

Abstract
Human cytomegalovirus (HCMV) is a common β-herpesvirus causing life-long latent infections. HCMV replication interferes with cell cycle regulation in host cells because the HCMV-encoded cyclin-dependent kinase (CDK) ortholog pUL97 extensively phosphorylates the checkpoint regulator retinoblastoma protein. pUL97 also interacts with cyclins B1, T1, and H, and recent findings have strongly suggested that these interactions influence pUL97 substrate recognition. Interestingly, here we detected profound mechanistic differences among these pUL97–cyclin interactions. Our study revealed the following. (i) pUL97 interacts with cyclins B1 and H in a manner dependent on pUL97 activity and HCMV-specific cyclin modulation, respectively. (ii) The phosphorylated state of both proteins is an important determinant of the pUL97–cyclin B1 interaction. (iii) Activated phospho-Thr-315 cyclin H is up-regulated during HCMV replication. (iv) Thr-315 phosphorylation is independent of intracellular pUL97 or CDK7 activity. (v) pUL97-mediated in vitro phosphorylation is detectable for cyclin B1 but not H. (vi) Mutual transphosphorylation between pUL97 and CDK7 is not detectable, and an MS-based phosphosite analysis indicated that pUL97 might unexpectedly not be phosphorylated in its T-loop. (vii) The binary complexes pUL97–cyclin H and CDK7–cyclin H as well as the ternary complex pUL97–cyclin-H–CDK7 are detectable in an assembly-based CoIP approach. (viii) pUL97 self-interaction can be bridged by the transcriptional cyclins T1 or H but not by the classical cell cycle–regulating B1 cyclin. Combined, our findings unravel a number of cyclin type–specific differences in pUL97 interactions and suggest a multifaceted regulatory impact of cyclins on HCMV replication.
Keywords: viral replication; cyclin; protein kinase; protein assembly; phosphorylation; reprogramming; cyclins B1, T1, and H; human cytomegalovirus; protein phosphorylation; regulatory interaction; viral kinase pUL97–cyclin complexes
Introduction
Human cytomegalovirus (HCMV)4 is a worldwide-distributed β-herpesvirus causing life-long latent infection in humans. In the immunocompetent host, HCMV may remain asymptomatic, whereas in immunosuppressed individuals, e.g. transplant recipients, tumor, and AIDS patients, HCMV infection can lead to severe symptoms and a life-threatening viral pathogenesis (1, 2). Most seriously, congenital HCMV infection represents a considerable risk for the unborn child to obtain developmental defects or cytomegalovirus inclusion disease (3, 4). Viral pathogenesis is closely linked to the efficiency of viral replication in individual tissues, a pronounced virulence and so far insufficiently understood determinant of virus–host interaction. On the molecular level, recent investigations stressed the importance of multiprotein complexes consisting of viral and host components (5–8). Notably, HCMV replication drastically interferes with cell cycle regulation, a process, in which the HCMV-encoded protein kinase pUL97 massively phosphorylates the checkpoint regulator retinoblastoma protein (Rb) (9–11). This initial Rb inactivation, followed by further viral regulatory steps of intervention, ultimately results in an early S-phase cell cycle arrest (1, 12, 13).
Typically, such events of virus–host interaction are regulated through higher-order protein–protein complexes and represent potential rate-limiting determinants of cytomegalovirus replication. The interaction between the HCMV-encoded protein kinase pUL97 and human cyclins of types B1, T1, and H has been described in our earlier reports (6, 14–17). These three cyclins obviously possess different affinities in terms of strength of pUL97 binding detected by CoIP-based analyses (6), as well as a requirement of pUL97 activity (cyclin B1) (16) or dependence on HCMV replication (cyclin H) (6). Recently published data indicate a substrate-bridging function of cyclin(s) for the binding of pUL97 to its substrate pp65, as determined with a pp65 mutant lacking a putative cyclin-docking motif (17). In this study, we present novel aspects of pUL97–cyclin interaction, which profoundly refine our picture of the differential mode of interaction between the viral kinase pUL97 and cellular cyclins B1, T1, and H.
Results
HCMV protein kinase pUL97 interacts with three different types of cyclins
The HCMV-encoded protein kinase pUL97 represents a CDK ortholog that is essential for efficient viral replication via phosphorylation of several viral and cellular substrates. A linear map of pUL97 and known substrate-binding regions are depicted in Fig. 1. Despite earlier data pointing to a cyclin-independent functional mechanism (9, 12), experimental evidence was provided for the occurrence of pUL97–cyclin complexes (14), which were detectable by several different methods. We demonstrated that at least three different types of cyclins, namely B1, T1, and H, can undergo pUL97 interaction (6, 15, 16) and that even a broader range of interactions, e.g. with cyclin A, may be possible, but that has not been consistently confirmed. Notably, this behavior places pUL97 in close relationship to CDKs binding multiple cyclins, such as CDK1 and CDK2, in contrast to single cyclin-binding CDKs, such as CDK7 (18). However, the various functional properties of pUL97 and related herpesviral UL-type kinases (13) show a unique combination of a number of CDK-specific phenotypes, as summarized by Table 1. This comparison shows at least seven characteristics, in which the mode of pUL97–cyclin interaction displays substantial differences between the three relevant types of cyclins. Hallmarks of this phenotypical variation have been demonstrated by our previous study (6), serving as a basis for present investigations (see summarizing illustration by Fig. 2). In all experiments performed so far, cyclin B1 strongly interacted with pUL97 in both plasmid-transfected (Fig. 2A) and HCMV-infected environments (Fig. 2B), thereby showing a strict dependence on pUL97 activity. In contrast to the strong cyclin B1 signals, T1 interaction comprised a relatively low signal level, which was independent of pUL97 activity. Finally, a very strong level of cyclin H interaction was detected, albeit exclusively when using proteins from HCMV-infected cells and not from ectopic expression.
Figure 1.

Schematic illustration of the modular structure and the so-far identified binding regions within viral CDK ortholog pUL97. The kinase domain is located between amino acids 337 and 706, as based on our biochemical validation (or 337 and 651, as based on sequence homologies). Lys-355 is an invariant lysine residue required for kinase activity. Expression of three pUL97 isoforms is determined by alternative translational initiation sites, i.e. isoforms M1, M74, and M157 (43). Two nuclear localization signals (NLS1 and NLS2) are contained in the N terminus, poorly structured portion of pUL97 (49, 50). Self-interaction/oligomerization of pUL97 is determined by amino acid region 231–280 (20). This region overlaps with a minimal binding region for cyclin T1 (14). Recent modeling approaches based on the in silico prediction of binding interfaces suggested extended binding interfaces for cyclins T1, B1, and H (6). Moreover, pUL97 is involved in the multiple regulatory steps during HCMV replication, as exerted through the phosphorylation of viral and cellular substrates (see horizontal bars for those binding regions within pUL97 that could be mapped so far), i.e. including the viral DNA polymerase cofactor pUL44 (19), viral RNA transport factor pUL69 (29), major tegument protein pp65 (51), nuclear egress core protein heterodimer pUL50–pUL53 (7, 27), cellular multiligand binding protein p32/gC1qR (5, 19), tumor suppressor protein Rb (9), nuclear lamins A/C (5, 28, 34, 35, 38), RNA polymerase II (52), translation factor EF-1δ (31, 32, 53), interferon-inducible protein IFI16 (54), and the therapeutically applied nucleoside analog ganciclovir (55, 56).
Table 1.
Comparison of the characteristics of interaction between three different cyclins and viral CDK ortholog pUL97
ND means not determined.
| Interaction characteristics | Cyclin B1 | Cyclin H | Cyclin T1 |
|---|---|---|---|
| Strong detectability of interaction by CoIP and MS/MS methods | + | + | ± |
| Interaction dependent on the activity of pUL97 | + | − | − |
| Interaction dependent on HCMV replication | − | + | − |
| Self-interaction/dimerization of pUL97 supported by cyclins | − | + | + |
| Intranuclear colocalization | + | + | + |
| Cytoplasmic cyclin relocalization during HCMV replication | + | − | − |
| Phosphorylated by pUL97 in vitro | + | − | ND |
Figure 2.

Interaction between pUL97 and cyclins B1, T1, and H depends on specific conditions provided by plasmid-transfected (A) or HCMV-infected cells (B). Note the strong interaction of cyclin B1 in both environments, which is strictly dependent on pUL97 activity, the relatively low signal of cyclin T1 interaction, which is independent of pUL97 activity, and the very strong cyclin H interaction, which is exclusively found within the environment of HCMV-infected cells. Note that Western blot splicing was performed to integrate the relevant lanes as indicated by vertical marking lines. This figure represents a refined illustration of previously published primary data (14). IP, immunoprecipitation;
To verify those features of cyclins B1, T1, and H in regard to their interaction with pUL97, their fine-localization in HCMV-infected primary human fibroblasts (HFF) was analyzed. On the one hand, we confirmed earlier findings that pUL97 localizes in intranuclear speckles representing prominent viral replication centers, which is demonstrated by the colocalization with appropriate marker proteins, e.g. the viral DNA polymerase processivity factor pUL44 (Fig. 3A) (19). On the other hand, a nuclear pUL97 colocalization could now be demonstrated for all three cyclins, i.e. B1 (Fig. 3B, panels 11–15), T1 (Fig. 3B, panels 1–5), and H (Fig. 3B, panels 6–10), as specifically reflected by the merge insets shown at right (Fig. 3B, panels 5, 10, and 15). In all cases, colocalization was most pronounced within viral replication centers as described previously for pUL97–cyclin B1 interaction (16). Viral replication centers typically grow from smaller early premature speckles to larger stains within the infected kidney-shaped nuclei. By characterizing the signals by an increase in size, pUL97–cyclin B1 colocalization was found dominant in early stages of viral replication centers (Fig. 3C, panels 6–10). Although for cyclin T1 and cyclin H, this pattern of speckled colocalization in viral replication centers was found in the majority of infected cells, such a speckled phenotype was less frequent for cyclin B1 (<10% of cells). In contrast, cyclins B2 and D1 did not show a similar pattern of pUL97 colocalization within viral replication centers, but basically maintained their even distribution in the cytoplasm and nucleus (Fig. 3B, panels 16–25), which was consistent with earlier data showing a lack of pUL97–cyclin B2 and D1 interaction (6).
