

Abstract
Genome replication and virion assembly of segmented RNA viruses are highly coordinated events, tightly regulated by sequence and structural elements in the UTRs of viral RNA. This process is poorly defined and likely requires the participation of host proteins in concert with viral proteins. In this study, we employed a proteomics-based approach, named RNA–protein interaction detection (RaPID), to comprehensively screen for host proteins that bind to a conserved motif within the rotavirus (RV) 3′ terminus. Using this assay, we identified ATP5B, a core subunit of the mitochondrial ATP synthase, as having high affinity to the RV 3′UTR consensus sequences. During RV infection, ATP5B bound to the RV 3′UTR and co-localized with viral RNA and viroplasm. Functionally, siRNA-mediated genetic depletion of ATP5B or other ATP synthase subunits such as ATP5A1 and ATP5O reduced the production of infectious viral progeny without significant alteration of intracellular viral RNA levels or RNA translation. Chemical inhibition of ATP synthase diminished RV yield in both conventional cell culture and in human intestinal enteroids, indicating that ATP5B positively regulates late-stage RV maturation in primary intestinal epithelial cells. Collectively, our results shed light on the role of host proteins in RV genome assembly and particle formation and identify ATP5B as a novel pro-RV RNA-binding protein, contributing to our understanding of how host ATP synthases may galvanize virus growth and pathogenesis.
Keywords: RNA virus, RNA-binding protein, virus assembly, virology, proteomics, ATP synthase, RNA–protein interaction, biotin proximity ligation, nonenveloped double-stranded RNA virus, rotavirus, viral gastroenteritis, virus–host interaction
Introduction
Viruses are obligate intracellular pathogens that usurp host cellular machinery for efficient replication and production of progeny infectious particles. Despite the availability of multiple genetic and biochemical tools that enable the examination of rotavirus–host interactions in a unbiased manner, including genome-wide siRNA screens (1, 2), interactome analysis of protein–protein interactions (3), and the recently developed CRISPR-Cas9 screens (4), the molecular interactions between rotaviral RNA and host proteins remain relatively poorly understood. RNA-binding proteins play many important roles in cellular functions (5). With the exception of cytoplasmic RNA sensors, RIG-I–like receptors, including RIG-I, MDA5, and LGP2, and effector molecules such as RNase L and protein kinase R (6–9), the identities of host proteins that specifically bind to virus RNA genome or other replication intermediates and by-products, have rarely been characterized. Lately, interrogation of stem-loop structures within the untranslated regions (UTRs) of Zika virus and dengue virus has led to the discovery of host proteins Musashi-1 and TRIM25 that play critical roles in the replication cycle of these flaviviruses (10, 11). It was also found that, for influenza virus and coxsackievirus, defective RNA secondary structures markedly diminish binding of host exosome complex and PCBP2, respectively, and result in reduced replication (12, 13).
Rotaviruses (RVs)4 are nonenveloped, segmented dsRNA viruses in the Reoviridae family (14). RV infections are the leading cause of infantile diarrhea and severe gastroenteritis, resulting in around 215,000 deaths annually worldwide (15). Similar to those of influenza virus and orthoreovirus, RV gene segments have to be precisely organized and packaged into newly synthesized icosahedral virus particles during genome assembly (16). A previous study of a highly-related bluetongue virus and recent studies of RV suggest that the smaller RV RNA segments may be sorted into the newly forming virion particles first, and then the larger RNA segments enter the particle via RNA–RNA interactions in a sequential and NSP2-dependent manner (17–19). Hydrodynamic studies that examined the “stiffness” of RV RNA segments in vitro also suggested that packaging of the viral RNA segments into the capsid likely necessitates intimate RNA–protein interactions (20). Given the precision required and the likely high-energy consumption needed for RNA packaging, we hypothesize that host RNA-binding proteins contribute to the correct assembly of RV gene segments.
For most organisms, ATP hydrolysis powered by the ATPase machinery is the single most important “energy currency” (21, 22). For bacteriophages and large DNA viruses like herpesvirus and poxviruses, a central component of the packaging motor that drives viral genome assembly is the ATPase subunit, provided by the viruses themselves (23–25). In contrast, RNA viruses, except bluetongue virus (26), by and large harbor a relative small genome and rarely encode viral ATPases (27), inviting the question whether or not these viruses hijack the host ATPase complex as an alternative strategy to obtain energy for genome packaging. Here, we employed a novel and powerful technique named RNA-Protein Interaction Detection (RaPID), recently developed to study RNA–protein interactions (28), to comprehensively profile the host factors that bind to a stretch of conserved sequences within 3′UTR of group A RV genomes. Surprisingly, we identified ATP5B, an integral part of the mitochondrial F1–F0-ATPase complex (29), as a cellular component that co-precipitated and co-localized with RV dsRNA during infection. Functional dissection using small interfering RNA (siRNA) knockdown and a panel of small-molecule pharmacological inhibitors suggested that ATP5B assists RV genome replication and virion assembly. Thus, our study systematically interrogated the host proteins that interact with the RV 3′ terminus and revealed a tractable method to rapidly identify host proteins that bind to viral RNA sequences of interest in living cells.
Results
Proteomic analysis reveals novel host proteins that interact with RV 3′UTR consensus sequences
For group A human and animal RVs, the last seven nucleotides within the mRNA 3′UTR, 5′-UGUGACC-3′, are highly conserved in all 11 gene segments (30–32) and distinct from those in group B (5′-UAUACCC-3′) and group C (5′-UGUGGCU-3′) RVs (Fig. 1). This short sequence forms a cis-acting signal that contributes to the efficient synthesis of minus strand RNA and is also conducive to viral gene expression in the host cells through interaction with RV protein NSP3 (33, 34). However, the nature and identity of host factors that bind to this important RNA region remain unknown.
Figure 1.

RNA sequence alignment of the 3′ terminus of 11 gene segments from different RV genogroups. RRV (rhesus RV) is a simian group A RV strain; Wa, adult diarrheal rotavirus (ADRV), and Bristol are all human RV strains from group A, B, and C, respectively.
To comprehensively identify host proteins that interact with RV 3′UTR consensus sequence, we took advantage of a new screening approach, named RaPID, that detects, with high sensitivity, any protein in the vicinity of the target RNA molecule (28). In brief, biotin–protein ligase BirA (36) is N-terminally fused to a 22-amino acid λN peptide that recognizes bacteriophage λ BoxB stem-loops with high affinity (37), flanking both 5′ and 3′ ends of an RNA of interest (Fig. 2A). Thus, BirA ligase is brought in close proximity to and biotinylates all proteins that directly or indirectly associate with target RNA. We then perform immunoprecipitation with streptavidin beads and used MS and bioinformatics analysis to identify interacting protein partners. As a proof of principle, we first tested the interaction between a pair of positive controls: a 15-nucleotide UG–rich EDEN15 motif (UGUUUGUUUGUUUGU) that is reported to bind to the CUG triplet repeat RNA-binding protein 1 (CUG-BP1) (38). As expected, specific immunoprecipitation of endogenous CUG-BP1 was only observed with EDEN15 and not with the negative control scrambled sequences (Fig. 2B). Next, we examined the well-documented interaction between the conserved RV 3′UTR and an RV nonstructural protein 3 (NSP3). Consistent with previous reports (34), we detected strong binding of the UGUGACC sequence with either ectopically expressed GFP-tagged NSP3 or endogenous NSP3 expressed during RV infection (Fig. 2, C and D), suggesting that this technique is well-suited to interrogate the host protein–binding partners of the RV 3′UTR sequences. In addition to examining binding proteins of 3′UTR RV monomers, we further tested whether a pentamer of RV 3′UTRs with two adenosine spacers between monomers would increase the sensitivity for detecting host protein binding to NSP3. However, our results indicated that a monomer was equally effective for pulling down NSP3 (data not shown). Therefore, for the rest of the study, we used the monomeric probes to reduce the possibility of creating artificial secondary structures.
Figure 2.

