

Abstract
Immunohistochemistry (IHC) for mismatch repair (MMR) proteins is used to identify MMR status: being diffusely positive (intact/retained nuclear staining) or showing loss of nuclear tumour staining (MMR protein deficient). Four colonic adenocarcinomas and a gastric adenocarcinoma with associated dysplasia that displayed heterogenous IHC staining patterns in at least one of the four MMR proteins were characterised by next‐generation sequencing (NGS). In order to examine a potential molecular mechanism for these staining patterns, the respective areas were macrodissected, analysed for microsatellite instability (MSI) and investigated by NGS and multiplex ligation‐dependent probe amplification (MLPA) analysis of MLH1, MSH2, MSH6 and PMS2 genes, including MLH1 methylation analysis. One colonic adenocarcinoma showed heterogenous MSH6 IHC staining and molecular analysis demonstrated increasing allelic burden of two MSH6 frameshift variants (c.3261delC and c.3261dupC) in areas with MSH6 protein loss compared to areas where MSH6 was retained. Two colonic adenocarcinomas with heterogenous MLH1 staining showed no differences in sequence variants. In one of these cases, however, MLH1 was hypermethylated in the area of MLH1 loss. Another colon carcinoma with heterogenous PMS2 staining (but with retained MSH6) showed both MSH6 c.3261dupC and 3260_3261dupCC where PMS2 protein was lost and only c.3261dupC where PMS2 was retained. The gastric carcinoma showed complete loss of MSH6 in dysplastic foci, while the underlying invasive carcinoma showed retention of MSH6. Both these areas, however, were MSI‐high and showed the same MSH6 variant: c.3261delC. The gastric dysplasia additionally showed MSH6 c.3261dupC. In four of the five cases where MMR protein was lost, these areas were MSI‐high. Heterogenous MMR IHC (focal and/or zonal within the same tumour or between invasive and dysplastic preinvasive areas) is not always due to artefact and is invariably related to MSI‐high status in the areas of loss. An interesting aspect to this study is the presence of MSH6 somatic mutations irrespective of whether MSH6 IHC staining was intact or lost.
Keywords: adenocarcinoma, colorectal, gastric, mismatch repair proteins, immunohistochemistry, mismatch repair genes, next‐generation sequencing
Introduction
In 1996, Leach et al developed monoclonal antibodies that detected the MSH2 protein in DNA mismatch‐proficient cell lines 1 and this led to a series of investigations on the utility of immunohistochemical detection of DNA mismatch repair (MMR) proteins in the identification of colorectal tumours with microsatellite instability (MSI). The fundamental defect underlying MSI has been found to be abnormal DNA MMR function secondary to either germline mutation of one of several MMR genes, namely, MLH1, PMS2, MSH2 and MSH6 or abnormal methylation of the promoter of MLH1.
In Lynch syndrome, the major underlying mechanism for the development of a tumour with MSI is the presence of a germline mutation in one allele of an MMR gene, coupled with a somatic mutation that leads to the loss of function of the corresponding non‐mutant allele. In sporadic colorectal cases, on the other hand, an abnormality in immunohistochemistry (IHC) is most often caused by abnormal methylation of the promoter region of the MLH1 gene, which results in transcriptional silencing and no detectable protein production 2, 3.
The identification of the MSI phenotype in colorectal cancers carries profound significance, not just identifying patients at risk of Lynch syndrome but also prognostic information as MSI has been shown to have a strong association with better outcome 4. Furthermore, there is growing evidence that MSI carries therapeutic implications as well; high‐frequency MSI cancers may not respond to fluorouracil‐based chemotherapy 5, 6.
The MSI phenotype can be detected by polymerase chain reaction (PCR)‐based molecular tests, but many studies have supported the use of IHC as the screening method of choice prior to mutation analysis of the MMR genes, citing its simplicity, ready availability, reproducibility and equally informative nature 7, 8, 9. The use of IHC has now been recommended as the first‐line screening method in the work‐up of patients suspected to have an MMR gene mutation in order to identify patients for mutation analysis for prognostic and therapeutic purposes.
When interpreting MMR IHC, loss of protein expression is defined as a complete absence of nuclear staining within the tumour 3, 10, 11. Several studies have described heterogeneity or discordance of staining in both colorectal and endometrial cancer, with most of these tumours containing areas of weak staining or no staining admixed with areas with strong and diffuse staining 12, 13, 14, 15. There are data suggesting that non‐truncating and/or even truncating mutations of MMR genes may result in impaired functional activity without a complete absence of MMR proteins, especially with MLH1 3, 16, 17. Others have ascribed the ‘discrepant’ immunohistochemical findings to MSI and more extensive and/or heterogenous methylation of MLH1 18, 19, 20.
There is insufficient data currently available to offer clear guidelines on the interpretation of heterogeneous or discordant MMR IHC and, indeed, if there is a molecular basis to the staining pattern encountered. Thus, the primary goal of this study is to interrogate the molecular changes, if any, of the heterogeneity of MMR protein IHC staining in five gastrointestinal adenocarcinomas by interrogating MSI – and Lynch genes – (by next‐generation sequencing [NGS]) status in these areas of heterogenous MMR protein expression.
Materials and methods
Patient information
Institutional ethical approval was granted for the performance of this study (University Health Network Research Ethics ID:16‐5382.2).
The cases were selected based on the unusual heterogenous patterns noted from all gastrointestinal tract cancers tested for MMR over the 3‐year period 2015–2017 inclusive; 389 colorectal cancer resections were performed. Of the 389 cases, 82 received neoadjuvant chemoradiation and were excluded from the study. This left 307 colorectal cancer resections, and 233 of these cases were stained for MMR proteins. During the study period, reflex MMR IHC testing of all colorectal cancer resections in patients younger than 70 years of age was not standard procedure as it is now at our institution. Clinical data, including patients' age, gender and anatomical location of tumour, were obtained from pathology and hospital information databases. Cases in which any combination of the MMR proteins were unequivocally and completely retained or lost were excluded from the study cohort.
Four colorectal adenocarcinomas were identified (cases 1–4), and a patient with synchronous pancreatic and gastric adenocarcinomas (case 5) formed the basis of the study. These cases were selected based on their striking heterogenous MMR immunohistochemical findings, which prompted further investigation.
Histological analysis
Haematoxylin and eosin (H&E)‐stained sections of all of the resection specimens were reviewed. Several parameters, including tumour type and grade/differentiation, were recorded. Cases were pathologically staged according to the American Joint Committee on Cancer (AJCC) 7th edition pTNM cancer staging manual.
