

Abstract
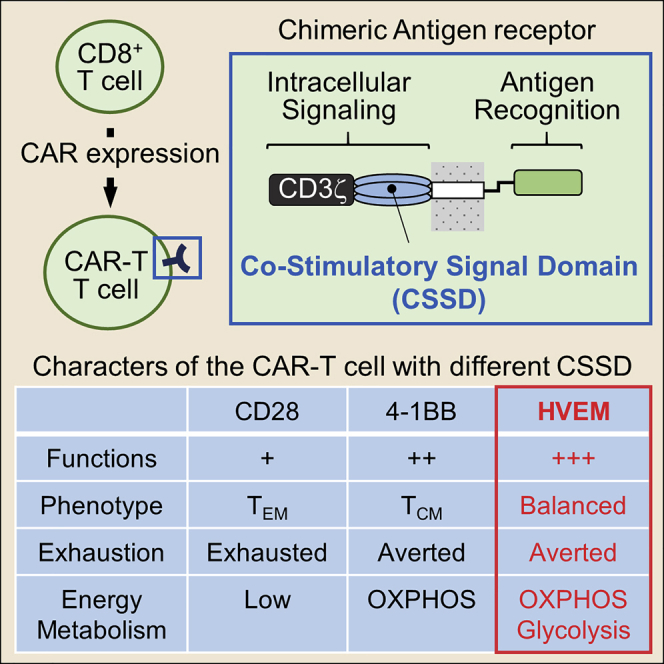
Chimeric antigen receptor (CAR) is a hybrid molecule consisting of an antigen-binding domain and a signal transduction domain. The artificial T cells expressing CAR (CAR-T cells) are expected to be a useful tool for treatment of various diseases, such as cancer. The addition of a co-stimulatory signal domain (CSSD) to CAR is shown to be critical for modulating CAR-T cell activities. However, the interplay among types of CSSDs, effector functions, and characteristics of CAR-T cells is largely unknown. To elucidate the interplay, we analyzed effector functions, differentiation to memory T cell subsets, exhaustion, and energy metabolism of the CAR-T cells with different CSSDs. Comparing to the CAR-T cells bearing a CD28- or 4-1BB-derived CSSD, which are currently used for CAR-T cell development, we found that the CAR-T cells with a herpes virus entry mediator (HVEM)-derived CSSD exhibited enhanced effector functions and efficient and balanced differentiation to both central and effector memory subsets, associated with an elevated energy metabolism and a reduced level of exhaustion. Thus, we developed the CAR-T cells bearing the CSSD derived from HVEM with high functional potency. The HVEM-derived CSSD may be useful for developing effective CAR-T cells.
Keywords: chimeric antigen receptor, T cell-mediated immunotherapy, co-stimulatory signal molecule, herpes virus entry mediator
Graphical Abstract
Introduction
Chimeric antigen receptor (CAR) comprises an extracellular antigen-binding domain combined with an intracellular signal transduction domain.1 The first-generation CAR-transduced T (CAR-T) cells that have CD3ζ as a signal transduction domain of the CAR often become anergic and fail to elicit a potent immune response.2 The second- and third-generation CAR-T cells have been developed by adding one and two co-stimulatory signal domains (CSSDs) in the CAR, respectively.1 These CAR molecules with the modular structure have been shown to successfully mimic the T cell receptor-mediated signal transduction upon cognate antigen stimulation, leading to proliferation and activation of CAR-T cells.3
The immunotherapy using CD19-targeted CAR-T cells demonstrated remarkable efficacy against B cell malignancy.4 To expand the possibilities to various clinical applications,5 it may be necessary to further improve CAR-T cell efficacy. Signaling assumed by the CSSD derived from co-stimulatory molecules is known be a key event for exhibiting potent CAR-T cell efficacy.1 However, the effects of CSSDs on CAR-T cell functions and characteristics are still largely unknown.
Co-stimulatory molecules can be classified into two major families: the CD28 family, including CD28 and inducible T cell co-stimulator (ICOS), and the tumor necrosis factor (TNF) receptor superfamily (TNFRSF), including 4-1BB and herpes virus entry mediator (HVEM). So far, the CSSD derived from CD28 or 4-1BB has commonly been used to construct CARs.6 A previous study showed that the second-generation CAR-T cells with a 4-1BB-derived CSSD persisted for more than 6 months in the blood of most patients, whereas the CAR-T cells with a CD28-derived CSSD became mostly undetectable after 3 months.7 Additionally, 4-1BB co-stimulation induced enhanced differentiation to the central memory subset with increased in vitro persistence.8 4-1BB co-stimulation has also been shown to increase mitochondrial biogenesis and oxidative metabolism for energy production and avert tonic signaling-induced T cell exhaustion.9 Therefore, the CSSD derived from the TNFRSF appears to function better than the one from the CD28 family in the context of second-generation CAR-T cells.
Accumulating reports have suggested potential roles of HVEM, another member of the TNFRSF, in effector function and memory development of CD8+ T cells. For example, HVEM deficiency in CD8+ T cells has been shown to profoundly impair effector CD8+ T cell survival and development of protective immunological memory.10 An interaction between HVEM expressed on CD8+ T cells and B- and T-lymphocyte attenuator has also been reported to promote survival and immunological memory generation in response to bacterial infection.11 Additionally, tumor cells that express anti-HVEM single-chain antibody induce a potent proliferation and cytokine production of co-cultured T cells.12 These findings have indicated that HVEM serves as a potent co-stimulatory molecule in T cells, suggesting that the CSSD derived from HVEM may also be useful in the context of CAR-T cells.
We generated the HIV Env-targeting CAR-T cells with CSSDs derived from CD28, 4-1BB, and HVEM, and we examined their effector functions using HIV Env-expressing target cells. The CAR-T cells with the HVEM-derived CSSD exhibited higher effector functions than those with CD28- and 4-1BB-derived CSSDs. Further analyses showed that the CAR-T cells with the HVEM-derived CSSD efficiently induced both central and effector memory subsets with significantly higher glycolysis and mitochondrial respiration while they averted exhaustion. Therefore, we demonstrate that the CAR-T cells with the HVEM-derived CSSD exhibit high functional potency.
