

Abstract
Mutation in leucine-rich repeat kinase 2 (LRRK2) is a common cause of familial Parkinson's disease (PD). Recently, we showed that a disease-associated mutation R1441H rendered the GTPase domain of LRRK2 catalytically less active and thereby trapping it in a more persistently “on” conformation. However, the mechanism involved and characteristics of this on conformation remained unknown. Here, we report that the Ras of complex protein (ROC) domain of LRRK2 exists in a dynamic dimer–monomer equilibrium that is oppositely driven by GDP and GTP binding. We also observed that the PD-associated mutations at residue 1441 impair this dynamic and shift the conformation of ROC to a GTP-bound–like monomeric conformation. Moreover, we show that residue Arg-1441 is critical for regulating the conformational dynamics of ROC. In summary, our results reveal that the PD-associated substitutions at Arg-1441 of LRRK2 alter monomer–dimer dynamics and thereby trap its GTPase domain in an activated state.
Keywords: leucine-rich repeat kinase 2 (LRRK2), Parkinson disease, GTPase, conformational change, molecular dynamics, conformational dynamics, disease mutation, enzyme activation, kinase, Ras of complex proteins (ROC)
Introduction
Age-associated neurodegeneration is one of the most desperate and challenging public health crises due to the rapidly aging population and the lack of effective treatments (1). Fortunately, discoveries of precise mutations in disease-associated genes in recent decades have provided new venues for developing mechanism-based therapeutic strategies. One of these genes is called leucine-rich repeat kinase 2 (LRRK2)3, mutations in which are common causes of Parkinson's disease (PD) (2–6). LRRK2 is a large multidomain protein (2,527 amino acids) consisting of seven putative domains (3), including a Ras-like GTPase domain called “Ras of complex proteins” (ROC) followed by a domain called C-terminal of ROC (COR), then a kinase domain (Kin). It remains unclear as to how perturbations of LRRK2 activities result in disease. However, the most common mutation in LRRK2-associated PD, G2019S in the kinase domain, shows higher kinase activity than that of the wildtype (WT); therefore, its over-activation might be associated with disease pathogenesis (7).
The tandem ROC-COR-Kin arrangement suggests that their activities might be coupled such that the GTPase activity of ROC might modulate the kinase activity of Kin. Indeed, several studies have shown that GTP binding to the ROC domain regulates the activity of the Kin domain (8, 9) and inhibition of the ROC domain affects the activity of the Kin domain (10). Moreover, PD-associated mutations in the ROC domain (R1441C) have been shown to have higher kinase activity (11), suggesting that mutations in the ROC domain might also up-regulate kinase activity. We previously showed that the Parkinson's disease-associated mutation R1441H locks the ROC domain in a prolonged activated state (12) and a recent report showed that a homologue of LRRK2, CtRoco, undergoes dimer–monomer transition upon GTP binding (13), which together suggest that perturbations in dimer–monomer dynamics might be the key factor leading to the reduction in GTPase activity observed in the PD-associated mutations at Arg-1441.
Here, we show that all the disease-associated mutations at Arg-1441 we examined (R1441H/R1441G/R1441C) showed decreased GTPase activity and a complete loss of monomer–dimer conformational dynamics. These data, taken together with prior reports, suggest that the mutations at Arg-1441 impair monomer–dimer dynamics leading to trapping the GTPase domain of LRRK2 in a more persistently “on” state.
Results and discussion
Arginine 1441 is uniquely essential for the GTPase activity of ROC
A detailed understanding of LRRK2 has been hampered by a lack of protein samples amenable for quantitative biochemical and biophysical studies. As such, most of the detailed insights into the structure and function of LRRK2 have been gleaned from the observations of the homologous proteins. We have recently created a stably-folded G-domain construct of LRRK2 consisting of residues 1329–1520 (Fig. 1) that we called ROCext, which enabled us to study quantitatively the GTPase activity of WT ROCext and the R1441H mutant (12). To further understand the critical effects of the disease-associated mutations and their mechanisms leading to the biochemical perturbations that we and others have observed (12, 14–16), we have created stably-folded ROCext constructs harboring the disease-associated mutations R1441G/R1441C (in addition to the R1441H, which we have previously described), and artificial mutants R1441K/R1441Y for detailed biochemical investigations.
Figure 1.