Immunohistochemical analysis
IHC for four MMR proteins (MLH1, PMS2, MSH2 and MSH6) was performed on formalin‐fixed, paraffin‐embedded tissue taken from representative sections of the resection specimens.
The IHC and clinical genetics laboratories at University Health Network are accredited, Ontario Ministry of Health‐approved reference laboratories for Lynch testing.
The antibody clones used were: MLH‐1 (clone ES05, Agilent Technologies Canada Inc.), MSH‐2 (clone G219‐1129, Becton Dickinson Biosciences, Canada), MSH‐6 (clone EPR3945, Abcam, Canada) and PMS‐2 (clone A16‐4, BD Biosciences). Pretreatment protocols were the same for all four antibodies: heat‐induced epitope retrieval and all stains were performed on a Dako Autostainer Link 48.
Normal colonic crypt epithelium adjacent to the tumour, lymphoid cells and stromal cells served as internal positive controls. In addition, on slide positive controls are a routine practice in our IHC laboratory. Technically, failed slides were excluded as the IHC process is automated and there are established standard operating procedures and quality assurance measures where every slide is vetted by a pathologist medical director and finally by the reporting pathologist. All cases in the study were internal to the institution and were fixed with 10% neutral‐buffered formalin and fixed for 24–48 h prior to sectioning as per departmental prescribed operating procedures. This ensured local uniform pre‐analytical standardisation (in the absence of universally accepted pre‐analytical guidelines for MMR IHC testing) for fixation and grossing protocols. Hence, the immunohistochemical findings were based on quality control‐verified bona fide stained cases for the MMR proteins.
Heterogeneous staining was defined according to the criteria established by Joost and colleagues as tumours showing intra‐glandular heterogeneity (strongly immunoreactive cells admixed with negative cells) and/or zonal loss (confluent areas of staining loss involving multiple adjacent glands) 14. IHC was repeated twice on each case using the same blocks. In all cases labelled as showing MMR heterogeneity according to the patterns described above, there was a distinct loss of nuclear staining in tumour cells, while normal stroma and lymphocytes showed strong nuclear staining in the same areas, thus excluding artefact and/or staining failure. An arbitrary cut‐off value of approximately 10% of the tumour showing either retention or loss of MMR proteins was used. This facilitated microdissection of the differently stained areas.
MSI molecular testing
For cases 1–4, MSI molecular testing was performed on normal tissue and on two areas of tumour in all colorectal adenocarcinoma cases. Areas of tumour with heterogeneous MMR IHC staining were separately macrodissected so that both IHC‐retained tumour and IHC‐lost tumour areas were separately isolated for MSI and NGS testing for each case. For case 5, molecular testing was performed on normal tissue, on the pancreatic tumour, on the gastric adenocarcinoma and on the gastric non‐invasive gastric dysplasia/adenocarcinoma. For all cases, a comparison of MSI molecular results was made between normal tissue and macrodissected areas of tumour.
The MSI molecular test was performed on genomic DNA extracted using a Maxwell 16 FFPE plus LEV DNA purification kit (Promega, Madison, WA, USA). The MSI molecular test consisted of a multiplex PCR of five microsatellite mononucleotide markers (NR‐21, BAT‐26, BAT‐25, NR‐24, MONO‐27) to assess MSI and two pentanucleotide markers (Penta C, Penta D) primarily used to detect potential contamination (MSI Analysis System Version 1.2; Promega). The amplified fragments were separated by capillary electrophoresis (ThermoFisher, Waltham, MA, USA) and analysed by GeneMapper software v4.1 (Applied Biosystems, Foster City, CA, USA). As described, the MSI molecular assay was performed on normal tissue and on two areas of tumour in all cases to assess MSI, with a comparison of marker repeats between normal tissue and macrodissected areas of tumour. MSI was defined as any marker with the highest peak shifted more than two base pairs when compared to the same marker in the normal sample. Instability in two or more of the five mononucleotide markers was classified as MSI‐high (MSI‐H), instability in one mononucleotide marker was classified as MSI‐low (MSI‐L) and absence of instability was classified as microsatellite‐stable (MSS).
Next‐generation sequencing
NGS was performed on normal tissue and on two areas of tumour for cases 1–4. Areas of tumour with heterogeneous MMR IHC staining were separately macrodissected so that both IHC‐retained tumour and IHC‐lost tumour areas underwent NGS testing.
NGS was performed on normal tissue, on the pancreatic tumour, on the gastric invasive tumour and on the gastric non‐invasive dysplastic mucosa for case 5. Areas of tumour with heterogeneous MMR IHC staining were separately macrodissected so that both IHC‐retained tumour and IHC‐lost tumour areas underwent NGS testing. Genomic DNA was extracted as described and NGS was performed on normal tissue and on two areas of tumour in all cases. Library preparation used a custom SureSelect XT probe library (Agilent, Santa Clara, CA, USA) targeting the exonic coding regions and flanking intronic regions (±5 bp) of 52 cancer‐related genes, including MLH1, MSH2, MSH6 and PMS2. Genomic DNA was sheared using focused ultrasonification (Covaris LE220, Woburn, MA, USA) and then indexed and enriched according to the SureSelectXT Target Enrichment System for paired‐end sequencing on the Illumina NGS platform (Illumina, San Diego, CA, USA). Libraries were visualised (TapeStation, Agilent) and sequencing was performed on the NextSeq500 (Illumina) platform.
NGS bioinformatics included alignment through the Burns‐Wheeler Alignment (BWA‐MEM), with processing and quality metrics by GATK and PICARD and variant calling by VarScan2. Variants not meeting laboratory‐defined quality metrics (read depth < 250, population frequency > 5% in the ExAC database, or variant allele fraction <5%) were removed from further analysis. Variant filtration was performed in the Alissa Clinical Informatics Platform (Agilent).
The interpretation of variants was according to prescribed guidelines 21. The predictions from the databases of a potential deleterious impact of the mutations detected in this study are highlighted in Table 1.
Table 1.
