

Abstract
Cyanobactin biosynthetic enzymes have exceptional versatility in the synthesis of natural and unnatural products. Cyanobactins are ribosomally synthesized and post-translationally modified peptides (RiPPs) synthesized by multistep pathways involving a broad suite of enzymes, including heterocyclases / cyclodehydratases, macrocyclases, proteases, prenyltransferases, methyltransferases, and others. Here, we describe the enzymology and structural biology of cyanobactin biosynthetic enzymes, aiming at the twin goals of understanding biochemical mechanisms and biosynthetic plasticity. We highlight how this common suite of enzymes may be utilized to generate a large array or structurally and chemically diverse compounds.
Keywords: natural products, cyanobactins, RiPP biosynthesis, cyanobactin enzymes
1. Introduction
Cyanobactins are common peptide natural products found in diverse cyanobacterial taxa (Figure 1) (Donia, Ravel, & Schmidt, 2008; Leikoski, Fewer, & Sivonen, 2009). They are distributed through many different genera in the cyanobacterial lineage and have been estimated to occur in perhaps one in five strains (Donia & Schmidt, 2011; Leikoski et al., 2009). They are found in cyanobacteria that form massive oceanic blooms (Sudek, Haygood, Youssef, & Schmidt, 2006), in terrestrial and freshwater cyanobacteria (Ziemert et al., 2008), in symbionts of marine animals (Schmidt et al., 2005), in food products (Donia & Schmidt, 2011) and elsewhere. Overall, they represent a common but as yet to be understood component of the Earth’s biochemistry.
Figure 1. Representative cyanobactins.
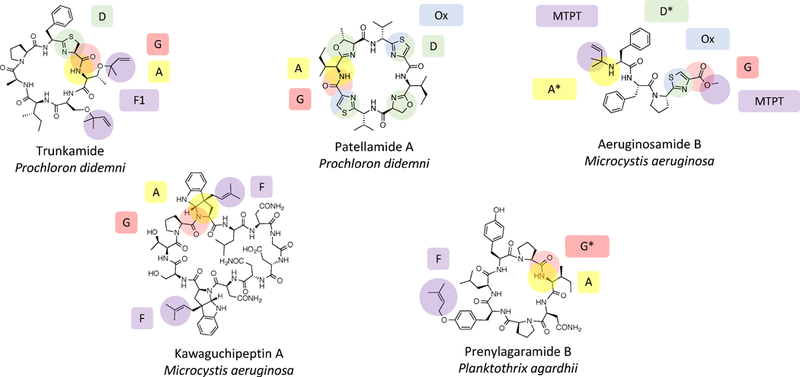
Posttranslational modifications are indicated by colors, tied to enzymes that catalyze each transformation. * indicates hypothetical reaction, all others are experimentally defined.
What are cyanobactins?
The cyanobactins are defined not by their structural features, but rather by similarities in their biosynthesis (Arnison et al., 2013). All cyanobactins are ribosomally synthesized and posttranslationally modified (RiPP) natural products, and, as such are derived from a ribosomally synthesized precursor peptide, which is then posttranslationally modified by enzymes to produce elaborate products. The precursor peptide contains a core sequence that is found in the final natural product, flanked by leader and/or follower sequences that are not retained. Usually, the larger precursor peptide undergoes proteolysis to release the mature core, the bioactive natural product. Cyanobactins are defined as derived from a peptide similar to PatE, the precursor from the patellamide (pat) biosynthetic pathway. In addition, cyanobactins are synthesized by at least one subtilisin-like protease from the PatG family. Initially, cyanobactins were defined as being N-C circular, via the action of PatG-like macrocyclizing proteases, but several linear members have also been identified.
How are cyanobactins made?
Despite this broad biochemical definition, cyanobactins share several structural features that make them recognizable. All known cyanobactins contain a heterocycle at the C-terminus of the core peptide (either proline or an azol(in)e derived from cysteine, serine or threonine) (Donia & Schmidt, 2010, 2011; Leikoski et al., 2013). Most members of the family are circular via ligation of their N and C termini. Commonly, they contain either heterocycles or isoprene units, and sometimes both. The azoline heterocycles may be further oxidized to thiazole or oxazole. A small subset of cyanobactins includes linear compounds that are terminated with isoprene on the N-terminus and methyl ester on the C-terminus (Leikoski et al., 2013). Other posttranslational modifications are known beyond this core set, and there are still several proteins and domains with no defined function. Together this common set of enzymes produces a vast array of structural diversity.
What are the applications of cyanobactins?
Because they are synthesized from genetic (i.e. ribosomal) precursors, RiPPs in general are thought to provide good platforms for biosynthetic engineering (Burkhart, Kakkar, Hudson, van der Donk, & Mitchell, 2017; Ozaki et al., 2017; Sardar & Schmidt, 2016; Zhang, Li, & Kelly, 2016). Indeed, there is a broad range of mutability evident in nature, and the tolerance of cyanobactin biosynthetic pathways for a range of substrates makes the system useful in biotechnology. Because of this large substrate tolerance, cyanobactins have seen many applications in the synthesis of designed, engineered compounds. Such biotechnological applications, and the underlying ideas about biosynthetic plasticity, have been reviewed recently (Gu & Schmidt, 2017).
What do cyanobactins do in nature?
Beyond the application to synthetic biology, cyanobactins are also of interest for their intrinsic biological activities (Donia & Schmidt, 2010). Several cyanobactins chelate metals (Bertram & Pattenden, 2007; Comba et al., 2017), while others block P-glycoprotein (Aller et al., 2009; Williams & Jacobs, 1993). Others are cytotoxic (Degnan et al., 1989), and some may even block potassium channels (Tianero et al., 2016). How, and indeed whether, these activities relate to the natural role(s) of cyanobactins remains a mystery.
2. Biosynthetic diversity of the cyanobactin family
General biosynthetic pathway.
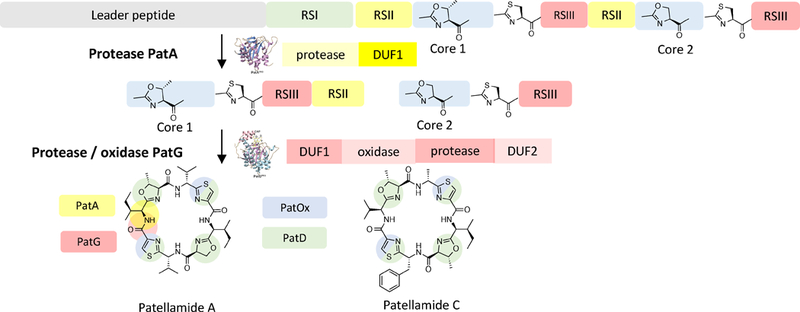
The first cyanobactin biosynthetic pathway to be characterized was that for patellamides A and C (pat), so the biosynthetic schemes for cyanobactins follow pat nomenclature (see Text Box) (Schmidt et al., 2005). The canonical biosynthetic pathway is thought to proceed in the following order (Figure 2). The precursor peptide, e.g. PatE, is first synthesized on the ribosome. Commonly, PatE and relatives contain more than one core peptide, flanked by enzyme recognition sequences (RSs), where the core peptides are known as “cassettes”, highlighting their repetitive organizational structure. The enzymatic modifications occur at the cassettes. For example, a PatE homolog (i.e., TruE3) containing three cassettes can encode three different natural products of different amino acid sequences. Subsequently, if the natural products contain heterocycles, the heterocyclase, e.g. PatD, acts on PatE to produce a linear peptide product that contains azolines. A subtilisin-like serine protease, e.g. PatA, then liberates free N-termini in each cassette of PatE. A second subtilisin-like serine protease, PatG, cleaves the C-termini of the core peptide in each cassette, usually in tandem with N-C circularization to yield cyclic peptides. Other enzymatic tailoring reactions usually take place after this core set of modifications is installed. In pat and related pathways, oxidation by the oxidase domain of PatG is one such potentially late-stage modification. Sometimes, the pathways may diverge from this order or exhibit greater complexity.
Text Box: Cyanobactin Terminology.
Substrates in Cyanobactin Biosynthesis
E (e.g., PatE): A precursor peptide encoding the substrates for cyanobactin biosynthetic enzymes.
Precursor Peptide: A peptide substrate in ribosomal peptide biosynthesis that encodes the following elements:
Leader Peptide: The portion of a precursor peptide that precedes the core peptide, and that is cleaved by proteolysis to afford the mature natural product.
Core Peptide: The portion of the precursor peptide that directly encodes the natural product.
Recognition Sequence (RS): An element found in some precursor peptides that binds to an enzyme and thus directs the biosynthetic process, but that is not part of the core peptide or the mature natural product.
Cassette: A core peptide and its flanking recognition sequences, used when multiple core peptides are present in a single precursor. For example, in cyanobactin biosynthesis a cassette consists of RSII-core peptide-RSIII.
Follower Peptide: An element found in some precursor peptides that follows a core peptide. This term is ambiguous in cyanobactin biosynthesis because of the presence of multiple cassettes in most precursor peptides, and thus “recognition sequence” is preferred.
Proteins in Cyanobactin Biosynthesis
A (e.g., PatA): Protease proteins responsible for cleaving the leader peptide and liberating free N-termini for cyclization. They recognize RSII and contain 2 domains: an S8A protease domain and a DUF.
B and C (e.g., PatB and PatC): Short proteins with no known function and no similarity to proteins of known function.
D (e.g., PatD): Proteins responsible for heterocyclizing cysteine (and in some cases serine/threonine) to produce azolines. They recognize RSI and contain 3 domains: a substrate recognition domain, an E1-like domain, and a YcaO domain.
F (e.g., PatF): Some variants of these proteins have no known function, while others are single domain ABBA prenyltransferases that react with serine/threonine, tyrosine, tryptophan, or the N-terminus.
G (e.g., PatG): Macrocyclases that recognize and cleave RSIII in tandem with circularization. The minimum domain set consists of N-terminal and C-terminal DUFs flanking an S8A protease responsible for circularization or hydrolysis of RSIII. Some variants also contain an oxidase domain that converts azolines to azoles, while others contain domains such as methyltransferases with no known function.
Figure 2. Canonical cyanobactin biosynthetic pathway.