Figure 3.

Intracellular colocalization between HCMV pUL97 and human cyclins determined by confocal imaging. HFF cells were infected with HCMV strain AD169 and harvested 4 days post-infection (dpi; A and B) or at several time points post-infection (C) and were subjected to indirect immunofluorescence analysis. A, triple-staining of cyclin T1, HCMV kinase pUL97, and the viral DNA polymerase processivity factor pUL44 using primary antibodies from mouse, rabbit, and goat species. B, costaining of several human types of cyclins with pUL97 showing a colocalization of pUL97 and cyclins T1, B1, and H in early viral subnuclear replication centers. C, localization of pUL97 and cyclin B1 during the time course of infection. Scale bars: 10 μm. DAPI, 4,6-diamidino-2-phenylindole; dpi, days post-infection.
Interaction between HCMV pUL97 and human cyclin B1
Previous data showed the dependence of pUL97–cyclin B1 binding on the catalytically active state of pUL97. Substantiating this point, we show here that the pUL97–cyclin B1 interaction is abrogated by dephosphorylation of both proteins (Fig. 4A). In reverse, the capability to interact was conferred to a catalytically inactive mutant of pUL97, containing a mutation on the essential amino acid in the potential ATP-binding site, K355M (Fig. 4B), through experimental transphosphorylation. λ-Phosphatase treatment was used to dephosphorylate proteins of total cell lysates containing endogenous cyclin B1 and transiently expressed pUL97 (Fig. 4A, left panel). Combinations of dephosphorylated and λ-phosphatase–untreated samples were coincubated as indicated and used for CoIP to analyze the requirement of phosphorylation for pUL97–cyclin B1 interaction (Fig. 4A, middle panel). Positive signals were obtained in those cases of λ-phosphatase treatment, which were restricted to one of the two samples (Fig. 4A, middle panel, lanes 1–3), whereas pUL97–cyclin B1 CoIP signals showed a drastic decrease when both proteins were dephosphorylated (lane 4). Note that a comparison of lanes 3 and 4 indicates that cyclin B1 phosphorylation might be more important than pUL97 phosphorylation for this interaction. A positive control reaction was performed with a phospho-specific antibody against PKCα to confirm the activity of λ-phosphatase (Fig. 4A, right panel). Next, we investigated whether a pUL97-specific transphosphorylation of the autophosphorylation-negative, catalytically inactive mutant K355M of pUL97 can confer the capability of cyclin B1 interaction (Fig. 4B). Thus, we cotransfected the inactive pUL97 mutant together with a larger-size version of active pUL97–GFP (WT–GFP). As expected, the mutant K355M showed only a marginally low signal of cyclin B1 interaction (Fig. 4B, lane 2), whereas WT pUL97 showed a massive CoIP signal (WT, lane 1). However, interaction of the mutant could be substantially increased by coexpression and thus transphosphorylation with the active WT–GFP version of pUL97 (Fig. 4B, lane 3). Because of differences in size and the antibody specificity, we could clearly distinguish between the K355M mutant (∼95 kDa) and the WT version tagged to GFP (∼125 kDa). This effect on the mutant K355M could be blocked by the addition of increasing concentrations of pUL97 inhibitor maribavir (MBV; Fig. 4B, lanes 4–6, 1st and 2nd upper panels). Similarly, the CoIP signal of WT pUL97–GFP was blocked by MBV (Fig. 4B, lanes 4–6, 2nd upper panel, 125-kDa band). In contrast, no interaction-conferring effect was induced by coexpression of an active version of PKCα (lane 7). It should be mentioned, however, that in this setting, the distinction between a requirement of solely phosphorylated pUL97 or solely phosphorylated cyclin B1 for interaction was not completely possible. Nevertheless, the combined data of dephosphorylation and transphosphorylation experiments emphasize the importance of a phosphorylated state of pUL97 and cyclin B1 for interaction.
Figure 4.

Interaction between pUL97 and cyclin B1 depends on the state of phosphorylation, as shown by dephosphorylation (A) and transphosphorylation (B) analyses. A, dephosphorylation of protein lysates from transfected 293T cells (3 days post-transfection) expressing pUL97 or vector control (mock) was performed using λ-phosphatase (λ-PP; 30 min, 30 °C). Immunoprecipitation (IP) of cyclin B1 was performed using combinations of input samples, either untreated or treated with λ-PP. A positive control reaction confirmed the λ-PP activity using a phospho-specific antibody against PKCα. B, transfection of 293T cells expressing the WT version of pUL97–FLAG (WT) or the inactive mutant K355M (KM), optionally cotransfected with a larger-size version pUL97–GFP (WT–GFP). PKCα-GFP was used as a reference kinase lacking pUL97 activity. The transphosphorylation samples were harvested at 3 days post-transfection and used for cyclin B1–specific CoIP. MBV, pUL97 inhibitor maribavir applied at the concentrations 0.2, 1, and 10 μm (lanes 4–6). Note that Western blotting splicing was performed to integrate the relevant lanes as indicated by vertical marking lines.
Next, we addressed the question whether a cyclin B1–binding negative mutant of pUL97 might either still retain its kinase activity as well as its ability to self-interact, or alternatively it would show an impairment of functionality. To this end, a series of pUL97 deletion and replacement mutants was generated and analyzed by CoIP and in vitro kinase assays (Table 2). As a central finding, pUL97 kinase activity proved to be consistently linked to pUL97–cyclin B1 interaction (Table 2, see middle and right columns under kinase-specific parameters). Thus, our data state that pUL97 kinase activity is mostly required for cyclin B1 interaction, with the only exception given by mutant S483A (Table 2, 8th line). This may be explained by the possibility that an active conformation of pUL97 may provide the structural prerequisite for cyclin B1 docking (16), which seems to be disrupted in most kinase-inactivating mutants, but it may be maintained in rare cases such as S483A. In contrast to the pUL97–cyclin B1 interaction, pUL97–pUL97 self-interaction remained preserved in all mutants negative for kinase activity, i.e. this property is obviously not activity-dependent (Table 2, left column). In this context, it seems worth mentioning that our earlier report described an opposite relationship for these two pUL97 properties of self-interaction and kinase activity, namely the finding that a pUL97 mutant defective in self-interaction was strongly impaired in kinase activity (20). Thus, it should be emphasized for the context of pUL97–cyclin B1 interaction that all attempts failed to identify a pUL97 mutant, which lost the ability to interact with cyclin B1 but still retained kinase activity. This confirmed our observation that pUL97 kinase activity is essential for the cyclin B1 interaction. Of note, the mutation of a putative cyclin-docking motif (RXL) in the C terminus (amino acids 702–704) had no influence on these parameters of self-interaction, kinase activity, or cyclin B1 binding, thus arguing against the functionality of this motif. In contrast, the deletion of the last five amino acids in mutant 1–702, including this putative binding motif, resulted in reduced self-interaction and cyclin B1 binding as well as a complete loss of kinase activity, suggesting a potential importance of the C terminus for proper pUL97 functionality (presumably based on a domain back-folding mechanism (16)). To define a putative cyclin B1–binding structural interface (independent from linear RXL cyclin-docking motifs) for pUL97, bioinformatic modeling was used based on the cyclin B1–CDK2 crystal structures (because no crystal structure of pUL97 is available until now). Three- and seven-fold amino acid replacements in this interface showed a strong impact on kinase activity and cyclin B1 interaction (Table 2, lowest four lines), stressing the interface's verification and relevance. A loss of proper folding was rendered improbable, because pUL97 self-interaction was still detectable. As CDK activity and cyclin binding strongly depend on the phosphorylation of a highly conserved threonine in the activation loop (T-loop), we mutated two serine residues, Ser-483 and Ser-485, in the putative pUL97 T-loop in the absence of any pUL97 T-loop threonine residues. This putative T-loop was predicted by bioinformatic sequence alignment (approximately amino acids 480–499) and was modeled in Steingruber et al. (16). The double mutant S483A/S485A was inactive by in vitro kinase analysis and negative for cyclin B1 interaction (whereas single mutants were not). Even though phosphomimetic mutants (S483D/S485D and S483E/S485E) could not rescue the pUL97 phenotype, these findings indicate a role of these serine residues for pUL97 activity. Interestingly, the single-site mutation S483A showed normal cyclin B1 interaction but lacked kinase activity, thus representing the first case in which pUL97 activity was not a prerequisite for cyclin B1 interaction.
Table 2.
Summarized characterization of the kinase-specific parameters of mutant versions of pUL97
++, positive reaction, very strong signal; +, positive reaction, clearly detectable signal; ±, weak signal; −, negative.
| Ectopic expression of fragments or replacement mutants | Kinase-specific parameters |
Phenotype of pUL97 | ||
|---|---|---|---|---|
| Self-interactiona | Kinase activityb | Cyclin B1 bindingc | ||
| 1–707 | ++ | ++ | + | pUL97 full-length, wildtype activity |
| 1–706 | ++ | + | + | C-terminal truncation, catalytically active |
| 1–702 | + | − | ± | C-terminal truncation, catalytically inactive |
| R702A/L704A | ++ | ++ | + | Mutation of putative cyclin-docking motif |
| K355M | ++ | − | − | Catalytically inactive mutant (ATP-binding site) |
| 1–707 + MBV | ++ | − | ± | pUL97 full-length, activity inhibited |
| S483A/S485A | ++ | − | − | Mutation of serine residues in putative T-loop |
| S483A | ++ | − | + | Mutation of serine residues in putative T-loop |
| S485A | ++ | + | + | Mutation of serine residues in putative T-loop |
| S483D/S485D | ++ | − | − | Mutation of serine residues in putative T-loop |
| S483E/S485E | + | − | − | Mutation of serine residues in putative T-loop |
| Q382A/H406A/R451A | ± | − | − | Mutation of putative cyclin-binding interface |
| Q382A/H406A/R645A | ± | − | − | Mutation of putative cyclin-binding interface |
| Q382A/H451A/R645A | ± | + | + | Mutation of putative cyclin-binding interface |
| Gln-382/His-406/His-448/Arg-451/Asp-490/Ser-643/Arg-645 | + | − | − | Mutation of putative cyclin-binding interface |
a Determined by CoIP analysis using two different tagged versions of pUL97.
b Determined by in vitro kinase assay measuring autophosphorylation and histone phosphorylation.
c Determined by CoIP analysis (interaction profiles of N-terminal truncations published in Steingruber et al. (16).