Prediction of deleterious potential of variants identified in this study using bioinformatics algorithms
| Variant | Polyphen‐2 | Mutation Taster | SIFT | Provean |
|---|---|---|---|---|
| PMS2 c.92T>C (p.Val31Ala) | Probably damaging | Disease causing | Deleterious | Deleterious |
| PMS2 c.1289C>T (p.Thr430Ile) | Benign | Polymorphism | Tolerated | Neutral |
| MSH6 c.95G>A (p.Gly32Asp) | Benign | Polymorphism | Tolerated | Neutral |
| MSH6 c.3261dupC (p.Phe1088Leufs*5) | – | Disease causing | – | – |
| MSH6 c.3261delC (p.Phe1088Serfs*2) | – | Disease causing | – | – |
| MSH6 c.3260_3261dupCC (p.Phe1088Profs*3) | – | Disease causing | – | – |
Polyphen‐2 – http://genetics.bwh.harvard.edu; SIFT – http://sift.bii.a‐star.edu.sg/; Provean – http://provean.jcvi.org/; Mutation Taster – http://www.mutationtaster.org
Sanger sequencing
Sanger sequencing was performed on two areas of tumour for two cases (cases 2 and 4) using 100 ng of genomic DNA with 0.2 mm of each primer, 0.5 U Platinum TAQ, ×10 PCR Buffer (Invitrogen, Burlington, Ontario, Canada) and 400 mm each dNTP. Cycling conditions were denaturation at 94 °C for 1 min, followed by 35 cycles of 30 s at 95 °C, 30 s at 55 °C and 1 min at 72 °C, with final elongation at 72 °C for 5 min. Sanger sequencing was performed using the BigDye Terminator v1.1 Cycle Sequencing Kit (ThermoFischer Scientific) and analysed using Sequence Analysis Software (Applied Biosystems). Primers used to confirm the MSH6 variant were forward (5′‐AGCCTCACTTTTACCCTCTCTTTT‐3′) and reverse (5′‐ACTGGCTGACTTTTATGTAACTGTG‐3′). Areas of tumour with heterogeneous MMR IHC staining were separately macrodissected, so both IHC‐positive tumour and IHC‐negative tumour areas underwent Sanger sequencing performed. Sanger sequencing was performed on the gastric invasive tumour and on the gastric dysplastic non‐invasive component from case 5.
MLPA and MLH1 methylation analysis
To observe exonic deletions and duplications, multiplex ligation‐dependent probe amplification (MLPA) was performed on the macrodissected tumour specimens, including methylation‐specific MLPA for MLH1 methylation analysis using the HhaI restriction enzyme. MLPA kits were purchased from MRC Holland and used according to manufacturer's recommendations. Analysis was performed using GeneMarker (v2.7.0) software. MLPA for MLH1 (P003) was used for cases 1 and 3, PMS2 (P008) for cases 1 and 2 and MSH6 (P072) for cases 4 and 5. MLH1 methylation (ME011) assessing MLH1 promoter regions was performed on cases 1 and 3.
As controls, patients with known methylation status (positive and negative) were enrolled.
Results
Patient information
Data regarding clinicopathological features are presented in Table 2.
Table 2.
Clinicopathological features
| Case | Age | Gender | Site | Histology | Differentiation |
|---|---|---|---|---|---|
| Case 1 | 85 | M | Caecum | Adenocarcinoma, mucinous differentiation (<50%) | Moderate |
| Case 2 | 84 | F | Ascending colon | Adenocarcinoma | Poor |
| Case 3 | 45 | F | Transverse colon | Adenocarcinoma | Moderate |
| Case 4 | 70 | M | Caecum | Adenocarcinoma mucinous differentiation (<50%) | Moderate |
| Case 5 | 62 | M | Head of pancreas; gastric antrum | Ductal pancreatic; gastric intestinal type | Both well differentiated |
The colorectal cancers occurred in two males and two females with an age range of 45–85 years (mean, 71 years; median, 77 years). None of the patients had a history of Lynch syndrome or family histories of cancer. None received preoperative neoadjuvant therapy.
The fifth patient (case 5; 62‐year‐old male) had two synchronous tumours: a pancreatic adenocarcinoma and a small gastric adenocarcinoma.
None of the patients had Lynch syndrome.
Histological analysis
Histological parameters and pTNM stage are outlined in Table 2. All four colorectal adenocarcinomas were right‐sided; three were well differentiated and one was poorly differentiated. Two cases had mucinous differentiation (with >10% but <50% of the tumour showing extracellular mucin in the stroma).
Case 5 comprised an invasive pancreatic ductal adenocarcinoma and a gastric adenocarcinoma, with associated overlying high‐grade dysplasia of the mucosal surface.
Immunohistochemical analysis
Heterogeneity of MMR IHC staining (with discordance in at least 10% of the tumour facilitating microdissection of the different areas) occurred in our series as a rare event, found in 2% of colorectal cancer resections (5 out of 233), which allowed for a more detailed case‐by‐case analysis.
Table 3 outlines the MMR immunohistochemical findings.
Table 3.
Mismatch repair immunohistochemistry
| Case | MLH1 | PMS2 | MSH2 | MSH6 |
|---|---|---|---|---|
| Case 1 | Heterogeneous* | Heterogeneous* | Retained | Retained |
| Case 2 | Complete loss | Heterogeneous* | Retained | Retained |
| Case 3 | Heterogeneous* | Retained | Retained | Retained |
| Case 4 | Complete loss | Complete loss | Retained | Heterogeneous* |
| Case 5: Pancreas cancer | Retained | Retained | Retained | Retained |
| Case 5: Gastric cancer | Complete loss | Complete loss | Retained | Retained |
| Case 5: Gastric dysplasia | Complete loss | Complete loss | Retained | Complete loss |
All cases with heterogeneous staining showed a combination of both intra‐glandular loss (strongly immunoreactive cells admixed with negative cells) and zonal loss (confluent areas of staining loss involving multiple adjacent glands.
Case 1 showed heterogeneous staining for MLH1 and PMS2, with intact staining of MSH2 and MSH6 proteins (Figure 1). The remaining three colon cases (cases 2, 3, 4) each demonstrated complete loss and/or heterogeneous staining for one MMR protein: either MLH1, PMS2, or MSH6, with intact staining of the other three MMR proteins. Case 2 showed complete loss of MLH1 and heterogeneous staining for PMS2 (Figure 2); case 3 heterogeneous staining for MLH1; and case 4 displayed complete loss of MLH1 and PMS2 and heterogeneous staining for MSH6 (Figure 3). The heterogeneous areas showed both intra‐glandular heterogeneity and zonal loss of MMR protein expression. Intra‐glandular heterogeneity was characterised by strongly immunoreactive cells admixed with negative cells, while zonal loss comprised confluent areas of staining loss involving multiple adjacent glands, accompanied by confluent areas of staining retention.
Figure 1.