RaPID identifies host proteins that bind to group A RV 3′UTR. A, schematics of a two-plasmid system to screen for RV RNA-binding host proteins. BirA fusion protein expression is driven by a cytomegalovirus promoter, and RNA expression is driven by a U6 promoter. B, HEK293T cells were co-transfected with a BirA expression plasmid and a pMotif-scrambled or EDEN15 plasmid for 24 h and incubated with biotin (50 μm) for 24 h. Immunoprecipitation was performed using magnetic streptavidin beads. Eluted lysates and input were examined by Western blotting using primary antibody against CUG-BP1. C, same experiment as B except that HEK293T cells were transfected with a GFP-NSP3 expression plasmid together with a pMotif plasmid encoding scrambled sequences or conserved RV 3′UTR. D, HEK293T cells were infected with RRV (m.o.i. = 3), transfected with pMotif-scrambled or 3′UTR plasmids at 1 hpi, and cultured in the presence of biotin at 24 hpi for another 24 h. Immunoprecipitation and Western blotting were performed to measure the levels of endogenous NSP3. Quantification was performed using ImageJ on the basis of three blots. E, screening results for RV 3′UTR binding host proteins. The x axis represents the fold change of specific host protein binding to 3′UTR sequences as compared with a scrambled sequence during RV infection. The y axis represents the SAINT score (ranging from 0 to 1.0), calculated based on the reproducibility between three independent immunoprecipitation-MS experiments. F, spectral counts of several ATP synthase complex subunits from a representative MS and bioinformatics analysis. (Scr, scrambled). For all figures, experiments were repeated at least three times with similar results. Experiment in E was performed three times independently.
By comparing a scrambled sequence (AUAGGCGUC) to an authentic conserved RV 3′UTR probe (GAUGUGACC) in the presence or absence of RV infection, we were able to identify host proteins that specifically immunoprecipitated with the 3′UTR consensus sequences (Table S1). We then used CRAPome filtering analysis (39) and a Significance Analysis of INTeractome (SAINT) threshold score of 0.9 to identify specific host proteins. Our analysis identified three high-confidence “hits”: the mitochondrial F1 complex β-polypeptide ATP5B; the RAS oncogene family member RAB1A; and the isoleucyl-tRNA synthetase IARS as interacting partners to the RV 3′UTR consensus sequences (Fig. 2E and Table S2). Of note, ATP5B, an ATP synthase β subunit (29), was found to be enriched at 100-fold in its interaction with 3′UTR probe over control (Fig. 2E). ATP5B was previously reported to promote human immunodeficiency virus (HIV)-1 replication and chikungunya virus infection (40, 41), prompting us to hypothesize that it may also act as a pro-RV host factor. Importantly, we also detected two other pivotal components of the ATP synthase complex, ATP5A1 and ATP5O, in the pulldown of 3′UTR sequence during RV infection (Fig. 2F). It is worth noting that all three ATP synthase subunits were found to interact with the RV 3′UTR probe only in the context of RV infection (Fig. 2F). Taken together, this new assay technique to detect host RNA-binding proteins allowed us to identify several novel proteins, including several ATP synthase subunits that had previously not been known to interact with viral RNA.
ATP5B immunoprecipitates and co-localizes with RV 3′UTR during infection
We next carried out a set of experiments to validate the association between ATP5B and the conserved RV 3′UTR sequences. Using streptavidin beads to directly pull down biotinylated host proteins in mock- or RV-infected cells, we observed strong binding of endogenous ATP5B with an RV 3′UTR probe, but only very weak interaction was detected with the scrambled probe (Fig. 3A). Importantly, the ATP5B–3′UTR interaction was observed exclusively during active RV infection (Fig. 3A), suggesting that such interaction might be regulated by other host factors, viral factors, or virus-induced cellular changes.
Figure 3.

ATP5B interacts and co-localizes with RV RNA during infection. A, HEK293T cells were infected with RRV (m.o.i. = 3), transfected with pMotif-scrambled (Scr) or 3′UTR (3′) plasmids at 1 hpi, and cultured in the presence of biotin at 24 hpi for another 24 h. Immunoprecipitation was carried out with streptavidin beads, and Western blotting was performed to measure the levels of endogenous ATP5B that interact with 3′UTR. B, HEK293T cells were infected with RRV (m.o.i. = 3) for 24 h and stained for nucleus (DAPI, blue), mitochondria (Mitotracker, green), ATP5B (red), and viral protein VP6 (gray). ATP5B (red) and mitochondria (green) co-localization generates the perinuclear yellow signals. Scale bar, 15 μm. C, MA104 cells were infected with RRV (m.o.i. = 1) for 16 h and stained for nucleus (DAPI, blue), viral RNA (FISH probes, green), and viral protein NSP2 (red). Co-localization of RV RNA with NSP2 is shown in yellow in merge. Scale bar, 15 μm. D, MA104 cells were uninfected (UI) or infected with RRV (m.o.i. = 1) for 16 h and stained for nucleus (DAPI, blue), viral RNA (FISH probes, green), and MAVS or ATP5B (red). Scale bar, 15 μm. Co-localization of RV RNA with ATP5B is highlighted by a red box of the enlarged inset panel, for which the scale bar is 25 μm. Quantification of co-localization between viral RNA and indicated host proteins was performed based on at least 10 micrographs using Volocity. For all figures, experiments were repeated at least three times with similar results.
We hypothesize that to physically interact with RV RNA, ATP5B is likely to co-localize with viral RNA in infected cells. We first determined ATP5B localization in the presence or absence of RV infection. In mock-infected cells, almost all ATP5B was found at the mitochondria (Fig. 3B, upper panel), consistent with previous reports (42) and its role as part of the ATP synthase complex. However, in RV-infected cells, we observed a significant condensation of mitochondrial organization in viral antigen VP6–positive cells (Fig. 3B, lower panel), recently noted by Green and Pelkmans (1) as well. To examine the subcellular localization of ATP5B relative to viral RNA, we designed specific fluorescent in situ hybridization (FISH) probes that detect RV RNA with high resolution. Our RV FISH probes were highly specific for viral RNA and only observed within the viroplasm, the distinct punctate viral factories that are positive for the nonstructural protein NSP2 (Fig. 3C). Importantly, viral RNA signal was also seen, at least partially, to overlap with ATP5B localization during infection (Fig. 3D and inset). In contrast, minimal co-localization was observed between RV RNA with mitochondrial antiviral-signaling protein (MAVS) (Fig. 3D). Therefore, data from both immunoprecipitation and immunofluorescence experiments are consistent with our screening results indicating that ATP5B is a bona fide RV 3′UTR–interacting protein.
ATP5B supports group A RV infection in a strain-independent manner
Having established ATP5B–rotaviral RNA interaction, we next sought to examine the functional role of ATP5B during RV infection. Because genetic depletion of ATP5B induces lethality (43), we utilized siRNA to knock down ATP5B expression levels. All three top candidates identified in our proteomics screen, ATP5B, RAB1A, and IARS, were effectively silenced with their specific siRNAs and examined by their mRNA levels by quantitative PCR (Fig. 4A). Both ATP5B and IARS siRNA also led to a marked reduction of respective protein levels, although the decrease was more significant with IARS than ATP5B (Fig. 4B). Importantly, in a multiple-round infection assay, at 24 h post-infection, knockdown of ATP5B led to 40% reduction in the mRNA levels of NSP5, an RV gene transcript representative of intracellular RV RNA synthesis (Fig. 4C), and we also observed an ∼40% decrease in the amount of infectious RVs in the supernatant of the ATP5B knockdown cells (Fig. 4D), suggesting that ATP5B facilitates RV replication in host cells.
Figure 4.

ATP5B siRNA knockdown specifically inhibits RV infection. A, HEK293 cells were transfected with control (ctrl) siRNA or siRNA against ATP5B, RAB1A, and IARS. At 48 h post-transfection, total RNA was harvested, and the expression levels of indicated genes were examined by RT-qPCR and normalized to that of GAPDH. B, same as A except that cell lysates were harvested and examined by Western blotting for the levels of indicated proteins. C and D, HEK293 cells were transfected with indicated siRNA for 48 h and infected with RRV (m.o.i. = 1) for 24 h in the presence of 0.05 μg/ml trypsin, which activates newly released viruses and allows for multiple rounds of infection. Total RNA was harvested to quantify the levels of viral NSP5 by RT-qPCR (C), and virus yield in the supernatant was measured by a focus-forming unit assay (D). E, same as C except that in addition to RRV (rhesus RV), a panel of animal and human RVs was used for infection (m.o.i. = 1). SA11 (simian), UK (bovine), OSU (porcine), ETD (murine), Wa and ST3 (human) RV strains were used. F, same as C except that VSV-GFP was used for infection (m.o.i. = 1). G, same as C except that two strains of influenza A viruses (H1N1 and H3N2) were used for infection (m.o.i. = 1). H, same as D except that colony-forming units (CFUs) were measured for S. typhimurium infection (m.o.i. = 10) at indicated time points. I, same as C except that reovirus T1L strain was used for infection (m.o.i. = 1). For all figures, experiments were repeated at least three times with similar results. n.s., not significant.