The question of cyclin phosphorylation by pUL97 was analyzed additionally by in vitro pUL97 kinase assays using recombinant cyclins. The labeled autoradiographs (Fig. 5, upper panels, [γ-33P]ATP) showed that cyclin B1 was phosphorylated by pUL97 as a substrate (Fig. 5A), whereas cyclin H was not (Fig. 5B). Note that for cyclin B1, a specific phospho-signal is seen in lanes 3 and 5, which is absent in control lanes 1, 2, and 4 (Fig. 5A), whereas for cyclin H, a signal similar to the specific lane 2 is seen in the control lane 4 (Fig. 5B), thus indicating the lack of pUL97-specific phosphorylation (obviously due to cyclin-associated contaminating kinase activity, which was reproducible in confirmatory settings). Additionally, the putative modulatory effect of cyclin B1 or H association on pUL97 activity was determined. In vitro kinase assay (IVKA) results indicated that cyclin binding is not a prerequisite for catalytic activity of pUL97 per se, because pUL97 showed activity in the absence of cyclin B1 (Fig. 5A, lane 4) and H (Fig. 5B, lane 1). Because of the lack of available recombinant cyclin T1, the question of T1 phosphorylation could not be addressed in parallel. Nevertheless, the finding further underlines differences in pUL97 interaction with cyclins. In addition, it should be mentioned that pUL97–cyclin H interaction is favored in HCMV-infected cells, possibly through a so-far unknown HCMV-specific cyclin modification. Interestingly, a recent mass spectrometry (MS)–based phosphosite analysis showed HCMV-specific phosphorylation of MAT1 (pThr-279), the CDK7–cyclin H assembly factor, a modification which might be involved in the HCMV-specific pUL97–cyclin H interaction (data not shown). Ongoing experimentation using phosphorylation-specific MS did not provide evidence that cyclin H was subject to hyperphosphorylation in HCMV-infected cells. So far, the only phosphosite detected was the prominent pThr-315 (data not shown). Referring to Figs. 2, 6, and 7A, further experiments will have to clarify whether additional, possibly uncharacterized phosphorylation sites may play a role in a postulated cyclin H modulation occurring in the course of HCMV replication.
Figure 5.

pUL97-specific IVKA determining the putative modulatory effect of cyclin B1 (A) or H (B) association on pUL97 activity. Transiently expressed pUL97–FLAG or pUL97(K355M)–FLAG (KM) were harvested 3 days post-transfection in a cyclin-avoiding fashion by applying high-stringency IP (500 mm NaCl buffer) to remove associated cyclins (for cyclin B1 monitored on WB). The two versions of pUL97 were subjected to IVKA reactions under standard conditions. Each reaction was supplemented by the addition of either human cyclin B1 (A, 2 μg), human cyclin H (B, 5 μg), or human histones (20 μg) as indicated. Upper panels, IVKA (autoradiogram) and detection of His–cyclin B1/His–cyclin H on the IVKA membrane (WB restaining); middle panel, detection of comparable pUL97 levels (precipitation control); lower panels, total input levels contained in cell lysates (expression control). Red fluorescent protein (RFP; A, lane 1; B, lane 5) was used as negative control. Note that Western blotting splicing was performed to integrate the relevant lanes as indicated by vertical marking lines.
Figure 6.

Cyclin H expression levels and the state of phosphorylation (pThr-315) in HCMV-infected cells. A, validation of the specificity of the phospho-cyclin H antibody by λ-phosphatase assay performed with HeLa cell lysates. B and C, HFF cells were infected with HCMV AD169 and harvested at different time points during infection. Protein levels of immediate early (IE1), early (pUL44), and late viral proteins (MCP) as well as cyclin H were detected by WB analysis using the antibodies as indicated.
Figure 7.

Protein assembly–based CoIP approach demonstrating the formation of a ternary complex pUL97–cyclin H–CDK7. A, scheme of possible binary and ternary complexes. HFFs were infected (blue) with HCMV expressing either pUL97–HA or pUL97–FLAG. 293T cells were transiently transfected (green) with either CDK7–HA or CDK7–FLAG (B). Cells were harvested at 48 h post-transfection or when the cytopathic effect of HCMV infection was observable, and combinations of cell lysates were coincubated for protein assembly as indicated and subsequently subjected to CoIP/Western blot analysis (C, ternary complexes framed). Note that the detectability of the ternary complex, in addition to the binary complexes (D), supports the conclusion that interactions of cyclin H with pUL97 and CDK may not occur in a competitive way. Note that Western blotting splicing was performed to integrate the relevant lanes as indicated by vertical marking lines.
Interaction between HCMV pUL97 and human cyclin H
First, we analyzed the expression levels of cyclin H via Western blotting by the use of antibodies against cyclin H and phospho-cyclin H (pThr-315), showing that both signals were up-regulated during HCMV replication (Fig. 6). This up-regulation was induced at ∼48 h post-infection and occurred in a fashion that was similarly detected for total cyclin H and cyclin pThr-315, indicating that Thr-315 phosphorylation of cyclin H might not be HCMV-specific. Next, we analyzed whether higher-order complexes may be assembled, including cyclin H, CDK7, and pUL97. To this end, we established an assembly-based CoIP approach, which was able to differentiate between binary and higher-order cyclin complexes (Fig. 7A). FLAG- and HA-tagged versions of both kinases were expressed in HCMV-infected (blue) or transiently plasmid-transfected (green) cells (Fig. 7B; note that although CDK7–HA could not be detected in the input material due to limited expression levels, this quantity proved to be sufficient for detection upon CoIP, see below). For the CoIP analysis, all four combinations of pUL97–HA/pUL97–FLAG and CDK7–HA/CDK7–FLAG samples were coincubated and used for protein assembly–based CoIP (Fig. 7C). The obtained signal pattern demonstrated that both binary complexes, pUL97–cyclin H and CDK7–cyclin H, as well as the ternary complex, pUL97–cycH–CDK7, were formed under these conditions (Fig. 7D). Although ternary complexes were only represented by weak signals (compare pUL97–, CDK–, and cyclin H–specific bands) in those samples in which pUL97 was immunoprecipitated (Fig. 7C, lanes 1 and 4), signal intensities were higher when CDK7 was immunoprecipitated (lanes 2 and 3). The result confirmed that ternary or even higher-order complexes can principally be formed, thus arguing against a competitive mode of binding between cyclin H and the two kinases. However, the limitation of detectability of the ternary complex as seen in weak signal intensities may point to a more multifaceted binding situation. Therefore, we cannot completely exclude competitive effects or a dynamic mode of competitive binding, which still might occur along the course of viral replication, possibly undetectable under these experimental conditions. Nevertheless, this previously established assembly-based CoIP approach proved to be a reliable and sensitive model system for the assessment of various protein–protein interactions.
Seen from the perspective of structural modeling, we suggest a scheme in which cyclin H bridges CDK7 and pUL97 (Fig. 8). The molecular basis of the different modes of cyclin–kinase interaction may be provided by a combination of globular domain interfaces (Fig. 8, A and B) and linear docking motifs present in the unstructured N terminus of pUL97 (Fig. 8C). This model also considers the postulated HCMV-specific modulation of cyclin H (Fig. 8C) and is compatible with the idea of higher-order complexes that may contain additional copies of some components, such as an additional cyclin H molecule bound to pUL97 via its globular domain interface (combination of options Fig. 8, C and B).
Figure 8.
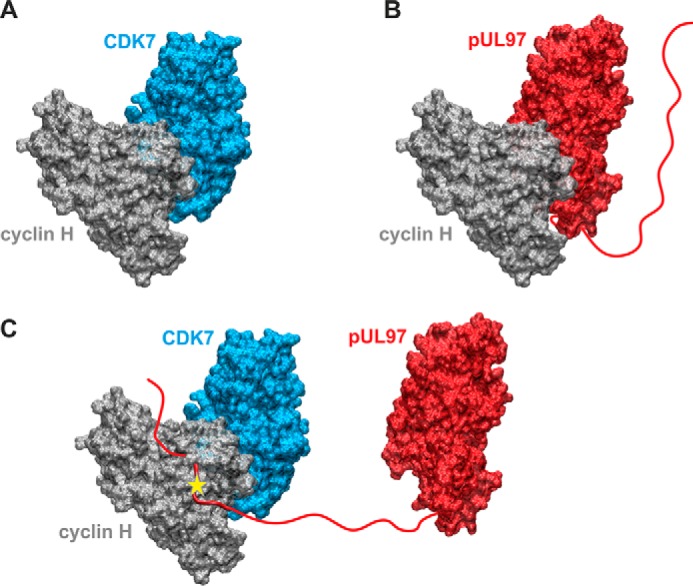
Structural model of the putative cyclin H-bridged interaction between pUL97 and CDK7 as presented in Fig. 7. A, model of the binary CDK7–cyclin H complex. B, model of the binary pUL97–cyclin H complex. Both models were generated based on the homologous CDK9–cyclin T1 complex crystal structure using the strategy described before (14) and show the canonical interaction via the large globular domain interfaces. The globular domain interfaces shown in A and B are overlapping, and thus only one partner can interact with cyclin H via this interface. C, model of a putative ternary CDK7–cyclin H-pUL97 complex, in which the long unstructured N-terminal region of pUL97 is supposed to interact with cyclin H via an alternative binding motif or interface. A yellow star denotes a yet undefined modification of cyclin H induced by the HCMV-specific infectious environment that is required for pUL97 binding. Note that this figure shows the minimal composition of such a ternary complex containing one copy of each component. However, it is possible that higher-order complexes may contain additional copies of some components (e.g. two cyclin H molecules bound to pUL97, one via its N-terminal binding motif, a second via its globular domain interface, see B).