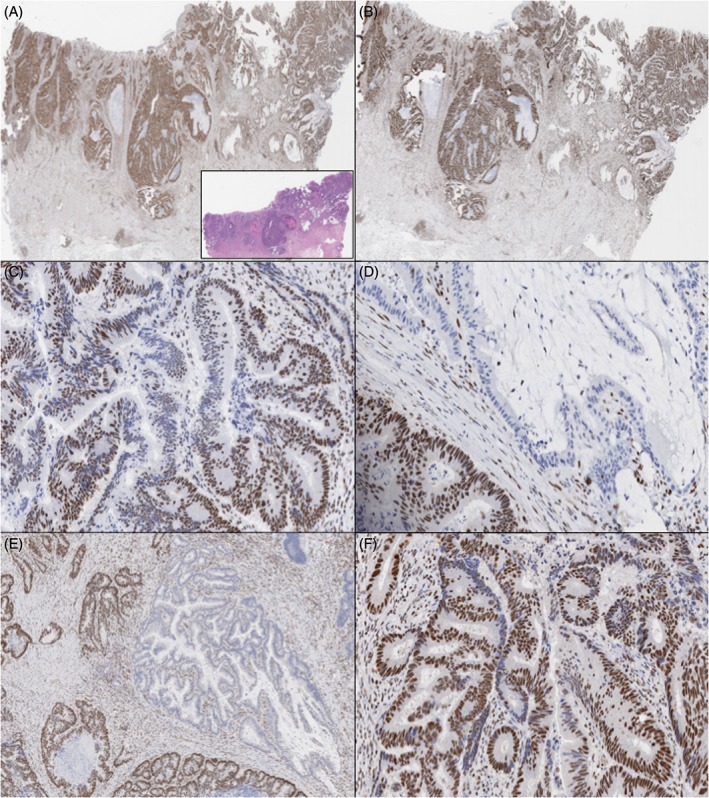
Case 1 (H&E, inset of A) showed intact MSH2 (A) and MSH6 (B), with heterogeneous staining of MLH1 (intra‐glandular loss in C and geographical/zonal loss in D) and PMS2 with both geographic or zonal loss (E) and intra‐glandular loss (F).
Figure 2.
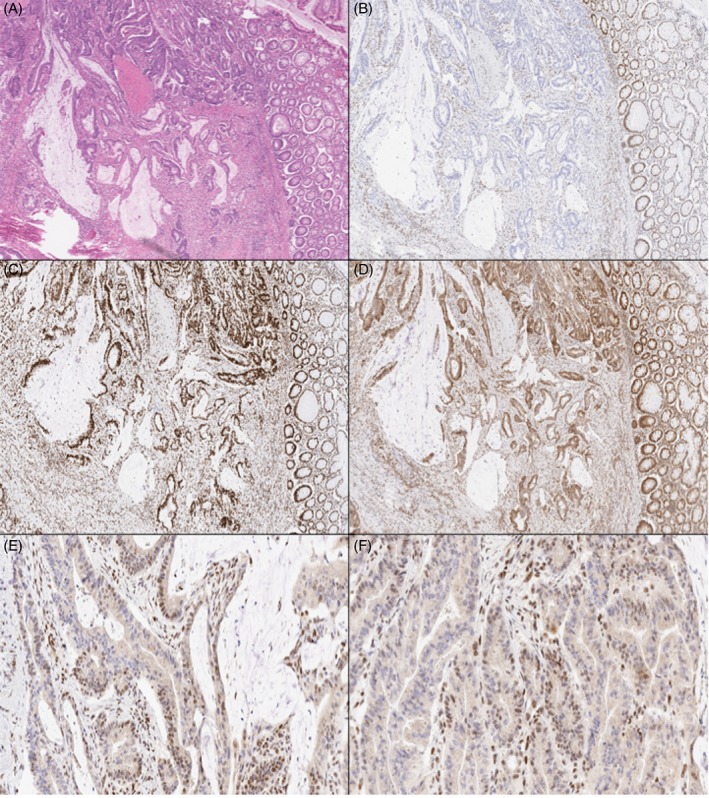
Case 2 (H&E, A) showed complete loss of MLH1 (B), retained MSH2 (C) and retained MSH6 (D), with heterogeneous staining of PMS2 with intra‐glandular (E) and geographical/zonal loss (F).
Figure 3.
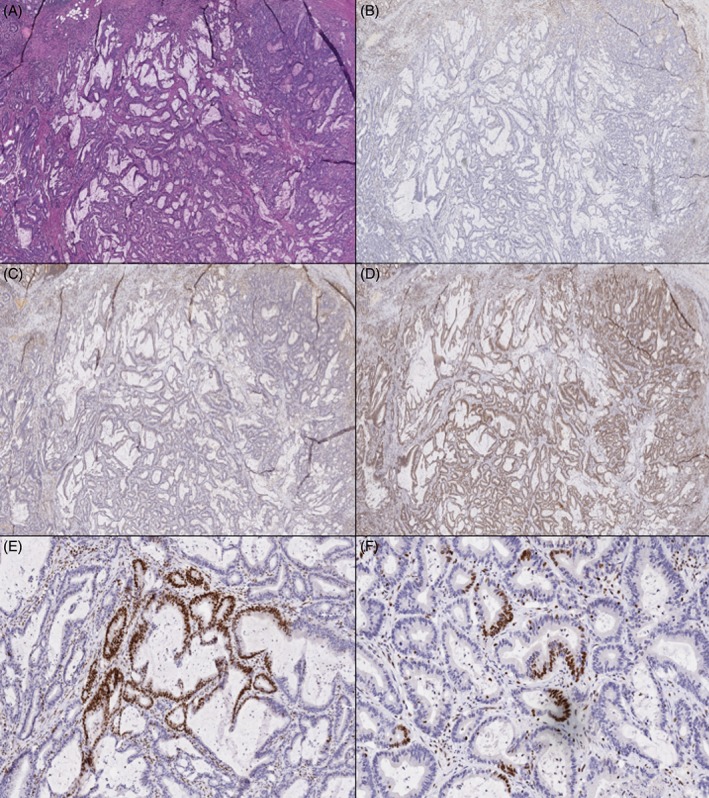
Case 4 (H&E, A) showed intact MLH1 (B), PMS2 (C) and MSH2 (D), with heterogeneous staining of MSH6 (E and F showing both geographic and intra‐glandular loss).