Results
CAR with the HVEM-Derived CSSD Is Efficiently Expressed in a Human T Cell Line
An sCD4, corresponding to 1–178 amino acids of human CD4, was reported to selectively target HIV-infected cells through binding to an HIV Env.13 To generate HIV Env-targeting CAR-T cells, we constructed lentiviral vectors expressing the CAR in combination with CSSDs derived from CD28, 4-1BB, and HVEM (Figure 1A). Flow cytometric analysis indicated that the transduction rates of Jurkat E6.1 cells with different lentiviral vectors were similar to each other (Figure S1). On the other hand, the levels of CAR expression on the cell surface of GFP+ cells differed considerably (Figure 1B), and they were the highest on the CAR-T cells with the HVEM-derived CSSD (Figure 1C). Western blot analysis also revealed that amounts of the CAR with the HVEM-derived CSSD were larger than those with CD28- and 4-1BB-derived CSSDs in whole-cell lysates (Figure 1D).
Figure 1.

CAR with the HVEM-Derived CSSD Is Efficiently Expressed in a Human T Cell Line
(A) Schematic representation of the CAR-expressing lentiviral vector constructs. The CARs contain sCD4 as antigen recognition domain and differ in CSSD. All lentiviral vector constructs express GFP under control of internal ribosome entry site (IRES). (B) Gating strategy for CAR expression analysis in the transduced human T cell line. GFP+-transduced cells were gated (bottom) to analyze CAR expression (top) with anti-c-myc tag antibody by flow cytometry. Typical dot plot and CAR histograms of each transduced cell are shown. Similar transduction efficiencies were achieved by lentiviral transduction (see also Figure S1). (C) Bar graph shows normalized CAR expression level in each CAR-transduced T cell line, which has the indicated CSSD. Each bar shows the mean with SDs (n = 5). **p < 0.05 (one-way ANOVA, Bonferroni post hoc). (D) CAR (50 kDa, top), endogeneous CD3ζ (18 kDa, middle), and actin (43 kDa, bottom) expression levels in the CAR-transduced T cells harboring different CSSDs were determined by western blot with anti-CD3ζ and anti-actin antibodies, respectively.
Human T Cell Line Expressing CAR with the HVEM-Derived CSSD Is Efficiently Activated upon Cognate Antigen Stimulation
To examine functions of the CAR-transduced Jurkat E6.1 cells with different CSSDs, CD3− target cells (Chinese hamster ovary [CHO]-GFP or CHO-Env-GFP) were co-cultured with CD3+ Jurkat E6.1 cells, which had been transduced with CAR-expressing lentiviral vectors (Figure S2, left). Then, the GFP− and successfully transduced GFP+ Jurkat E6.1 cells were examined for CD69 upregulation, an indicator of T cell activation (Figure S2, middle and right). As shown in Figure 2A, no significant activation was observed in the GFP− and GFP+ Jurkat E6.1 cells upon co-cultivation with control CHO-GFP cells. The GFP+, but not GFP−, Jurkat E6.1 cells were efficiently activated when CHO-Env-GFP cells were used as target cells (Figure 2A). Also, measurement of interleukin-2 (IL-2) in the culture supernatants showed that IL-2 secretion by the CAR-transduced Jurkat E6.1 cells was induced upon co-cultivation with CHO-Env-GFP cells, but not with control CHO-GFP cells (Figure 2B). These data demonstrated that human T cells transduced with the CAR-expressing vectors constructed in this study were specifically activated upon cognate antigen stimulation. Linear regression analysis demonstrated that the percentages of activated T cells upon cognate antigen stimulation and IL-2 secretion were correlated with the levels of CAR expression on the cell surface (Figures 2C and 2D).
Figure 2.

Human T Cell Line Expressing CAR with the HVEM-Derived CSSD Is Efficiently Activated upon Cognate Antigen Stimulation
(A) The activated (CD69+) cell percentages in the transduced (CD3+GFP+) or untransduced (CD3+GFP−) T cells were determined by the co-culture assay, with target cells expressing GFP (CHO-GFP) or HIV Env and GFP (CHO-Env-GFP) (see also Figure S2). Each bar shows the mean with SDs from the co-culture assay using the CAR-T cells with the CD28-, 4-1BB-, and HVEM-derived CSSDs (n = 6). **p < 0.05 (two-way ANOVA, Bonferroni post hoc). (B) IL-2 secretion in the co-culture assay was measured by ELISA. Each bar shows the mean with SDs from the co-culture assay using the CAR-T cells with the CD28-, 4-1BB-, and HVEM-derived CSSDs (n = 5). **p < 0.05 (two-way ANOVA, Bonferroni post hoc). (C) Linear regression analysis between percentages of activated CAR-T cells (y axis) and the levels of normalized CAR expression (x axis). (D) Linear regression analysis between IL-2 secretion from antigen-stimulated CAR-transduced T cells (y axis) in the co-culture assay and the levels of normalized CAR expression (x axis).
Human Primary T Cell-Derived CAR-T Cells with the HVEM-Derived CSSD Exhibit Potent Effector Functions
To compare the CAR-T cells with different CSSDs in more detail, we transduced human primary CD8+ with the CAR-expressing lentiviral vectors, and we analyzed for their effector functions. Similar to the observations in Jurkat E6.1 cells (Figures 1B–1D), the CAR expression levels on the cell surface and in whole-cell lysate were the highest for the CAR-T cells with the HVEM-derived CSSD and the lowest for those with the CD28-derived CSSD (Figures 3A–3C and S3). We also compared the effector functions of the CAR-T cells with different CSSDs. Although all of the three CAR-T cells led to antigen-specific cytotoxic activities against HIV Env-expressing target cells, the CAR-T cell with the HVEM-derived CSSD exhibited the highest activity (Figure 3D). Similarly, the highest levels of cytokines, such as IL-2, TNF-α, and interferon (IFN)-γ, were secreted from the CAR-T cells with the HVEM-derived CSSD compared to the other CAR-T cells tested in this study (Figure 3E). Comparable with the observations in Jurkat E6.1 cells (Figures 2C and 2D), effector functions of the CAR-T cells measured by cytotoxic activities and cytokine secretions were correlated with the levels of CAR expression on the cell surface (Figures 3F and S4). Thus, the CAR-T cells with the HVEM-derived CSSD appeared to be potent.