The CDK7–cyclin-H–MAT1 complex has been characterized as the CDK-activating kinase (CAK), primarily responsible for a site-specific activating phosphorylation of CDKs, irrespective of whether they belong to the group of cell cycle–associated, transcription-associated, or unconventional CDKs (18, 21). Mutual transphosphorylation between pUL97 and CDK7 was investigated under the conditions of IVKAs (Fig. 9). Two different settings were investigated in parallel: pUL97 was either immunoprecipitated from transiently transfected cells (Fig. 9A) or coimmunoprecipitated with cyclin H and CDK7 from HCMV-infected cells (Fig. 9B). Immunoprecipitates were combined with a commercially available CDK7–cyclin-H–MAT1 complex produced by recombinant expression, as multicomponent setting for IVKA reactions. Notably, none of these conditions and sources of samples showed a CDK7-specific pUL97 phosphorylation signal (also addition of CDK7 inhibitor LDC4297 had no effect on pUL97-specific signals; Fig. 9A, lane 4, compare lane 3 and 6B, lane 1 compare with lane 5). As a control, a C-terminal fragment of the retinoblastoma protein (Rb-CTF) was strongly phosphorylated by pUL97 as well as CDK7–cyclin-H–MAT1 (Fig. 9, A, lanes 1 and 3–6; 9B, lanes 1, 5 and 7). CDK7-specific phosphorylation took place in a fashion independent of pUL97 activity (Fig. 9A, lane 5, MBV; lane 6, catalytically inactive mutant), whereas the phosphorylation of all substrates of the CDK7–cyclin-H–MAT1 complex (including His–CDK7 autophosphorylation and His–cyclin-H–MAT1 phosphorylation) seemed to be increased in samples derived from HCMV-infected cells (Fig. 9B). Combined, these in vitro kinase assays did not provide an indication for mutual transphosphorylation between pUL97 and CDK7.
Figure 9.

Analyses of CDK7 and pUL97 activities applying in vitro kinase assays. FLAG-tagged pUL97 and its inactive mutant pUL97(K355M)–FLAG (KM) were transiently transfected in 293T cells and harvested 48 h post-transfection (A); HFFs were infected with HCMV or remained uninfected (mock) and harvested 120 h post-infection (B). Total cell lysates were subjected to CoIP and IVKA. Two different NaCl concentrations were used in the CoIP buffer (A, 500 mm NaCl; B, 150 mm NaCl) to achieve optimized stringency of CoIP. Each IVKA reaction was supplemented by addition of either CDK7–cyclin-H–MAT1 complex (200 ng), Rb-CTF (1 μg), CDK7 inhibitor LDC4297 (1 μm), or pUL97 inhibitor MBV (3 μm) as indicated. Upper panel, IVKA (autoradiogram) and detection of CDK7, cyclin H, and MAT1 on the IVKA membrane (Western blot staining); lower panels, total input levels of different proteins as expression control. Note that Western blot splicing was performed to integrate the relevant lanes as indicated by vertical marking lines.
To further analyze the phosphorylation status of pUL97 during HCMV replication (including the events of autophosphorylation and possibly transphosphorylation by other, so far undefined kinases), we performed a MS-based phosphosite analysis. Immunoprecipitated pUL97 from HCMV-infected cells was subjected to phosphopeptide enrichment using titanium dioxide (TiO2) for MS analyses. Importantly, pUL97 was not found to be phosphorylated in its T-loop residues Ser-483 and Ser-485 (Table 3, analysis I; Table S1). This is contrary to cellular CDKs, which are subject to activating phosphorylation on a conserved T-loop threonine (22–24). Interestingly, Bigley et al. (25) could detect slight phosphorylation signals in the T-loop of pUL97 at amino acids Tyr-482 and Ser-483 after phosphopeptide enrichment by TiO2 and MS analysis from pUL97–HA and pUS27–HA cotransfected U2OS cells. However, a number of other phosphosites could be identified, which confirmed published phosphosites found in several analyses performed by different research groups (see Table 3). Additionally, some of the phosphosites identified here have not been described before such as Ser-121, Ser-139, Ser-142, Ser-386, and Thr-504 (Table 3, shown in bold). All of these phosphosites were detected in at least two parallel runs of our analyses I and II, with the exception of Ser-139 and Thr-504, which were only found in analysis I/run II. To be able to distinguish between autophosphorylation and phosphorylation by other kinases, we used virion-associated pUL97 derived from parental HCMV AD169/HB15 or the defective HCMV mutant UL97ΔK355 (Table 3, analysis II). Remarkably, Ser-121 was the only site found phosphorylated in samples derived from the UL97ΔK355 mutant virus, suggesting that Ser-121 might be independent from autophosphorylation. It should be mentioned that phosphorylated Ser-121 was found largely enriched in the HB15 WT sample compared with UL97ΔK355, indicating an additional involvement of pUL97 in Ser-121 phosphorylation. Altogether, pUL97 carries a variety of phosphosites referring to both autophosphorylation or transphosphorylation by cellular kinases. So far, we have no direct evidence that CDKs, especially CAK activity provided by CDK7–cyclin-H–MAT1, are involved in pUL97 phosphorylation.
Table 3.
Phosphosites of HCMV kinase pUL97
| Phosphositea | Analysis Ib | Analysis IIc | Previously publishedd |
|---|---|---|---|
| Ser-2 | + | ||
| Ser-3 | + | ||
| Ser-11 | + | ||
| Ser-13 | + | ± | + |
| Thr-16 | + | ||
| Thr-18 | + | ||
| Ser-121 | + | + | |
| Ser-133 | ± | + | |
| Thr-134 | + | ||
| Ser-135 | + | + | |
| Ser-136 | ± | + | |
| (Ser-139) | + | ||
| Ser-142 | + | + | |
| Thr-177 | + | + | |
| Ser-180 | + | + | + |
| Ser-183 | + | + | |
| Ser-185 | ± | + | + |
| Ser-187 | ± | + | |
| Thr-190 | + | ||
| Ser-232 | + | + | |
| Ser-235 | + | ||
| Ser-239 | + | + | |
| Tyr-326 | + | ||
| Ser-386 | + | ||
| Thr-482 | + | ||
| Ser-483 | + | ||
| (Thr-504) | ± | ||
| Thr-580 |
a Boldface shows newly detected sites (sites detected only once are shown in parentheses). Detection levels were defined as follows; ±, <75% probability; +, ≥75% probability.
b Samples for analysis I were prepared from HFFs infected with HCMV AD169 for 5 days; pUL97 was immunoprecipitated from total lysates by the use of a pool of two specific polyclonal and monoclonal antibodies.
c Samples for analysis II were prepared from gradient-purified HCMV parental AD169/HB15 or mutant UL97ΔK355 virions, respectively; input material was adjusted by SDS-PAGE separation of the samples followed by Coomassie/silver staining.
Interaction between HCMV pUL97 and human cyclin T1
Previous mapping analysis provided a perfect overlap of the minimal region required for pUL97–cyclin T1 interaction and the region identified for pUL97–pUL97 self-interaction (amino acids 231–280; see Fig. 1) (14). This coincidence led us to the assumption that cyclin T1 might be involved in pUL97 self-interaction, i.e. the ability to form dimers and oligomers, which was found to be important for full kinase activity (20). To address this question, an assembly-based CoIP was performed (see flowchart of individual steps in Fig. 10), and the results indeed provided first experimental evidence that pUL97–pUL97 self-interaction can be bridged in vitro by transcriptional cyclins T1 or H but not by the classical cell cycle–regulating B1 (Fig. 11). To achieve technically an efficient depletion of cyclins from total cell lysates, we performed repeated steps of cyclin-specific immunoprecipitation, as monitored by control staining on Western blotting (Fig. 11, B and D, see labeling by dashed frames). The differentially tagged versions of pUL97 (FLAG or HA tags) were derived from lysates of HFFs infected with recombinant HCMVs. IP samples (Fig. 11C), optionally taken after cyclin depletion, were subsequently assembled with second lysates containing alternatively tagged pUL97 (Fig. 11D). The signal strength of CoIP bands representing self-interaction was quantitated by AIDA, and values were normalized toward the quantity of first step IP of pUL97 (Fig. 11E, orange labeling). As a central finding, self-interaction was reduced to 19% when cyclin T1 had been depleted in both assembly samples compared with control samples without depletion (Fig. 11E, lane 3). A quantitation of the data derived from five independent cyclin T1 depletion experiments was performed (Table 4). This comparison confirmed that cyclin T1 depletion in one or both assembly samples reduced self-interaction markedly, albeit to varying degrees, with the strongest effect seen in samples with cyclin T1 depletion in both lysates, resulting in pUL97–pUL97 signals of 53 ± 21% (mean value 44 ± 22% derived from the first three experiments using identical settings). This result clearly confirmed the functional importance of cyclin T1 for pUL97 self-interaction.
Figure 10.