For case 5, the MMR IHC was as follows: pancreatic adenocarcinoma showed retention of all four MMR proteins; the gastric invasive adenocarcinoma demonstrated loss of MLH1 and PMS2 with retention of MSH2 and MSH6 (Figure 4), while the gastric dysplastic foci displayed loss of MLH1, PMS2 and MSH6, with retention of MSH2 (Figure 5).
Figure 4.
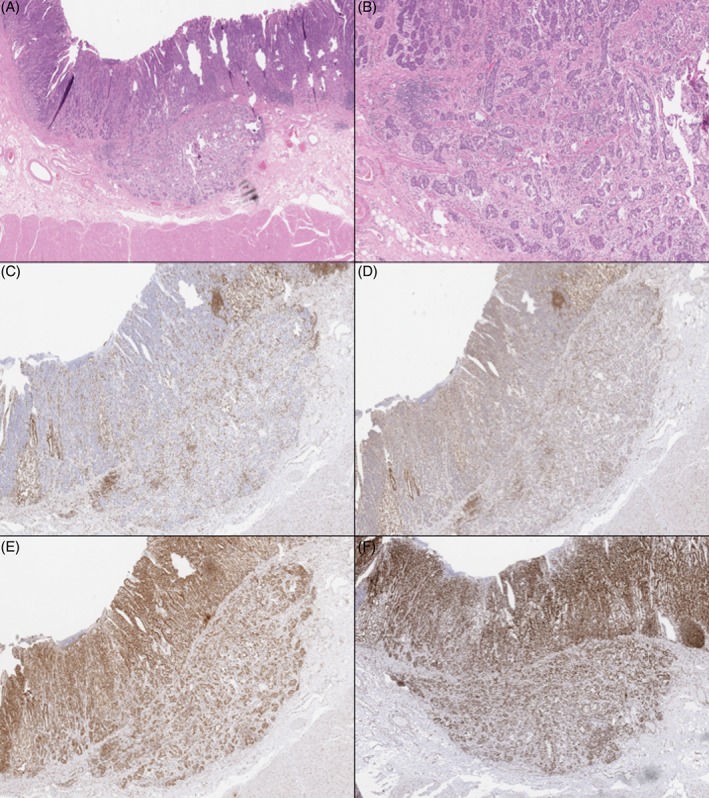
Case 5: The gastric invasive adenocarcinoma (H&E, A and B) showed complete loss of MLH1 (C) and PMS2 (D) and retention of MSH2 (E) and MSH6 (F). Occasional lymphoid aggregates are positive.
Figure 5.
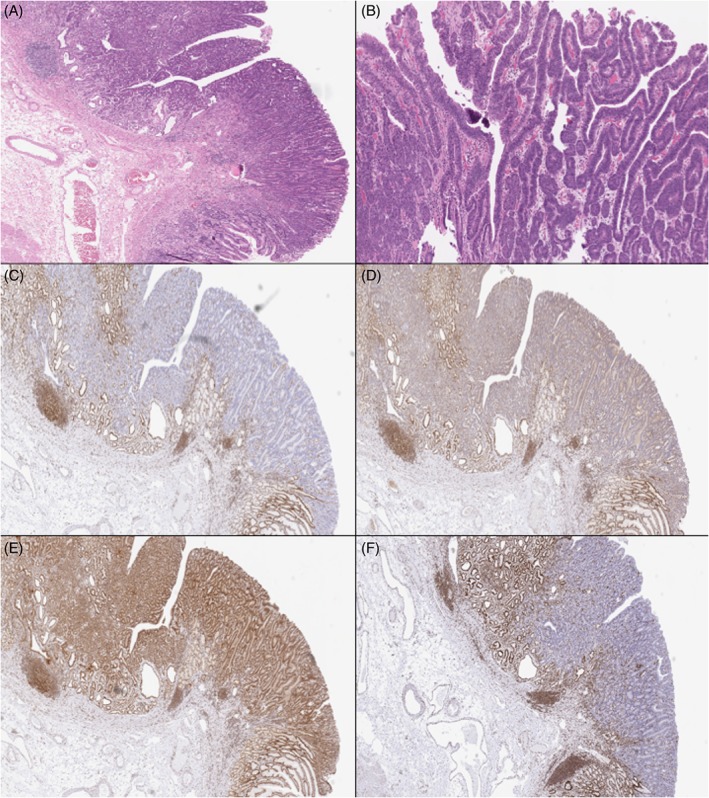
Case 5: The gastric dysplasia (H&E, A and B) showed complete loss of MLH1 (C), PMS2 (D) and MSH6 (F) and retention of MSH2 (E).
MSI testing
MSI testing results are presented in Table 4. In case 1 (heterogeneous MMR IHC for MLH1 and PMS2), the cancer was microsatellite stable (MSS) in IHC‐retained areas but MSI‐H in IHC‐lost areas. Cases 2 and 4 (heterogeneous MMR IHC for PMS2 and MSH6, respectively) were MSI‐H in both IHC‐retained and ‐lost areas.
Table 4.
MSI status and NGS analysis of MLH1, PMS2, MSH2 and MSH6 genes in FFPE tumour tissue and MLH1 methylation findings
| Case | MSI in MMR‐retained areas | MSI in MMR‐lost areas | NGS in MMR IHC‐retained regions | NGS in MMR IHC‐lost regions |
|---|---|---|---|---|
| Case 1 | MSS | MSI‐H (5/5) | No variants* | No variants*
MLH1 promoter hypermethylated (45%) |
| Case 2 | MSI‐H (5/5) | MSI‐H (5/5) |
MSH6 c.3261dupC (p.Phe1088Leufs*5) Allele Fraction: 19% PMS2 c.1289C>T (p.Thr430Ile) Allele Fraction: 24% PMS2 c.92 T>C (p.Val31Ala) Allele Fraction 28% |
MSH6 c.3260_3261dupCC (p.Phe1088Profs*3) Allele Fraction: 7% MSH6 c.3261dupC (p.Phe1088Leufs*5) Allele Fraction: 3% |
| Case 3 | MSS | MSS | No variants* | No variants* |
| Case 4 | MSI‐H (3/5) | MSI‐H (5/5) |
MSH6 c.3261delC (p.Phe1088Serfs*2) Allele Fraction:20% MSH6 c.3261dupC (p.Phe1088Leufs*5) Allele Fraction:10% |
MSH6 c.3261delC (p.Phe1088Serfs*2) Allele Fraction:34% MSH6 c.3261dupC (p.Phe1088Leufs*5) Allele Fraction:35% |
| Case 5: Pancreas cancer | MSI‐L (1/5) | Not applicable | No variants* | No variants* |
| Case 5: Gastric cancer | MSS | MSI‐H (5/5) | No variants* |
MSH6 c.3261delC (p.Phe1088Serfs*2) Allele Fraction: 22% |
| Case 5: Gastric dysplasia | MSS | MSI‐H (4/5) | No variants* |
MSH6 c.3261delC (p.Phe1088Serfs*2) Allele fraction: 17% MSH6 c.3261dupC (p.Phe1088Leufs*5) Allele Fraction: 13% MSH6 c.95G>A (p.Gly32Asp) Allele fraction:14% |
MSI‐H with number of markers showing instability out of a total of five possible markers indicated.