Figure 3.

CAR-T Cells with the HVEM-Derived CSSD Exhibit Potent Effector Functions
(A) Comparison of cell surface CAR expression among the CAR-T cells with different CSSDs. CAR expression in GFP+-transduced primary human CD8 T cells was analyzed with anti-c-myc tag antibody by flow cytometry. Representative histograms of CAR expression (x axis) in GFP+ cells are shown. The number in each histogram shows MFI of the CAR. (B) Bar graph shows normalized CAR expression level in each CAR-T cell with the indicated CSSD. Each bar shows the mean with SDs (n = 6). **p < 0.05 (one-way ANOVA, Bonferroni post hoc). (C) CAR (50 kDa, top), endogeneous CD3ζ (18 kDa, middle), and actin (43 kDa, bottom) expression levels in the CAR-T cells with different CSSDs were determined by western blot with anti-CD3ζ and anti-actin antibodies, respectively. (D) The CAR-T cells with different CSSDs were co-cultured with CHO-GFP (open circle) or CHO-Env-GFP (filled circle) cells at effector-to-target (E:T) ratios of 10:1, 5:1, and 1:1. The lactate dehydrogenase release into the culture supernatant was measured, and the cytotoxicity (% lysis) was calculated. The results show the mean of two separate experiments with primary CD8 T cells from two different healthy human donors (n = 6). (E) Bar graph shows IL-2 (left), TNF-α (middle), and IFN-γ (right) secretion measured by ELISA at an E:T ratio of 10:1 in the co-culture assay (n = 6). **p < 0.05 (one-way ANOVA, Bonferroni post hoc). (F) Linear regression analysis between TNF-α secretion (y axis) in the co-culture assay and the levels of normalized CAR expression (x axis) among the CAR-T cells with different CSSDs. (See also Figure S4 for linear regression analysis of IL-2, IFN-γ, and cytotoxicity.)
CAR-T Cells with the HVEM-Derived CSSD Efficiently Differentiate to Both Central and Effector Memory Subsets
Relative proportions of memory subsets in CAR-T cells have been suggested to affect their function and persistence in vivo.14, 15 Particularly important memory subsets for protective immunity are known as central memory (TCM, CD45RO+CCR7+) and effector memory (TEM, CD45RO+CCR7−) T cells.16 We thus analyzed proportions of memory T cell subsets by surface expression of CD45RO and CCR7 (Figure 4A). Whereas the control T cells dominantly contained the TEM subset, the CAR-T cells with different CSSDs showed larger percentages of the TCM subset (Figure 4A). In comparison among the CAR-T cells with different CSSDs, the CAR-T cells with CD28- and 4-1BB-derived CSSDs dominantly contained the TEM and TCM subsets, respectively (Figure 4B). Interestingly, the CAR-T cells with the HVEM-derived CSSD contained equivalent percentages of TCM and TEM subsets (Figure 4B), suggesting that the HVEM-derived CSSD efficiently induced both TEM and TCM subsets.
Figure 4.

CAR-T Cells with the HVEM-Derived CSSD Efficiently Differentiate to Both Central and Effector Memory Subsets
(A) Representative plots shows the cell surface expression of CD45RO (x axis) and CCR7 (y axis) on the CAR-T cells with different CSSDs. Control shows CD8 T cells transduced only with GFP. (B) Bar graph shows T cell subsets, including naive (N, CCR7+CD45RO−), central memory (CM, CCR7+CD45RO+), effector memory (EM, CCR7−CD45RO+), and terminally differentiated effector memory (TEMRA, CCR7−CD45RO−) among the CAR-T cells with different CSSDs. The mean with SDs of two separate experiments with primary CD8 T cells from two different donors are shown (n = 6). **p < 0.05 (two-way ANOVA, Bonferroni post hoc).
CAR-T Cells with the HVEM-Derived CSSD Avert T Cell Exhaustion
Prolonged stimulation of T cells has been known to cause exhaustion characterized by decreased proliferation, lowered levels of cytokine production, high rates of apoptosis, and expression of inhibitory receptors, such as programmed cell death 1 (PD-1) and lymphocyte activation gene 3 (LAG-3).17, 18 To determine the effect of CSSDs on T cell exhaustion, we examined the percentages of PD-1+, LAG-3+, and PD-1+/LAG-3+ exhausted T cell populations (Figures 5A and 5B). The results showed that the CAR-T cells with the CD28-derived CSSD contained relatively large percentages of exhausted T cells, whereas those with the 4-1BB-derived CSSD contained significantly lower percentages of exhausted T cells at days 9 and 16 after transduction (Figures 5C and 5D). The LAG-3+ population, but not the PD-1+/LAG3+ population, was induced in the CAR-T cells with the HVEM-derived CSSD compared to control T cells at day 9 after transduction (Figure 5C). Moreover, the CAR-T cells with the HVEM-derived CSSD contained the lowest percentage of exhausted T cells among the CAR-T cells tested in this study at day 16 after transduction (Figure 5D). Interestingly, the percentages of PD-1+ and LAG-3+ cells were larger and smaller, respectively, for the CAR-T cells with the HVEM-derived CSSD than those with the 4-1BB-derived CSSD at day 16 after transduction (Figure 5D). Taken together, these data suggested that the CAR-T cells with the HVEM-derived CSSD averted T cell exhaustion.
Figure 5.

CAR-T Cells with the HVEM-Derived CSSD Avert CAR-T Cell Exhaustion
(A and B) Representative plots shows cell surface expression of PD-1 (x axis) and LAG-3 (y axis) on the CAR-T cells with different CSSDs at days 9 (A) and 16 (B). The numbers in each plot show percentages of each population in the CAR-T cells with different CSSDs. Control shows CD8 T cells transduced only with GFP. (C and D) Bar graph shows the percentages of the CAR-T cell populations that express one or two inhibitory receptors (PD-1 and LAG-3) at days 9 (C) and 16 (D). The mean with SDs of two separate experiments with primary CD8 T cells from two different healthy human donors are shown (n = 6). **p < 0.05 (one-way ANOVA, Bonferroni post hoc).