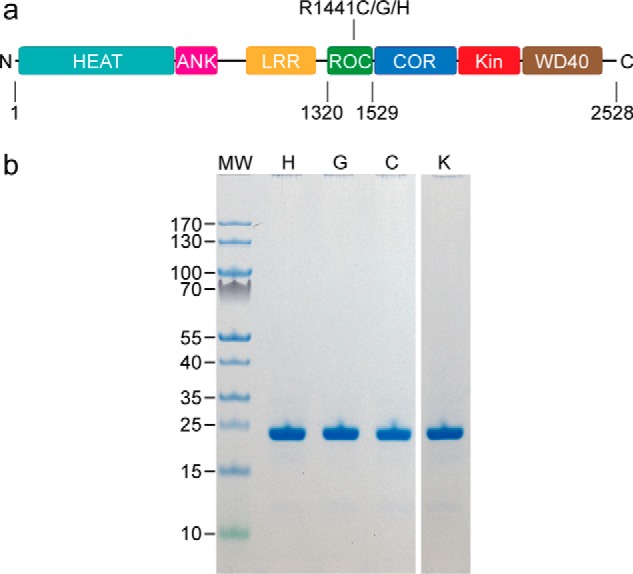
The ROC constructs of LRRK2 under investigation. a, a schematic depiction of LRRK2 showing the boundaries of ROCext with respect to the other domains, and the sites of PD-associated mutations studied. b, SDS-PAGE of purified ROCext carrying the PD-associated mutations R1441H (H), R1441G (G), and R1441C (C), and a synthetic mutant R1441K (K).
We first measured the GTP hydrolysis activities of the R1441H, R1441G, and R1441C constructs and observed that the specific GTPase activity (pmol mg−1 min−1) was 9.0 ± 0.8, 12.6 ± 0.8, and 12.9 ± 0.8, respectively, which are ∼4-fold lower than that of the WT (48.6 ± 0.8) (Fig. 2a). We found it notable that despite having very different side chain properties, all the mutants showed a similar reduction in activity, which suggests that the arginine residue at position 1441 might be specifically essential for the activity of ROCext. To test this, we made an artificial mutant substituting arginine 1441 for lysine and measured its GTPase activity. Similarly, we observed that the GTPase activity of the R1441K mutant (SA = 10.5 ± 2.5) to be about 4-fold lower than that of the WT (Fig. 2a). These data further supports the notion that arginine at position 1441 is uniquely important for ROCext activity; thus, understanding its role would provide insights into the mechanism of this atypical G-domain and its role in disease pathogenesis.
Figure 2.

Biochemical properties of PD-associated mutations in the ROC domain of LRRK2 compared with the WT. a, GTPase activity, phosphate release against time, of WT (black line) ROCext and various mutants R1441C (orange), R1441G (gray), R1441H (blue), and R1441K (green). b, fluorescence polarization-based GDP binding assay of WT ROCext (black) and the PD-associated mutants R1441C/R1441G/R1441H (orange, gray, and blue, respectively). Affinity is shown in parentheses. c, fluorescence polarization-based GTP-binding assay of WT ROCext (black) and the PD-associated mutants R1441C/R1441G/R1441H (orange, gray, and blue, respectively). Affinity is shown in parentheses. d, fluorescence-based thermal denaturation assay of ROCext (black), R1441C (orange), R1441G (gray), and R1441H (blue). Melting temperature of shown in parentheses.
GTPase activity and thermostability
We reported previously that the R1441H mutant has a higher affinity for GTP compared with that of WT, which we suggested might be responsible, in part, for the aberrant activity of LRRK2 (12). To determine whether or not this is a general property of mutations at Arg-1441, we measured the affinity of the other mutants, R1441G and R1441C, for GDP and GTP using a fluorescence polarization assay we previously described (12). We reconfirmed that the R1441H mutant has higher affinity for GTP (Kd = 3.0 ± 0.4 μm) than that of WT (Kd = 4.1 ± 0.3 μm); however, they are not replicated in the R1441G and R1441C mutants, which have similar GTP affinities (Kd = 4.5 ± 0.7 and 4.9 ± 0.8 μm, respectively) to the WT. Similarly, there are no consistent trends for GDP affinity, where the R1441C mutant has lower affinity for GDP (Kd = 2.5 ± 0.4 μm) compared with that of WT (Kd = 1.2 ± 0.1 μm); whereas the R1441G (Kd = 1.8 ± 0.3 μm) and R1441H (Kd = 1.9 ± 0.3 μm) mutants bind GDP with affinities comparable with the WT (Fig. 2b). These data indicate that the higher affinity for GTP observed in the R1441H mutant is not a commonly shared property among the disease-associated mutations at position 1441; thus, it is probably not a key feature responsible for the reduction in GTPase activity observed in the mutants.