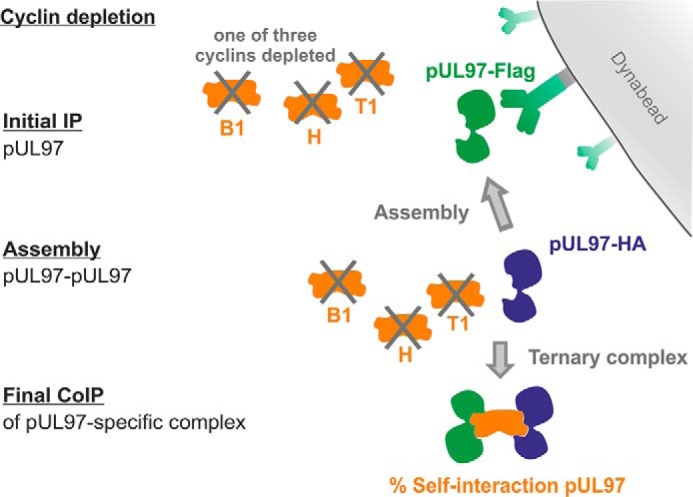
Flow chart of the individual steps of assembly-based CoIP as presented in Fig. 11. Two flagged versions of pUL97 were expressed separately using different recombinant HCMVs for the infection of primary fibroblasts (HFFs). Total lysates of both infected cell samples were subjected to an immunoprecipitation-mediated depletion of optionally one of the three relevant cyclins. Initial pUL97-specific IP was performed using the first tag antibody coupled to Dynabeads. Thereafter, another pUL97-containing lysate (likewise optionally cyclin-depleted) was added and incubated on the IP samples to assemble dimeric/oligomeric pUL97–pUL97 complexes. Finally, CoIP of the assembled complexes was performed by Dynabeads-mediated separation from the residual lysates, before the percentage of pUL97 self-interaction was semi-quantitatively determined by applying an antibody against the second tag.
Figure 11.

Cyclins T1 and H are able to bridge pUL97–pUL97 self-interaction. A, scheme for putative cyclin-bridged ternary self-interaction. For samples in the left and middle panels (cyclin T1 and H), HFFs were infected with recombinant HCMV AD169 encoding pUL97–HA (lanes labeled with H) or pUL97–FLAG (lanes labeled with F; pUL97–FLAG was expressed in the variant version Mx4, representing the functionally intact large pUL97 isoform (43)). Four and 7 days post-infection, one of the cyclin types was specifically removed by a three-step depletion (B) so that the indicated combinations of samples (FH) could be combined and subjected to assembly-based CoIP/Western blot analysis (C–E; see flow chart of this procedure given in Fig. 10). For samples in the right panels (cyclin B1), 293T cells were transfected with HA- and FLAG-tagged pUL97, respectively, followed by cyclin B1 depletion and assembly-based CoIP performed 2 days post-transfection. For a further illustration of the efficiency of cyclin depletion, residual samples of cyclin T1 (on CoIP WB, left panel) or cyclin H (in CoIP supernatants, middle panel) were additionally immunostained using the indicated antibodies (F). Note that the effect of cyclin bridging on pUL97–pUL97 self-interaction was calculated from the signal reduction of pUL97 CoIP obtained upon cyclin depletion (E, orange numbers, values were derived from this experiment by duplicate densitometric determinations of pUL97 normalized for the amount of initially immunoprecipitated pUL97 in C and are expressed as percentage relative to the reference value without cyclin depletion; also to be compared with the entire data set derived from five independent experiments, Table 4). *, cross-reactive irrelevant band.
Table 4.
Quantities of pUL97–pUL97 self-interaction determined by Western blot densitometry using proteins from five different experiments
The signal intensities of Western blot bands were quantitated with AIDA software, and values were normalized against the quantity of pUL97 immunoprecipitated under control conditions (T1|T1, no cyclin T1 depletion); ND means not determined; tagged versions of pUL97 derived from HCMV-infected are underlined, and plasmid-transfected cells are in italics.
| Tagged versions of pUL97 for assembly | T1|T1 | T1|− | −|− | −| T1 |
|---|---|---|---|---|
| Derived from HCMV-infected cells | ||||
| pUL97–FLAG|pUL97–HA | 100% | 30% | 19% | 23% |
| pUL97–FLAG|pUL97–HA | 100% | 93% | 57% | 57% |
| pUL97–FLAG|pUL97–HA | 100% | 98% | 56% | 36% |
| Mean | 100 ± 0% | 74 ± 38% | 44 ± 22% | 39 ± 17% |
| Derived from plasmid-transfected cells | ||||
| pUL97–HA|pUL97–FLAG | 100% | 88% | 59% | 55% |
| Derived from infected and transfected cells | ||||
| pUL97–HA |pUL97–FLAG | 100% | 63% | 76% | ND |
For cyclins H and B1, a similar role in pUL97 self-interaction was investigated in parallel. A specific effect on pUL97 self-interaction could also be observed for cyclin H depletion (at least 3-fold; Fig. 11, middle panels), whereas cyclin B1 depletion did not markedly influence the self-interaction (Fig. 11, right panels). These findings point to functional difference between the transcriptional cyclins T1 and H compared with the classical cell cycle–regulating cyclin B1.
Discussion
To our knowledge, this study represents the first molecular analysis comparing the interaction between a herpesviral CDK-like protein kinase and three different types of cyclins. Our data provide novel insights into the individual modes of interaction, which differ between cyclins B1, H, and T1. Central conclusions derived from these data are as follows. (i) pUL97 interacts with cyclins B1 and H in a manner dependent on pUL97 kinase activity and phosphorylation or HCMV-specific cyclin modulation. (ii) pUL97-mediated in vitro phosphorylation of cyclins is measurable for type B1 but not H, and no evidence is provided for mutual transphosphorylation between pUL97 and CDK7. (iii) A cyclin T1/H–mediated bridging mechanism of pUL97 self-interaction is supported by the data of this study.
Possibly, cyclin B1, which requires active pUL97 for interaction, regularly undergoes pUL97-specific phosphorylation (similar to CDK-specific phosphorylation), whereas cyclins H and T1, both showing independence of pUL97 activity for interaction, may not serve as pUL97 substrates but may exert phosphorylation-independent regulatory effects. As a main outcome of our study, we present a cyclin bridging concept of the viral CDK ortholog pUL97. The concept anticipates that cyclin binding is not a prerequisite for the catalytic activity of pUL97 per se, which stands in contrast to the typical CDK–cyclin binding that confers activity to the complex. Rather, the binding of one of the three cyclins has an impact on pUL97 substrate recognition, self-interaction, autophosphorylation, and/or the recruitment of CDKs into ternary or higher-order pUL97–cyclin–CDK complexes. This concept points out differences as well as common features of the pUL97-associated cyclins. In addition to the cyclin-bridging mechanism, cyclin-independent situations of pUL97 substrate binding are also conceivable. A recently published example is the pUL97 binding and phosphorylation of the viral core nuclear egress complex, pUL50–pUL53, which is bridged through the multiligand-binding protein p32/gC1qR (5, 7, 8, 26–28) but not through cyclins according to available data.
pUL97 shares a number of substrates typically phosphorylated by CDKs, such as Rb, nuclear lamins, RNAP II, EF-1δ, cyclin B1, as well as viral pUL69 and pUL50 (9, 29–35). This intimate link between pUL97, CDKs, and substrate proteins may arise from the formation of ternary complexes, an option that is substantiated by the findings in this report, and may result in the dual phosphorylation of a single substrate by the two types of protein kinases. In some examples, the phosphorylation at identical sites by CDKs and pUL97 has been proven, such as Ser-22 phosphorylation of nuclear lamin A/C (35–37) as well as the complex pattern of Rb phosphorylation (10, 11, 38).
In conclusion, these findings indicate that the multifunctional nature of the viral kinase pUL97 is finely regulated by alternative events of human cyclin binding. It is conceivable that within one HCMV-infected cell various combinations of pUL97–cyclin complexes may exist simultaneously or in a timely coordinated fashion so that individual activities of pUL97, including the selective substrate binding along the course of viral replication, may be regulated by undergoing alternative pUL97–cyclin complexes. Future studies will have to define the functional modulation arising from this sophisticated mode of pUL97–cyclin interaction.
Experimental procedures
Antibodies
Antibodies used for immunoprecipitation are as follows: pAb–cyclin B1 (sc-752, Santa Cruz Biotechnology); pAb–cyclin T1 (sc-10750, Santa Cruz Biotechnology); pAb–cyclin H (sc-609, Santa Cruz Biotechnology; LS-C331195, LSBio); mAb–CDK7 (sc-56284, Santa Cruz Biotechnology); pAb–UL97 (kindly provided by D. M. Coen, Harvard Medical School, Boston, MA); mAb–FLAG (F1804, Sigma); pAb–FLAG (F7425, Sigma); mAb–HA (clone 7, H9658, Sigma); pAb–Fc (rabbit Fc fragment, 011-000-008, Dianova); mAb–Fc (mouse Fc fragment, 015-000-008, Dianova).
Antibodies used for WB detection are as follows: mAb–UL97 (kindly provided by T. Lenac and S. Jonick; Department of Histology and Embryology, University of Rijeka, Croatia); mAb–cyclin B1 (sc-7393, Santa Cruz Biotechnology); GNS11 (ThermoFisher Scientific); pAb–cyclin B1 (sc-752, Santa Cruz Biotechnology; ABIN 3174614, antibodies online), mAb–cyclin T1 (sc-271348, Santa Cruz Biotechnology); pAb–cyclin T1 (sc-10750, Santa Cruz Biotechnology); pAb–cyclin H (sc-609, Santa Cruz Biotechnology; LS-C331195, LSBio); mAb–β-actin (AC-15, Sigma); pAb–phospho-PKC (γThr-514, 9379, Cell Signaling); mAb–FLAG (F1804, Sigma); mAb–GFP (11814460001, Roche Applied Science); mAb–His6 (MA1-21315, ThermoFisher Scientific); mAb–IE1p72 (63-27, kindly provided by W. Britt, University of Alabama, Birmingham, AL); mAb–UL44 (BS510, kindly provided by B. Plachter, University of Mainz, Germany); mAb–major capsid protein and mAb–pp65 (kindly provided by T. Stamminger, University of Ulm, Germany); pAb–phospho-cyclin H (pThr-315, 11689, Signalway); mAb–HA (clone 7, H9658, Sigma); mAb–CDK7 (sc-56284, Santa Cruz Biotechnology).