CAR-T Cells with the HVEM-Derived CSSD Exhibit Reprogrammed Energy Metabolism
It has recently been shown that T cell exhaustion was accompanied by deficient energy metabolism.19, 20 Thus, we compared metabolic states of the CAR-T cells with different CSSDs at basal condition and following the serial addition of reagents to analyze relative contribution of mitochondrial and non-mitochondrial mechanisms of oxygen consumption (Figure 6A). Mitochondrial respiration and glycolysis can be measured by oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), respectively. The CAR-T cells with the HVEM-derived CSSD showed the highest level of basal OCR, followed by those with the 4-1BB-derived CSSD and the control T cells; those with the CD28-derived CSSD showed the lowest level of basal OCR (Figure 6B). We also measured ATP-linked respiration by adding an ATP synthase inhibitor oligomycin and maximal OCR levels by adding carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP; uncoupling of oxygen consumption from ATP production) (Figure 6A). The results indicated that ATP-linked respiration and maximal OCR levels in the different CAR-T cells were similarly elevated as basal OCR levels (Figures 6C and 6D). Interestingly, basal ECAR was significantly increased only for the CAR-T cells with the HVEM-derived CSSD (Figure 6E). These data indicated that the CAR-T cells with the CD28-derived CSSD exhibited a low-energy state, whereas those with the 4-1BB-derived CSSD induced higher mitochondrial respiration. Moreover, the CAR-T cells with the HVEM-derived CSSD induced enhanced glycolysis as well. These results suggested that the HVEM-derived CSSD could lead to reprogramming of energy metabolism.
Figure 6.

CAR-T Cells with the HVEM-Derived CSSD Exhibit Reprogrammed Energy Metabolism
(A) The OCRs of the CAR-T cells with different CSSDs under basal metabolic conditions and in response to mitochondrial inhibitors, as specified in the Materials and Methods. The data are the summary of two experiments performed with the CAR-T cells made from two different healthy human donors and plotted as mean with SDs. Control shows CD8 T cells transduced only with GFP. (B–E) Basal OCR levels (B), basal ECAR levels (C), ATP-linked respiration (D), and maximal OCR levels (E) are shown. Bar graph shows the mean with SDs of two separate experiments with the CAR-T cells made from two different healthy human donors (n = 5). **p < 0.05 (one-way ANOVA, Bonferroni post hoc).
Discussion
We developed lentiviral vectors expressing the CAR consisting of sCD4 as an antigen recognition domain combined with CSSDs derived from CD28, 4-1BB, and HVEM. Transducing the human T cell line and primary T cells with these vectors, we demonstrated that the CSSD in CAR construct was a crucial determinant for the levels of CAR expression on the cell surface, which appeared to be correlated with the activities and effector functions of the CAR-T cells. It was also shown in this study that T cell exhaustion, energy metabolism, and induction of the memory T cell subsets were also affected by the CSSD in CAR constructs. Among the CSSDs examined in this study, the HVEM-derived CSSD led to the highest level of CAR expression, most potent effector functions of CAR-T cells, evasion of exhaustion, and balanced induction of both central and effector memory T cell subsets, associated with elevated glycolysis and mitochondrial respiration. The results of this study suggest that the HVEM-derived CSSD may be useful for generating effective CAR-T cells in a certain context.
It is unknown yet how the CSSD controls the level of CAR expression on the cell surface. Since amounts of CAR molecules in whole-cell lysate were correlated to the level of the cell surface expression, CAR synthesis rather than trafficking might be affected by the CSSD. Among other possibilities, different CSSDs might activate different signaling pathways, resulting in distinct levels of gene expression. For example, CD28-mediated co-stimulation induces the phosphoinositide 3-kinase (PI3K)-Akt pathway, whereas 4-1BB-mediated co-stimulation primarily activates c-Jun N-terminal kinase (JNK) and p38 through TNFR-associated factors.21, 22 It has also been shown that 4-1BB and HVEM are capable of activating alternative nuclear factor κB (NF-κB) pathways.23 These differences in signaling pathways may affect the CAR expression at a transcriptional or translational level.24
Antigen-independent tonic signaling during ex vivo expansion has been shown to cause exhaustion of the CAR-T cells with the CD28-derived CSSD, whereas those with the 4-1BB-derived CSSD are relatively refractory to exhaustion.9 Our studies not only confirmed these observations but also demonstrated that the HVEM-derived CSSD could avert CAR-T cell exhaustion. In addition, our data suggested that CSSDs in CAR constructs could affect the degrees of T cell exhaustion through differential effects on the inhibitory receptor expression (Figure 5B). Since 4-1BB and HVEM are classified in the TNFRSF, it is possible that the CSSD derived from the TNFRSF, but not from the CD28 family, might allow CAR-T cells to avoid exhaustion.
Mitochondrial dynamics have been suggested to control T cell fate through metabolic programming.25 In addition, 4-1BB-mediated co-stimulation has been reported to enhance mitochondrial respiration with increased central memory subsets.8 Our results were compatible with these previous studies, and they also demonstrated that the HVEM-derived CSSD enhanced both glycolysis and mitochondrial respiration and yielded relatively low levels of T cell exhaustion as well. It has also been shown that various T cell subsets require distinct metabolic programs to support their function.26, 27, 28 Our data suggested that co-stimulation signals impacted differentiation of the CAR-T cells to central and effector memory T cell subsets and that the HVEM-derived CSSD induced efficient and balanced differentiation. Different co-stimulatory signals are likely to affect metabolic programs in distinct manners to differentiate specific memory T cell subsets.
Development of new anti-HIV drugs and progress in anti-retroviral therapy (ART) dramatically improved the prognosis of HIV-infected patients. However, it has not been successful for ART to completely eliminate HIV from the patient’s body due to persistence of latently infected cells. To achieve this goal, the so-called “shock-and-kill” approach, which combines pharmacological reactivation of the latently infected cells and following immunotherapies with HIV-specific cytotoxic T lymphocytes (CTLs) or broadly neutralizing antibodies (bNAbs), has been proposed.29 In this protocol, CD4-based CAR-T cells targeting the conserved region of HIV-1 Env might be more useful than T cell receptor (TCR)-based or bNAb-based CAR-T cells, which fail to recognize the antigens with escape mutations. In fact, a recent study reported promising results of the CAR-T cells harboring a 4-1BB-derived CSSD in the in vitro and in vivo models of HIV treatment.30 The results of our study strongly suggest that the CD4-based CAR-T cells with the HVEM-derived CSSD, which showed more potent effector functions than those with the CD28- and 4-1BB-derived CSSDs, may also be a useful tool for the shock-and-kill treatment of HIV infection.