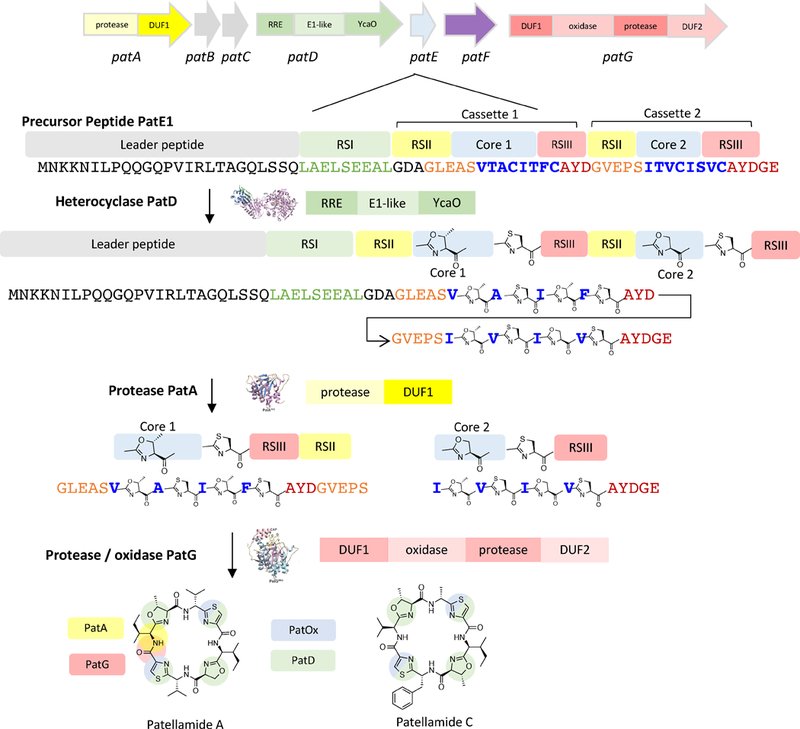
Top: The pat pathway to patellamides, with an expansion showing the amino acid sequence of PatE1, the substrate for biosynthetic enzymes. The precursor peptide substrate may be demarcated as described in the Text Box. The order of the modification occurs such that heterocyclization occurs first, followed by proteolysis and macrocyclization. Late stage tailoring may further elaborate the modified peptides.
Recognition sequences (RSs) in precursor peptides.
RSs are short sequence elements in PatE-like precursor peptides that are paired with each enzyme (Figure 3). As recruitment of the precursor depends only on the conserved RS, rather than the hypervariable core sequence, the modification enzymes are tolerant of diverse core sequences. In patellamide biosynthesis, Recognition sequence I (RSI) binds to and directs the PatD heterocyclase (Donia & Schmidt, 2011; Koehnke et al., 2015), while RSII and RSIII direct the activities of PatA and PatG proteases, respectively (Lee, McIntosh, Hathaway, & Schmidt, 2009). During the course of biosynthesis, the conserved RS elements are excised leaving a natural product peptide that consists only of the hypervariable sequence. For example, of the 29 variants of PatE that were initially described, all had identical sequences except in the cores, resulting in products of different chemical structures (Donia et al., 2006). The model of RS binding is accurate but oversimplified due to the intrinsic binding energy of the core peptide.
Figure 3. Recognition sequences and precursor peptides.
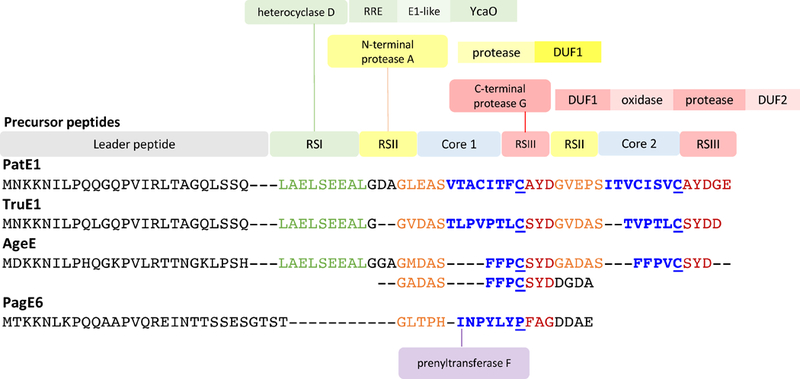
Shown are example sequences of cyanobactin precursors, with color-coded RSs tied to modifying enzymes. The domain architecture of each enzyme is laid out adjacent to the enzyme name. Prenyltransferase does not require a recognition sequence. Many cyanobactin precursor peptides contain multiple core sequences that are processed into different products.
Proteins of unknown function.
Even within the well-characterized pat pathway, there are several proteins or domains with no known function (Agarwal, Pierce, McIntosh, Schmidt, & Nair, 2012; Lee et al., 2009; Schmidt et al., 2005). PatA and PatG proteases contain homologous C-terminal domains of unknown function (DUFs). Additionally, PatG and relatives contain an uncharacterized N-terminal domain that flanks the oxidase domain. patF encodes an ABBA prenyltransferase-like protein that is not required for biosynthesis in vitro but is required for heterologous expression in Escherichia coli (Bent et al., 2013; Donia et al., 2006). PatB and PatC are two further proteins that do not prevent synthesis in vivo or in vitro but negatively affect synthesis in E. coli if removed (by reducing yield, for example) (Donia et al., 2006; Houssen et al., 2014). B-, C-, and F-like proteins are common in cyanobactin pathways.
Two basic biosynthetic subdivisions.
Following pat, many other types of cyanobactin gene clusters have been described (Donia et al., 2008; Donia & Schmidt, 2011). The major division between cyanobactin subtypes is those with heterocyclases and those that lack heterocyclases. When the heterocyclase is present, precursor peptides are cleaved on the C-terminal side at azol(in)e heterocycles derived from cysteine, serine, or threonine. This heterocycle is required for cleavage or macrocyclization by PatG-like enzymes. The structure of the precursor peptide reflects this division: in pathways containing the heterocyclase, RSI is found in the precursor peptide, where it directs heterocyclization of the core (Figure 3). However, in pathways where the heterocyclase is absent, the precursor peptide lacks RSI. The site of macrocyclization is still a heterocycle, but instead of azol(in)e, it is the amino acid proline.
Cassette duplication in precursor peptides.
PatE relatives vary in sequence, RS content, and in the variability of the core peptide (Donia et al., 2006; Donia & Schmidt, 2011). In addition, other PatE-like precursor peptides may vary in number of cassettes, from as few as one to as many as ten (Leikoski et al., 2013), and in copy number of PatE-like sequences in the genome. When multiple cassettes are present, they can each encode a different natural product, or potentially multiple copies of the same natural product, or mixtures thereof. When multiple precursors are present, they usually contain different cores, and several may be nonfunctional pseudogenes (Donia & Schmidt, 2011; Leikoski et al., 2009). This potentially reflects a duplication-divergence-recombination pathway to functional variation in cyanobactin natural products. For example, multiple precursor peptides may recombine to generate highly conserved RS sequences paired with new cores.
Other enzymes.
Cyanobactin pathways also often include prenyltransferases, methyltransferases, and/or other enzymes. PatF-like prenyltransferases add dimethylallyl pyrophosphate (DMAPP) or, occasionally, geranyl pyrophosphate (GPP) to the side chains of the core peptide (Leikoski et al., 2012). The prenyltransferases are known to prenylate in either the forward or reverse orientation in respect to DMAPP. TruF is specific to serine and threonine O-prenylation (Donia et al., 2008; Sardar, Lin, & Schmidt, 2015; Tianero, Donia, Young, Schultz, & Schmidt, 2012; Tianero et al., 2016), while other variants prenylate tyrosine (Hao et al., 2016; McIntosh, Donia, Nair, & Schmidt, 2011; McIntosh, Lin, Tianero, & Schmidt, 2013) or tryptophan (Parajuli et al., 2016). While each prenyltransferase appears to be relatively selective for the amino acid, orientation, and isoprene donor, there is much less selectivity as far as peptide structure, and no known associated RS (Hao et al., 2016; McIntosh et al., 2011). One variant homolog carries out prenylation of the N-terminus, rather than the side chain, on peptide substrates (Leikoski et al., 2013; Sardar et al., 2017).
Some pathways contain oxidase enzymes that convert thiazoline, and sometimes also oxazoline, into thiazole and oxazole, respectively (Donia & Schmidt, 2010). Methyltransferases are also very common within the genetic organization of cyanobactin pathways, but thus far only one has been characterized (Donia & Schmidt, 2011; Leikoski et al., 2009; Leikoski et al., 2013; Sardar et al., 2017).
Unusual cyanobactin pathways.
The tru biosynthetic pathway to trunkamide has several peculiarities in comparison to other cyanobactin pathways (Donia, Fricke, Ravel, & Schmidt, 2011; Donia et al., 2008). Like pat, tru is found in symbiotic Prochloron living in animals. The products of pat pathway are heterocyclic at cysteine, serine and threonine, whereas the tru products are heterocyclic only at cysteine and reverse prenylated on serine and threonine. tru is nearly identical to pat on the 5’- and 3’-ends of the cluster, and often the clusters are 100% identical in those regions. These nearly identical genes and intergenic sequences include pat/truA, pat/truB, pat/truC, the N-terminal domain of pat/truD, and the protease and DUF of pat/truG. Where the pathways are not identical, the resulting enzyme activities explain the differences in chemistry. Notably, in tru, the PatF-like protein TruF1 from the tru pathway carries out O-prenylation on serine and threonine residues, whereas PatF itself shows no activity (Sardar, Lin, et al., 2015; Tianero et al., 2016).
Another divergent pathway is that involved in the biosynthesis of linear cyanobactins, such as aeruginosamides (Leikoski et al., 2013; Sardar et al., 2017). In the age pathway, the PatG homolog AgeG produced a linear peptide product, rather than performing macrocyclization. A bifunctional methyl/prenyl transferase protects the N- and C- termini with prenyl and methyl groups, respectively.
Stereochemistry.
Cyanobactins characterized to date contain all L-amino acids, except in or adjacent to the C=N bond of azoline heterocycles, where epimerization can occur. This process is known to be spontaneous (e.g., not enzyme catalyzed), and occurs after azoline formation and macrocyclization (Donia et al., 2008; Donia, Ruffner, Cao, & Schmidt, 2011; McKeever & Pattenden, 2003; Tianero et al., 2012; Tianero et al., 2016). It is likely caused by two factors: 1) the intrinsic chemistry of azoline heterocycles; 2) the thermodynamics of the macrocycle. The C=N bond makes adjacent protons more acidic due to the potential for transient double bond migration. Macrocyclic cyanobactins are known to adopt specific shapes (Donia & Schmidt, 2010). It is likely that epimerization allows the macrocycles to adopt the thermodynamically preferred conformation.
It was long presumed that epimerization is spontaneous, rather than enzymatic (Wipf & Uto, 2000; Zabriskie, Foster, Stout, Clardy, & Ireland, 1990). Because of the facile inversion of configuration that occurs adjacent to thiazoline, the epimerization process has caused some vexation in the field, notably in the assignment of the phenylalanine configuration in trunkamide (Wipf & Uto, 2000). Evidence in favor of non-enzymatic epimerization was achieved during heterologous expression of trunkamide and certain patellins (Donia et al., 2008; Donia, Ruffner, et al., 2011; Tianero et al., 2012; Tianero et al., 2016). Notably, in trunkamide, the L-Phe isomer is observed first, whereupon it gradually transforms into the D-isomer over hours to days either in vivo or after organic extraction (Donia et al., 2008; Donia, Ruffner, et al., 2011). Similarly, subsequent in vitro studies of patellamide biosynthesis revealed an identical process, where heterocyclization occurs following thiazoline formation and macrocyclization but before oxidation to thiazole (Houssen et al., 2014). During the expression of hundreds of analyzed trunkamide and patellin derivatives, a doubling of HPLC-MS derived extracted ion chromatographic peaks is commonly observed that later resolves into single peaks, presumably because spontaneous epimerization converts L to a D-L mixture, followed by all D adjacent to thiazoline (Ruffner, Schmidt, & Heemstra, 2015; Tianero et al., 2012). It is also possible that, in some substrates, epimerization may be fast enough to occur on the macrocyclase.