Continuing our search for the fundamental properties that rendered the Arg-1441 mutants less active, we measured the thermal stability of each construct using a thermofluor-based protein denaturation assay (17). We observed that the R1441H ROCext mutant is as thermally stable as the WT with a melting temperatures (Tm) of 54.3 ± 0.1 and 54.3 ± 0.1 °C, respectively (Fig. 2c); whereas the Tm for the R1441G (Tm = 50.5 ± 0.1 °C) and R1441C (Tm = 51.5 ± 0.1 °C) mutants are about 3 °C lower than that of the WT (Fig. 2d). However, the GTPase activity of the R1441H mutant (despite having a higher Tm) is similar to that of R1441C and R1441G, thus it is unlikely that the lowered thermal stability of the mutants is responsible for the reduction in GTPase activity mentioned above.
The PD-associated mutations at residue 1441 disrupt dimer formation
We next examined whether the mutations at position 1441 cause structural perturbation by using CD spectroscopy. We found no significant differences in the CD spectra of the mutants compared with that of the WT, indicating that the mutations have no significant impact to the secondary structure of ROCext (Fig. 3a).
Figure 3.

Conformations of PD-associated mutations in the ROC domain of LRRK2 compared with the WT. a, CD spectrometry of WT (black line) ROCext and various mutants R1441C (orange), R1441G (gray), R1441H (blue). b, SEC-MALS of WT ROCext dimer (solid black line), WT monomer (dotted black line) along with the PD-associated mutants R1441C/R1441G/R1441H (orange, gray, and blue, respectively).
Previously we reported that WT ROCext existed in solution in a monomer–dimer equilibrium (12), which was also recently described for a LRRK2 homolog CtRoco, where the authors suggested that the disease-associated mutations in the ROC domain might disrupt the dimer–monomer dynamics (13). To investigate the effects of PD-associated mutations on the dimerization of ROC, we used multiangle light scattering coupled to size-exclusion chromatography (SEC-MALS). We found that all the mutants (R1441G/R1441C/R1441H) occurred as only monomers (Fig. 3b). This result might suggest that the monomeric form of ROC might be catalytically inactive. However, in a previous study, we showed that the monomeric form of ROC is catalytically active. Moreover, we showed that the addition of GTP to the GTPase reaction renders ROC conversion from dimer to monomer. Therefore, it is more likely that the dimer–monomer inter-conversion or cycling is essential for GTP hydrolysis as recently suggested for CtRoco (13).
Conformational dynamics is essential for GTPase activity
Because all the disease-associated mutations at residue Arg-1441 abolished ROCext dimerization, we investigated whether or not position 1441 is critical for regulating the dimer–monomer dynamics. Through systematic substitution mutations at position 1441, we found that a ROCext containing a tyrosine residue at 1441 forms a constitutive dimer with no observable monomers (Fig. 4a). Interestingly, we found that the R1441Y dimer is catalytically inactive (Fig. 4b). A similar observation was recently made in a ROC homolog CtRoco (13), where a constitutively dimeric mutant showed reduced GTPase activity. Taken together, our data suggest that the conformational dynamics that occur in the monomer–dimer transition is essential for the GTPase activity of LRRK2 and that the disease-associated mutations at Arg-1441 disrupt this activity through interfering with the dimer-monomer dynamics.
Figure 4.

Oligomeric states and GTPase activity of R1441Y. a, size-exclusion chromatography of the WT dimer (solid black line), WT monomer (dotted black line), and the R1441Y mutant (green line). b, GTPase activity, phosphate release against time, of the R1441Y mutant (green) compared with that of the WT (black).
The remarkable similarities between LRRK2 ROCext and CtRoco in their dimer–monomer dynamics suggests that their mechanism of actions might be conserved. However, a critical difference between CtRoco and our LRRK ROCext is that CtRoco dimerization is driven by interactions in its COR domain (13, 18), whereas our LRRK2 ROCext construct readily forms dimers in the absence of the COR domain. This result suggests that the dimerization mechanism of LRRK2 ROC might be different from that of CtRoco. Indeed, as mentioned above, the R1441Y mutation in ROCext locks it in a constitutive homodimeric state; however, the equivalent WT position in CtRoco, residue 558, is natively tyrosine. Moreover, mutations in CtRoco at Tyr-558 mimicking the LRRK2 R1441G/R1441C/R1441H did not affect its dimerization, whereas these mutations in LRRK2 ROCext abolished dimer formation as shown above. These similarities and differences between CtRoco and LRRK2 suggest that the subtle variability in the mechanisms of action observed in the typical Ras and Rab families of GTPases might also occur in the Roco family of GTPases (19).