No Lynch syndrome‐associated germline variants were detected in the normal tissue from any of the five cases.
Case 3 (heterogeneous MMR IHC for MLH1) was MSS in both IHC‐retained and ‐lost areas. In case 5, the pancreatic ductal adenocarcinoma was MSI‐L; the invasive gastric adenocarcinoma and the gastric high‐grade dysplastic foci were MSS in the MMR IHC retained areas but MSI‐H in the MMR‐lost areas.
Variants identified by NGS
Table 4 outlines the results of NGS of MLH1, PMS2, MSH2 and MSH6 and MLH1 promoter methylation analysis. NGS did not identify any Lynch syndrome‐associated germline variants in the normal tissue from any of our five cases.
In case 1, no variants were detected in the tumours (heterogeneous MLH1 and PMS2 protein staining; MLH1 promoter hypermethylation was found in the area where MLH1 immunohistochemical staining was lost).
In case 2, an MSH6 variant (c.3261dupC; p.Phe1088Leufs*5) and two PMS2 missense variants (PMS2 c.1289C>T, p.Thr430Ile and PMS2 c.92T>C, p.Val31Ala) were present in the tumour area with retained expression of PMS2, which was MSI‐H. In the area with PMS2 expression loss (also MSI‐H), the MSH6 variant (c.3261dupC; p.Phe1088Leufs*5) and an additional MSH6 variant in the same location (c.3260_3261dupCC; p.Phe1088Profs*3) were also found. No evidence of the PMS2 variants was found in this region. In addition, no exonic deletions or duplications of PMS2 gene were detected by MLPA analysis (data not shown).
In case 3, MLH1 promoter methylation analysis was normal in both MMR protein‐retained and ‐lost foci.
In case 4, two MSH6 variants (MSH6 c. c.3261delC; p.Phe1088Serfs*2) and MSH6 c. (c.3261dupC; p.Phe1088Leufs*5) were present in both IHC‐positive and IHC‐negative tumour areas (heterogeneous MSH6 IHC staining pattern, both regions MSI‐H).
In case 5, the same MSH6 variant (MSH6 c.3261delC; p.Phe1088Serfs*2) was present in both the gastric adenocarcinoma and the gastric dysplasia, but an additional MSH6 variant (MSH6 c.95G>A; p.Gly32Asp) was also present in the surface dysplasia (both MSI‐H). No MSH6 variants were detected in the pancreatic invasive ductal adenocarcinoma (MSI‐L).
Sanger sequencing
Sanger sequencing confirmed the presence of the MSH6 variants in cases 2, 4 and 5 and is shown in Figure 6.
Figure 6.
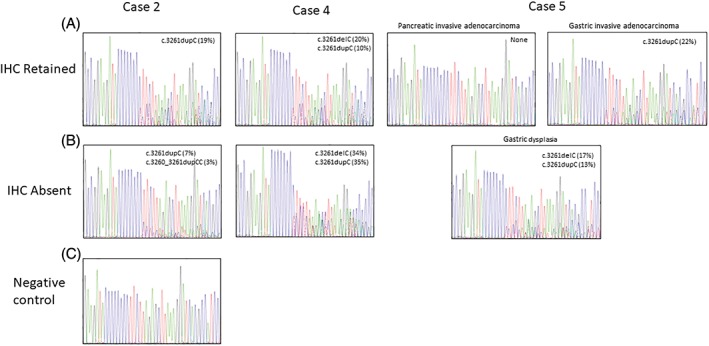
Sanger sequencing electropherograms of exon 5 variants found in MSH6 for cases 2, 4 and 5. Variant allele frequencies from NGS results are displayed in the top right corners of each case. (A) Sequencing results from regions with retained immunohistochemical staining. (B) Sequencing results from regions with lost immunohistochemical staining. (C) Normal FFPE control does not show any variants.
Discussion
MMR deficiency occurs ostensibly via 3 main mechanisms: (1) by somatic hypermethylation of MMR genes, most commonly of MLH‐1; (2) an inherited germline mutation of the MMR genes, typified by Lynch syndrome; and (3) double somatic mutations in MMR genes. The identification of the MSI phenotype in colorectal cancers carries clinical significance as it helps in the identification of patients at risk of Lynch syndrome, as well as providing both prognostic and therapeutic information for patients with sporadic tumours 4. Colorectal cancer patients with MSI‐H colorectal cancers have been shown to have improved prognosis compared to patients with MSS cancers.
IHC has been recommended as a first‐line screening method in the evaluation of the MMR status as well as serving as an excellent surrogate marker of MSI.
MMR IHC staining heterogeneity has been described previously, with most cases demonstrating areas of weak or no staining coexisting with areas with strong and/or diffuse staining 12, 13, 14, 15. These cases have, in general, been interpreted as retained MMR proteins. However, little attention has been paid to such heterogenous or discordant staining patterns, with no real molecular explanation for this phenomenon or its possible implications and several researchers have regarded such staining patterns as artefactual. It is important to ensure that pre‐analytical and IHC protocol standardisation caution has been exercised. While there are no internationally accepted guidelines for pre‐analytical handling and fixation of specimens, standardisation within each institution reduces the risk of IHC staining vagaries. All cases in this study were from within our institution, were fixed for 12–24 h in 10% neutral‐buffered formalin before grossing and were analysed by automated IHC with on‐slide positive and negative controls. Repetition of all cases with heterogenous IHC was also performed to mitigate artefact and false IHC staining.
The results of the molecular interrogation in our cases showed that the immunohistochemically heterogenous areas harboured different, and sometimes unique, molecular aberrations.