Given that all group A RVs share consensus sequences of the ATP5B-binding site at their 3′UTR ends (Fig. 1), we next examined whether the pro-RV role of ATP5B is conserved across different RV strains. Consistent with our hypothesis, the infectivity of a panel of cell culture adapted group A human and animal RVs was inhibited by siRNA depletion of ATP5B (Fig. 4E). In contrast, the replication of several other RNA viruses, including vesicular stomatitis virus and two strains of influenza A viruses, which do not possess 3′-UGUGACC sequences, was not affected by ATP5B knockdown (Fig. 4, F and G). ATP5B knockdown also did not influence the replication of Salmonella typhimurium, an intracellular bacterial pathogen or even reovirus, another Reoviridae family member with many properties similar to RVs (Fig. 4, H and I), thereby in support of a highly-specific pro-RV role of ATP5B.
ATP5B contributes to RV genome assembly
To mechanistically determine at which step in the RV replication cycle in which ATP5B is involved, we undertook a series of experiments to target various molecular processes within a single RV replication cycle. First, initial viral adsorption and subsequent endocytosis, assayed by input viral RNA levels 4 and 37 °C post-incubation, respectively, and using EDTA wash as a negative control, were not affected by ATP5B siRNA, when compared with control siRNA (Fig. 5, A and B). These two pieces of experimental evidence strongly suggest that early events in RV infection are not perturbed by the loss of ATP5B.
Figure 5.

ATP5B positively regulates RV virus assembly. A, HEK293 cells were transfected with control (ctrl) or ATP5B siRNA for 48 h, then incubated with RRV (m.o.i. = 20) at 4 °C for 1 h, and washed with PBS to remove unbound viruses. As positive controls, cells were washed with EDTA to completely remove bound viruses. After a PBS wash, cells were then harvested for RT-qPCR analysis of input viral RNA levels to quantify RV attachment to the cell monolayer at 4 °C. B, same as A except that after a 1-h incubation on ice, cells were shifted to 37 °C, incubated for another 1 h, and washed with EDTA to remove unendocytosed RV particles prior to the harvest. C, HEK293 cells were transfected with control or ATP5B siRNA for 48 h and infected with RRV (m.o.i. = 1) for 8 h. The levels of ATP5B and indicated viral mRNA were measured by RT-qPCR and normalized to that of GAPDH. D, same as C except that the levels of viral structural proteins VP2 and VP6 were examined by Western blotting. E, HEK293 cells were transfected with control or ATP5B siRNA for 48 h, infected with RRV (m.o.i. = 1) for the indicated time points, and stained for nucleus (DAPI, blue), ATP5B (red), and NSP2 (green). Scale bar, 15 μm. F, same as E except that RV VP4, VP6, and NSP2 were stained with monoclonal antibodies. Quantification of co-localization between ATP5B and indicated viral proteins was performed based on at least 10 micrographs using Volocity. Scale bar, 15 μm. G, MA104 cells were infected with RRV (m.o.i. = 1) for 16 h and stained for nucleus (DAPI, blue), ATP5B (red), NSP2 (green), and viral RNA (FISH probes, gray). Scale bar, 15 μm. Co-localization of ATP5B with NSP2, and RV RNA is highlighted by a red box of the enlarged inset panel, for which the scale bar is 42 μm. H, quantification of newly synthesized virions, stained by a conformation-specific antibody against trimeric VP7 (mAb 159), on the basis of over 20 micrographs using Volocity. I, HEK293 cells transfected with control or ATP5B siRNA for 48 h were infected with RRV (m.o.i. = 1) for 2 or 8 h and harvested for a standard plaque assay to titrate the amount of infectious virus particles. J, focus-forming unit assay was performed to determine the amount of infectious RVs in the supernatant of RRV-infected HEK293 cells (m.o.i. = 1) at 8 hpi. For all figures, experiments were repeated at least three times with similar results. n.s., not significant.
Next, we measured viral RNA by quantitative PCR and protein levels using polyclonal antibody that recognizes RV double-layered particles (44) at 8 h post-infection within a single virus replication cycle. We found that both levels were comparable between control and ATP5B siRNA-transfected cells (Fig. 5, C and D). Thus, we focused our further attention on the formation of viroplasms, the specialized electron-dense inclusions within the cytoplasm where assembly of immature virus particles is initiated (45). RV viroplasms, detected by a mAb against viral protein NSP2 (46), were also similar with or without ATP5B siRNA silencing (Fig. 5E). Importantly, at 8 h post-infection, we found significant co-localization of ATP5B with NSP2, further supporting our previous immunofluorescence analysis on ATP5B and viral RNA (Fig. 3D). ATP5B was found to co-localize only with NSP2 but not with the two other RV proteins VP4 and VP6 (Fig. 5F). This finding was further validated using co-staining of ATP5B, NSP2, and RV RNA in the same infected cells (Fig. 5G). Using a conformation-dependent mAb that specifically recognizes the trimeric form of VP7 on mature RV particles (47), we correlated and estimated the number of fully assembled RV particles. At 8 h post-infection, there was a significant 4-fold decrease in ATP5B-depleted cells (Fig. 5H). Consistent with a role of ATP5B in the later steps in the RV replication cycle, the amount of infectious RVs and virus progeny released into the supernatant was also reduced by ATP5B knockdown (Fig. 5, I and J). Taken together, these data point to a supportive role of ATP5B for RV genome assembly, which is likely to be a high- energy–consuming process and occurs following viroplasm formation and prior to virus egress.
Inhibition of ATP synthase complex restricts RV infection
Using available crystal structures of both the ATP synthase complex (21) and the RV polymerase VP1 binding to UGUGACC (48), we extracted the viral RNA structure and attempted to computationally model the interaction between ATP5B and RV 3′UTR sequences. We applied HDOCK (49), an on-line bioinformatics tool that combinatorially adopts template-based modeling and free docking, to simulate the biochemical association between ATP5B and the RV 3′UTR. This algorithm calculates the possibility and potential affinity of a protein–RNA ligand interaction and assigns a root-mean-square deviation (RMSD) score. The lower the score, the more likely this interaction represents a biologically meaningful binding (50). Of note, compared with the negative control RV protein NSP4, which localizes to the endoplasmic reticulum and does not bind viral RNA (51, 52), the validated UGUGACC-binding protein NSP3 had a much lower RMSD value (Fig. 6A). Importantly, the ATP5B–3′UTR value was predicted to be similar to our positive control NSP3 value and much lower than cullin-3, a host protein that interacts with RV NSP1 but not viral RNA that we used as a negative control (Fig. 6A) (3). In addition, we found that the inclusion of ATP5A1, another component of ATP synthase complex and also identified by our proteomics screen (Fig. 2F), to ATP5B significantly favored the interaction with RV 3′UTR by 24% (Fig. 6A). It is noteworthy that the ATP synthase–viral RNA interaction is predicted to be weaker than that between viral polymerase VP1 and RV RNA, suggesting that ATP5B–RNA binding may not be direct and possibly require additional host/viral factors. We further predicted the spatial position of UGUGACC, which inserts into the canal of the headgroup of F0–F1-ATP synthase complex, consisting of three ATP5A1 subunits and three ATP5B subunits (Fig. 6B). Such conformation would allow the Arg-286 of ATP5A1 and Pro-276 of ATP5B to create a docking site for the negatively charged RV 3′UTR sequences (Fig. 6C).
Figure 6.

Three-dimensional modeling of inter-molecular ATP synthase complex and RV 3′UTR interaction. A, estimated binding strength between UGUGACC sequences and selected viral or host proteins using HDOCK algorithm. Low RMSD score favors strong binding between the ligand (RNA) and target protein. Cul3 stands for cullin-3, a host E3 ligase protein that does not interact with RNA molecules. B, top view of PyMOL modeling of RV 3′UTR sequences sticking into the head domain of ATP synthase complex (trimeric ATP5A1-ATP5B subunits). C, close-up details of B revealing the nucleotides and possible interacting amino acids.
Consistent with the hypothesis that the full structure of the ATP motor is needed for RV infection, we found that siRNA knockdown of both ATP5A1 and ATP5O phenocopied ATP5B silencing and suppressed RV infection (Fig. 7A), suggesting that the pro-RV role of ATP5B depends on the integrity of the holoenzyme. In line with this observation, the concurrent knockdown of several ATP synthase complex subunits led to an even more prominent reduction in RV infection (Fig. 7A). In addition to siRNA-mediated genetic depletion, a specific small-molecule ATP synthase inhibitor, isoapoptolidin (53), potently down-regulated intracellular RV RNA levels and virus yield in the supernatant (Fig. 7, B and C). Several other pharmacological inhibitors of ATP synthase activity also inhibited RV replication in a similar manner (Fig. 7D). These inhibitors did not induce cytotoxicity, just like control and ATP5B siRNA (Fig. 7E). Further corroborated by the fact that the replication of other pathogens was not affected by ATP5B knockdown (Fig. 4, F–I), our data demonstrate that reduction in RV infection was not accounted for by the unhealthy cellular environment created by ATP5B inhibition. Collectively, our results support an important function of the ATP synthase complex in promoting efficient RV replication by enhancing viral genome assembly.