PD-associated mutations abolish the guanine nucleotide-dependent dimer–monomer interconversions
We and others have previously shown that incubating dimeric ROC with GTP renders it monomeric (12, 20), which suggest that guanine nucleotides might regulate the dynamics of ROC oligomerization. However, in our previous investigation, we observed that both GTP and GDP were equally effective for converting dimeric ROC to its monomeric conformation (12). Through further investigations, we observed that the EDTA used in the nucleotide exchange itself was sufficient to convert all the dimers into monomers. Based on this, we hypothesized that the monomerization effects of EDTA might have masked the potentially differential effects of GTP and GDP. To investigate this, we incubated ROCext (93% dimers) with varying concentrations of GDP or GTP without EDTA and analyzed their proportions of dimers and monomers using size-exclusion chromatography. As expected, we observed a GTP concentration-dependent conversion of dimeric ROCext to monomers with an equilibrium proportion of dimers and monomers at 5 mm GTP to be 15 and 85%, respectively (Fig. 5a). In contrast, GDP caused a significantly smaller shift in the dimer–monomer equilibrium with a ratio of dimer:monomer at 71:29% in 5 mm GDP (Fig. 5b). These results indicate that GTP drives ROC dimers toward monomers and, reciprocally, GDP shifts the equilibrium toward the dimeric conformation. To investigate this, we developed a method to detect a dynamic conversion from dimer to monomer and then back to dimer in the same protein sample. We did so by first starting with a protein sample consisting of mostly the dimeric form of ROCext (∼85%), then we incubated it with GTP, which converts all the dimers to monomers, and then followed by exchanging it with GDP. We analyzed the dimeric state of the protein sample at each step and found that dimeric ROCext dissociated into monomers upon GTP binding and then about 18% reassembled into dimers upon exchanging for GDP, but none converted to dimers when GTP was added instead at this stage (Fig. 5c). In contrast, the PD-associated mutation R1441G entirely abolished the dimer–monomer dynamics, where it was trapped in a monomeric conformation similar to the GTP-bound samples (Fig. 5d). These results unambiguously demonstrate a reversible dynamic interconversion between dimers and monomers that is mediated by GDP and GTP binding, respectively, and that the PD-associated mutation abolishes this dynamic process.
Figure 5.

Nucleotide-dependent dimer–monomer equilibrium of ROC. a, size-exclusion chromatography of the WT ROCext (solid black line) incubated with 2.5 mm GTP (orange dashed line) and 5.0 mm GTP (red dashed line). b, the same experiment as a, but with 2.5 mm GDP (light green dashed line) and 5.0 mm GDP (green dashed line). c, dimer–monomer interconversion experiment showing that the same protein sample converting from mostly dimers (solid black line) to nearly completely monomers upon binding excess GTP (dashed black line) and then reverts back to forming some dimers upon binding to GDP (green dashed line), but the same treatment with GTP did not produce any dimers (red dashed line). d, the same experiment as c for the ROCext carrying the PD-associated mutation R1441G, showing a complete loss of nucleotide-dependent dimer–monomer interconversion.
Conformational changes in ROC regulate LRRK2 subcellular localization
To investigate the potential significance of the observed dimer–monomer dynamics of ROC in the context of the full-length LRRK2 in live cells, we examined the subcellular localization of LRRK2 carrying the mutations that gave rise to monomeric and dimeric ROCext. We used our previously described method to characterize interactions of LRRK2 with a binding partner RAB29 (21) in HEK293FT cells. Under normal conditions LRRK2 is localized to the cytosol, but when co-transfected with RAB29 it re-localizes in clusters that overlap with the trans-Golgi network. Upon over-expression of the mutants with RAB29 in HEK293FT cells, we observed a significant increase in LRRK2 re-localization to the trans-Golgi network with all the Arg-1441 mutations compared with WT (Fig. 6). Although the biological function of LRRK2 and its normal subcellular localization are still unclear, our results clearly demonstrate differential effects in subcellular localization between the different mutations that are shown dimeric and monomeric in our in vitro assays. Taken together, the results suggest that Arg-1441 is critical for the conformational dynamics of ROC, which modulates the activity and potentially the subcellular localization of LRRK2.
Figure 6.

Effects of PD-associated mutations on LRRK2. a, cell-based subcellular localization assay showing that the disease-associated mutations (R1441H/R1441C/R1441G) that perturbed ROC dimerization cause accumulation of full-length LRRK2 at the trans-Golgi network. The symbols ** and *** denote p values 0.001 to 0.01 and 0.0001 to 0.001, respectively. b, representative images of the HEK293FT cells collected in the trans-Golgi localization assay shown in a. The 50 μm scale shown in the bottom right panel is representative of all other panels in the figure.