3. Heterocyclase
a. Discovery and initial characterization.
The prevalence of peptide-based secondary metabolites that contain oxazole and thiazole heterocycles was appreciated from the initial discovery and structural characterization of several classes of natural products that bear such modification. The thiopeptide antibiotics nosiheptide (Pascard, Ducruix, Lunel, & Prange, 1977) and thiostrepton (Dutcher & Vandeputte, 1955) were among the earliest azolic compounds discovered, and metabolic labeling studies were consistent with a ribosomally synthesized peptide as the biosynthetic precursor (Mocek et al., 1989). Studies on the DNA gyrase inhibitor microcin B17, guided by identification of the corresponding biosynthetic gene cluster, identified an unmodified, genetically encoded peptide as the precursor for this molecule (Davagnino, Herrero, Furlong, Moreno, & Kolter, 1986). Subsequent characterization of the post-translational modifications (Genilloud, Moreno, & Kolter, 1989; San Millan, Kolter, & Moreno, 1985), and partial in vitro reconstitution studies (Li, Milne, Madison, Kolter, & Walsh, 1996) identified a heterotrimeric complex of three polypeptides (McbB/C/D) that carried out post-translational modifications on Cys, Ser, or Thr residues on the precursor peptide to install the thiazole/oxazole heterocycles found in the final product. Briefly, McbB (a member of the E1-ubiquitin activating-like gene family) and McbD (an ortholog of the E. coli YcaO gene product) were proposed to promote the heterocyclization of nucleophile-containing residues in the precursor peptide, in an ATP-dependent manner, to yield the corresponding azolines. More robust reconstitution experiments firmly established that the YcaO protein catalyzed the ATP-dependent backbone amide activation to facilitate heterocyclization on the peptide substrate engaged by the E1-like protein (Dunbar, Melby, & Mitchell, 2012).
Patellamide cyanobactins are heterocyclic at cysteine, serine, and threonine. The patellamide (pat) pathway was identified by sequencing and expressed in E. coli (Schmidt et al., 2005). Although the patD gene lacked similarity to previously characterized proteins, it was predicted to encode an adenylating enzyme–hydrolase hybrid based upon a short series of ~10 residues similar to the ATP grasping region of an otherwise unrelated protein from microcin C7 biosynthesis (Gonzalez-Pastor, San Millan, Castilla, & Moreno, 1995). It is now known that PatD represents a class of three domain heterocyclases found in many different types of organisms (Lee et al., 2008). Despite the absence of sequence similarity, PatD and relatives are fusions of the homologous microcin B17 heterocyclase subunits, McbB and McbD (Li et al., 1996). PatG also contains an oxidase domain that bears sequence similarity to microcin B17 oxidase McbC (Schmidt et al., 2005). A second cyanobactin variation of PatD, represented by TruD in trunkamide and patellin biosynthesis, heterocyclizes primarily cysteine, and not serine and threonine (Donia et al., 2008). Both PatD and TruD were expressed as part of cyanobactin pathways in E. coli, but as removal of PatD or TruD from the biochemical pathway led to a loss of detectable products, their biochemical roles could not be elucidated in vivo (Donia et al., 2006; Donia et al., 2008). However, the totality of evidence strongly supported their proposed function as heterocyclases. Crucially, work with the Streptomyces goadsporin pathway involved a homologous enzyme (Onaka, Nakaho, Hayashi, Igarashi, & Furumai, 2005).
In 2010, PatD and TruD were purified and biochemically analyzed, confirming their roles as heterocyclases (Figure 4) (McIntosh, Donia, & Schmidt, 2010). This work demonstrated that the heterocyclases dictated which residues were cyclic in the final product, and that the chemical differences resulting from tru were not due to other factors such as blocking by prenyltransfer. TruD in particular has since been extensively characterized, as have several other cyanobactin heterocyclases that are similar to either PatD or TruD (Koehnke et al., 2015; McIntosh & Schmidt, 2010; Sardar, Lin, et al., 2015; Sardar, Pierce, McIntosh, & Schmidt, 2015). A recent review on YcaO enzymes provides excellent background and comparison between cyanobactin heterocyclases and those from other types of biochemical pathways (Burkhart, Schwalen, Mann, Naismith, & Mitchell, 2017).
Figure 4. Heterocyclase reactions.
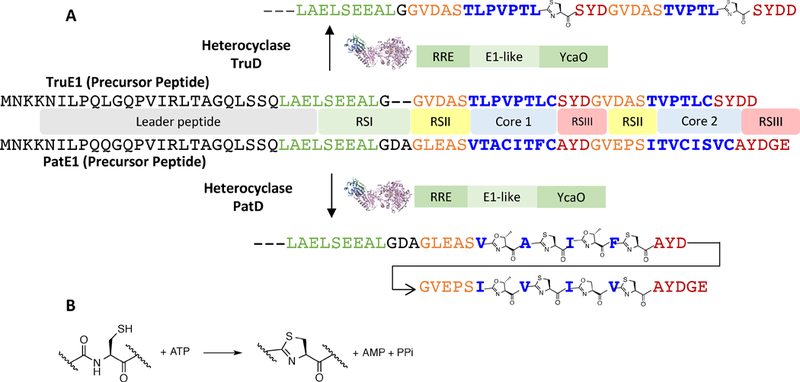
A. TruD and PatD bind to RSI and catalyze heterocycle formation. TruD, ~100% identical to PatD in the RRE and E1-like domain, is 77% identical to PatD in YcaO domain. PatD synthesizes thiazoline and oxazoline, while TruD is a thiazoline specialist. As defined by Walsh, thiazoline is energetically more favorable (Belshaw et al., 1998). B. TruD converts ATP to AMP and PPi, whereas some other heterocyclases such as those involved in linear azol(in)e-containing peptides (LAP) or thiopeptide biosynthesis yield ADP and Pi as the products.
b. Substrates and products.
Heterocyclases use the PatE-like precursor peptide and ATP (or to a minor degree, GTP) as substrates (McIntosh, Donia, et al., 2010; McIntosh & Schmidt, 2010). ATP hydrolysis accompanies heterocyclization in a 1:1 manner, in analogy to other YcaO enzymes (McIntosh & Schmidt, 2010). Unlike earlier characterized heterocyclases, which generate ADP and phosphate, cyanobactin enzymes hydrolyze ATP to AMP and pyrophophosphate in tandem with heterocyclization (Koehnke, Bent, et al., 2013; Koehnke et al., 2015). An early report indicating ADP as the product of the D reaction was likely due to a minor amount of contaminating AMP kinase (unpublished data).
Heterocyclase is thought to act at the first step of the pathway, on the unmodified precursor peptide (McIntosh, Donia, et al., 2010). This was based upon the initial observation that all PatE-like proteins contain the RSI when a heterocyclase is present in the gene cluster (Donia & Schmidt, 2011). Since the subsequent step in the pathway would involve proteolytic removal of RSI, this biochemical step likely proceeds first in vivo. Additionally, initial in vitro data suggested that heterocyclization requires the intact precursor peptide, although in some cases in vitro substrates absent RSI are still heterocyclized (Y. Goto, Ito, Kato, Tsunoda, & Suga, 2014; Houssen et al., 2014; Koehnke, Bent, et al., 2013; Koehnke et al., 2015; McIntosh, Donia, et al., 2010; McIntosh & Schmidt, 2010; Sardar, Lin, et al., 2015; Sardar, Pierce, et al., 2015). Substantial data described below delineate the interaction between RSI and the RiPP recognition element (RRE) present in the heterocyclases (Koehnke et al., 2015; Sardar, Pierce, et al., 2015).
Upon binding RSI, heterocyclase catalyzes the conversions of one or more cysteine, serine, and / or threonine into the corresponding azoline (McIntosh & Schmidt, 2010). Different heterocyclases are chemoselective, with a subset capable of heterocyclizing both sulfur and oxygen nucleophiles (e.g., PatD and relatives) and another subset specializing in sulfur nucleophiles (e.g., TruD and relatives). It should be emphasized that in artificial substrates, some oxygen heterocyclization was detected in vitro with TruD when serine was in the ideal position for heterocyclization, indicating that oxygen based heterocyclization is likely much slower, rather than forbidden. The theoretical underpinning of this difference was described in early work by Walsh et al. on microcin biosynthesis (Belshaw, Roy, Kelleher, & Walsh, 1998; Kelleher, Hendrickson, & Walsh, 1999; Li et al., 1996).
PatE-like precursors contain multiple heterocyclizable residues, but only those within core peptides are heterocyclized (Y. Goto et al., 2014; McIntosh, Donia, et al., 2010; McIntosh & Schmidt, 2010). This indicates a positional dependence for heterocyclase, both in relation to RSI and to other sequence elements. Interestingly, PatD and homologs catalyze heterocyclization of appropriate residues present in multiple cassettes on a single precursor peptide.
The native substrates for PatD are at least 29 different natural precursor peptides, which contain identical RSs and hypervariable core sequences (Donia et al., 2006). At least six natural TruD substrates exhibit identical properties (Donia et al., 2008). Several types of heterocyclases have been identified that process multiple different precursor peptides with single cores (e.g., ThcD), where the RS elements are not identical (Donia & Schmidt, 2011; Koehnke et al., 2015; Sardar, Pierce, et al., 2015). Thus, the characterized PatD-like enzymes are broadly accepting of diverse substrates.
Extensively modified variants of the precursor peptide itself have been examined experimentally. In analogy with work on lanthipeptides (Oman, Knerr, Bindman, Velasquez, & van der Donk, 2012), RSI has been fused with TruD, accelerating the enzymatic reaction rate on short cassette peptides (Sardar et al., 2017). Portions of the precursor pared down to a minimum containing just RSI have been designed and fused to various unnatural derivatives (Y. Goto et al., 2014; Koehnke et al., 2015; Sardar, Lin, et al., 2015; Sardar, Pierce, et al., 2015). In some cases, even cassettes alone without RSI have been successfully cyclized (Koehnke et al., 2015; Sardar, Pierce, et al., 2015). For example, PatD has been used to synthesize up to 18 heterocycles on a single “cassette” (Y. Goto et al., 2014). Mutants of the RSII also resulted in fully heterocyclized products (Y. Goto et al., 2014). Although PatD tolerates noncanonical C-terminus RSIII, it still influenced the catalytic efficiency to some extent (Yuki Goto & Suga, 2016).