Figure 7.

Inhibition of ATP synthase activity restricts RV virus assembly. A, HEK293 cells were transfected with siRNA against different components of ATP synthase complex for 48 h and infected with RRV (m.o.i. = 1) for multiple cycles of virus replication. At 24 h post-infection, expression of RV NSP5, ATP5A1, ATP5B, and ATP5O was examined by RT-qPCR and normalized to that of GAPDH. B, HEK293 cells were treated with isoapoptolidin (2 μm) for 24 h and infected with RRV (m.o.i. = 1) for another 24 h. Expression of RV NSP5 was examined by RT-qPCR and normalized to that of GAPDH. C, same as B except that virus yield in the supernatant was measured by a focus-forming unit assay. D, same as B except that several other ATP synthase inhibitors (2 μm for 24 h) were used instead of isoapoptolidin. E, inhibitor-treated (24 h) or siRNA-transfected (48 h) HEK293 cells were left uninfected (UI) or infected with RRV (m.o.i. = 1) for 8 h and were subject to a cell survival assay to measure the induction of cell death. Puromycin treatment (1 μg/ml) for 24 h was used as a positive control. For all figures, experiments were repeated at least three times with similar results. n.s., not significant.
ATP synthase supports RV infection in human enteroids
Finally, we extended our findings and tested the role of ATP synthase activity in RV replication in human intestinal enteroids, a primary human intestinal epithelial cell (IEC) three-dimensional culture system that faithfully recapitulates the IEC population diversity in vivo (54, 55). We found that isoapoptolidin treatment of human intestinal enteroids significantly decreased plaque-forming units while not affecting RV RNA and protein levels in a one-step replication cycle (Fig. 8). The intracellular NSP5 levels and percentage of viral antigen-positive cells were comparable in mock and ATP inhibitor treatment (Fig. 8, A and B, respectively). However, blocking ATP synthase activity significantly reduced the formation of progeny infectious RV focus-forming units (Fig. 8C), further supporting a role for ATP5B in RV assembly.
Figure 8.

ATP synthase inhibitor impedes RV assembly in human intestinal enteroids. A, ileum intestinal enteroids were treated with isoapoptolidin for 24 h and infected with human RV WI61 strain (m.o.i. = 5) for 12 h. Expression of RV NSP5 was measured by RT-qPCR and normalized to that of GAPDH. B, ileum intestinal enteroids were treated with isoapoptolidin for 24 h, infected with human RV WI61 strain (m.o.i. = 5) for 12 h, and stained for nucleus (DAPI, blue), viral antigen VP6 (green), and actin (phalloidin, gray). Scale bar, 100 μm. Note that the actin staining is stronger on the inside of the enteroids, indicative of the apical luminal side. C, same as A except that whole-cell lysates were harvested for a standard plaque assay to titrate the amount of infectious RV particles. For all figures, experiments were repeated at least three times with similar results. n.s., not significant.
Discussion
Mechanisms of genome replication and particle assembly of segmented icosahedral RNA viruses are poorly understood. In the case of RVs, a prototypical enteric viral pathogen with 11 discrete dsRNA segments, the majority of what we know derives from studies on viral replication intermediates and cis-acting signals on viral RNAs. The current model states that all 11 RV (+) RNA segments are present in the viroplasm and that the viral VP1 polymerase is recruited and interacts with a highly structured region toward the end of a conserved 3′UTR UGUGACC, which forms a panhandle structure (31, 48, 56–60). The additional binding of VP3 with the 5′UTR end facilitates the formation of an RNA(+)–VP1–VP3 complex (61). Subsequently, different segments are assorted via a not well-characterized RNA–RNA interaction and packaged in a VP2-dependent manner (18, 62, 63). However, whether any and which host factors might participate in these processes remains largely unexplored.
In this study, we utilized a recently developed method (RaPID) (28) to identify RNA-binding proteins to study RV–host interaction. Compared with the traditional tools to study RNA–protein interaction (64–66), this new method has several advantages including the following: 1) using whole-infected cells rather than lysates to enable RNA–protein binding to take place in its “natural” cellular environment; 2) bypassing the need for cross-linking and reversal; and 3) enhanced sensitivity based on biotin protein ligase BirA that promiscuously biotinylates proteins in a proximity-dependent fashion. Compared with other methods of analyzing essential host factors of virus infection, RaPID offers a different and complementary approach, as ATP5B knockout is lethal according to the recent haploid screens for survival genes (43), which probably explains why ATP5B was not identified as a major hit in our recent CRISPR-Cas9 screen for pro-RV host factors (4). Of note, RaPID technique can be easily adopted to examine the RV gene 11 loop regions and helical junctions (19) or a 6-nucleotide-long pyrimidine-rich tract shared by several RV genes (67). This method can also be used to study the structural elements of other RNA viruses, such as the stem-loop regions and subgenomic viral RNA sequences.
In addition to ATP5B, our screen also identified IARS and RAB1A as rotaviral RNA-binding proteins. It is intriguing that these proteins exhibit distinct subcellular localization: ATP5B at mitochondria where the key adaptor protein for cytosolic RNA-sensing pathway MAVS is localized (68); RAB1A at endoplasmic reticulum; and IARS in the cytosol. Because we did not observe significant effects on RV replication from the siRNA knockdown of ATP5B in a single virus replication cycle, it is possible that their interactions with viral RNA do not have direct effect on virus replication but rather serve as modulators of innate immune signaling in the cytoplasm. Interestingly, upon examination of whether the interferon pathway is potentiated or dampened in the absence of ATP5B during RV infection, we found that both IFN-λ3 (IFNL3) expression and secretion into the supernatant were significantly reduced in RRV-infected ATP5B knockdown cells as compared with the control siRNA-transfected cells (Fig. 9). We will explore this new direction further in future studies.
Figure 9.
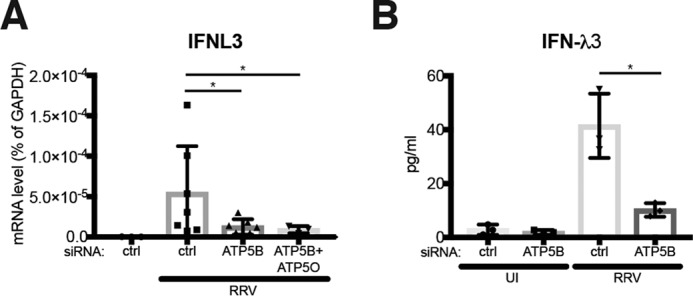
ATP5B positively regulates IFN induction in response to RV infection. A, HEK293 cells were transfected with the indicated siRNA and infected with RRV (m.o.i. = 0. 1) for 12 h. Expression of IFNL3 was measured by RT-qPCR and normalized to that of GAPDH. B, same as A except that IFNL3 (IFN-λ3) secretion was measured by an ELISA. For all figures, experiments were repeated at least three times with similar results.
An important aspect of this study that remains to be further examined is whether ATP5B directly or indirectly binds to RV 3′UTR during infection. Our bioinformatics modeling approach suggests that the ATP5A1–ATP5B head complex could potentially interact with the conserved UGUGACC sequence at a strength comparable with NSP3. However, considering the even higher affinity between the RV polymerase VP1 and 3′UTR (Fig. 6A), one might expect that ATP5B–3′UTR binding is also dependent on the presence of selected RV proteins, such as VP1. This hypothesis is consistent with our data that the interaction between 3′UTR and all three subunits of ATP synthase complex (5A1, 5B, and 5O) is greatly enhanced during RV infection (Fig. 2F) and that several attempts to directly test ATP5B–RV RNA interaction using electrophoretic mobility shift assay have not been successful (data not shown). It is tempting to speculate that the on and off status of ATP5B may provide critical energy when the ensemble of VP1–VP3–RNA and VP2 building blocks comes together to encapsulate the RNA segments into the newly-forming capsid.