Conclusion
Detailed investigations of LRRK2 has been stymied by a lack of protein samples in the quality and quantity required for quantitative studies. Recently, we reported the construction of a ROC domain and a method to obtain it in a form amenable for quantitative studies, which showed that disease-associated mutation R1441H rendered it catalytically less active and thereby, trapping it more persistently on conformation, although we were unable to precisely define the “on conformation” at the time (12).
Here, in this report, we showed that the other disease-associated mutations, R1441G and R1441C, also perturb the GTPase activity of ROC, as well as abolishing their ability to form dimers. This revealed that a single substitution mutation at residue 1441 is sufficient to cause a significant conformational change in the ROC domain. We further showed that the GDP-bound form of ROC exists in an equilibrium of dimers and monomers, but favoring the dimeric conformation in a ratio of about 7:3; however, upon binding GTP, the ratio is shifted to 100% monomers. This indicated that GDP and GTP might regulate a cycle of dimer–monomer conformational dynamics in the ROC domain of LRRK2. Indeed, we were able to demonstrate in the same protein sample that ROCext dimers convert to monomers upon GTP binding and then cycled back to the dimeric form upon binding to GDP. This result clearly shows that the ROCext dimer–monomer conformational change is in a dynamic equilibrium that is oppositely driven by GDP and GTP binding, and that the disease-associated mutations at residue 1441 impair this dynamic and shift the equilibrium 100% to the GTP-bound-like monomeric conformation (Fig. 7). Moreover, the results presented herein revealed that arginine 1441, whose mutation is associated with PD, is critical for regulating the conformational dynamics of ROC. We have recently described another PD-associated mutation in ROC, N1437H, which also caused perturbation in its dimer–monomer dynamics (22), thus this might be a common effect of disease-associated mutations in the GTPase domain of LRRK2.
Figure 7.
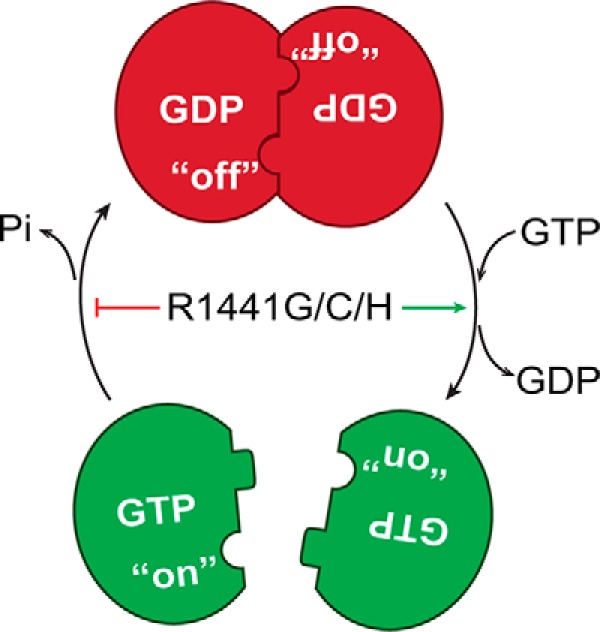
Model of ROC dimer–monomer dynamic equilibrium summarizing our observations that GTP binding to dimeric ROC led to its dissociation into monomers. GDP binding, either by GTP hydrolysis or nucleotide exchange to GDP shifts the equilibrium to the dimeric conformation. The PD-associated mutations perturbs its GTPase activity and results in a prolonged on state.
Experimental procedures
Protein expression and purification
An extended GTPase domain of LRRK2 consisting of residues 1329–1520 (ROCext) was subcloned into a pETDuet-1 vector (Novagen, Darmstadt, Germany) using PCR cloning techniques. The resulting protein consisting of an N-terminal hexahistidine tag was expressed from Rosetta2 (DE3) Escherichia coli (Novagen) by inducing with 0.5 mm isopropyl β-d-thiogalactopyranoside for 16 h at 20 °C. Cells were harvested by centrifugation and lysed by sonication in a buffer containing 30 mm HEPES (pH 7.4), 250 mm NaCl, 10 mm MgCl2, 10 mm glycine, 20 mm imidazole, 10 μm GDP, and 10% (v/v) glycerol. Cell debris was cleared by ultracentrifugation at 14,000 × g (35,000 rpm, Beckman 45 Ti rotor). The supernatant was incubated with nickel-nitrilotriacetic acid-agarose (Qiagen, Hilden, Germany) for 2 h at 4 °C, then washed with lysis buffer (detailed above) and eluted with buffer containing 30 mm HEPES (pH 7.4), 250 mm NaCl, 10 mm MgCl2, 10 mm glycine, 300 mm imidazole, 1 mm DTT, 10 μm GDP, and 10% glycerol. The purified protein was then “polished” by passing through a size-exclusion column (Superdex 200, GE Healthcare) in buffer containing 30 mm HEPES (pH 7.4), 150 mm NaCl, 10 mm MgCl2, 10 mm glycine, 1 mm DTT, and 10% glycerol. The purified protein was then concentrated to ∼15 mg/ml, flash frozen in liquid nitrogen, and stored at −80 °C.
