-0093-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yimit A., Adebali O., Sancar A., and Jiang Y. (2019) Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat. Commun. 10, 309 10.1038/s41467-019-08290-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kemp M. G., and Sancar A. (2016) ATR kinase inhibition protects non-cycling cells from the lethal effects of DNA damage and transcription stress. J. Biol. Chem. 291, 9330–9342 10.1074/jbc.M116.719740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., and Gingeras T. R. (2013) STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liao Y., Smyth G. K., and Shi W. (2014) featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 54. Love M. I., Huber W., and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-Seq data in this study have been deposited in Gene Expression Omnibus (GEO) with accession number GSE125370. All code used in this study is available at https://github.com/yuchaojiang/damage_repair/tree/master/NT2.4