Antibodies used for immunofluorescence analysis are as follows: pAb–cyclin T1 (sc-10750, Santa Cruz Biotechnology); pAb–UL97 (kindly provided by D. M. Coen, Harvard Medical School, Boston); mAb–pUL97 (produced and kindly provided by T. Lenac Rovis/S. Jonjic, Dept. Histology and Embryology, Univ. Rijeka, Croatia); mAb–UL44 (BS 510, inventory of the Plachter laboratory); pAb–cyclin H (sc-609, Santa Cruz Biotechnology); pAb–cyclin B1 (sc-752, Santa Cruz Biotechnology); pAb–cyclin B2 (sc-22776, Santa Cruz Biotechnology); and pAb–cyclin D1 (sc-753, Santa Cruz Biotechnology).
Plasmids and transfection
293T cells were transfected via polyethyleneimine reagent as described (16) using the following plasmids: pcDNA–UL97–FLAG, pcDNA–UL97–HA, and pcDNA–UL97(K355M)–FLAG (39); pcDNA–UL97(1–706)–FLAG and pcDNA–UL97(1-702)–FLAG (16); pcDNA–PKCα–FLAG (33); pEGFP–N1–UL97 (ORF-UL97 cloned into vector pEGFP–N1, Clontech, by using restriction sites EcoRI and SalI); and SRα–CDK7–HA (40).
Expression constructs coding for pUL97 carrying amino acid replacements, pcDNA–UL97(R702A/L704A)–FLAG, pcDNA–UL97(S483A/S485A)–FLAG, pcDNA–UL97(S483D/S485D)–FLAG, pcDNA–UL97(S483E/S485E)–FLAG, pcDNA–UL97(S483A)–FLAG, pcDNA–UL97(S485A)–FLAG, pcDNA–UL97(Q382A/H406A/R451A)–FLAG, pcDNA–UL97(Q382A/H406A/R645A)–FLAG, pcDNA–UL97(Q382A/H451A/R645A)–FLAG, and pcDNA–UL97(Q382A/H406A/H448A/R451A/D490A/S643A/R645A)–FLAG were generated using the GeneArt site-directed mutagenesis system (ThermoFisher Scientific) according to the manufacturer's protocol. Site-directed mutagenesis PCR was performed with pcDNA–UL97–FLAG as a template and oligonucleotide primers with nucleotides differing (bold letters) from the WT sequence (purchased from Biomers.net). UL97–R702A/L704A, forward, 5-CTTGACGGTGACTGCGCCCAAGCGTTCCCCGAGGACTAC-3, and UL97–R702A/L704A, reverse 5-GTAGTCCTCGGGGAACGCTTGGGCGCAGTCACCGTCAAG-3; UL97–S483A/S485A forward, 5-GCGCTGTGCGATTACGCCTCGCCGAGCCCTATCCGGATTAC-3, and UL97–S483A/S485A reverse, 5-GTAATCCGGATAGGGCTCGGCGAGGGCGTAATCGCACAGCGC-3.
A C-terminally tagged version of CDK7 (pcDNA–CDK7–FLAG) was cloned by PCR of SRα–CDK7–HA with primers obtained by Biomers.net using Vent DNA polymerase (New England Biolabs) under standard conditions (20). CDK7–FLAG forward, 5-TACGGATCCATGAATTCATGGCTCTGGACGTGAAGTCTCGG-3, and CDK7–FLAG reverse, 5-GGATTGCCCAAGAAACTAATTTTTGACTACAAAGACGATGACGACAAGTAACTCGAGGTA-3 (FLAG sequence underlined). The PCR product was subsequently inserted into pcDNA3.1+ (Invitrogen) via EcoRI and XhoI. pDsRed1-N1 (Clontech) expressing red fluorescent protein was used as a control.
Cell culture and HCMV infection
Primary HFF (derived from clinical samples, Children Hospital, Erlangen, Germany) and human embryonic kidney epithelial cells (HEK293T and CRL-3216, ATCC) were maintained at 37 °C, 5% CO2 and 80% humidity using minimal essential medium (21090022, ThermoFisher Scientific) and Dulbecco's modified Eagle's medium (11960044, ThermoFisher Scientific), respectively. Cell culture media were supplemented with 1× GlutaMAXTM (35050038, ThermoFisher Scientific), 10 μg/ml gentamicin, and 10% fetal bovine serum (F7524, Sigma). For infection, HFFs were inoculated with stocks of HCMV strain AD169 (41) or AD169-derived recombinant viruses (AD169–GFP (42); AD169 UL97ΔK355, AD169 UL97–HA, or AD169 UL97(Mx4)–FLAG (43) using a low volume (5 ml per T750 or 1 ml per 6-well). After a 90-min incubation at 37 °C, the supernatant was removed, and fresh medium was added to the cells. Cells were harvested at the indicated time points for immunofluorescence staining, MS analyses, or immunoprecipitation methods. Virions of HCMV AD169 HB15 and UL97ΔK355 were prepared as published earlier (44).
Indirect immunofluorescence assay and confocal laser-scanning microscopy
HFFs were grown on coverslips and used for infection with HCMV strain AD169. At indicated time points post-infection, cells were fixed with 4% paraformaldehyde solution (10 min, room temperature) and permeabilized by incubation with 0.2% Triton X-100 solution (15 min, 4 °C). Nonspecific staining was blocked by incubation with 2 mg/ml human γ-globulin (Cohn fraction II, Sigma; 30 min, 37 °C). Indirect immunofluorescence staining was performed by stepwise incubation with primary antibodies as indicated for 60–90 min each at 37 °C, followed by incubation with dye-conjugated secondary antibodies (Alexa 488, Alexa 555, and Alexa 647 from goat or donkey species) for 30–60 min at 37 °C. Cell samples were mounted with Vectashield Mounting Medium containing 4,6-diamidino-2-phenylindole and analyzed using a DMI6000 B microscope and a ×63 HCX PL APO CS oil immersion objective lens (Leica Microsystems). Confocal laser-scanning microscopy was performed with a TCS SP5 microscope (Leica Microsystems). Images were processed using the LAS AF software (version 2.6.0 build 7266; Leica Microsystems).
CoIP and IVKA
CoIP was performed as described (16) using lysates from plasmid-transfected 293T cells or HCMV-infected HFFs. Antibody-coupled Dynabeads (25 μg/ml, 10002D, ThermoFisher Scientific) were used to obtain specific immunoprecipitates, and CoIP samples were further analyzed by Western blotting, MS, or IVKA (see detailed description in Refs. 6, 16 for pUL97 kinase conditions). Optionally, recombinantly produced human cyclin B1 (2 μg; ab128445, Abcam) and cyclin H (5 μg, ab95351, Abcam) were added to the IVKA reactions. For CDK7 kinase conditions, immunoprecipitate samples were resuspended in 40 μl of IVKA buffer (30), and optionally 0.5 μl of a purified histone mix H1–4 (Roche Applied Science), retinoblastoma C-terminal fragment (Rb-CTF; 1000 ng, ProQinase), MBV (3 μm, ChemScene), LDC4297 (1 μm (45)), or CDK7–cyclin-H–MAT1 (ProQinase) was added. The kinase reaction was then performed at 30 °C (900 rpm) and was stopped after 30 min by adding 15 μl of 4× boiling mix followed by a denaturation for 10 min at 95 °C. The γ-33P-labeled proteins were separated with SDS-PAGE and immunoblotted to nitrocellulose membranes, which were analyzed by autoradiography using the phosphorimager (Dürr Medical CR35BIO).
Cyclin depletion and assembly-based CoIP
Cyclin depletion from total cell lysates was performed by three steps of cyclin-specific immunoprecipitation using 200 μl of Sepharose Protein A beads (25 mg/ml, Sigma). Each depletion step was carried out under rotation for 1.5 h at 4 °C and repeated with fresh depletion beads under identical conditions. Subsequently, CoIP was performed using Dynabeads Protein A (25 μl, Invitrogen) for further analysis of protein–protein interaction. Signal intensities of Western blotting bands of interacting proteins were quantitated using Aida Image Analyzer version 4.22 and were normalized to the levels of control pUL97 immunoprecipitation. To analyze the bridging function of cyclins, assembly-based CoIP experiments were performed using the respective proteins carrying HA and FLAG tags expressed separately. One of the two proteins was immunoprecipitated with the tag-specific antibody in an initial IP for 1–2 h. After washing, the sample containing the second, alternatively tagged protein was added for assembly, and a final CoIP was performed for 1.5–2 h or overnight. Finally, beads were washed, denaturated in 2× protein loading buffer (62.5 mm Tris/HCl, pH 6.8, 1 mm EDTA, 10% glycerol, 2% SDS, 5% β-mercaptoethanol, 0.005% bromphenol blue) before the samples were analyzed by 12.5% SDS-PAGE/Western blotting.
Phosphatase assay
To analyze the impact of the phosphorylation state of pUL97 and cyclin B1 on their interaction properties, a phosphatase assay was performed using λ protein phosphatase (λ-PP, Mn2+-dependent activity toward phosphorylated serine, threonine, and tyrosine residues; New England Biolabs, P0753S). For phosphatase reaction, total cell samples were lysed in CoIP buffer without EDTA (50 mm Tris/HCl, pH 8.0, 150 mm NaCl, 0.5% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml, aprotinin, 2 μg/ml, leupeptin, and 2 μg/ml pepstatin) containing 200 units of λ-PP under specified buffer conditions (1× NEBuffer metallophosphatases, 1 mm MnCl2, New England Biolabs), and samples were cleared from cell debris by centrifugation (14000 rpm, 4 °C, 10 min). For analyzing protein interaction properties, λ-PP–treated and –untreated samples were subject to assembly reactions at different combinations and rotated at 4 °C for 1 h. Antibody-coupled protein A beads were added and used for CoIP at 4 °C for 1.5–2 h. Optionally, a second dephosphorylation step was performed after CoIP (Fig. 4A, control IP λ-PP). Each dephosphorylation reaction was stopped by washing with EDTA containing CoIP buffer and subsequent boiling in 2× loading buffer, and all samples were analyzed by SDS-PAGE/Western blotting.