Our results showed that CAR expression levels were clearly associated with effector functions in Jurkat T cells (Figures 2C and 2D) and CD8 T cells (Figures 3F and S4) under similar transduction efficiencies (Figures S1 and S3), suggesting that CAR expression level could be the major contributor for the higher effector functions. Since CAR expression was different among CAR-T cells with different CSSDs, it is very hard to determine whether the CAR expression or the biological functions of CSSDs mainly affect CAR-T cell effector functions and characteristics using this experimental system. However, the biological functions of CSSDs might affect CAR-T cell characteristics, such as T cell exhaustion and memory phenotype, because the type of CSSD, but not CAR expression level, was associated with these CAR-T cell characteristics (Figures 4 and 5).
To further determine this issue, adjusting CAR expression levels in CAR-T cells with different CSSDs may be necessary in future studies using the appropriate experimental system. In addition, while our data indicated the usefulness of the HVEM-derived CSSD in the context of CAR with sCD4 as an antigen-binding domain, it is unknown yet whether the HVEM-derived CSSD is universally versatile. Further studies will be necessary to examine the effects of the HVEM-derived CSSD on activities and functions of CAR-T cells with various antigen specificities. Also, the CD4-based CAR-T cells have been shown to have no potential toxicities in a humanized mouse model31 and clinical trials.32 However, it may also be necessary to test, in the future, the potential toxicities of the CD4-based CAR-T cells with the HVEM-derived CSSD, because they secreted a relatively larger amount of cytokines than those with the CD28- and 4-1BB-derived CSSDs.
In summary, our results demonstrate that the CSSD in CAR is a crucial determinant for effector functions and characteristics of CAR-T cells, indicating that the CSSD in CAR is important for designing more potent CAR-T cells. Moreover, the HVEM-derived CAR-T cells may be a promising candidate for generating effective CAR-T cells.
Materials and Methods
Study Approval
This study was conducted according to the principles of the Declaration of Helsinki and with the approval of the Scientific Ethics Committee of the Dokkyo Medical University. Written informed consent was obtained from all subjects after oral explanation of this study.
Cell Culture
The culture media, minimum essential medium (MEM), DMEM, and RPMI (Thermo Fisher Scientific, Waltham, MA), were supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific), 2 mM glutamine (Thermo Fisher Scientific), 10 U/mL penicillin, and 10 μg/mL streptomycin (Thermo Fisher Scientific) and named shortly M10, D10, and R10, respectively. CHO cells and their transfectants were maintained in M10 supplemented with non-essential amino acid (M10-NEAA). 293FT cells were cultured in D10, and Jurkat E6.1 cells, obtained from European Collection of Authenticated Cell Cultures through DS Pharma, were maintained in R10. Human peripheral blood mononuclear cells (PBMCs) from healthy donors were prepared by Ficoll-Paque (GE Healthcare Life Sciences, Pittsburgh, PA) density gradient and cultured in AIM-V (Thermo Fisher Scientific) supplemented with 5% FBS and 10 mM HEPES (shortly, complete AIM-V) overnight to remove plastic-adherent monocytes. Monocyte-depleted PBMCs were used for transduction experiments. All cells were grown at 37°C with 5% CO2.
Vector Constructions
To introduce multiple restriction enzyme sites, a DNA linker was introduced into lentiviral vector plasmid pTK643-CMV-IRES-GFP/blastcidine (BSD). The DNA linker containing XbaI-XhoI-BsiWI-BstBI-BamHI restriction enzyme sites was made by incubating two oligonucleotides, L1 (5′-CTAGACTCGAGCGTACGTTCGAAG-3′) and L2 (5′-GATCCTTCGAACGTACGCTCGAGT-3′), at a molar ratio of 1:1 at 70°C for 10 min and left at room temperature for 2 h. The linker DNA was inserted into XbaI/BamHI-digested pTK643-CMV-IRES-GFP/BSD (resulting in pTK643-CMV-MCS-IRES-GFP/BSD).
For making the sCD4-encoding DNA fragment, total RNA extracted from Jurkat E6.1 cells by ISOGEN (Nippon Gene, Tokyo, Japan) was used for cDNA synthesis with ReverTra Ace (Toyobo, Osaka, Japan), according to the manufacturer’s instructions. The sCD4 DNA fragment containing XbaI/EcoRI sites was amplified using KOD-FX (Toyobo) with the following primers: sCD4 forward, 5′-GAATCTAGAGCCACCATGAACCGGGGAGTC-3′; and sCD4 reverse, 5′-GATCTTGAATTCAGCTAGCACCACGATGTC-3′. Purified DNA fragment was incubated with Ampli Taq (Thermo Fisher Scientific, Waltham, MA) at 72°C for 10 min to add A tails. The A-tailed DNA fragment was ligated into pGEM-T easy vector (Promega, Madison, WI). The sequence was verified with BigDye Terminator version (v.)3.1 Cycle Sequencing Kit (Thermo Fisher Scientific).
The cDNA fragments encoding CSSDs derived from CD28, 4-1BB, and HVEM were artificially synthesized by GenScript (Piscataway, NJ). To make sCD4-CAR-expressing lentiviral vectors, the XbaI/EcoRI-digested sCD4 DNA fragment and each of the EcoRI/BamHI-digested CSSD DNA fragments were ligated into XbaI/BamHI-digested pTK643-EF1a-IRES-GFP/BSD.
For making the HIV Env- (NL4-3 strain) expressing lentiviral vector plasmid, XbaI/XhoI-digested pRE11-NL43, kindly provided by Dr. Noriaki Hosoya,33 was ligated into XbaI/XhoI-digested pTK643-CMV-MCS-IRES-GFP/BSD.