The order of biosynthetic steps catalyzed by heterocyclases has been measured in various ways. In initial characterization, the enzymes were determined to be distributive when using multi-cassette substrates (McIntosh, Donia, et al., 2010; McIntosh & Schmidt, 2010). As found in microcin B17 synthetase (Kelleher et al., 1999), TruD may sometimes release the substrate between heterocyclization reactions. As a result, both the partially heterocyclized products and the fully heterocyclized products can be detected by MS spectrometry, SDS-PAGE electrophoresis, or NMR methods (Koehnke, Bent, et al., 2013; McIntosh, Donia, et al., 2010; McIntosh & Schmidt, 2010). When a single-cassette substrate was employed, TruD exhibited a strict directionality (Koehnke, Bent, et al., 2013). This indicates a potential complex mechanism and reaction order underlying heterocyclase activity, which is perhaps similar to that rigorously characterized in prochlorosin lanthipeptide synthesis (Thibodeaux, Ha, & van der Donk, 2014).
The diversity of naturally occurring core sequences that can be modified has been exploited for the synthesis of many derivatives (Houssen et al., 2014; Koehnke, Morawitz, et al., 2013; McIntosh, Donia, et al., 2010; Ruffner et al., 2015; Sardar, Lin, et al., 2015; Tianero et al., 2012). Artificial substrates can be categorized as those that utilize the native precursor peptide elements, and those with modified precursor peptide arrangements. Using native (or nearly native) precursors, sequence-diverse derivatives have been produced in vivo and in vitro by modifying core peptide sequences. E. coli expression libraries encoding millions of derivatives have been prepared and partially characterized (Ruffner et al., 2015). Non-proteinogenic amino acids have been incorporated into backbone peptides in vivo and in vitro (Koehnke, Morawitz, et al., 2013; Tianero et al., 2012). Intriguingly, unnatural azolines have been prepared, including selenazoline (Koehnke, Morawitz, et al., 2013).
c. Enzyme structure and function.
A sequence similarity diagram of fused heterocyclases shows the expected distribution of thiopeptide, LAP, and trifolitoxin like biosynthetic clusters across various genera (Figure 5). These fusions contain only the E1-like and YcaO proteins and lack the N-terminal extension that harbors the RRE. Likewise, the cyanobactin pathways also cluster together (cluster 4), presumably due to the presence of the RRE in these proteins. Two noteworthy features are evident from this analysis. First, actinobacteria contain a large clade of sequences (cluster 3) that contain a polypeptide with the RRE/E1-like/YcaO architecture but their corresponding pathways are missing all other biosynthetic genes. The products of these pathways have yet to be determined. Secondly, the cyanobactin cluster also contains polypeptides with the similar RRE/E1-like/YcaO architecture, but the corresponding pathways contain a large protein (~1900 amino acids) in place of all of the other cyanobacterial biosynthetic genes. Such biosynthetic clusters are all found in cyanobacteria but the products of these pathways are likewise enigmatic for now.
Figure 5. Sequence similarity analysis of heterocyclases.
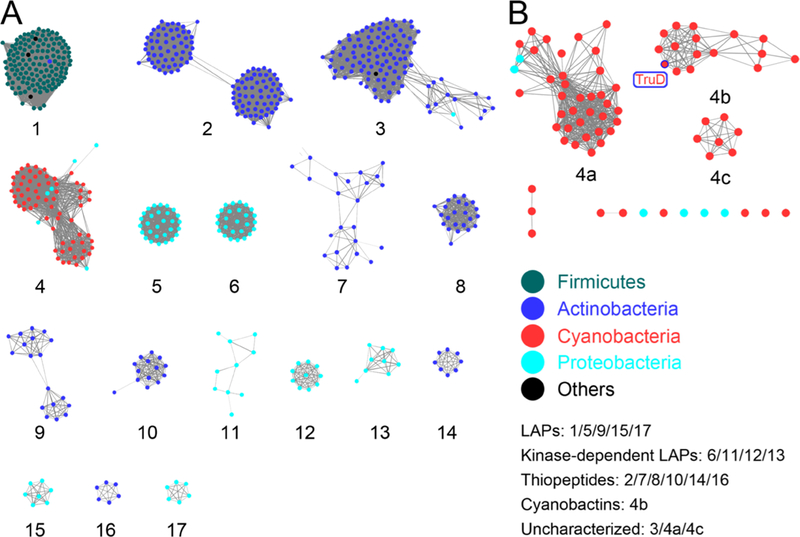
A. All heterocyclase domain-containing proteins. B. Expansion of cyanobacterial heterocyclase group and close relatives. The cyanobactin group is found in 4b of this branch.
Experiments using isotopic labeling and substrate analogs firmly establish that the E1-like protein serves as a docking scaffold, while the YcaO protein catalyzes the ATP-dependent amide backbone activation to facilitate heterocyclization (Dunbar et al., 2012). Structural studies of TruD, in the absence of any bound ligands, reveal a tripartite domain organization, in which domains 1 and 2 share modest structural similarity with the MccB adenylase involved in microcin C7 biosynthesis (Figure 6) (Regni et al., 2009). However, topological and sequence differences result in the elimination of residues that are necessary for nucleotide binding by MccB, and the binding site for ATP could not be reconciled. Subsequently, the crystal structure of E. coli YcaO, an orphan family member that lacks a corresponding docking domain, in complex with a bound ATP analog, identified a new ATP-binding motif (Dunbar et al., 2014) and provided conclusive evidence for the YcaO as the catalytic component of the heterocyclase. In 2015, the structure of LynD, the heterocyclase from the Lyngbya sp. aestuaramide pathway, was determined in complex with nucleotide analogs and a synthetic precursor derived from a patellamide cassette (Koehnke et al., 2015). This confirmed the YcaO domain as the site for nucleotide binding, and engagement of the leader sequence by the RRE, as first observed in the cocrystal structure of the dehydratase involved in lanthipeptide biosynthesis (Ortega et al., 2015). Moreover, in this work, a fusion of RSI with the RRE domain led to in trans synthesis of derivatives, in analogy to previous work with fusion of lanthipeptide leader peptides to biosynthetic enzymes (Oman et al., 2012).
Figure 6. Structures of the cyanobactin heterocyclase.
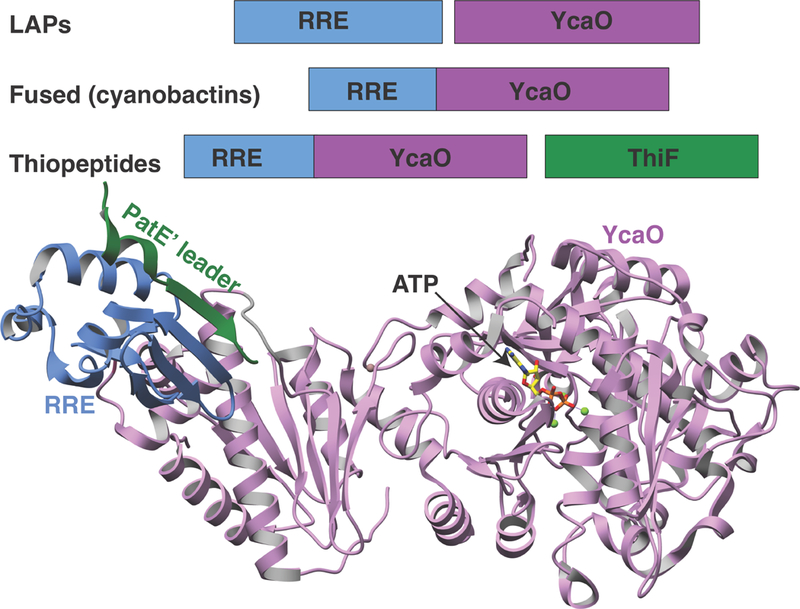
Crystal structures of LynD (PDB code 4V1T) showing RSI from the PatE precursor peptide (in green) bound to the RRE (in blue) and ATP in the YcaO active site (right, in pink).
More recent studies have resulted in the identification of single chain heterocyclase fusion enzymes present in the biosynthetic pathways for linear azol(in)e-containing peptides (Cox, Doroghazi, & Mitchell, 2015), and thiopeptides (Morris et al., 2009; Wieland Brown, Acker, Clardy, Walsh, & Fischbach, 2009; Yu et al., 2009). A distinguishing feature of the fused heterocyclase from cyanobactin pathways is that the RiPP precursor peptide recognition element (RRE), which recruits the substrate peptide (Burkhart, Hudson, Dunbar, & Mitchell, 2015), is universally at the N-terminus of the heterocyclases from cyanobactin clusters, while the enzymes from thiopeptide and LAP pathways require an auxiliary peptide binding protein (Dunbar, Tietz, Cox, Burkhart, & Mitchell, 2015).
4. Proteases
a. Discovery and initial characterization.
Proteases PatA and PatG were first characterized in the discovery of the pat gene cluster (Figure 7) (Schmidt et al., 2005). Heterologous expression of the gene cluster composed of genes patA-patG in an E. coli strain demonstrated that this cluster carried out the biosynthesis of patellamide. Embedded within the cluster was a gene (patE) whose product bore sequences for patellamide C and A, flanked by leader and follower sequences that are not found in the final product. Analyses of the gene cluster failed to identify traditional candidates (such as condensation domains, thioesterase domains, or ATP grasp enzymes) that could facilitate the necessary α-amino to α-carboxylate transamidation reaction necessary to produce the macrocyclic product. Rather, the patA and patG genes encoded large polypeptides, each of which contained domains that resembled subtilisin-related (family S8A) serine proteases (Figure 8). Based on this sequence homology, both PatA and PatG were proposed to function in the cleavage and/or maturation of the PatE precursor peptide (Schmidt et al., 2005). Identification of the trunkamide pathway (tru) and characterization of the pathway products isolated from a heterologous system further fortified the hypothesis that homologs of the PatA and PatG proteases are involved in the maturation of all cyclic cyanobactins (Donia et al., 2008).
Figure 7. Macrocyclization by PatA and PatG.
PatA frees an N-terminus, which is then available for macrocyclization by PatG.
Figure 8. Sequence similarity diagram of bacterial S8A proteases.