SEC-MALS
To determine the absolute molecular weight of Rocext in solution, we used multiple angle light scattering. Our experimental setup includes an AKTA FPLC (GE Healthcare Biosciences) with a silica-based size exclusion chromatography column (WTC-030S5, Wyatt Technology Corporation, Santa Barbara, CA) as liquid chromatography unit. Down from the liquid chromatography units is a refractive index detector (Optilab T-rEX, Wyatt Technology) followed by a multiple light scattering detector (Dawn HeleosII, Wyatt Technology) for determining protein concentration and particle size, respectively. Each sample injection consisted of ∼1 mg of purified ROCext in buffer containing 30 mm HEPES (pH 7.4), 0.15 m NaCl, 10 mm MgCl2, 10 mm glycine, 1 mm DTT, and 10% glycerol. The flow rate was set at 0.4 ml/min and data were collected in a 1-s interval. Data processing and analysis were performed using the ASTRA software (Wyatt Technology).
Dimer–monomer inter-conversion assay
To determine the effect of GDP/GTP cycles on the dynamic oligomeric states, we incubated purified ROCext with GDP or GTP in buffer containing 30 mm HEPES (pH 7.4), 0.15 m NaCl, 10 mm MgCl2, 10 mm glycine, 1 mm DTT, and 10% glycerol. This incubation was done without addition of EDTA. To test the dimeric to monomeric conversion, the dimeric protein samples (∼15 mg/ml, 0.6 mm) were incubated in room temperature with 16 mm of either GDP or GTP (×25 to Roc) for 6 h. To test the monomeric to dimeric conversion, the monomeric protein samples were first obtained by GTP incubation and desalting purification (Zeba spin desalting column, Thermo Fisher). The monomeric samples were then incubated with 16 mm GDP or GTP for 6 h. The ratios of monomeric and dimeric forms of the incubated samples were then determined by size exclusion chromatography (Superdex 75, GE Healthcare).
Circular dichroism spectroscopy
CD spectra were collected on a Biologic Science Instruments MOS450 AF/CD spectrometer with the slit width of 1.0 mm and data acquisition of 1.0 s. The protein samples with concentrations ranging from 0.46 to 0.86 mg/ml (based on absorbance at 280 nm) were dissolved in the buffer containing 10 mm Tris-HCl (pH 7.4), 150 mm NaCl, 5 mm MgCl2, 1 mm DTT, and 5% glycerol. Data were analyzed with Dichroweb and plotted with Prism 7.
Fluorescence polarization nucleotide-binding assay
To estimate the binding affinity of guanine nucleotides BODIPY-FL-GTPγS (100 nm) or BODIPY-FL-GDP (150 nm) (Molecular Probes) were titrated with ROCext (starting at 0.1 μm) until saturation was reached (15 and 10 μm, respectively). Fluorescence polarization signals were read using an EnVison 2102 Multilabel Plate Reader (PerkinElmer Life Sciences) with excitation at 485 nm and emission at 535 nm. Experiments were performed at 25 °C in buffer containing 30 mm HEPES (pH 7.4), 150 mm NaCl, 10 mm MgCl2, 10 mm glycine, 4 mm EDTA, 1 mm DTT, 10% glycerol. Data were analyzed using Prism 7 (GraphPad Software).
GTPase activity assay
GTPase activity of ROCext was assessed by using the Enzcheck assay kit (Invitrogen) according to the manufacturer's instructions. Briefly, Rocext (30 μm) was incubated with 2 mm GTP in buffer containing 30 mm HEPES (pH 7.4), 150 mm NaCl, 10 mm MgCl2, 10 mm glycine, 1 mm DTT, and 10% glycerol at 25 °C. Absorbance at 360 nm was recorded every 3 min for 3 h using a microplate reader. The amount of Pi released from GTP hydrolysis at each time point was determined by extrapolation using a phosphate standard curve. Data analysis and curve fitting were done with GraphPad Prism 7.