Molecular modeling
Originally, the homology model of pUL97 was prepared using human CDK2 (PDB code 2JGZ) as a template (16). In this study, molecular models of the binary complexes CDK7–cyclin H and pUL97–cyclin H were generated based on the homologous CDK9–cyclin T1 complex crystal structure using the strategy described before (6).
Phosphosite identification by nanoLC-MS/MS analysis
Proteins were prepared and in-gel digested as described previously (5). Phosphopeptides were enriched using TiO2 beads and analyzed by nanoliquid chromatography coupled to tandem MS according to the protocol described previously (7). Peptides and proteins were identified using Mascot (version 2.6) and filtered using Proline software.
Author contributions
M. S., F. H., H. S., and M. M. conceptualization; M. S., L. K., E. S., S. F., A.-M. H., N. B., H. S., and M. M. data curation; M. S., L. K., E. S., S. F., A.-M. H., and M. M. formal analysis; M. S., Y. C., F. H., B. P., H. S., and M. M. supervision; M. S., L. K., E. S., S. F., A.-M. H., Y. C., F. H., N. B., B. P., H. S., and M. M. validation; M. S., L. K., E. S., S. F., A.-M. H., N. B., and M. M. investigation; M. S., F. H., H. S., and M. M. visualization; M. S., L. K., E. S., S. F., A.-M. H., Y. C., F. H., N. B., B. P., H. S., and M. M. methodology; M. S., F. H., B. P., H. S., and M. M. writing-original draft; M. S., Y. C., F. H., B. P., H. S., and M. M. writing-review and editing; Y. C., B. P., H. S., and M. M. funding acquisition.
Supplementary Material
Acknowledgments
We are grateful to all members of the Marschall laboratory, especially to Deborah Horsch for reading the manuscript; M. M. thanks Laura Graf, Corina Hutterer, Rike Webel, Eric Sonntag, and Jens Milbradt for scientific support along the duration of the long-term project and Christina Wangen and Hanife Strojan for excellent technical assistance. We also thank Thomas Stamminger and group members (Virology, FAU Erlangen-Nürnberg/University of Ulm, Germany) for scientific discussion and long-term cooperation.
This work was supported by Deutsche Forschungsgemeinschaft Grants MA1289/8-1, STI155/5-1, and PL236/7-1) and the proteomic experiments were supported in part by the ProFl Grant ANR-10-INBS-08-01. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1.
- HCMV
- human cytomegalovirus
- CDK
- cyclin-dependent kinase
- HFF
- human foreskin fibroblast
- CoIP
- coimmunoprecipitation
- IP
- immunoprecipitation
- λ-PP
- λ-phosphatase
- IVKA
- in vitro kinase assay
- WB
- Western blot
- CAK
- CDK-activating kinase
- Rb
- retinoblastoma protein
- Rb-CTF
- retinoblastoma C-terminal fragment
- PKC
- protein kinase C
- MBV
- maribavir.
References
- 1. Mocarski E. S., Shenk T., Griffiths P. D., and Pass R. F. (2013) in Fields Virology, (Knipe D. M. and Howley P. M., eds) 6th Ed., pp. 1960–2014, Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Griffiths P., and Reeves M. (2017) in Clinical Virology (Richman D. D., Whitley R. J., and Hayden F. G., eds) 4th Ed., pp. 481–510, ASM Press, Washington, D. C. [Google Scholar]
- 3. Tsutsui Y. (2009) Effects of cytomegalovirus infection on embryogenesis and brain development. Congenit. Anom. 49, 47–55 10.1111/j.1741-4520.2009.00222.x [DOI] [PubMed] [Google Scholar]
- 4. Buxmann H., Hamprecht K., Meyer-Wittkopf M., and Friese K. (2017) Primary human cytomegalovirus (HCMV) infection in pregnancy. Deut. Arztebl. Int. 114, 45–52 10.3238/arztebl.2017.0045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Milbradt J., Kraut A., Hutterer C., Sonntag E., Schmeiser C., Ferro M., Wagner S., Lenac T., Claus C., Pinkert S., Hamilton S. T., Rawlinson W. D., Sticht H., Couté Y., and Marschall M. (2014) Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol. Cell. Proteomics 13, 2132–2146 10.1074/mcp.M113.035782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steingruber M., Kraut A., Socher E., Sticht H., Reichel A., Stamminger T., Amin B., Coute Y., Hutterer C., and Marschall M. (2016) Proteomic interaction patterns between human cyclins, the cyclin-dependent kinase ortholog pUL97 and additional cytomegalovirus proteins. Viruses 8, 219 10.3390/v8080219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sonntag E., Milbradt J., Svrlanska A., Strojan H., Häge S., Kraut A., Hesse A. M., Amin B., Sonnewald U., Couté Y., and Marschall M. (2017) Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J. Gen. Virol. 98, 2569–2581 10.1099/jgv.0.000931 [DOI] [PubMed] [Google Scholar]
- 8. Marschall M., Muller Y. A., Diewald B., Sticht H., and Milbradt J. (2017) The human cytomegalovirus nuclear egress complex unites multiple functions: recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Virol. 27, e1934 10.1002/rmv.1934 [DOI] [PubMed] [Google Scholar]
- 9. Hume A. J., Finkel J. S., Kamil J. P., Coen D. M., Culbertson M. R., and Kalejta R. F. (2008) Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320, 797–799 10.1126/science.1152095 [DOI] [PubMed] [Google Scholar]
- 10. Iwahori S., Hakki M., Chou S., and Kalejta R. F. (2015) Molecular determinants for the inactivation of the retinoblastoma tumor suppressor by the viral cyclin-dependent kinase UL97. J. Biol. Chem. 290, 19666–19680 10.1074/jbc.M115.660043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iwahori S., Umaña A. C., VanDeusen H. R., and Kalejta R. F. (2017) Human cytomegalovirus-encoded viral cyclin-dependent kinase (v-CDK) UL97 phosphorylates and inactivates the retinoblastoma protein-related p107 and p130 proteins. J. Biol. Chem. 292, 6583–6599 10.1074/jbc.M116.773150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hume A. J., and Kalejta R. F. (2009) Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div. 4, 1 10.1186/1747-1028-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marschall M., Feichtinger S., and Milbradt J. (2011) Regulatory roles of protein kinases in cytomegalovirus replication. Adv. Virus Res. 80, 69–101 10.1016/B978-0-12-385987-7.00004-X [DOI] [PubMed] [Google Scholar]
- 14. Graf L., Webel R., Wagner S., Hamilton S. T., Rawlinson W. D., Sticht H., and Marschall M. (2013) The cyclin-dependent kinase ortholog pUL97 of human cytomegalovirus interacts with cyclins. Viruses 5, 3213–3230 10.3390/v5123213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Graf L., Feichtinger S., Naing Z., Hutterer C., Milbradt J., Webel R., Wagner S., Scott G. M., Hamilton S. T., Rawlinson W. D., Stamminger T., Thomas M., and Marschall M. (2016) New insight into the phosphorylation-regulated intranuclear localization of human cytomegalovirus pUL69 mediated by cyclin-dependent kinases (CDKs) and viral CDK orthologue pUL97. J. Gen. Virol. 97, 144–151 10.1099/jgv.0.000337 [DOI] [PubMed] [Google Scholar]
- 16. Steingruber M., Socher E., Hutterer C., Webel R., Bergbrede T., Lenac T., Sticht H., and Marschall M. (2015) The interaction between cyclin B1 and cytomegalovirus protein kinase pUL97 is determined by an active kinase domain. Viruses 7, 4582–4601 10.3390/v7082834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. König P., Büscher N., Steingruber M., Socher E., Sticht H., Tenzer S., Plachter B., and Marschall M. (2017) Dynamic regulatory interaction between cytomegalovirus major tegument protein pp65 and protein kinase pUL97 in intracellular compartments, dense bodies and virions. J. Gen. Virol. 98, 2850–2863 10.1099/jgv.0.000939 [DOI] [PubMed] [Google Scholar]
- 18. Malumbres M. (2014) Cyclin-dependent kinases. Genome Biol. 15, 122 10.1186/gb4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marschall M., Freitag M., Suchy P., Romaker D., Kupfer R., Hanke M., and Stamminger T. (2003) The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 311, 60–71 10.1016/S0042-6822(03)00147-8 [DOI] [PubMed] [Google Scholar]
- 20. Schregel V., Auerochs S., Jochmann R., Maurer K., Stamminger T., and Marschall M. (2007) Mapping of a self-interaction domain of the cytomegalovirus protein kinase pUL97. J. Gen. Virol. 88, 395–404 10.1099/vir.0.82393-0 [DOI] [PubMed] [Google Scholar]
- 21. Kaldis P. (1999) The cdk-activating kinase (CAK): from yeast to mammals. Cell. Mol. Life Sci. 55, 284–296 10.1007/s000180050290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jeffrey P. D., Russo A. A., Polyak K., Gibbs E., Hurwitz J., Massagué J., and Pavletich N. P. (1995) Mechanism of CDK activation revealed by the structure of a cyclinA–CDK2 complex. Nature 376, 313–320 10.1038/376313a0 [DOI] [PubMed] [Google Scholar]
- 23. Morgan D. O. (1995) Principles of CDK regulation. Nature 374, 131–134 10.1038/374131a0 [DOI] [PubMed] [Google Scholar]
- 24. Morgan D. O. (1997) Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13, 261–291 10.1146/annurev.cellbio.13.1.261 [DOI] [PubMed] [Google Scholar]
- 25. Bigley T. M., Reitsma J. M., and Terhune S. S. (2015) Antagonistic relationship between human cytomegalovirus pUL27 and pUL97 activities during infection. J. Virol. 89, 10230–10246 10.1128/JVI.00986-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sonntag E., Hamilton S. T., Bahsi H., Wagner S., Jonjic S., Rawlinson W. D., Marschall M., and Milbradt J. (2016) Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. J. Gen. Virol. 97, 1676–1685 10.1099/jgv.0.000495 [DOI] [PubMed] [Google Scholar]