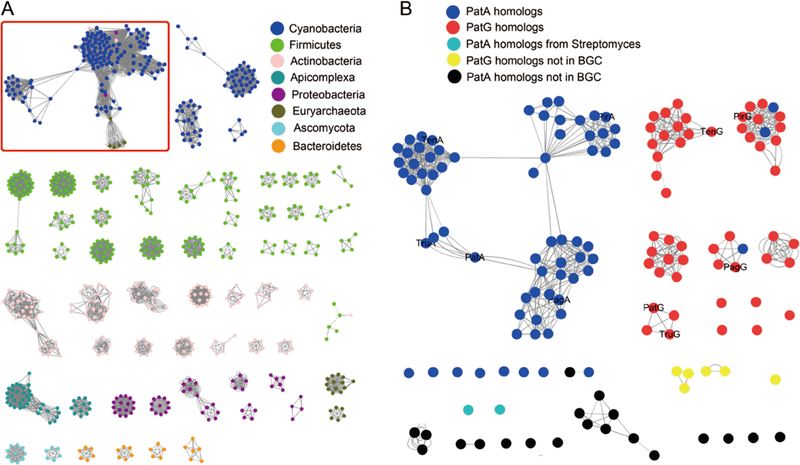
A. Proteases from a broad swath of bacteria. B. Expansion of red box from panel A. Showing the close relationship between PatA- and PatG-like proteases. BGC: biosynthetic gene cluster.
Direct evidence for the specific roles of the two proteases was established through in vitro biochemical studies of various constructs of recombinantly expressed and purified PatA and PatG (Lee et al., 2009). Specifically, the isolated protease domain of PatA processed PatEdm, an artificial precursor peptide substrate with two cassettes flanked by leader and follower sequences (later named RSII and RSIII), to liberate linear peptides corresponding to the embedded cassettes and a follower sequence (RSIII). Mutation of the active site Ser residue abolished proteolytic activity, confirming that PatA processed precursor peptide to remove the N-terminal leader sequence (Lee et al., 2009). Thus, macrocyclization was proposed to proceed via a transpeptidation mechanism with precedent in artificial subtilisin-like enzymes, such as subtiligase (Braisted, Judice, & Wells, 1997; Chang, Jackson, Burnier, & Wells, 1994).
Several other PatA and PatG homologs have been characterized, and all function similarly, although the recognition sequences differ. The major exception to this rule is the aeruginosamide (age) pathway, where AgeG performs C-terminal hydrolysis to liberate a linear product, in place of macrocyclization (Leikoski et al., 2013; Sardar et al., 2017).
b. Substrates and products.
A directed effort aimed at identifying the diversity of macrocyclic peptides produced by cyanobacterial symbionts revealed a collection of related, but chemically distinct patellamide-like products (Donia et al., 2006; Donia et al., 2008). Notably, sequence analysis of genes encoding PatE-like precursor identified an obvious pattern wherein the amino acid residues found in the final product were highly divergent but the flanking sequences (encoding the leader RSII and follower RSIII) were essentially identical.
PatA and related proteases cleave after RSII, releasing the free N-terminus of peptides. PatA recognizes several related RSII derivatives, including AVLAS-X, GVDAS-X, GLEAS-X, and GVEPS-X, embedded within longer peptides (-X indicates the position of cleavage) (Lee et al., 2009; McIntosh, Donia, et al., 2010; Sardar, Pierce, et al., 2015). So far, out of hundreds of peptides attempted of various sizes and sequences, only one containing a correct RSII sequence has failed to react (unpublished data). Otherwise, the enzyme has exhibited highly relaxed specificity, as might be expected for a subtilisin-like protease. Because of the simple reaction catalyzed by PatA, few homologs have been investigated, including ThcA, which recognized the sequence AVLAS-X (Sardar, Pierce, et al., 2015). By sequence gazing there are other quite different enzyme-RSII pairs, such as GLTPH-X in the prenylagaramide pathway, but experimental data are largely lacking (Donia & Schmidt, 2011).
Macrocyclase rule 1: Substrates must contain C-terminal recognition sequences.
PatG and related proteases cleave before RSIII (Figure 9). Both PatG and TruG are capable of recognizing the C-terminal sequences X-AYD-COOH, X-SYD-COOH, X-AYDG-COOH, X-AYDGE-COOH, X-SYDD-COOH, and relatives (X- indicates the position of cleavage) (Lee et al., 2009; McIntosh, Robertson, et al., 2010; Oueis et al., 2015; Oueis, Jaspars, Westwood, & Naismith, 2016; Oueis, Stevenson, Jaspars, Westwood, & Naismith, 2017; Sardar, Lin, et al., 2015). In addition, they recognize slightly longer sequences resulting from cleavage of multiple cassettes, such as X-AYDGLEAS-COOH (Lee et al., 2009). Thus far, they have not cleaved sequences with C-terminal longer peptides, although interestingly 8-amino-3,6-dioxaoctanoicacid has been appended to the end of the X-AYD sequence leading to successful macrocyclization (Oueis, Nardone, Jaspars, Westwood, & Naismith, 2017). This may prove useful in improving solubility of otherwise challenging substrates.
Figure 9. Macrocyclase rules.
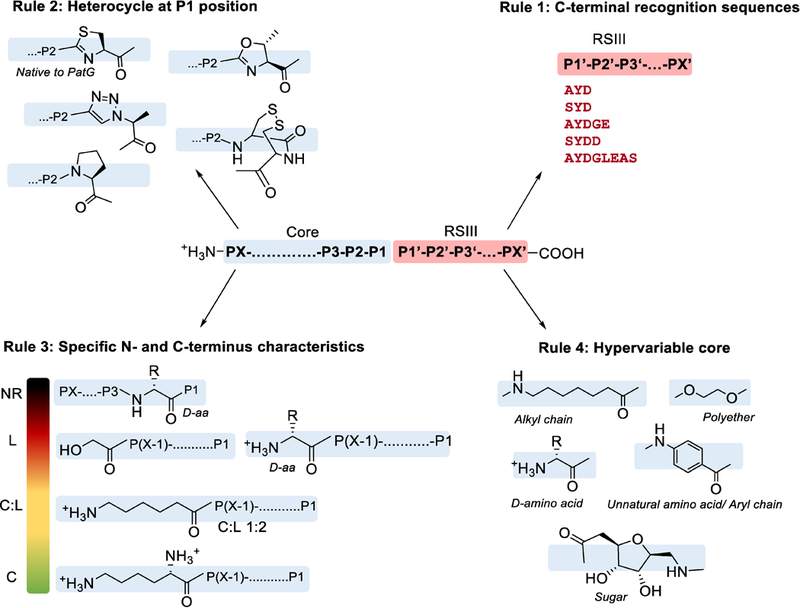
Macrocyclase PatG is broadly tolerant of substrates and is capable of generating circular, or sometimes linear products. The above rules are summarized from hundreds of substrates that have been analyzed. Recognition “rules” are useful in synthetic biology and in understanding enzyme mechanism, but there are always exceptions. For example, glycine is disfavored in position P2, but there are examples in which it is successfully used. In rule 3, NR = no reaction, L = linear product, C:L = both linear and cyclic product, and C = cyclic product.
Macrocyclase rule 2: Substrates must contain heterocycle in the P1 position.
PatG catalyzes a more complex reaction than PatA and has more complex substrate requirements. In addition to RSIII, a heterocycle is required in the P1 position (P1 site as per the nomenclature of Schecter and Berger) (Schechter & Berger, 1967), e.g. heterocycle-SYD, etc. In the case of PatG and relatives the natural heterocycle is thiazoline or oxazoline, while in proteins such as PagG it is apparently proline (Donia & Schmidt, 2011). A peptide that lacked the necessary heterocycle at the P1 position failed to react entirely with PatG (McIntosh, Robertson, et al., 2010). Even so, PatG accepts substrates with proline substituting for thiazoline, as well as triazoles and, interestingly, heterocycles comprised of disulfide linked cysteine-cysteine (e.g., CC-SYD, etc.) (Oueis et al., 2016; Oueis, Stevenson, et al., 2017). These are synthetically useful alternatives in the creation of large molecules. Genome mining approaches identified a cryptic cyanobactin gene cluster in Oscillatoria sp. PCC 6506, and substrate tolerance studies using the protease domain of the corresponding OscG enzyme revealed that this enzyme could still process substrates with the (Cys)- and (Ser)-pseudoprolines in the P1 position while PatG cannot, further expanding the potential macrocyclase substrates (Alexandru-Crivac et al., 2017). The minimal substrate requirement for macrocyclization by PatG (i.e. minimal three residue follower sequence preceded by a heterocycle) has prompted several efforts focused on utilizing the isolated protease domain as a biotechnology tool (Alexandru-Crivac et al., 2017; McIntosh, Robertson, et al., 2010; Oueis et al., 2016; Oueis, Nardone, et al., 2017; Oueis, Stevenson, et al., 2017; Sardar, Lin, et al., 2015). For example, the macrocyclase was predicted to be useful in producing discrete compounds such as peptides that mimic hormones or neuropeptides, or libraries of natural unnatural products.
Macrocyclase rule 3: For macrocyclization to occur, the N-terminal and C-terminal residues have specific characteristics.
McIntosh and co-workers tested the tolerance of the PatG protease domain against 23 different synthetic peptides containing five-residue follower sequence but with variable cassette residues (McIntosh, Robertson, et al., 2010). At the N-terminus, peptides terminating in a D-amino acid or in glycolate (OH nucleophile) in place of an amine nucleophile were hydrolyzed and linearized, instead of circularized. In addition, substrates were partially linearized and partially cyclized through the side chain when the terminal residue contained side-chain amines (such as diaminopropionate or lysine). These results showed that the position and nucleophilicity of the N-terminus is crucial in the circularization process.
At the C-terminus, a peptide that lacked the necessary heterocycle at the P1 position failed to react entirely, as described above. The P2 position is also somewhat fastidious, in that substrates containing D-amino acids or glycine in that position were not substrates for PatG (McIntosh, Robertson, et al., 2010). A later study suggested that the L-amino acid is critical for correct positioning of the peptide in the active site (Oueis, Stevenson, et al., 2017).
Macrocyclase rule 4: The central residues of PatG interact with a large pocket; specific directionality or hydrogen bonds are not required, explaining the broad substrate tolerance of the enzyme.
In the McIntosh work (McIntosh, Robertson, et al., 2010), the 23 substrates included D-amino acids in the central residues. While the C- and N-termini could not tolerate such modifications, in the central residues D-amino acids were readily accommodated. Aminohexanoic acid could be placed at the N-terminus or in the middle of the peptide, revealing that even hydrogen bonds to the peptide backbone are not required. PatG tolerated D-amino acids in the interior of the peptide, but not at the N-terminal or two C-terminal amino acids of the core peptide. The semi-synthetic approach was further refined to carry out the macrocyclic peptide hybrids, which incorporated synthetic chemical building blocks such as aryl rings, alkyl chains, sugars, and polyethers (Oueis, Nardone, et al., 2017). The ring size synthesized by G enzymes is very flexible, ranging from 5–22 amino acids for PatG and 6–30 for OscG (Alexandru-Crivac et al., 2017; Sardar, Lin, et al., 2015). Because non-proteinaceous compounds can also be included, another way to describe the size variability is between 15 to 90 atoms. Taken together, it is clear that PatG and related enzymes exhibit broad tolerance for changes in length and composition of interior residues. Other wild-type cyanobactins such as anacyclamide have been discovered to encompass 7–20 amino acids and have potentially different macrocyclization rules (Leikoski et al., 2010), indicating that the natural cyclases should be capable of affording a wide range of products.