Thermofluor assay
Solutions of 12.5 μl of ×10 Sypro Orange (prepared from a ×5,000 stock concentrate, Molecular Probes) in buffer containing 30 mm HEPES (pH 7.4), 0.15 m NaCl, 10 mm MgCl2, 10 mm glycine, 1 mm DTT, 4 mm GDP or Gpp(NH)p, 2 μm lauryl maltose neopentyl glycol, and 10% glycerol, and 12.5 μl of 25 μm ROCext or mutants were added to a 96-well thin-wall PCR plate. The plate was heated in the Real-Time PCR Detection System (Mastercycler realplex, Eppendorf) from 20 to 85 °C and fluorescence was recorded in increments of 0.4 °C. The emission wavelength was set at 550 nm.
Subcellular localization of full-length LRRK2 mutants
Mutations in ROC were cloned into 3× FLAG-LRRK2 using the QuikChange II XL Site-directed Mutagenesis kit (Agilent Technologies). The resulting plasmids were co-transfected with 2×Myc-RAB29 into HEK293FT cells using Lipofectamine 2000 (ThermoFisher). Proteins were labeled using primary antibodies to FLAG (F1804 Sigma, 1:500), myc (MCA1929 Bio-Rad, 1:500), and TGN46 (AHP500G Bio-Rad, 1:1,000). Alexa Fluor secondary antibodies donkey anti-mouse 488, donkey anti-sheep 568, and donkey anti-rat 647 (ThermoFisher) were used at 1:500. Hoescht 33342 (H3572 ThermoFisher, 1:10,000) was used as a nuclear dye. Cells were imaged, and overlap of 3× FLAG-LRRK2 with TGN46 was quantified using Cellomics ArrayScanVTI HCS Reader (Thermo Scientific) and HCS Studio version 6.6.0.
Author contributions
C.-X. W. and Q. Q. H. formal analysis; C.-X. W. validation; C.-X. W., J. L., X. R., V. A. E., and N. C. H. investigation; C.-X. W., Y. P., X. R., Y. T., M. W., and Q. Q. H. methodology; C.-X. W. and Q. Q. H. writing-original draft; C.-X. W., J. L., Y. P., X. R., S. M. J., M. R. C., and Q. Q. H. writing-review and editing; J. L., Y. P., X. R., V. A. E., N. C. H., R. S., and Q. Q. H. data curation; S. M. J., M. W., M. F., R. J. N., and M. R. C. resources; M. F. and Q. Q. H. funding acquisition; R. J. N., M. R. C., and Q. Q. H. conceptualization; Q. Q. H. supervision; Q. Q. H. project administration.
This work was supported by National Institutes of Health Grants R01GM111639 and R01GM115844 and the Michael J. Fox Foundation(to Q. Q. H.), Grant R01GM111695 and U.S. National Science Foundation Grant MCB-1157688 (to Y. T.), the Intramural Research Program of the National Institutes of Health, National Institute on Aging (to M. R. C.), National Institutes of Health Grant R01GM120350 (to S. M. J.), and funding from the Travel and Learn program of the Indiana Association of Chinese-Americans (IACA) (to N. C. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LRRK2
- leucine-rich repeat kinase 2
- PD
- Parkinson's disease
- ROS
- Ras of complex proteins
- COR
- C-terminal of ROC
- Kin
- kinase domain
- SEC-MALS
- multiangle light scattering coupled to size-exclusion chromatography
- Gpp(NH)p
- guanosine 5′-(β,γ-imido)triphosphate.