Two MSH6 variants (c.3261dupC and c.3260_3261dupCC) were present in the IHC‐lost tumour areas of case 2 (heterogeneous PMS2 by IHC, MSI‐H), while only the MSH6 c.3261dupC variant was detected in the IHC‐retained tumour areas (also MSI‐H). Two additional PMS2 variants (PMS2 c.1289C>T, p.Thr430Ile and PMS2 c.92T>C, p.Val31Ala) were present in the IHC‐retained tumour areas of case 2.
Within the same genomic loci of MSH6, variants c.3261delC and c.3261dupC were present in IHC‐retained and IHC‐lost tumour areas (both MSI‐H) of case 4 (heterogeneous MSH6 IHC).
In case 5, an MSH6 variant (c.3261delC; p.Phe1088Serfs*2) was identified in both the gastric invasive adenocarcinoma and the gastric dysplastic areas, with the dysplasia demonstrating an additional MSH6 variant (c.95G>A; p.Gly32Asp). Neither MSH6 variant was detected in the pancreatic‐invasive adenocarcinoma.
Although the MSH6 variants identified in the patients in this report were all somatic variants (as they were not identified in testing of normal adjacent tissue), these variants have also been reported previously in the germline context. MSH6 c.3261delC (p.Phe1088Serfs*2) has been described in multiple families in the literature 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31 and MSH6 c.3260_3261dupCC, p.Phe1088Profs*5 is listed as pathogenic in the germline context in ClinVar (1 submission). MSH6 c.3261dupC has been reported in three cases with Lynch‐associated cancers and other cancers (breast, colon, endometrial and renal cancers and colonic polyps) 14. In addition, in the somatic context, Akiyama et al identified MSH6 c.3261dupC in the somatic context in a patient with multiple colorectal tumours (confirmed not to be germline) 32, and Terui and colleagues reported it in one patient with colorectal cancer, with loss of MSH2 and MSH6 protein expression by IHC 33.
We postulate that the MSH6 c.3261delC and MSH6 c.3261dupC variants in the surface gastric dysplastic areas seen in case 5 resulted in the loss of MSH6 protein expression, while MSH6 IHC expression was maintained in the invasive component as only one variant (MSH6 c.3261delC) was present.
Case 3, which showed heterogenous MLH‐1 IHC but was microsatellite stable and did not show any variants by NGS, is more difficult to explain. IHC was repeated and heterogenous staining (intra‐glandular mainly with focal zonal loss) was consistently present. The NGS interrogation was limited to Lynch variants and other MMR genes not included in the panel may be operative in this case.
This study has demonstrated that the assumption that tumours with a heterogeneous MMR IHC staining pattern (in other words, retained and lost protein areas coexisting in the same tumour) represent retention of MMR proteins and, by extrapolation, intact MMR gene status and also artefactual IHC staining, is not necessarily true. These cases may indeed have variants in MMR genes, some of which may be clinically significant and which correlate with unstable status on MSI testing (MSI‐H). It is known that the MMR genes contain up to nine large intronic homopolymer sequences (over 10mer in length) that are in the proximity of exons and around splice sites 34.
Furthermore, it has been observed that approximately 5% of colorectal cancers that display retention of all four MMR proteins may indeed be MSI‐H 35. The rationale advanced to explain this discordance between IHC and molecular analysis is that the genes in question are abrogated (due to missense mutations), but the proteins emanating from these genes, although still detected by IHC, are functionally suboptimal and/or non‐functional.
It has been suggested that mutations, as evidenced in the consensus positions of the MSH6 gene in our series, generate aberrant transcripts resulting in protein expression loss and a consequent pathogenic affect. The MSH6 variants seen in this study in the homopolymeric region were confirmed by Sanger sequencing not to be an alignment or other sequencing error. In the instance where MLH1 and PMS2 proteins are lost and there are NO variants or methylation of MLH1, it is possible that other mechanisms are responsible and/or that areas not targeted by NGS are involved, such as in the promoter or intronic regions of MLH1.
MSI in a broader cancer context other than in colorectal cancer has been noted within several other types of cancer, most commonly endometrial and gastric adenocarcinomas. Furthermore, MSI‐H colorectal tumours have been shown to be more susceptible to immune‐enhancing therapies, like the programmed cell death 1‐inhibitor (PD‐1) drug pembrolizumab, which has been advocated for use in MSI‐H and/or MMR‐deficient unresectable and metastatic cancers. MSI‐H tumours have been shown to respond best to treatment with PD‐1 inhibitors, with good patient response and statistically significantly improved overall survival 36.
We advocate that tumours showing heterogenous areas of staining (loss of MMR IHC staining admixed with areas of strong and diffuse retained MMR protein expression) should not automatically be regarded as MMR IHC‐intact/‐retained cases or artefactual. For the purposes of excluding Lynch syndrome, if MSH2 protein is intact (and we have not observed heterogenous staining with this protein), then Lynch syndrome is extremely unlikely. After ensuring the fidelity of the IHC staining, these cases should be further investigated by molecular analysis, including MSI and potentially sequencing. The clinical significance of identifying heterogenous IHC staining patterns and tumour areas with differing MSI and molecular status needs further study but may define new tumour profiles and molecular mechanisms with therapeutic and prognostic implications.
In conclusion, we feel that the heterogeneity of MMR protein expression, especially with MSH6, is seen every so often by many who interpret MMR proteins. The aphoristic, gestalt reaction when confronted by such staining is to invoke an artefact effect and ask for the stain to be repeated. If the on‐slide and in‐built controls (stroma and lymphocytes) show retained staining, then the artefact is removed from the equation and the results of our study explain some of these cases with heterogenous staining. Clearly, not all the answers and explanations are at hand, but this does open new vistas for MMR IHC interpretation and implications.
Author contributions statement
AJM, J‐M C‐C, TS, SK‐R, SS, PS and RC interpreted the data (experimental and clinical) and contributed to the writing of the manuscript. SG carried out the experiments. TS performed the bioinformatics. All authors were involved in writing the paper and had final approval of the submitted and published versions.
No conflicts of interest were declared.