Much has been learned about TruG (macrocyclase domain nearly identical to PatG) via expression in E. coli. A heterologous production system from the tru pathway was modified with genes encoding for orthogonal tRNA/aaRS pairs, in order to incorporate non-proteinogenic amino acids within the macrocylic core (Tianero et al., 2012). As proof of concept, the non-natural amino acid p-acetylPhe was incorporated into the TruE precursor peptide. Modified, macrocyclic patellins with the non-proteinogenic amino acids was subsequently manufactured through differential induction for the aaRSs, resulting in the production of patellin derivatives with substituted Phe derivatives and trunkamide with p-bromoPhe (Tianero et al., 2012).
The genetic nature of both the precursor peptide and the modification enzymes renders the cyanobactin pathways as ideal toolkits for combinatorial production of small molecule libraries. A combinatorial approach was utilized to generate a series of random double and quadruple variants of the TruE precursor peptide in the heterologous tru system (Ruffner et al., 2015). This approach resulted in the production of more than 300 new macrocyclic hexapeptides. More importantly, analysis of variants that failed to produce the desired products provided guidelines for selectivity, which will guide rational design of new libraries. These studies demonstrate the potential of the system for the production of libraries of cyclized small molecules. Owning to the tolerance of the macrocyclase for residues within the cassette, PatG could also be employed to generate cyclic peptides that mimic hormones or neuropeptides.
Macrocyclase rules: Summary.
With this large amount of selectivity data now collected for PatG (and TruG / OscG), we propose an overarching scheme explaining substrate tolerance in the G series. A large number of substrates have been used in these studies, more than can be described in this review. Taken together, these data indicate that the appropriate heterocycle and adjacent L-amino acid at the C-terminal side, in addition to an appropriately located nitrogen nucleophile at the N-terminus or side chain, are the basic required elements. These C-terminal elements enable binding to the enzyme active site and reaction with the serine oxyanion, while the N-terminal elements are required to position the nucleophile appropriately for reaction. Between these two elements, a vast array of different amino acids and synthetic linkers have been used successfully, indicating a lack of a requirement for preorganization or for precise hydrogen bonding resulting from theses intermediate elements. While there are clearly preferences for some amino acids above others, especially immediately adjacent to the heterocycle, substrate tolerance is very broad.
c. Enzyme structure and function.
Both the PatA and PatG were first identified as large polypeptides that housed the respective protease domains (Schmidt et al., 2005). Full-length PatA consists of a C-terminal domain of unknown function (DUF), which encompass roughly 300 amino acids (Lee et al., 2009). A linker region of approximately 120 residues, for which no function has yet been assigned, separates the protease and DUF domains. The domain architecture of full-length PatG consists of two functionally annotated domains: a flavin-dependent oxidase domain (265 residues) followed by the protease domain (~350 residues), flanked by 210 residue N-terminal and 280 residue C-terminal DUFs. The oxidase domain serves to carry out dehydrogenation of thiazoline residues installed by the heterocyclase to yield the corresponding thiazoles and is functionally similar to the oxidases that function in LAP biosynthesis (Li et al., 1996).
Incubation of the PatA reaction product (namely, the linear peptide cassette followed by the follower sequence) with the PatG protease domain produced two fragments: the linear follower peptide and a macrocycle containing the cassette sequence (Lee et al., 2009). Incubation of PatG with intact PatEdm, containing both the leader and follower sequences flanking the cassette, did not produce any proteolytic product. Hence, the order by which the linear precursor peptide is processed into the macrocyclic product occurs through the removal of the N-terminal leader sequence by PatA to produce a linear peptide product bearing the cassette and follower sequence. Modification of the C-terminal linear product by PatG excises the five-residue linear follower peptide concomitant with the macrocyclization of the cassette sequence to yield the final cyanobactin product. Metagenomic analysis of patellamide pathways shows that PatG can process 29 different substrates to form macrocycles that are 7 or 8 residues in size (Donia et al., 2006). The cyclic peptides produced in nature are diverse in sequence but all contain a heterocyclic residue (either a Pro, or an azol(in)e derived from Ser/Cys/Thr) immediately preceding the follower peptide at the scission site. Consequently, substrates that conform to these requirements can be processed by PatG to produce engineered macrocyclic natural products (Donia et al., 2006).
Based on bioinformatics analyses that identified sequence similarities to subtilisin-like S8A proteases, macrocyclization of the cassette sequences was proposed to occur via the intermediacy of an acyl-enzyme adduct (McIntosh, Robertson, et al., 2010). Formation of the macrocycle is afforded via the attack of the α-amine onto the intermediate, rather than through hydrolysis as is typical for canonical proteases. The importance of the PatG protease domain in the N-C transamidation is further supported through mutational analysis, in which alteration of residues in the requisite catalytic triad resulted in a loss of activity. The mechanism is conceptually analogous to off-loading thioesterase domains that catalyze macrolactam formation of linear substrates that are bound as activated thio- or oxo-esters (Agarwal et al., 2012).
The protease domains of PatA and PatG share greater than 40% sequence identity, which is intriguing given that the two enzymes catalyze very different reactions. PatA recognizes and cleaves after the leader sequence preceding the cassette to yield two linear products, while PatG cleaves before the follower sequence to produce a linear product (the follower peptide) and a macrocyclic cassette. Details into this discrepancy were afforded through structural elucidation of the protease domains of PatA and PatG (Agarwal et al., 2012), as a complex of the PatG macrocyclase bound to a substrate mimic (Koehnke et al., 2012). While the structure of the PatA protease domain recapitulates the canonical α/β hydrolase fold common among S8A proteases (Agarwal et al., 2012; Houssen et al., 2012), the structure of the PatG protease domain reveals a similar fold decorated with a helix-turn-helix (‘capping helices’) positioned directly above the active site (Figure 10). The cocrystal structure with the substrate mimic reveals that the heterocyclic residue at the P1 position adopts a cis conformation, which helps to direct the cassette peptide away from the enzyme (Koehnke et al., 2012). Based upon structural and mutational analysis, the capping helix binds with RSIII and orients the α-amine back towards the active site Ser. This binding cap is essential, since otherwise subtilisin proteases are known to recognize the N-terminal side of the substrate, rather than the C-terminal side as in PatG (Figure 11) (Chang et al., 1994).
Figure 10. Crystal structures of PatA and PatG protease domains.
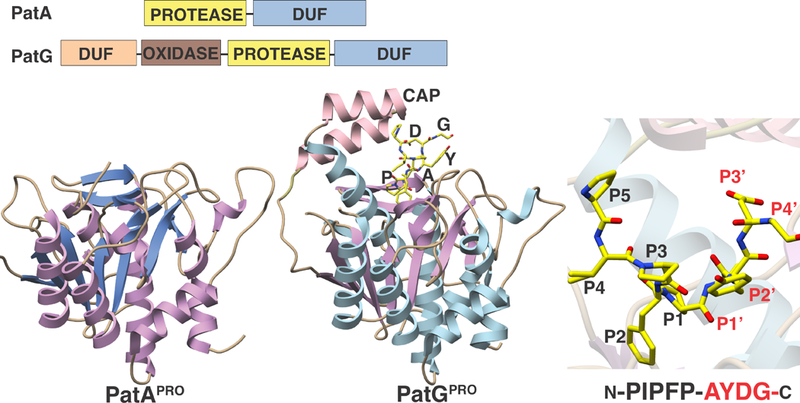
The proteins have very similar architectures except for a cap in PatG, shown here interacting with an artificial substrate (shown as yellow sticks). The panel on the right shows a close-up view of the PatG active site, in the vicinity of the scissile bond.
Figure 11. Mechanism of macrocyclase.
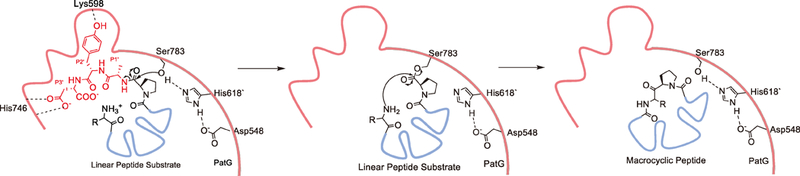
The cap (wavy red) interacts with RSIII (red) to hold the substrate in place. A catalytic triad typical of S8A proteases forms a covalent adduct with P1 at Ser783; this is displaced by an internal nucleophilic nitrogen to afford a macrocycle, while in most proteases hydrolysis would occur instead. The participation of individual residues in catalysis (other than Ser783) remains speculative.
The mechanism of macrocyclization versus hydrolysis has been discussed (Agarwal et al., 2012; Booth et al., 2017; Koehnke et al., 2012; Sardar et al., 2017). Certain substrates (e.g., those with terminal D-amino acids or hydroxyl rather than amine) are linearized rather than cyclized (McIntosh, Robertson, et al., 2010). PatG also naturally catalyzes transamidation of linear peptides in the presence of glycylglycine buffer (Agarwal et al., 2012). From these and other data, it is likely that PatG functions by preferentially activating amines, and not hydroxyls, to cleave the intermediate enzyme-bound ester. Such a mechanism has precedent in subtiligase, where two mutations in subtilisin switch the protease from being OH- to amine-preferring (Braisted et al., 1997). A second proposed mechanism involves exclusion of water by the unusual capping helix. This latter idea is controversial amongst workers in the field.
One issue with PatG and its analog so far is their exceptionally slow rate, both in vitro (Lee et al., 2009; Sardar et al., 2017; Sardar, Lin, et al., 2015) and in vivo (Tianero et al., 2016), although this may be an artifact of experimental conditions. In the first characterization of PatG, the best conditions still led to a rate of ~1 turnover per enzyme molecule per day, and the yield was optimized to 75% with a 3:1 substrate to enzyme ratio (Lee et al., 2009). The reaction could be completed in 1 day with a 50% catalyst load (McIntosh, Robertson, et al., 2010). This work used an artificial substrate with proline at the macrocyclization C-terminus, which is far from the ideal PatG substrate. The rate was improved by optimizing the enzymatic reaction solution (Koehnke et al., 2012). Ideal substrates terminating in thiazoline react more readily and do not show such a striking buffer dependence (Sardar et al., 2017; Sardar, Lin, et al., 2015). Thiazoline-containing substrates were quantitatively converted to the cyclic product in 1 day with a 20% catalyst load (Sardar, Lin, et al., 2015). Similarly, PatA was challenging to use, especially in the in vitro synthesis of patellamides (Houssen et al., 2014). This problem was demonstrated to be caused by an interesting redox problem (Sardar, Lin, et al., 2015). While PatE-like substrates need to be reduced, and enzymes such as TruD are accelerated in the presence of dithiothreitol (DTT) reductant, PatA is greatly inhibited by DTT. By manipulating DTT concentration, it was possible to use PatA efficiently as a synthetic tool. Perhaps a similarly simple problem hinders PatG; alternatively, perhaps an unknown role of the DUFs, or enzyme complex formation, is required.