Recombinant Lentivirus Production
Recombinant lentivirus was produced as previously described34 with some modification. Briefly, 293FT cells were cultured on a collagen-coated 10-cm dish (Iwaki, Shizuoka, Japan) with 80%–90% confluency. The culture medium was replaced to D10 containing 25 μM chloroquine (Sigma, Darmstadt, Germany) without antibiotics. Regarding vectors packed with ΔNRF, the following plasmid amounts were used: 15 μg lentiviral vector plasmid, 10 μg ΔNRF, and 5 μg pMD.G. The plasmids were co-transfected into 293FT cells with polyethyleneimine (PEI) MAX (Polysciences, Warrington, PA) at a DNA-to-PEI ratio of 2:1. The supernatant was replaced with D10 containing 5 mM sodium butylate (Wako Pure Chemicals Industries, Osaka, Japan) and 10 μM forskolin (Tokyo Chemical Industry, Tokyo, Japan). The culture supernatants containing recombinant lentiviruses were harvested at 48 h after transfection and cleared by centrifugation and 0.45-μm filtration (Millipore, Burlington, MA). The recombinant lentiviruses were concentrated by high-speed centrifugation at 18,000 rpm for 3 h using Himac CR21N (Hitachi Koki, Tokyo, Japan). The lentiviral titers were measured on HeLa cells based on the percentage of GFP-positive cells by flow cytometry.
Transduction with Lentiviral Vectors
The recombinant lentivirus-containing supernatant was used to transduce CAR and GFP genes into Jurkat E6.1 cells and HIV Env gene into CHO cells, respectively. Briefly, 2 million Jurkat E6.1 cells or semi-confluent CHO cells in a 6-well plate were exposed to 1 mL unconcentrated lentivirus-containing supernatant in the presence of polybrane at 8 μg/mL. The cells were centrifuged at 5,500 rpm for 3 h at 22°C to enhance viral infection. After removal of the supernatant, the cells were cultured at 37°C in a CO2 incubator for 48 h. The culture medium was replaced with the media supplemented with 10 μg/mL BSD. Thereafter, transduced cells were maintained in the media with 10 μg/mL BSD until the following assays were performed.
Human primary CD8 T cells were isolated from monocyte-depleted PBMCs with anti-human CD3-allophycocyanin (APC), CD4-phycierythrin (PE), and CD8-PE/Cy7 antibodies (all from BioLegend, San Diego, CA) by cell sorting on a FACS Aria II (BD Biosciences, Franklin Lakes, NJ), and they routinely achieved >95% purity. The purified CD8 T cells were activated with anti-CD3/CD28 beads (Thermo Fisher Scientific) at a bead-to-cell ratio of 3:1 in complete AIM-V supplemented with 40 U/mL recombinant human IL-2 (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, from Dr. Maurice Gately, Hoffmann-La Roche) for 3 days. After removal of the anti-CD3/CD28 beads, activated CD8 T cells were transduced with concentrated recombinant lentivirus on days 3 and 4 using Retronectin- (Takara Bio, Shiga, Japan) coated plates, according to the manufacturer’s instructions. The cells transduced only with GFP were used as a control. Thereafter, the cells were cultured for 16 days with fresh media containing 300 U/mL IL-2 replenished every 2–3 days. Successfully transduced GFP+ cells were sorted at 16 days after the initial anti-CD3/CD28 bead stimulation for energy metabolism analysis.
Flow Cytometry
Antibodies used in flow cytometry were obtained from BioLegend otherwise indicated. CAR expression on the transduced Jurkat E6.1 cells was analyzed with anti-c-myc tag antibody (Santa Cruz Biotechnology, Dallas, TX), followed by anti-mouse immunogloburins (Igs)-PE antibody (Agilent Technologies, Santa Clara, CA). Activation of the CAR-transduced Jurkat E6.1 cells in the co-culture assay was analyzed with anti-human CD3-APC antibody and CD69-PE antibody (see Figure S1 for gating strategy). CAR expression on CAR-transduced primary CD8+ T cells was analyzed with biotinylated anti-c-myc tag antibody followed by streptavidin-PE (Tonbo Biosciences, San Diego, CA). For normalizing CAR expression by GFP expression, we divided the mean fluorescence intensity (MFI) of CAR by the MFI of GFP. This value was represented as normalized CAR expression. Exhaustion of the CAR-T cells was analyzed with anti-human PD-1-APC antibody and anti-human LAG-3-PE/Cy7 antibody (Thermo Fisher Scientific). Memory phenotypes of the CAR-T cells were analyzed with CD45RO-PE, CD8-PE-Cy7, and CCR7-APC. The centrifuged cells were resuspended with antibody diluted in fluorescence-activated cell sorting (FACS) buffer (PBS containing 2% FBS and 0.02% sodium azide) and incubated on ice for 30 min. After washing with ice-cold FACS buffer, the cells were fixed with 1% paraformaldehyde in PBS and analyzed with a FACS Calibur (BD Biosciences).
Western Blot
Cells (5 × 106) were washed once with ice-cold PBS, resuspended in 150 μL radioimmunoprecipitation assay (RIPA) buffer (10 mM TrisHCl [pH 7.4], 1% NP-40, 0.1% Sodium Deoxychorate, 0.1% SDS, 0.15 M NaCl, and 1 mM EDTA), supplemented with Complete Mini (Roche, Mannheim, Germany) and 1 mM PMSF, and incubated on ice for 30 min. The cell lysate cleared by centrifugation (20 μL) was mixed with NuPAGE LDS sample buffer (Thermo Fisher Scientific) and 0.1 M DTT and boiled at 95°C for 5 min. Boiled samples were separated by electrophoresis in a NuPAGE 10% Bis-Tris gel (Thermo Fisher Scientific) and transferred to an Immobilon P membrane (Millipore, Burlington, MA). The membrane was blocked with the blocking buffer (5% non-fat dried milk and 0.1% Tween-20 in Tris-buffered saline [TBS]) for 30 min. Blocked membrane was incubated at 4°C overnight with anti-actin antibody (I-19) or anti-CD3zeta antibody (F-3) (Santa Cruz Biotechnology, Dallas, TX) diluted in a blocking buffer. The membrane was then incubated for 1 h with anti-goat immunoglobulin G (IgG)-horseradish peroxidase (HRP) (Millipore) or anti-mouse IgG-HRP (GE Healthcare, Buckinghamshire, UK) diluted in a blocking buffer. The membrane was washed three times with 0.1% Tween-20/TBS after all antibody incubation steps. Immunoreactivity was visualized with a Lumi-Light PLUS (Roche) as a substrate and detected with Light-Capture II (ATTO, Tokyo, Japan).