Experimental procedures
Cells and plasmids
HEK293 (CRL-1573) and HEK293T (CRL-3216) cells were purchased from American Type Culture Collection (ATCC) and cultured in complete DMEM. MA104 (CRL-2378.1) cells were obtained from ATCC and cultured in complete M199 medium. All three cell lines are not listed as commonly misidentified cell lines maintained by International Cell Line Authentication Committee (ICLAC), and they tested negative for mycoplasma contamination.
The pMotif vector that drives the transcription of target RNA (scrambled sequences, EDEN15 motifx5, and RV-3′UTR) was digested with BsmBI and ligated as described previously (28). For each Western blotting experiment, 2 μg of pMotif and 0.3 μg of pRaPID with optimized λN sequence were co-transfected into one well of a 6-well plate pre-seeded with HEK293T cells using 5 μl of Lipofectamine 2000 (Invitrogen). At 24 h post-transfection, biotin (Sigma, catalog no. B4501, 50 μm) was added to media and incubated for 18 h. For each MS experiment, a single 10-cm dish was used instead of a 6-well plate with 12 μg of pMotif RNA and 2 μg of pRaPID transfected with 30 μl of Lipofectamine 2000. The T7 vector that encodes gene 11 from simian RV SA11 strain was purchased from Addgene (catalog no. 89172).
RaPID
On the day of harvest, cells were washed with cold PBS (one time) and lysed with 200 μl of lysis buffer at room temperature. 20 μl of 25% Triton X-100 was then added and mixed thoroughly. Subsequently, lysates were supplemented with 220 μl of cold 50 mm Tris, pH 7.4, sonicated for 10 s, and centrifuged at 4 °C for 10 min. Free biotin was removed by Macrosep Advance Spin Filter 3K MWCO 20 ml (VWR 89131-974). Protein concentration in each sample was determined using spectrophotometer with the Pierce protein quantitation assay (ThermoFisher Scientific). Protein concentration across samples was normalized, and 40 μl of the sample was aliquoted and marked as IP input. Pulldown of biotinylated proteins was performed with magnetic beads MyOneTM Streptavidin C1 (Life Technologies, Inc., catalog no. 65002) and sample was rotated with beads for 2 h at room temperature. Samples were washed three times, and beads were boiled in Elution Buffer for 45 min at 98 °C and labeled as IP. Both input and IP were loaded and resolved on pre-cast Bio-Rad gels, and SDS-PAGE was conducted as described previously (69) using the following primary antibodies: ATP5B (HPA001520, Sigma), CUG-BP1 (3B1, catalog no. sc-20003, Santa Cruz Biotechnology), GAPDH (catalog no. 631402, BioLegend), and NSP3 (kindly provided by Drs. Susana Lopez and Didier Poncet). Secondary incubation was performed with anti-rabbit (CST, catalog no. 7074) or anti-mouse (CST, catalog no. 7076) IgG horseradish peroxidase–linked antibodies. Protein bands were visualized with Clarity ECL substrate (Bio-Rad, catalog no. 170-5061), Amersham Biosciences Hyperfilm (GE Healthcare), and STRUCTURIX X-ray film processor (GE Healthcare).
For sample preparation for LC-MS/MS, streptavidin magnetic beads were washed with 500 μl of 50 mm ammonium bicarbonate three times. Beads were resuspended in 200 μl of 50 mm ammonium bicarbonate supplemented with DTT to a final concentration of 5 mm, incubated on a heat block at 50 °C for 5 min followed by head-over-head rocking for 30 min at room temperature. Alkylation was performed by the addition of propionamide to a final concentration of 10 mm and head-over-head rocking for 30 min at room temperature. 250 ng of trypsin/LysC (Promega) was added to each sample and digested overnight at room temperature in the head-over-head shaker followed by the addition of formic acid to 1%. Peptides were removed and washed with 50 μl of 0.1% formic acid water. The acidified peptide pools were purified with C18 STAGE tip 37 (NEST group) microspin columns and dried in a speed vac. For LC-MS/MS, peptide pools were reconstituted and injected onto a C18 reversed phase analytical column. All MS/MS data were first analyzed in Preview to provide recalibration criteria and then reformatted to .MGF before full analysis with Byonic version 1.4 (ProteinMetrics). Analyses used Uniprot canonical .fasta files for human, concatenated with common contaminant proteins. Data were searched at 10 ppm mass tolerances for precursors, with 0.4-Da fragment mass tolerances assuming up to two missed cleavages and allowing for N-ragged tryptic digestion, and were validated at a 1% false discovery rate using typical reverse-decoy techniques. The resulting identified peptide spectral matches and assigned proteins were then exported for further analysis using custom tools developed in MatLab (MathWorks) to provide visualization and statistical characterization. SAINT scores were generated with spectral counts from experimental samples and controls using the on-line CRAPome tool (39) to minimize the MS background.
Virus infections
All human and animal RV strains used in this study were propagated in MA104 cells, and RV infection was performed as described previously (3). Recombinant VSV (strain Indiana) expressing GFP was a kind gift from Dr. Jack Rose (Yale University) and used as described previously (70). Influenza A viruses (H1N1 A/California/7/2009, H3N2 A/Victoria/261/2011) were used as described previously (71). Reovirus T1L strain was kindly provided by Dr. Carlos Arias (Universidad Nacional Autónoma de México). For all virus infection experiments, the initial inoculum was removed after 1 h of incubation at 37 °C and washed with complete or serum-free medium to avoid unsynchronized infection. For virus adsorption assay, cells were incubated with RVs at 4 °C for 1 h and washed with ice-cold PBS three times. For virus endocytosis assay, cells were infected with RVs at 37 °C for 1 h and washed with serum-free medium containing 5 mm EDTA to remove cell-bound virus at the surface. For multiple rounds of RV infection, 0.05 μg/ml trypsin was added to the serum-free medium for outer capsid protein VP4 cleavage, and infection lasted for at least 24 h.
Quantitative RT-PCR
Total RNA was harvested in RLT buffer supplemented with β-mercaptoethanol and extracted as described previously (72). Random hexamer was used for reverse transcription reaction. qPCR was performed with the Stratagene Mx3005P (Agilent) with a 25-μl reaction consisting of 50 ng of cDNA, 12.5 μl of Power SYBR Green master mix (Applied Biosystems), and 200 nm both forward and reverse primers. All SYBR Green primers (Table S3) have been validated with both dissociation curves and DNA electrophoresis of the correct amplicon size.
Western blotting and immunofluorescence
Western blotting and confocal imaging analysis was performed as described previously (73). In brief, HEK293 cells were fixed with 4% paraformaldehyde for 10 min at room temperature, permeabilized, and stained with the following primary antibodies or fluorescent dyes: ATP5B (HPA001520, Sigma), DAPI (P36962, ThermoFisher Scientific), MAVS (E3, sc-166583, Santa Cruz Biotechnology), NSP2 (mAb 191), VP4 (mAb HS2), VP6 (mAb 1E11), and trimeric VP7 (mAb 159). Stained cells were washed with PBS, mounted with Antifade Mountant with DAPI (ThermoFisher Scientific, P36962), and imaged with Zeiss LSM 710 confocal microscope. Co-localization quantification was analyzed by Volocity version 5.2 (PerkinElmer Life Sciences) using Pearson's coefficient value.
siRNA transfection
HEK293 cells in 24-well plates were transfected with 6 pmol of control or specific siRNAs against host genes (see information in Table S3) with RNAiMAX (Invitrogen) using a reverse transfection method (3). Cells were cultured for at least 48 h prior to qPCR analysis or virus infections. The transfection was performed in antibiotic-free medium and cells cultured for 48 h prior to virus infection, qPCR or Western blot analysis.
Focus-forming unit assay
HEK293 cells were transfected with control or ATP5B siRNA and infected with RRV at m.o.i. = 0.5 in 24-well plates. At different time points post-infection, supernatants were collected and applied to freshly seeded MA104 cells in a 10× dilution series as described previously (71). In brief, infected cells were fixed overnight and incubated with primary antibodies against rabbit anti-DLP at 37 °C for 1 h, and secondary incubation was performed with anti-rabbit IgG at 37 °C for 1 h, with the subsequent addition of AEC substrate (SK-4200, Vector Laboratories). Color developing was observed, and the number of fluorescent colonies was counted. Two independent experiments were performed with triplicate infections.