5. Prenyltransferases
a. Discovery and initial characterization.
The PatF family of prenyltransferases, often called “F enzymes”, comprises a group of small ABBA-type proteins that prenylate or geranylate serine, threonine, tyrosine, or tryptophan residues in the forward or reverse orientations (McIntosh et al., 2011). Recent characterization also identifies a novel F enzyme that can also prenylate the α-amine of peptide substrates (Sardar et al., 2017). Other F enzymes cluster in groups related to the amino acid that is prenylated, with the network analysis indicating potentially novel prenylation chemistry that has yet to be characterized (Figure 12).
Figure 12. Sequence similarity diagram of PatF-like prenyltransferases.
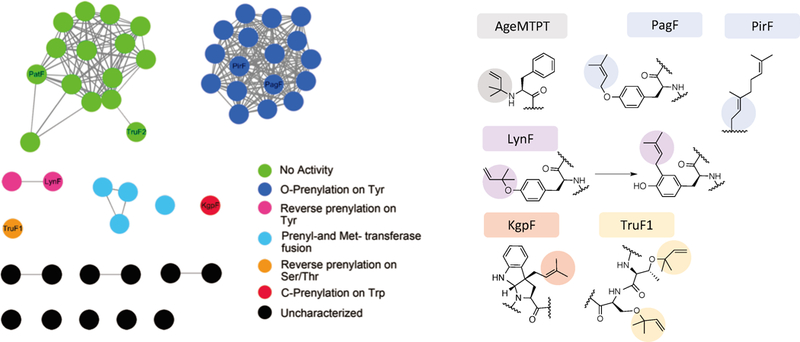
Note that sequence clusters shown in Network diagram (left) so far fairly accurately predict the chemistry catalyzed by the enzymes within each cluster (right).
As far as has been described, all such prenyltransferases act at the final step of biosynthesis, after the precursor peptide has been cyclized (McIntosh et al., 2011). Potential exceptions to this rule include the aforementioned AgeMTPT, which prenylates the N-terminus of the linear peptide substrate after cleavage by AgeA (Leikoski et al., 2013; Sardar et al., 2017). While PatF was the first in the cyanobactin F-group to be identified, it was not characterized as a prenyltransferase because of its lack of sequence similarity to other characterized proteins and the absence of any isoprene moieties in patellamides (Schmidt et al., 2005). When the tru pathway was identified, one of the major differences with pat was the presence of two PatF-like proteins, TruF1 and TruF2 (Donia et al., 2008). As expression of the tru pathway in E. coli lead to products that were prenylated in the reverse orientation on serine and threonine, it was proposed that these proteins exercised prenyltransferase activity. Subsequently, the LynF protein was characterized as a tyrosine prenyltransferase, with the substrates being DMAPP and the tyrosine residue on a circular peptide from the lyn pathway (McIntosh et al., 2011). The crystallization of inactive PatF revealed that the family adopts a modified ABBA prenyltransferase fold (Bent et al., 2013), while the structures of active prenyltransferase PagF with and without substrates revealed the mechanistic basis of broad substrate tolerance (Hao et al., 2016).
b. Substrates and products.
Cyanobactin prenyltransferases appear to be highly promiscuous regarding peptide sequence and structure, but highly specific with regards to the donor isoprene and the amino acid site of prenylation on the peptide substrate (Figure 13). As such, they are good tools for the prenylation of specific residues on diverse peptide substrates. All characterized prenyltransferases use DMAPP (or potentially IPP in some cases) as the substrate, and so these will be the subjects of discussion here. An additional family of geranylating prenyltransferases exists, but the product of the pathway has been characterized only via genetic means and mass spectrometry, and therefore the chemistry is not yet certain (Leikoski et al., 2012).
Figure 13. Prenylation by cyanobactin enzymes.
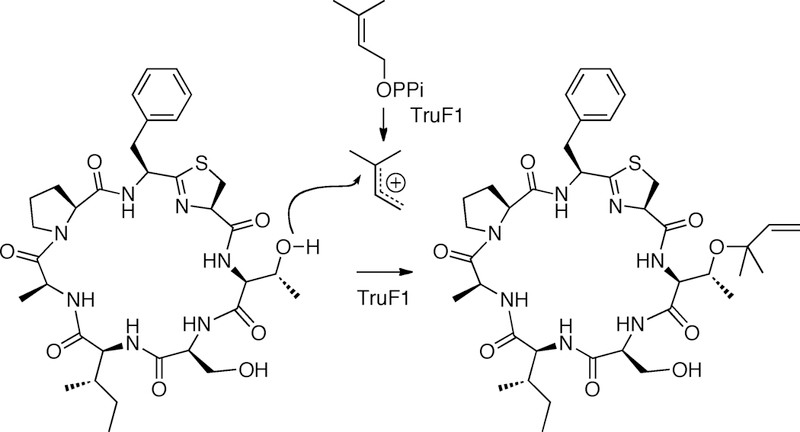
Shown is a single prenylation of the trunkamide precursor peptide by TruF1.
The prenyltransferases known so far can be broadly classified as those that naturally utilize cyclic substrates (most known F proteins), and those that use linear substrates (aeruginosamide pathway) (Leikoski et al., 2013). Amongst those enzymes that use circular substrates, in vitro studies demonstrate a capability of prenylation on both individual amino acids and close variants of those amino acids (e.g., various phenols in the case of tyrosine prenylating enzymes) (McIntosh et al., 2011). They also can prenylate many different variants of linear peptides, but show no activity on amino acids.
Threonine-serine prenylation.
Several natural 6–8 residue, macrocylic peptides from marine Prochloron symbiotic cyanobacteria were known to be prenylated on serine and threonine (Carroll et al., 1996). All of these products were shown to originate in the tru pathway, which uses hypervariable substrates and identical enzymes (Donia et al., 2008). The tru pathway was expressed in E. coli, leading to production of the natural products, with the isoprene group shown by feeding studies to originate from DMAPP (Tianero et al., 2016). Since the tru pathway contained two potential prenyltransferases, TruF1 and TruF2, genetic studies in E. coli were required to demonstrate activities of the enzyme (Tianero et al., 2016). TruF2 could be deleted without untoward effect, while deletion of TruF1 led only to macrocyclic peptide products lacking prenylation on serine or threonine. Thus, TruF1 was active as a peptide prenyltransferase, while TruF2 showed no such activity.
TruF1 was functional in vitro, but its low activity and solubility has precluded detailed studies. Using heterologous expression in E. coli, prenylation is shown to be the last step, with accumulation of unprenylated macrocycle in the cytoplasm, all of which could undergo prenylation upon subsequent addition of DMAPP (Donia, Ruffner, et al., 2011; Tianero et al., 2016). Although the substrate selectivity of TruF1 has not been addressed in vitro, the tru pathway has been used to create thousands of derivatives in E. coli (Ruffner et al., 2015; Tianero et al., 2012). A limitation of these studies is that they address the selectivity of all pathway enzymes, as well as stability and compatibility in E. coli, and thus are not specific to prenyltransferase activity. Nonetheless, a few potential rules were gleaned: 1) TruF1 is absolutely selective for serine and threonine above other amino acids; 2) TruF1 is highly promiscuous with regards to the choice of peptide substrate; 3) TruF1 has been demonstrated to prenylate up to three times on a single substrate; 4) TruF1 seems to prefer certain flanking sequences adjacent the modified serine or threonine, although this was only rigorously tested in one set of substrates. These sequences contained the amino acids arginine, leucine, isoleucine, methionine, serine, or threonine.
Tyrosine prenylation.
Many cyanobactins are prenylated on tyrosine, and such molecules contain a diversity of amino acid sequences and ring sizes (Donia & Schmidt, 2011; Leikoski et al., 2009). The characterized natural representatives are all forward prenylated on tyrosine. Identification of these biosynthetic pathways strongly implicated a PatF-like enzyme in prenylation. Characterization of products from a reconstituted prenylagaramide (pag) gene cluster led to identification of the F enzymes from this pathway (PagF) as bona fide tyrosine prenyltransferases (Donia & Schmidt, 2011).
Notably, biochemical characterization of a homologous enzyme from Lyngbya aesturaii showed that LynF prenylates tyrosine on oxygen in the reverse orientation (McIntosh et al., 2011; McIntosh et al., 2013). This leads to a product with the isoprene group poised to undergo a Claisen rearrangement to yield forward C-prenylated tyrosine. The rearrangement occurs spontaneously, and the rate of the rearrangement varies depending upon substrate and takes place within a few minutes using boc-tyrosine but requires potentially up to weeks in some of the natural products, as both O- and C-prenylated variants can be isolated from the cyanobacteria. By contrast, PagF forward O-prenylates tyrosine on oxygen. In the forward orientation, the products are poor substrates for the Claisen rearrangement, and do not under any further rearrangements. Most members of F- family of prenyltransferases are likely to be forward O-prenylating as found in PagF.
The substrate scopes of LynF and PagF have been investigated in vitro (Hao et al., 2016; McIntosh et al., 2011). LynF used boc-D- and boc-L-tyrosine, as well as a variety of cyclic peptides containing tyrosine. Linear biosynthetic intermediates were not modified, but other linear peptides proved to be reasonable substrates. PagF exhibited a similar substrate scope. In contrast to LynF, PagF accepted linear peptides with vastly different amino acid sequences than found in the parent compound, making it potentially useful for biotechnology.
Tryptophan prenylation.
Kawaguchipeptins A and B are ten-membered ring macrocyclic peptides originally isolated from Microcystis aeruginosa NIES-88 (Ishida, Matsuda, Murakami, & Yamaguchi, 1997), which contain prenylation on two tryptophan residues. Sequencing of the producing strain suggested that both of the products are RiPPs that are produced by a single cyanobactin pathway, which was confirmed by heterologous expression of the constituent genes in E. coli (Parajuli et al., 2016). A single enzyme, KgpF, functions on a range of cyclic and linear peptide substrates and prenylates C3 on the amino acid tryptophan, which is a unique modification in RiPP products. Notably, upon prenylation, a subsequent reaction forms a second reaction five-membered ring with the backbone amide nitrogen. Consequently, the KpgF enzyme does not accept the isolated amino acid L-tryptophan as a substrate. The stereospecificity of the modification was confirmed by comparison with synthetic standards (Okada et al., 2016). Both DMAPP, and to a lesser extent IPP, were reported to be used by KpgF, but GPP could not be used as an isoprene donor.
N-terminal prenylation.