- 27. Sharma M., Bender B. J., Kamil J. P., Lye M. F., Pesola J. M., Reim N. I., Hogle J. M., and Coen D. M. (2015) Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J. Virol. 89, 523–534 10.1128/JVI.02426-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marschall M., Marzi A., aus dem Siepen P., Jochmann R., Kalmer M., Auerochs S., Lischka P., Leis M., and Stamminger T. (2005) Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J. Biol. Chem. 280, 33357–33367 10.1074/jbc.M502672200 [DOI] [PubMed] [Google Scholar]
- 29. Thomas M., Rechter S., Milbradt J., Auerochs S., Müller R., Stamminger T., and Marschall M. (2009) Cytomegaloviral protein kinase pUL97 interacts with the nuclear mRNA export factor pUL69 to modulate its intranuclear localization and activity. J. Gen. Virol. 90, 567–578 10.1099/vir.0.005827-0 [DOI] [PubMed] [Google Scholar]
- 30. Rechter S., Scott G. M., Eickhoff J., Zielke K., Auerochs S., Müller R., Stamminger T., Rawlinson W. D., and Marschall M. (2009) Cyclin-dependent kinases phosphorylate the cytomegalovirus RNA export protein pUL69 and modulate its nuclear localization and activity. J. Biol. Chem. 284, 8605–8613 10.1074/jbc.M805693200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawaguchi Y., Kato K., Tanaka M., Kanamori M., Nishiyama Y., and Yamanashi Y. (2003) Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1δ. J. Virol. 77, 2359–2368 10.1128/JVI.77.4.2359-2368.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kawaguchi Y., Matsumura T., Roizman B., and Hirai K. (1999) Cellular elongation factor 1δ is modified in cells infected with representative alpha-, beta-, or gammaherpesviruses. J. Virol. 73, 4456–4460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Milbradt J., Auerochs S., Sticht H., and Marschall M. (2009) Cytomegaloviral proteins that associate with the nuclear lamina: components of a postulated nuclear egress complex. J. Gen. Virol. 90, 579–590 10.1099/vir.0.005231-0 [DOI] [PubMed] [Google Scholar]
- 34. Milbradt J., Webel R., Auerochs S., Sticht H., and Marschall M. (2010) Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 285, 13979–13989 10.1074/jbc.M109.063628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamirally S., Kamil J. P., Ndassa-Colday Y. M., Lin A. J., Jahng W. J., Baek M. C., Noton S., Silva L. A., Simpson-Holley M., Knipe D. M., Golan D. E., Marto J. A., and Coen D. M. (2009) Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 5, e1000275 10.1371/journal.ppat.1000275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Milbradt J., Hutterer C., Bahsi H., Wagner S., Sonntag E., Horn A. H., Kaufer B. B., Mori Y., Sticht H., Fossen T., and Marschall M. (2016) The prolyl isomerase Pin1 promotes the herpesvirus-induced phosphorylation-dependent disassembly of the nuclear lamina required for nucleocytoplasmic egress. PLoS Pathog. 12, e1005825 10.1371/journal.ppat.1005825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kochin V., Shimi T., Torvaldson E., Adam S. A., Goldman A., Pack C. G., Melo-Cardenas J., Imanishi S. Y., Goldman R. D., and Eriksson J. E. (2014) Interphase phosphorylation of lamin A. J. Cell Sci. 127, 2683–2696 10.1242/jcs.141820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuny C. V., Chinchilla K., Culbertson M. R., and Kalejta R. F. (2010) Cyclin-dependent kinase-like function is shared by the β- and γ-subset of the conserved herpesvirus protein kinases. PLoS Pathog. 6, e1001092 10.1371/journal.ppat.1001092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marschall M., Stein-Gerlach M., Freitag M., Kupfer R., van Den Bogaard M., and Stamminger T. (2001) Inhibitors of human cytomegalovirus replication drastically reduce the activity of the viral protein kinase pUL97. J. Gen. Virol. 82, 1439–1450 10.1099/0022-1317-82-6-1439 [DOI] [PubMed] [Google Scholar]
- 40. Fisher R. P., and Morgan D. O. (1994) A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 78, 713–724 10.1016/0092-8674(94)90535-5 [DOI] [PubMed] [Google Scholar]
- 41. Rowe W. P., Hartley J. W., Waterman S., Turner H. C., and Huebner R. J. (1956) Cytopathogenic agent resembling human salivary gland virus recovered from tissue cultures of human adenoids. Proc. Soc. Exp. Biol. Med. 92, 418–424 10.3181/00379727-92-22497 [DOI] [PubMed] [Google Scholar]
- 42. Marschall M., Freitag M., Weiler S., Sorg G., and Stamminger T. (2000) Recombinant green fluorescent protein-expressing human cytomegalovirus as a tool for screening antiviral agents. Antimicrob. Agents Chemother. 44, 1588–1597 10.1128/AAC.44.6.1588-1597.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Webel R., Hakki M., Prichard M. N., Rawlinson W. D., Marschall M., and Chou S. (2014) Differential properties of cytomegalovirus pUL97 kinase isoforms affect viral replication and maribavir susceptibility. J. Virol. 88, 4776–4785 10.1128/JVI.00192-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reyda S., Büscher N., Tenzer S., and Plachter B. (2014) Proteomic analyses of human cytomegalovirus strain AD169 derivatives reveal highly conserved patterns of viral and cellular proteins in infected fibroblasts. Viruses 6, 172–188 10.3390/v6010172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hutterer C., Eickhoff J., Milbradt J., Korn K., Zeitträger I., Bahsi H., Wagner S., Zischinsky G., Wolf A., Degenhart C., Unger A., Baumann M., Klebl B., and Marschall M. (2015) A novel CDK7 inhibitor of the pyrazolotriazine class exerts broad-spectrum antiviral activity at nanomolar concentrations. Antimicrob. Agents Chemother. 59, 2062–2071 10.1128/AAC.04534-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baek M. C., Krosky P. M., and Coen D. M. (2002) Relationship between autophosphorylation and phosphorylation of exogenous substrates by the human cytomegalovirus UL97 protein kinase. J. Virol. 76, 11943–11952 10.1128/JVI.76.23.11943-11952.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marschall M., Stein-Gerlach M., Freitag M., Kupfer R., van den Bogaard M., and Stamminger T. (2002) Direct targeting of human cytomegalovirus protein kinase pUL97 by kinase inhibitors is a novel principle for antiviral therapy. J. Gen. Virol. 83, 1013–1023 10.1099/0022-1317-83-5-1013 [DOI] [PubMed] [Google Scholar]
- 48. Oberstein A., Perlman D. H., Shenk T., and Terry L. J. (2015) Human cytomegalovirus pUL97 kinase induces global changes in the infected cell phosphoproteome. Proteomics 15, 2006–2022 10.1002/pmic.201400607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Webel R., Milbradt J., Auerochs S., Schregel V., Held C., Nöbauer K., Razzazi-Fazeli E., Jardin C., Wittenberg T., Sticht H., and Marschall M. (2011) Two isoforms of the protein kinase pUL97 of human cytomegalovirus are differentially regulated in their nuclear translocation. J. Gen. Virol. 92, 638–649 10.1099/vir.0.026799-0 [DOI] [PubMed] [Google Scholar]
- 50. Webel R., Solbak S. M., Held C., Milbradt J., Gross A., Eichler J., Wittenberg T., Jardin C., Sticht H., Fossen T., and Marschall M. (2012) Nuclear import of isoforms of the cytomegalovirus kinase pUL97 is mediated by differential activity of NLS1 and NLS2 both acting through classical importin-α binding. J. Gen. Virol. 93, 1756–1768 10.1099/vir.0.040592-0 [DOI] [PubMed] [Google Scholar]
- 51. Becke S., Fabre-Mersseman V., Aue S., Auerochs S., Sedmak T., Wolfrum U., Strand D., Marschall M., Plachter B., and Reyda S. (2010) Modification of the major tegument protein pp65 of human cytomegalovirus inhibits virus growth and leads to the enhancement of a protein complex with pUL69 and pUL97 in infected cells. J. Gen. Virol. 91, 2531–2541 10.1099/vir.0.022293-0 [DOI] [PubMed] [Google Scholar]
- 52. Baek M. C., Krosky P. M., Pearson A., and Coen D. M. (2004) Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology 324, 184–193 10.1016/j.virol.2004.03.015 [DOI] [PubMed] [Google Scholar]
- 53. Romaker D., Schregel V., Maurer K., Auerochs S., Marzi A., Sticht H., and Marschall M. (2006) Analysis of the structure–activity relationship of four herpesviral UL97 subfamily protein kinases reveals partial but not full functional conservation. J. Med. Chem. 49, 7044–7053 10.1021/jm060696s [DOI] [PubMed] [Google Scholar]
- 54. Dell'Oste V., Gatti D., Gugliesi F., De Andrea M., Bawadekar M., Lo Cigno I., Biolatti M., Vallino M., Marschall M., Gariglio M., and Landolfo S. (2014) Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J. Virol. 88, 6970–6982 10.1128/JVI.00384-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ruiz-Carrascoso G., Romero-Gómez M. P., Plaza D., and Mingorance J. (2013) Rapid detection and quantitation of ganciclovir resistance in cytomegalovirus quasispecies. J. Med. Virol. 85, 1250–1257 10.1002/jmv.23570 [DOI] [PubMed] [Google Scholar]
- 56. Chou S. (2008) Cytomegalovirus UL97 mutations in the era of ganciclovir and maribavir. Rev. Med. Virol. 18, 233–246 10.1002/rmv.574 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.