Salmonella invasion assay
Salmonella enterica serovar Typhimurium strain SL1344 was grown on an LB agar plate at 37 °C. A single colony was used to inoculate an LB broth culture and grown overnight with shaking at 37 °C. Bacteria were subcultured 1:20 into high-salt LB (300 mm NaCl) and grown statically at 37 °C in a 5% CO2 incubator for 2–3 h. Bacteria were pelleted and resuspended in DMEM, 10% FBS at an m.o.i. of 10 and added to wells of HEK293 cells previously transfected with ATP5B or control siRNA. After 30 min of infection, bacteria were removed, and DMEM, 10% FBS with 100 μg/ml gentamicin were added to kill all extracellular bacteria. After 90 min, media were changed to 10 μg/ml. Intracellular bacteria were quantified at 2 or 8 h post-infection. Infected cells were lysed with 1% Triton X-100 in PBS for 5 min at room temperature, serially diluted, and then plated on LB agar plates to determine the number of colony-forming units.
Cytotoxicity assay
Cell survival rate of ATP synthase inhibitor-treated or siRNA-transfected HEK293 cells were measured by a luminescence-based CellTiter-Glo® 2.0 Assay (Promega, G9241) according to the manufacturer's instructions. In brief, ATP was serially diluted from 100 μm to 0.01 nm to formulate a standard curve. Cell lysates were harvested, and intracellular ATP levels were measured and normalized to the standard curve.
RNA–protein modeling
Interaction prediction between RV 3′UTR sequences and host/viral proteins were performed using HDOCK hybrid algorithm (http://hdock.phys.hust.edu.cn/)5 (49). The crystal structures were obtained from RCSB database in Protein Data Bank format: UGUGACC (2R7R; chain X) (48); ATP synthase complex (2XND; chains A–F for trimeric structure of ATP5A1 and ATP5B) (74); Cullin-3 (4HXI; chain B); NSP3 (1KNZ; chains A-D, I-N) (75); and NSP4 (3MIW; chain A) (76). Proteins were used as “receptor” and RNA molecule as “ligand.” No specific binding sites have been assumed before the calculation. 3D conformation of ATP synthase complex and UGUGACC was visualized by MacPyMOL (PyMOL version 1.7.4.4).
RNA fluorescent in situ hybridization
A custom set of 48 Stellaris FISH probes to detect the RRV genome is directly conjugated to Quasar 670 dye (LGC Biosearch Technologies). Mock- or RV-infected cells were fixed, permeabilized, and hybridized with FISH probes to visualize viral RNA molecules according to the manufacturer's instructions. Some samples were also stained with anti-ATP5B and anti-NSP2 antibodies to measure co-localization.
Statistical analysis
The bar graphs are displayed as mean ± S.D. Statistical significance in Figs. 4, A and E–I, 5, C and H–J, 7, B and C, and 8, A and C, was calculated by Student's t test using Prism 7.0c (GraphPad). Statistical significance in Figs. 3D, 4, C and D, 5, A, B, and F, 7, A, D, and E, and 9, A and B, was calculated by pairwise analysis of variance using Prism 7.0. p values are indicated on each figure (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001). All experiments, unless otherwise noted, have been repeated at least three times. Mass spectrometry experiments in Fig. 2E were performed precisely three times.
Author contributions
L. R., S. D., Y. S., B. L., and J. C. investigation; S. D., M. R., P. A. K., and H. B. G. conceptualization; S. D., M. R. A., P. A. K., and H. B. G. supervision; S. D. writing-original draft; S. D. and H. B. G. writing-review and editing; P. A. K. and H. B. G. funding acquisition.
Supplementary Material
Acknowledgments
We are grateful to Dr. Susana Lopez (Universidad Nacional Autónoma de México, Mexico) and Dr. Didier Poncet (Institut de Biologie Intégrative de la Cellule, France) for providing precious anti-RRV NSP3 antibodies (35) that greatly facilitated this study. We thank all members of the Greenberg lab for helpful discussions. We appreciate Drs. Christopher Adams and Ryan Leib and the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University Mass Spectrometry, for help with MS. The Mass Spectrometry experiment was supported by National Institutes of Health Award S10RR027425 from the NCRR.
This work was supported by an Early Career Award (Thrasher Research Fund), Instructor Support Program (Stanford Maternal and Child Health Research Institute), National Institutes of Health Grant K99 AI135031 (to S. D.), and National Institutes of Health Grants R01 AR49737 and R01 AR43799 (to P. A. K.), National Institutes of Health Grants R01 AI021362, R56 AI021362, and U19 AI116484 and VA Merit Review Grant GRH0022 (to H. B. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- RV
- rotavirus
- RaPID
- RNA–protein interaction detection
- m.o.i.
- multiplicity of infection
- qPCR
- quantitative PCR
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- DAPI
- 4′,6-diamidino-2-phenylindole
- IP
- immunoprecipitation
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- FISH
- fluorescent in situ hybridization
- RMSD
- root-mean-square deviation
- IEC
- intestinal epithelial cell
- SAINT
- Significance Analysis of INTeractome
- hpi
- hours post-infection
- MAVS
- mitochondrial antiviral-signaling protein
- VSV
- vesicular stomatitis virus.
References
- 1. Green V. A., and Pelkmans L. (2016) A systems survey of progressive host-cell reorganization during rotavirus infection. Cell Host Microbe 20, 107–120 10.1016/j.chom.2016.06.005 [DOI] [PubMed] [Google Scholar]
- 2. Silva-Ayala D., López T., Gutiérrez M., Perrimon N., López S., and Arias C. F. (2013) Genome-wide RNAi screen reveals a role for the ESCRT complex in rotavirus cell entry. Proc. Natl. Acad. Sci. U.S.A. 110, 10270–10275 10.1073/pnas.1304932110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ding S., Mooney N., Li B., Kelly M. R., Feng N., Loktev A. V., Sen A., Patton J. T., Jackson P. K., and Greenberg H. B. (2016) Comparative proteomics reveals strain-specific β-TrCP degradation via rotavirus NSP1 hijacking a host cullin–3-Rbx1 complex. PLoS Pathog. 12, e1005929 10.1371/journal.ppat.1005929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ding S., Diep J., Feng N., Ren L., Li B., Ooi Y. S., Wang X., Brulois K. F., Yasukawa L. L., Li X., Kuo C. J., Solomon D. A., Carette J. E., and Greenberg H. B. (2018) STAG2 deficiency induces interferon responses via cGAS-STING pathway and restricts virus infection. Nat. Commun. 9, 1485 10.1038/s41467-018-03782-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hentze M. W., Castello A., Schwarzl T., and Preiss T. (2018) A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 19, 327–341 10.1038/nrm.2017.130 [DOI] [PubMed] [Google Scholar]
- 6. Loo Y. M., and Gale M. Jr. (2011) Immune signaling by RIG-I–like receptors. Immunity 34, 680–692 10.1016/j.immuni.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sánchez-Tacuba L., Rojas M., Arias C. F., and López S. (2015) Rotavirus controls activation of the 2′–5′-oligoadenylate synthetase/RNase L pathway using at least two distinct mechanisms. J. Virol. 89, 12145–12153 10.1128/JVI.01874-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Silverman R. H. (2007) Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 81, 12720–12729 10.1128/JVI.01471-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uzri D., and Greenberg H. B. (2013) Characterization of rotavirus RNAs that activate innate immune signaling through the RIG-I–like receptors. PLoS One 8, e69825 10.1371/journal.pone.0069825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chavali P. L., Stojic L., Meredith L. W., Joseph N., Nahorski M. S., Sanford T. J., Sweeney T. R., Krishna B. A., Hosmillo M., Firth A. E., Bayliss R., Marcelis C. L., Lindsay S., Goodfellow I., Woods C. G., and Gergely F. (2017) Neurodevelopmental protein Musashi-1 interacts with the Zika genome and promotes viral replication. Science 357, 83–88 10.1126/science.aam9243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Manokaran G., Finol E., Wang C., Gunaratne J., Bahl J., Ong E. Z., Tan H. C., Sessions O. M., Ward A. M., Gubler D. J., Harris E., Garcia-Blanco M. A., and Ooi E. E. (2015) Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350, 217–221 10.1126/science.aab3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lévêque N., Garcia M., Bouin A., Nguyen J. H. C., Tran G. P., Andreoletti L., and Semler B. L. (2017) Functional consequences of RNA 5′-terminal deletions on coxsackievirus B3 RNA replication and ribonucleoprotein complex formation. J. Virol. 