Co-culture Assay
Activation and IL-2 secretion of the CAR-transduced Jurkat E6.1 cells were determined by co-culturing with target cells (CHO-GFP or CHO-Env-GFP). Target cells (1 × 105) were seeded in each well of a 96-well flat-bottom plate. CAR-transduced Jurkat E6.1 cells (2 × 105) were added and co-cultured overnight. On the next day, the culture supernatants including cells were harvested and centrifuged to separate cells and cell-free supernatants. IL-2 secretion into the supernatant was measured using Human IL-2 ELISA MAX Deluxe (BioLegend), according to the manufacturer’s instructions. The remaining cells were used to determine CD69 expression by flow cytometry.
To determine cytotoxicity of the CAR-T cells, target cells (1 × 104) were seeded in each well of a 96-well flat-bottom plate. The CAR-T cells were added at different effector-to-target ratios (see also in the figure legend) and co-cultured in phenol red-free R10 with NEAA. After overnight incubation, the cell-free supernatant was collected by centrifugation. Lactate dehydrogenase release in the supernatant was assayed using CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega). The cytotoxicity (% lysis) was calculated according to the manufacturer’s instructions. In cases of unequal percentages of CAR-transduced cells in the cultures, untransduced T cells were added to ensure that both the number of CAR+ T cells and the total number of T cells remained consistent across the CAR-T cell groups. Collected supernatants were also used to determine IL-2, TNF-α, and IFN-γ secretion using human IL-2, TNF-α, and IFN-γ ELISA MAX Deluxe (all from BioLegend), according to the manufacturer’s instructions.
Analysis of Energy Metabolism
Mitochondrial function of the CAR-T cells was analyzed with an extracellular flux analyzer XFp (Agilent Technologies, Santa Clara, CA). Each well of the cell culture microplate was coated with CellTak (BD Biosciences), according to the manufacturer’s instructions. To assay mitochondrial function, the sorted CAR-T cells without further culture were resuspended in XF RPMI medium supplemented with 5.5 mM Glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate, and they were seeded at 3 × 105 cells/well. The plate was centrifuged at 200 × g for 1 min and incubated at 37°C in a non-CO2 incubator for 30–60 min. During incubation, the instrument XFp and its assay cartridges were calibrated according to the manufacturer’s instructions. OCRs were measured under basal conditions and following treatment with 1 μM oligomycin (an inhibitor of ATP synthase), 1 μM FCCP (uncoupling of oxygen consumption from ATP production), and 1 μM rotenone and antimycin A (inhibitor for complexes I and III of the electron transport chain, respectively) (XFp Cell Mito Stress Kit, Agilent Technologies). Four measurements of basal condition and after each treatment were performed. ATP-linked respiration was defined as (last rate measurement before oligomycin addition) − (minimum rate measurement after oligomycin addition). Maximal OCR was measured after FCCP treatment.
Statistics
Statistical analysis was described previously.35, 36 Unpaired one-way or two-way ANOVA with Bonferroni multiple comparison test and linear regression analysis were performed, and R2 value was calculated using GraphPad Prism (GraphPad, San Diego, CA). A p value < 0.05 was considered statistically significant.
Author Contributions
J.N. and L.S. conceived the research projects. J.N. designed, planned, and performed the experiments. J.N., M.M., C.Y., and L.S. discussed the results and wrote the paper.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Yuki Shinozaki, Satomi Yoshida, and Kayoko Kobayashi for technical and secretarial assistance; Yasuko Nonaka for cell sorting; and Takashi Namatame for DNA sequencing. We also thank members of Department of Microbiology, Dokkyo Medical University for critical discussion of the paper and their input and assistance. This work was supported in part by Dokkyo Medical University Investigator-Initiated Research Grant (2014-03 and 2017-03), the Science Research Promotion Fund for young investigators, and JSPS KAKENHI Grant-in-Aid for Young Scientists (B) JP16K19617 and Grant-in-Aid for Scientific Research (C) JP18K07272 to J.N.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2019.03.002.
Contributor Information
Jun-ichi Nunoya, Email: nunoya@dokkyomed.ac.jp.
Lishan Su, Email: lishan_su@med.unc.edu.