References
- 1. Leach FS, Polyak K, Burrell M, et al Expression of the human mismatch repair gene hMSH2 in normal and neoplastic tissues. Cancer Res 1996; 56: 235–240. [PubMed] [Google Scholar]
- 2. Kim HC, Kim CN, Yu CS, et al Methylation of the hMLH1 and hMSH2 promoter in early‐onset sporadic colorectal carcinomas with microsatellite instability. Int J Colorectal Dis 2003; 18: 196–202. [DOI] [PubMed] [Google Scholar]
- 3. Salahshor S, Koelble K, Rubio C, et al Microsatellite instability and hMLH1 and hMSH2 expression analysis in familial and sporadic colorectal cancer. Lab Invest 2001; 81: 535–541. [DOI] [PubMed] [Google Scholar]
- 4. Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite‐unstable colorectal cancer for pathologists. Diagn Pathol 2017; 12: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawakami H, Zaanan A, Sinicrope FA. Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol 2015; 16: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sargent DJ, Marsoni S, Monges G, et al Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil‐based adjuvant therapy in colon cancer. J Clin Oncol 2010; 28: 3219–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beamer LC, Grant ML, Espenschied CR, et al Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow‐up of abnormal results. J Clin Oncol 2012; 30: 1058–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kastrinos F, Syngal S. Screening patients with colorectal cancer for Lynch syndrome: what are we waiting for? J Clin Oncol 2012; 30: 1024–1027. [DOI] [PubMed] [Google Scholar]
- 9. Shia J, Ellis NA, Klimstra DS. The utility of immunohistochemical detection of DNA mismatch repair gene proteins. Virchows Arch 2004; 445: 431–441. [DOI] [PubMed] [Google Scholar]
- 10. Rodriguez‐Bigas MA, Boland CR, Hamilton SR, et al A National Cancer Institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997; 89: 1758–1762. [DOI] [PubMed] [Google Scholar]
- 11. Ward R, Meagher A, Tomlinson I, et al Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut 2001; 48: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graham RP, Kerr SE, Butz ML, et al Heterogenous MSH6 loss is a result of microsatellite instability within MSH6 and occurs in sporadic and hereditary colorectal and endometrial carcinomas. Am J Surg Pathol 2015; 39: 1370–1376. [DOI] [PubMed] [Google Scholar]
- 13. Halvarsson B, Lindblom A, Rambech E, et al Microsatellite instability analysis and/or immunostaining for the diagnosis of hereditary nonpolyposis colorectal cancer? Virchows Arch 2004; 444: 135–141. [DOI] [PubMed] [Google Scholar]
- 14. Joost P, Veurink N, Holck S, et al Heterogenous mismatch‐repair status in colorectal cancer. Diagn Pathol 2014; 9: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Renkonen E, Zhang Y, Lohi H, et al Altered expression of MLH1, MSH2, and MSH6 in predisposition to hereditary nonpolyposis colorectal cancer. J Clin Oncol 2003; 21: 3629–3637. [DOI] [PubMed] [Google Scholar]
- 16. Raevaara TE, Vaccaro C, Abdel‐Rahman WM, et al Pathogenicity of the hereditary colorectal cancer mutation hMLH1 del616 linked to shortage of the functional protein. Gastroenterology 2003; 125: 501–509. [DOI] [PubMed] [Google Scholar]
- 17. Wahlberg SS, Schmeits J, Thomas G, et al Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ‐line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res 2002; 62: 3485–3492. [PubMed] [Google Scholar]
- 18. Kato A, Sato N, Sugawara T, et al Isolated loss of PMS2 immunohistochemical expression is frequently caused by heterogenous MLH1 promoter hypermethylation in Lynch syndrome screening forendometrial cancer patients. Am J Surg Pathol 2016; 40: 770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pai RK, Plesec TP, Abdul‐Karim FW, et al Abrupt loss of MLH1 and PMS2 expression in endometrial carcinoma. Molecular and morphologic analysis of 6 cases. Am J Surg Pathol 2015; 39: 993–999. [DOI] [PubMed] [Google Scholar]
- 20. Watson N, Grieu F, Morris M, et al Heterogeneous staining for mismatch repair proteins during population‐based prescreening for hereditary nonpolyposis colorectal cancer. J Mol Diagn 2007; 9: 472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baglietto L, Lindor NM, Dowty JG, et al Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst 2010; 102: 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dymerska D, Serrano‐Fernández P, Suchy J, et al Combined iPLEX and TaqMan assays to screen for 45 common mutations in Lynch syndrome and FAP patients. J Mol Diagn 2010; 12: 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lavoine N, Colas C, Muleris M, et al Constitutional mismatch repair deficiency syndrome: clinical description in a French cohort. J Med Genet 2015; 52: 770–778. [DOI] [PubMed] [Google Scholar]
- 24. Meric‐Bernstam F, Brusco L, Daniels M, et al Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann Oncol 2016; 27: 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Plaschke J, Engel C, Krüger S, et al Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German hereditary nonpolyposis colorectal cancer consortium. J Clin Oncol 2004; 22: 4486–4494. [DOI] [PubMed] [Google Scholar]
- 26. Richards S, Aziz N, Bale S, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sjursen W, Haukanes BI, Grindedal EM, et al Current clinical criteria for Lynch syndrome are not sensitive enough to identify MSH6 mutation carriers. J Med Genet 2010; 47: 579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Susswein LR, Marshall ML, Nusbaum R, et al Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next‐generation cancer panel testing. Genet Med 2016; 18: 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tavakkol Z, Keller JJ, Furmanczyk PS, et al Germline mutation in MSH6 associated with multiple malignant neoplasms in a patient with Muir‐Torre syndrome. J Clin Oncol 2012; 30: e195–e198. [DOI] [PubMed] [Google Scholar]
- 30. van der Post RS, Kiemeney LA, Ligtenberg MJ, et al Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet 2010; 47: 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wijnen J, de Leeuw W, Vasen H, et al Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat Genet 1999; 23: 142–144. [DOI] [PubMed] [Google Scholar]
- 32. Akiyama Y, Sato H, Yamada T, et al Germ‐line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res 1997; 57: 3920–3923. [PubMed] [Google Scholar]
- 33. Terui H, Tachikawa T, Kakuta M, et al Molecular and clinical characteristics of MSH6 germline variants detected in colorectal cancer patients. Oncol Rep 2013; 30: 2909–2916. [DOI] [PubMed] [Google Scholar]
- 34. Castillejo MI, Castillejo A, Barbera VM, et al Analytical challenges in the genetic diagnosis of Lynch syndrome‐difficult detection of germ‐line mutations in sequences surrounding homopolymers. J Int Fed Clin Chem Lab Med 2016; 27: 77–83. [Google Scholar]
- 35. Ryan E, Sheahan K, Creavin B, et al The current value of determining the mismatch repair status of colorectal cancer: a rationale for routine testing. Critical Rev Oncol Hematol 2017; 116: 38–57. [DOI] [PubMed] [Google Scholar]
- 36. Le DT, Uram JN, Wang H, et al PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 2015; 372: 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]