91, e00423–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rialdi A., Hultquist J., Jimenez-Morales D., Peralta Z., Campisi L., Fenouil R., Moshkina N., Wang Z. Z., Laffleur B., Kaake R. M., McGregor M. J., Haas K., Pefanis E., Albrecht R. A., Pache L., Chanda S., Jen J., Ochando J., Byun M., Basu U., Garcia-Sastre A., Krogan N., van Bakel H., and Marazzi I. (2017) The RNA exosome syncs IAV-RNAPII transcription to promote viral ribogenesis and infectivity. Cell 169, 679–692.e14 10.1016/j.cell.2017.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Estes M. K. G., HB. (2013) in Rotaviruses (Knipe D. M., Howley P. M., et al., eds) Fields Virology, 6th Ed., pp. 1347–1401. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 15. Tate J. E., Burton A. H., Boschi-Pinto C., Parashar U. D., and World Health Organization–Coordinated Global Rotavirus Surveillance Network (2016) Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000–2013. Clin. Infect. Dis. 62, Suppl. 2, S96–S105 10.1093/cid/civ1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Long C. P., and McDonald S. M. (2017) Rotavirus genome replication: some assembly required. PLoS Pathog. 13, e1006242 10.1371/journal.ppat.1006242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fajardo T. Jr, Sung P. Y., and Roy P. (2015) Disruption of specific RNA–RNA interactions in a double-stranded RNA virus inhibits genome packaging and virus infectivity. PLoS Pathog. 11, e1005321 10.1371/journal.ppat.1005321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fajardo T., Sung P. Y., Celma C. C., and Roy P. (2017) Rotavirus genomic RNA complex forms via specific RNA–RNA interactions: disruption of RNA complex inhibits virus infectivity. Viruses 9, 167 10.3390/v9070167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borodavka A., Dykeman E. C., Schrimpf W., and Lamb D. C. (2017) Protein-mediated RNA folding governs sequence-specific interactions between rotavirus genome segments. eLife 6, e27453 10.7554/eLife.27453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kapahnke R., Rappold W., Desselberger U., and Riesner D. (1986) The stiffness of dsRNA: hydrodynamic studies on fluorescence-labeled RNA segments of bovine rotavirus. Nucleic Acids Res. 14, 3215–3228 10.1093/nar/14.8.3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abrahams J. P., Leslie A. G., Lutter R., and Walker J. E. (1994) Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628 10.1038/370621a0 [DOI] [PubMed] [Google Scholar]
- 22. Noji H., Yasuda R., Yoshida M., and Kinosita K. Jr. (1997) Direct observation of the rotation of F1-ATPase. Nature 386, 299–302 10.1038/386299a0 [DOI] [PubMed] [Google Scholar]
- 23. Hilbert B. J., Hayes J. A., Stone N. P., Duffy C. M., Sankaran B., and Kelch B. A. (2015) Structure and mechanism of the ATPase that powers viral genome packaging. Proc. Natl. Acad. Sci. U.S.A. 112, E3792–E3799 10.1073/pnas.1506951112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nadal M., Mas P. J., Blanco A. G., Arnan C., Solà M., Hart D. J., and Coll M. (2010) Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc. Natl. Acad. Sci. U.S.A. 107, 16078–16083 10.1073/pnas.1007144107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rao V. B., and Feiss M. (2008) The bacteriophage DNA packaging motor. Annu. Rev. Genet. 42, 647–681 10.1146/annurev.genet.42.110807.091545 [DOI] [PubMed] [Google Scholar]
- 26. Stäuber N., Martinez-Costas J., Sutton G., Monastyrskaya K., and Roy P. (1997) Bluetongue virus VP6 protein binds ATP and exhibits an RNA-dependent ATPase function and a helicase activity that catalyze the unwinding of double-stranded RNA substrates. J. Virol. 71, 7220–7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodríguez P. L., and Carrasco L. (1993) Poliovirus protein 2C has ATPase and GTPase activities. J. Biol. Chem. 268, 8105–8110 [PubMed] [Google Scholar]
- 28. Ramanathan M., Majzoub K., Rao D. S., Neela P. H., Zarnegar B. J., Mondal S., Roth J. G., Gai H., Kovalski J. R., Siprashvili Z., Palmer T. D., Carette J. E., and Khavari P. A. (2018) RNA–protein interaction detection in living cells. Nat. Methods 15, 207–212 10.1038/nmeth.4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. von Ballmoos C., Cook G. M., and Dimroth P. (2008) Unique rotary ATP synthase and its biological diversity. Annu. Rev. Biophys. 37, 43–64 10.1146/annurev.biophys.37.032807.130018 [DOI] [PubMed] [Google Scholar]
- 30. Chen D., and Patton J. T. (2000) De novo synthesis of minus strand RNA by the rotavirus RNA polymerase in a cell-free system involves a novel mechanism of initiation. RNA 6, 1455–1467 10.1017/S1355838200001187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patton J. T. (1996) Rotavirus VP1 alone specifically binds to the 3′ end of viral mRNA, but the interaction is not sufficient to initiate minus-strand synthesis. J. Virol. 70, 7940–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wentz M. J., Patton J. T., and Ramig R. F. (1996) The 3′-terminal consensus sequence of rotavirus mRNA is the minimal promoter of negative-strand RNA synthesis. J. Virol. 70, 7833–7841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patton J. T., Wentz M., Xiaobo J., and Ramig R. F. (1996) cis-Acting signals that promote genome replication in rotavirus mRNA. J. Virol. 70, 3961–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Poncet D., Laurent S., and Cohen J. (1994) Four nucleotides are the minimal requirement for RNA recognition by rotavirus nonstructural protein NSP3. EMBO J. 13, 4165–4173 10.1002/j.1460-2075.1994.tb06734.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gratia M., Vende P., Charpilienne A., Baron H. C., Laroche C., Sarot E., Pyronnet S., Duarte M., and Poncet D. (2016) Challenging the roles of NSP3 and untranslated regions in rotavirus mRNA translation. PLoS One 11, e0145998 10.1371/journal.pone.0145998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Austin R. J., Xia T., Ren J., Takahashi T. T., and Roberts R. W. (2002) Designed arginine-rich RNA-binding peptides with picomolar affinity. J. Am. Chem. Soc. 124, 10966–10967 10.1021/ja026610b [DOI] [PubMed] [Google Scholar]
- 38. Graindorge A., Le Tonquèze O., Thuret R., Pollet N., Osborne H. B., and Audic Y. (2008) Identification of CUG-BP1/EDEN-BP target mRNAs in Xenopus tropicalis. Nucleic Acids Res. 36, 1861–1870 10.1093/nar/gkn031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W. H., Goudreault M., Lin Z. Y., Badillo B. G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A. J., Choi H., Gstaiger M., Mohammed S., Cristea I. M., Bennett K. L., Washburn M. P., Raught B., Ewing R. M., Gingras A. C., and Nesvizhskii A. I. (2013) The CRAPome: a contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 10, 730–736 10.1038/nmeth.2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou H., Xu M., Huang Q., Gates A. T., Zhang X. D., Castle J. C., Stec E., Ferrer M., Strulovici B., Hazuda D. J., and Espeseth A. S. (2008) Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4, 495–504 10.1016/j.chom.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 41. Fongsaran C., Jirakanwisal K., Kuadkitkan A., Wikan N., Wintachai P., Thepparit C., Ubol S., Phaonakrop N., Roytrakul S., and Smith D. R. (2014) Involvement of ATP synthase β subunit in chikungunya virus entry into insect cells. Arch. Virol. 159, 3353–3364 10.1007/s00705-014-2210-4 [DOI] [PubMed] [Google Scholar]
- 42. Yang W., Nagasawa K., Münch C., Xu Y., Satterstrom K., Jeong S., Hayes S. D., Jedrychowski M. P., Vyas F. S., Zaganjor E., Guarani V., Ringel A. E., Gygi S. P., Harper J. W., and Haigis M. C. (2016) Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell 167, 985–1000.e21 10.1016/j.cell.2016.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blomen V. A., Majek P., Jae L. T., Bigenzahn J. W., Nieuwenhuis J., Staring J., Sacco R., van Diemen F. R., Olk N., Stukalov A., Marceau C., Janssen H., Carette J. E., Bennett K. L., Colinge J., Superti-Furga G., and Brummelkamp T. R. (2015) Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 10.1126/science.aac7557 [DOI] [PubMed] [Google Scholar]
- 44. Feng N., Vo P. T., Chung D., Vo T. V., Hoshino Y., and Greenberg H. B. (1997) Heterotypic protection following oral immunization with live heterologous rotaviruses in a mouse model. J. Infect. Dis. 175, 330–341 10.1093/infdis/175.2.330 [DOI] [PubMed] [Google Scholar]