References
- 1. Petsko G. A. (2006) The next epidemic. Genome Biol. 7, 108 10.1186/gb-2006-7-5-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cookson M. R. (2010) The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat. Rev. Neurosci. 11, 791–797 10.1038/nrn2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mata I. F., Wedemeyer W. J., Farrer M. J., Taylor J. P., and Gallo K. A. (2006) LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 29, 286–293 10.1016/j.tins.2006.03.006 [DOI] [PubMed] [Google Scholar]
- 4. Giasson B. I., and Van Deerlin V. M. (2008) Mutations in LRRK2 as a cause of Parkinson's disease. Neurosignals 16, 99–105 10.1159/000109764 [DOI] [PubMed] [Google Scholar]
- 5. Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., Stoessl A. J., Pfeiffer R. F., Patenge N., Carbajal I. C., Vieregge P., et al. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 10.1016/j.neuron.2004.11.005 [DOI] [PubMed] [Google Scholar]
- 6. Paisán-Ruiz C., Jain S., Evans E. W., Gilks W. P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A. M., Khan N., Johnson J., Martinez J. R., Nicholl D., Carrera I. M., Pena A. S., et al. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44, 595–600 10.1016/j.neuron.2004.10.023 [DOI] [PubMed] [Google Scholar]
- 7. Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M. P., Beilina A., Blackinton J., Thomas K. J., Ahmad R., Miller D. W., Kesavapany S., Singleton A., Lees A., et al. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Disease 23, 329–341 10.1016/j.nbd.2006.04.001 [DOI] [PubMed] [Google Scholar]
- 8. Guo L., Gandhi P. N., Wang W., Petersen R. B., Wilson-Delfosse A. L., and Chen S. G. (2007) The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp. Cell Res. 313, 3658–3670 10.1016/j.yexcr.2007.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ito G., Okai T., Fujino G., Takeda K., Ichijo H., Katada T., and Iwatsubo T. (2007) GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry 46, 1380–1388 10.1021/bi061960m [DOI] [PubMed] [Google Scholar]
- 10. Oueslati A., Fournier M., and Lashuel H. A. (2010) Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog. Brain Res. 183, 115–145 10.1016/S0079-6123(10)83007-9 [DOI] [PubMed] [Google Scholar]
- 11. West A. B., Moore D. J., Choi C., Andrabi S. A., Li X., Dikeman D., Biskup S., Zhang Z., Lim K. L., Dawson V. L., and Dawson T. M. (2007) Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 16, 223–232 10.1093/hmg/ddl471 [DOI] [PubMed] [Google Scholar]
- 12. Liao J., Wu C. X., Burlak C., Zhang S., Sahm H., Wang M., Zhang Z. Y., Vogel K. W., Federici M., Riddle S. M., Nichols R. J., Liu D., Cookson M. R., Stone T. A., and Hoang Q. Q. (2014) Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc. Natl. Acad. Sci. U.S.A. 111, 4055–4060 10.1073/pnas.1323285111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deyaert E., Wauters L., Guaitoli G., Konijnenberg A., Leemans M., Terheyden S., Petrovic A., Gallardo R., Nederveen-Schippers L. M., Athanasopoulos P. S., Pots H., Van Haastert P. J. M., Sobott F., Gloeckner C. J., Efremov R., Kortholt A., and Versées W. (2017) A homologue of the Parkinson's disease-associated protein LRRK2 undergoes a monomer-dimer transition during GTP turnover. Nat. Commun. 8, 1008 10.1038/s41467-017-01103-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li X., Tan Y. C., Poulose S., Olanow C. W., Huang X. Y., and Yue Z. (2007) Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J. Neurochem. 103, 238–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Y., Dunn L., Greggio E., Krumm B., Jackson G. S., Cookson M. R., Lewis P. A., and Deng J. (2009) The R1441C mutation alters the folding properties of the ROC domain of LRRK2. Biochim. Biophys. Acta 1792, 1194–1197 10.1016/j.bbadis.2009.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lewis P. A., Greggio E., Beilina A., Jain S., Baker A., and Cookson M. R. (2007) The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem. Biophys. Res. Commun. 357, 668–671 10.1016/j.bbrc.2007.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pantoliano M. W., Petrella E. C., Kwasnoski J. D., Lobanov V. S., Myslik J., Graf E., Carver T., Asel E., Springer B. A., Lane P., and Salemme F. R. (2001) High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen 6, 429–440 10.1177/108705710100600609 [DOI] [PubMed] [Google Scholar]
- 18. Gotthardt K., Weyand M., Kortholt A., Van Haastert P. J., and Wittinghofer A. (2008) Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. EMBO J. 27, 2352 10.1038/emboj.2008.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liao J., and Hoang Q. Q. (2018) Roco proteins and the Parkinson's disease-associated LRRK2. Int. J. Mol. Sci. 19, 4074 10.3390/ijms19124074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Z., Mobley J. A., DeLucas L. J., Kahn R. A., and West A. B. (2016) LRRK2 autophosphorylation enhances its GTPase activity. FASEB J. 30, 336–347 10.1096/fj.15-277095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beilina A., Rudenko I. N., Kaganovich A., Civiero L., Chau H., Kalia S. K., Kalia L. V., Lobbestael E., Chia R., Ndukwe K., Ding J., Nalls M. A., International Parkinson's Disease Genomics, C., North American Brain Expression, C., Olszewski M., et al. (2014) Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 111, 2626–2631 10.1073/pnas.1318306111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang X., Wu C., Park Y., Long X., Hoang Q. Q., and Liao J. (2018) The Parkinson's disease-associated mutation N1437H impairs conformational dynamics in the G domain of LRRK2. FASEB J. 10.1096/fj201802031R 10.1096/fj201802031R [DOI] [PMC free article] [PubMed] [Google Scholar]