Supplemental Information
References
- 1.Dotti G., Gottschalk S., Savoldo B., Brenner M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014;257:107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kershaw M.H., Westwood J.A., Parker L.L., Wang G., Eshhar Z., Mavroukakis S.A., White D.E., Wunderlich J.R., Canevari S., Rogers-Freezer L. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maus M.V., Grupp S.A., Porter D.L., June C.H. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625–2635. doi: 10.1182/blood-2013-11-492231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude S.L., Teachey D.T., Porter D.L., Grupp S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125:4017–4023. doi: 10.1182/blood-2014-12-580068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maldini C.R., Ellis G.I., Riley J.L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018;18:605–616. doi: 10.1038/s41577-018-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller B.C., Maus M.V. CD19-Targeted CAR T Cells: A New Tool in the Fight against B Cell Malignancies. Oncol. Res. Treat. 2015;38:683–690. doi: 10.1159/000442170. [DOI] [PubMed] [Google Scholar]
- 7.Zhang T., Cao L., Xie J., Shi N., Zhang Z., Luo Z., Yue D., Zhang Z., Wang L., Han W. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: a meta-analysis. Oncotarget. 2015;6:33961–33971. doi: 10.18632/oncotarget.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawalekar O.U., O’Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Jr., Patel P.R., Guedan S., Scholler J., Keith B. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 9.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flynn R., Hutchinson T., Murphy K.M., Ware C.F., Croft M., Salek-Ardakani S. CD8 T cell memory to a viral pathogen requires trans cosignaling between HVEM and BTLA. PLoS ONE. 2013;8:e77991. doi: 10.1371/journal.pone.0077991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinberg M.W., Huang Y., Wang-Zhu Y., Ware C.F., Cheroutre H., Kronenberg M. BTLA interaction with HVEM expressed on CD8(+) T cells promotes survival and memory generation in response to a bacterial infection. PLoS ONE. 2013;8:e77992. doi: 10.1371/journal.pone.0077992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park J.J., Anand S., Zhao Y., Matsumura Y., Sakoda Y., Kuramasu A., Strome S.E., Chen L., Tamada K. Expression of anti-HVEM single-chain antibody on tumor cells induces tumor-specific immunity with long-term memory. Cancer Immunol. Immunother. 2012;61:203–214. doi: 10.1007/s00262-011-1101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaudhary V.K., Mizukami T., Fuerst T.R., FitzGerald D.J., Moss B., Pastan I., Berger E.A. Selective killing of HIV-infected cells by recombinant human CD4-Pseudomonas exotoxin hybrid protein. Nature. 1988;335:369–372. doi: 10.1038/335369a0. [DOI] [PubMed] [Google Scholar]
- 14.Sommermeyer D., Hudecek M., Kosasih P.L., Gogishvili T., Maloney D.G., Turtle C.J., Riddell S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Busch D.H., Fräßle S.P., Sommermeyer D., Buchholz V.R., Riddell S.R. Role of memory T cell subsets for adoptive immunotherapy. Semin. Immunol. 2016;28:28–34. doi: 10.1016/j.smim.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boots A.M., Maier A.B., Stinissen P., Masson P., Lories R.J., De Keyser F. The influence of ageing on the development and management of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013;9:604–613. doi: 10.1038/nrrheum.2013.92. [DOI] [PubMed] [Google Scholar]
- 17.Virgin H.W., Wherry E.J., Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 18.Wherry E.J. T cell exhaustion. Nat. Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 19.Fisicaro P., Barili V., Montanini B., Acerbi G., Ferracin M., Guerrieri F., Salerno D., Boni C., Massari M., Cavallo M.C. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017;23:327–336. doi: 10.1038/nm.4275. [DOI] [PubMed] [Google Scholar]
- 20.Bengsch B., Johnson A.L., Kurachi M., Odorizzi P.M., Pauken K.E., Attanasio J., Stelekati E., McLane L.M., Paley M.A., Delgoffe G.M., Wherry E.J. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity. 2016;45:358–373. doi: 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim H.H., Kwack K., Lee Z.H. Activation of c-jun N-terminal kinase by 4-1BB (CD137), a T cell co-stimulatory molecule. Mol. Cells. 2000;10:247–252. [PubMed] [Google Scholar]
- 22.Cannons J.L., Choi Y., Watts T.H. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J. Immunol. 2000;165:6193–6204. doi: 10.4049/jimmunol.165.11.6193. [DOI] [PubMed] [Google Scholar]
- 23.Hauer J., Püschner S., Ramakrishnan P., Simon U., Bongers M., Federle C., Engelmann H. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc. Natl. Acad. Sci. USA. 2005;102:2874–2879. doi: 10.1073/pnas.0500187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehta M.M., Weinberg S.E., Chandel N.S. Mitochondrial control of immunity: beyond ATP. Nat. Rev. Immunol. 2017;17:608–620. doi: 10.1038/nri.2017.66. [DOI] [PubMed] [Google Scholar]
- 25.Buck M.D., O’Sullivan D., Klein Geltink R.I., Curtis J.D., Chang C.H., Sanin D.E., Qiu J., Kretz O., Braas D., van der Windt G.J. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Araki K., Turner A.P., Shaffer V.O., Gangappa S., Keller S.A., Bachmann M.F., Larsen C.P., Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearce E.L., Walsh M.C., Cejas P.J., Harms G.M., Shen H., Wang L.S., Jones R.G., Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao R.R., Li Q., Odunsi K., Shrikant P.A. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halper-Stromberg A., Lu C.L., Klein F., Horwitz J.A., Bournazos S., Nogueira L., Eisenreich T.R., Liu C., Gazumyan A., Schaefer U. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell. 2014;158:989–999. doi: 10.1016/j.cell.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leibman R.S., Richardson M.W., Ellebrecht C.T., Maldini C.R., Glover J.A., Secreto A.J., Kulikovskaya I., Lacey S.F., Akkina S.R., Yi Y. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. 2017;13:e1006613. doi: 10.1371/journal.ppat.1006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhen A., Kamata M., Rezek V., Rick J., Levin B., Kasparian S., Chen I.S., Yang O.O., Zack J.A., Kitchen S.G. HIV-specific Immunity Derived From Chimeric Antigen Receptor-engineered Stem Cells. Mol. Ther. 2015;23:1358–1367. doi: 10.1038/mt.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scholler J., Brady T.L., Binder-Scholl G., Hwang W.T., Plesa G., Hege K.M., Vogel A.N., Kalos M., Riley J.L., Deeks S.G. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teeranaipong P., Hosoya N., Kawana-Tachikawa A., Fujii T., Koibuchi T., Nakamura H., Koga M., Kondo N., Gao G.F., Hoshino H. Development of a rapid cell-fusion-based phenotypic HIV-1 tropism assay. J. Int. AIDS Soc. 2013;16:18723. doi: 10.7448/IAS.16.1.18723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cockrell A.S., Ma H., Fu K., McCown T.J., Kafri T. A trans-lentiviral packaging cell line for high-titer conditional self-inactivating HIV-1 vectors. Mol. Ther. 2006;14:276–284. doi: 10.1016/j.ymthe.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Nunoya J., Washburn M.L., Kovalev G.I., Su L. Regulatory T cells prevent liver fibrosis during HIV type 1 infection in a humanized mouse model. J. Infect. Dis. 2014;209:1039–1044. doi: 10.1093/infdis/jit548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li G., Nunoya J.I., Cheng L., Reszka-Blanco N., Tsao L.C., Jeffrey J., Su L. Regulatory T Cells Contribute to HIV-1 Reservoir Persistence in CD4+ T Cells Through Cyclic Adenosine Monophosphate-Dependent Mechanisms in Humanized Mice In Vivo. J. Infect. Dis. 2017;216:1579–1591. doi: 10.1093/infdis/jix547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.