- 45. Fabbretti E., Afrikanova I., Vascotto F., and Burrone O. R. (1999) Two nonstructural rotavirus proteins, NSP2 and NSP5, form viroplasm-like structures in vivo. J. Gen. Virol. 80, 333–339 10.1099/0022-1317-80-2-333 [DOI] [PubMed] [Google Scholar]
- 46. Bass D. M., Mackow E. R., and Greenberg H. B. (1990) NS35 and not vp7 is the soluble rotavirus protein which binds to target cells. J. Virol. 64, 322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shaw R. D., Vo P. T., Offit P. A., Coulson B. S., and Greenberg H. B. (1986) Antigenic mapping of the surface proteins of rhesus rotavirus. Virology 155, 434–451 10.1016/0042-6822(86)90205-9 [DOI] [PubMed] [Google Scholar]
- 48. Lu X., McDonald S. M., Tortorici M. A., Tao Y. J., Vasquez-Del Carpio R., Nibert M. L., Patton J. T., and Harrison S. C. (2008) Mechanism for coordinated RNA packaging and genome replication by rotavirus polymerase VP1. Structure 16, 1678–1688 10.1016/j.str.2008.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yan Y., Zhang D., Zhou P., Li B., and Huang S. Y. (2017) HDOCK: a web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 45, W365–W373 10.1093/nar/gkx407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bagaria A., Jaravine V., Huang Y. J., Montelione G. T., and Güntert P. (2012) Protein structure validation by generalized linear model root-mean-square deviation prediction. Protein Sci. 21, 229–238 10.1002/pro.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ball J. M., Mitchell D. M., Gibbons T. F., and Parr R. D. (2005) Rotavirus NSP4: a multifunctional viral enterotoxin. Viral Immunol. 18, 27–40 10.1089/vim.2005.18.27 [DOI] [PubMed] [Google Scholar]
- 52. Hyser J. M., Collinson-Pautz M. R., Utama B., and Estes M. K. (2010) Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. MBio. 1, e00265–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pennington J. D., Williams H. J., Salomon A. R., and Sulikowski G. A. (2002) Toward a stable apoptolidin derivative: identification of isoapoptolidin and selective deglycosylation of apoptolidin. Org. Lett. 4, 3823–3825 10.1021/ol026829v [DOI] [PubMed] [Google Scholar]
- 54. Foulke-Abel J., In J., Kovbasnjuk O., Zachos N. C., Ettayebi K., Blutt S. E., Hyser J. M., Zeng X. L., Crawford S. E., Broughman J. R., Estes M. K., and Donowitz M. (2014) Human enteroids as an ex-vivo model of host–pathogen interactions in the gastrointestinal tract. Exp. Biol. Med. 239, 1124–1134 10.1177/1535370214529398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Saxena K., Blutt S. E., Ettayebi K., Zeng X. L., Broughman J. R., Crawford S. E., Karandikar U. C., Sastri N. P., Conner M. E., Opekun A. R., Graham D. Y., Qureshi W., Sherman V., Foulke-Abel J., In J., Kovbasnjuk O., Zachos N. C., Donowitz M., and Estes M. K. (2016) Human intestinal enteroids: a new model to study human rotavirus infection, host restriction, and pathophysiology. J. Virol. 90, 43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boudreaux C. E., Kelly D. F., and McDonald S. M. (2015) Electron microscopic analysis of rotavirus assembly-replication intermediates. Virology 477, 32–41 10.1016/j.virol.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57. Boudreaux C. E., Vile D. C., Gilmore B. L., Tanner J. R., Kelly D. F., and McDonald S. M. (2013) Rotavirus core shell subdomains involved in polymerase encapsidation into virus-like particles. J. Gen. Virol. 94, 1818–1826 10.1099/vir.0.052951-0 [DOI] [PubMed] [Google Scholar]
- 58. Gallegos C. O., and Patton J. T. (1989) Characterization of rotavirus replication intermediates: a model for the assembly of single-shelled particles. Virology 172, 616–627 10.1016/0042-6822(89)90204-3 [DOI] [PubMed] [Google Scholar]
- 59. McDonald S. M., and Patton J. T. (2011) Assortment and packaging of the segmented rotavirus genome. Trends Microbiol. 19, 136–144 10.1016/j.tim.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen D., Barros M., Spencer E., and Patton J. T. (2001) Features of the 3′-consensus sequence of rotavirus mRNAs critical to minus strand synthesis. Virology 282, 221–229 10.1006/viro.2001.0825 [DOI] [PubMed] [Google Scholar]
- 61. Patton J. T., and Chen D. (1999) RNA-binding and capping activities of proteins in rotavirus open cores. J. Virol. 73, 1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gridley C. L., and Patton J. T. (2014) Regulation of rotavirus polymerase activity by inner capsid proteins. Curr. Opin. Virol. 9, 31–38 10.1016/j.coviro.2014.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li W., Manktelow E., von Kirchbach J. C., Gog J. R., Desselberger U., and Lever A. M. (2010) Genomic analysis of codon, sequence and structural conservation with selective biochemical-structure mapping reveals highly conserved and dynamic structures in rotavirus RNAs with potential cis-acting functions. Nucleic Acids Res. 38, 7718–7735 10.1093/nar/gkq663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bachler M., Schroeder R., and von Ahsen U. (1999) StreptoTag: a novel method for the isolation of RNA-binding proteins. RNA 5, 1509–1516 10.1017/S1355838299991574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McHugh C. A., Russell P., and Guttman M. (2014) Methods for comprehensive experimental identification of RNA–protein interactions. Genome Biol. 15, 203 10.1186/gb4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Oeffinger M. (2012) Two steps forward–one step back: advances in affinity purification mass spectrometry of macromolecular complexes. Proteomics 12, 1591–1608 10.1002/pmic.201100509 [DOI] [PubMed] [Google Scholar]
- 67. De Lorenzo G., Drikic M., Papa G., Eichwald C., Burrone O. R., and Arnoldi F. (2016) An inhibitory motif on the 5′UTR of several rotavirus genome segments affects protein expression and reverse genetics strategies. PLoS One 11, e0166719 10.1371/journal.pone.0166719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ding S., Zhu S., Ren L., Feng N., Song Y., Ge X., Li B., Flavell R. A., and Greenberg H. B. (2018) Rotavirus VP3 targets MAVS for degradation to inhibit type III interferon expression in intestinal epithelial cells. eLife 7, e39494 10.7554/eLife.39494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bolen C. R., Ding S., Robek M. D., and Kleinstein S. H. (2014) Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 59, 1262–1272 10.1002/hep.26657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. van den Pol A. N., Ding S., and Robek M. D. (2014) Long-distance interferon signaling within the brain blocks virus spread. J. Virol. 88, 3695–3704 10.1128/JVI.03509-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li B., Ding S., Feng N., Mooney N., Ooi Y. S., Ren L., Diep J., Kelly M. R., Yasukawa L. L., Patton J. T., Yamazaki H., Shirao T., Jackson P. K., and Greenberg H. B. (2017) Drebrin restricts rotavirus entry by inhibiting dynamin-mediated endocytosis. Proc. Natl. Acad. Sci. U.S.A. 114, E3642–E3651 10.1073/pnas.1619266114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ding S., Khoury-Hanold W., Iwasaki A., and Robek M. D. (2014) Epigenetic reprogramming of the type III interferon response potentiates antiviral activity and suppresses tumor growth. PLoS Biol. 12, e1001758 10.1371/journal.pbio.1001758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhu S., Ding S., Wang P., Wei Z., Pan W., Palm N. W., Yang Y., Yu H., Li H. B., Wang G., Lei X., de Zoete M. R., Zhao J., Zheng Y., Chen H., Zhao Y., Jurado K. A., Feng N., Shan L., Kluger Y., Lu J., Abraham C., Fikrig E., Greenberg H. B., and Flavell R. A. (2017) Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546, 667–670 10.1038/nature22967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Watt I. N., Montgomery M. G., Runswick M. J., Leslie A. G., and Walker J. E. (2010) Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. U.S.A. 107, 16823–16827 10.1073/pnas.1011099107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Deo R. C., Groft C. M., Rajashankar K. R., and Burley S. K. (2002) Recognition of the rotavirus mRNA 3′ consensus by an asymmetric NSP3 homodimer. Cell 108, 71–81 10.1016/S0092-8674(01)00632-8 [DOI] [PubMed] [Google Scholar]
- 76. Bowman G. D., Nodelman I. M., Levy O., Lin S. L., Tian P., Zamb T. J., Udem S. A., Venkataraghavan B., and Schutt C. E. (2000) Crystal structure of the oligomerization domain of NSP4 from rotavirus reveals a core metal-binding site. J. Mol. Biol. 304, 861–871 10.1006/jmbi.2000.4250 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.