AgeMTPT, from the aeruginosamide pathway, is the only known prenyltransferase that modified the α-amino moiety of peptides (Leikoski et al., 2013). As described above, aeruginosamides stand out from other cyanobactins because they are linear, rather than cyclic products produced by the G-group protease AgeG. Thus, proteolysis liberates a free N-terminus that can be forward prenylated. AgeMTPT is not named with “F” nomenclature because it is a didomain protein consisting of a C-terminal, F-type prenyltransferase (PT) fused with an N-terminal methyltransferase (MT). AgeMTPT uses DMAPP to prenylate the N-terminus with a relatively restricted substrate scope, possibly requiring an N-terminal phenylalanine residue to react (Sardar et al., 2017). Additionally, the protein prenylates SAM and other adenosine analogs, presumably due to the close placement of the active site in the MT in comparison to that of PT.
Potentially non-prenylating PatF homologs.
A large group of PatF homologs, including PatF itself, have no known activity (Donia et al., 2006; Houssen et al., 2014; Tianero et al., 2016). They have not been shown to bind to DMAPP or other isoprene donors. A comparison of the structure of PatF (Bent et al., 2013) with that of the fully functional PagF (Hao et al., 2016) shows that the former enzyme lacks many critical residues. Nonetheless, PatF is essential for the production of patellamide in E. coli heterologous system (Donia et al., 2006). The only other nonfunctional F variant that has been characterized in any way is TruF2, which also lacks key residues necessary for functionality (Tianero et al., 2016). Unlike PatF, TruF2 can be deleted from the heterologous production system without loss of patellin production. Notably, there are cyanobactin biosynthetic clusters (such as that for trichamide) (Sudek et al., 2006) that lack any F homolog, suggesting that these enzymes are not necessary for the production of heterocycles and/or macrocycles. Hence, while PatF is necessary for patellamide biosynthesis, the general role for other non-functional homologs has yet to be elucidated. Hypotheses about their potential roles include a chaperone/carrier function for shuttling intermediates or products, a role in multi-protein complex formation, as well as a number of other possibilities.
c. Enzyme structure and function.
The crystal structure of PatF revealed an overall architectural similarity to the ABBA-prenyltransferase family, but the basis for substrate selectivity could not be reconciled as PatF is not a functional enzyme (Bent et al., 2013). Subsequent structure determination of PagF, in complex with both linear and cyclic peptide substrates, and structure-guided mutational analysis provided the context for functional analysis (Figure 14) (Hao et al., 2016). The enzyme shares the same ABBA-fold (Kuzuyama, Noel, & Richard, 2005; Metzger et al., 2009), but unlike other ABBA-type enzymes that function of small molecule substrates, lacks a hydrophobic cavity for productive isoprene transfer. Notably, binding of the peptide substrate itself provided the suitable hydrophobic pocket to form a catalytically competent active site. The degree of surface area buried by the substrate corresponded directly with the catalytic efficiency, explaining why isolated amino acids are not turned over, but cyclic peptides that can suitably occlude the solvent exposed active site are kinetically competent. The structure also provides a rationale for the lack of function for PatF as this homolog has mutations at several active site residues that are shown to be critical in PagF for binding to the isoprene donor.
Figure 14. Structure of PagF bound to substrate.
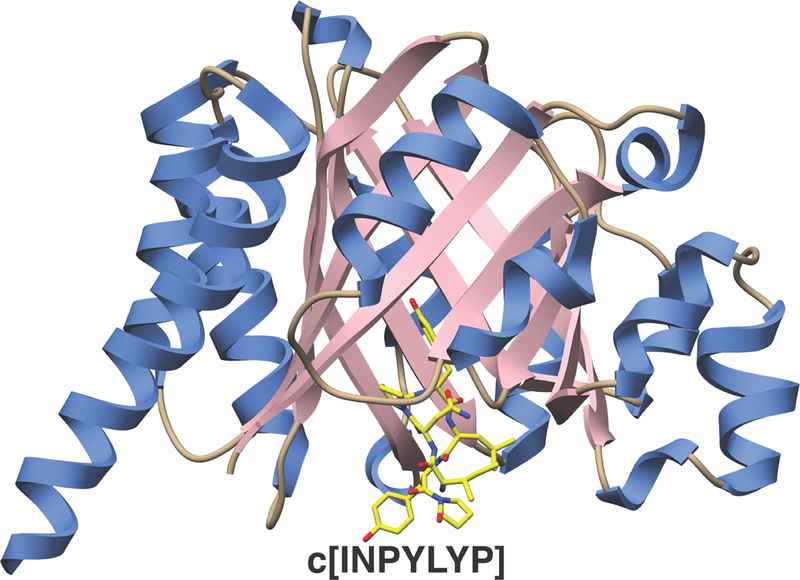
While most ABBA prenyltransferases contain a hydrophobic active site that precludes solvent, the structure of PagF reveals a solvent exposed cavity. Binding of the peptide substrate is necessary to form a solvent excluded cavity where productive prenyl transfer can occur.
6. Other enzymes
a. Discovery and initial characterization.
Beyond the core enzymes described above, two classes of enzymes are relatively common in cyanobactin biosynthesis: oxidases and methyltransferases (Donia & Schmidt, 2011). In addition, a number of proteins and domains of unknown function are commonly found amongst several cyanobactin clusters but are not typically prevalent across all pathways.
The oxidase domain was first identified as the N-terminal domain in the polypeptide that also harbored the PatG protease domain (Schmidt et al., 2005). It was the only domain or protein in the pathway with obvious homology to characterized RiPP proteins, showing similarity to the microcin B17 oxidase involved in the oxidative conversion of azoline moieties to the aromatic azoles (Li et al., 1996). While most cyanobactin oxidases are found as fusions with the corresponding G protease, there are also instances of standalone oxidases that carry out the same function, as inferred from the structures of natural products associated with those pathways. Expression of the pat pathway in E. coli led to the natural products that contained both oxidized thiazoles but also unmodified oxazolines, validating the sufficiency of the gene cluster for the oxidation reaction (Donia et al., 2006; Schmidt et al., 2005). Moreover, a pathway lacking the oxidase domain, tru, does not contain oxidized heterocycles and instead contain thiazoline, both in the producing organisms, and also when the pathway is heterologously expressed in E. coli (Donia et al., 2008). Oxidase ThcOxi from Cyanothece cyanobactin pathway only oxidizes thiazoline-containing cyclic peptides in vitro, but a homologous enzyme Apoxi from a microcin biosynthetic pathway Arthrospira pathway, was able to oxidize the thiazoline on both cyclic and linear cyanobactin substrates (Houssen et al., 2014).
Another class of ubiquitous modification enzymes that are commonly found in cyanobactin biosynthetic clusters is methyltransferases (Donia & Schmidt, 2011). In sequenced pathways they are often fused with other functional proteins, such as homologs of PatG. However, the function is known for only one such fusion, AgeMTPT, which is consistent with the known natural product associated with its pathway. The age pathway for aeruginosmides was reconstituted in vitro and shown to yield linear cyanobactin natural products in which the terminal α-carboxylate is methyl esterified (Figure 15) (Sardar et al., 2017). Biochemical analysis with the purified protein showed that AgeMTPT carried methylation of the C-terminus using SAM as the cofactor and could also prenylate the α-amine of the same peptide using DMAPP. Other similar enzymes that contain SAM-binding motifs have not yet been characterized, and much remains to be learned about the role of methyltransferases in cyanobactin biosynthesis.
Figure 15.
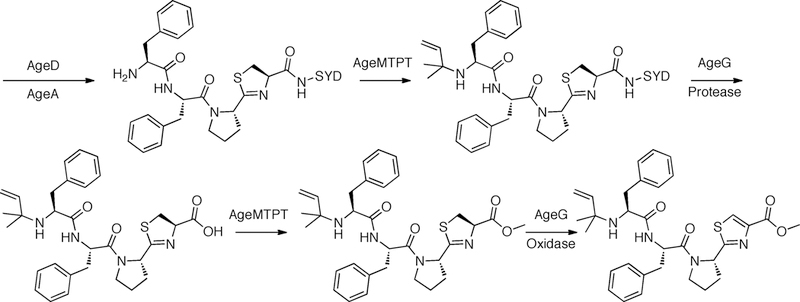
Aeruginosamide biosynthesis via the age pathway.
Several proteins and domains, including PatB and PatC and the C-terminal DUFs on PatA and PatG, are virtually universally found in cyanobactin pathways but have no known function (Donia et al., 2006). PatB and PatC are not required for synthesis in vitro, and they can be deleted from the pathway in vivo when proteins are under control of individual promoters (albeit with low yields). However, they are apparently essential in expressing the pat and tru operons in E. coli. Similarly, the DUFs can be deleted from PatA and PatG with retention of protein function in vitro (Lee et al., 2009). No published data exists concerning their essentiality in vivo. Crystal structures are available for the C-terminal DUFs, but functional studies failed to elucidate any binding between this domain and the PatE precursor peptide (Mann et al., 2014). Finally, an N-terminal DUF is present in PatG and homologs; its function and structure is unknown, but it is not essential for macrocyclase activity in vitro (Lee et al., 2009). Because these proteins and domains are universally found in cyanobactin biosynthesis, it is certain that they are essential, or at least play an important biological role in the natural cyanobacterial producers.
b. Substrates and products.
A wide range of thiazole-, and occasionally oxazole-containing cyanobactins have been isolated, but relatively little biosynthetic data exist. Probably the best studied in terms of substrate scope is the patellamide oxidase embedded in PatG (Schmidt et al., 2005). Since about 50 natural hepta- and octapeptide substrates are known to be modified by identical PatG oxidase, this implies that the oxidase is catalytically competent on a variety of substrates. Although many cyanobactins contain oxazole, PatG oxidase is apparently incapable of oxidizing oxazolines. Thcoxi only modified the thiazolines on cyclic substrates. The specificity for thiazoline versus oxazoline was previously explained by Walsh and co-workers in their study of the homologous microcin B17 oxidase, where oxidation to thiazole is intrinsically faster (Belshaw et al., 1998).
The AgeMTPT methyltransferase natively modifies a series of short linear peptides with closely related structures (Sardar et al., 2017). AgeMTPT was tested with 17 linear peptide substrates in vitro, but only modified a close relative of the native substrate: isoprene-FFP-thiazoline, a presumed intermediate in the natural pathway. Interestingly, the methyltransferase only modified the thiazoline carboxylate, and not the thiazole carboxylate, implying that methylation precedes oxidation.
Enzyme structure and function.
Relatively little has been reported concerning the structures of these enzymes. Key exceptions are that structures of the PatG DUF and ThcG oxidase were reported (Bent et al., 2016; Mann et al., 2014). No biological insight was gained from the DUF structure, but the crystal structure of the Cyanothece oxidase structure revealed the site of binding of the flavin mononucleotide and showed a probable biologically relevant dimer. The structure also revealed an N-terminal extension consisting of twin RRE domains but the functional relevance of these RRE domains has not yet been experimentally verified.
7. In vitro construction of artificial cyanobactins
Several principles are essential for pathway engineering: 1) Modularity. Enzymes from different pathways can be mixed and assembled to make different cyanobactins; 2) Hypervariability.In a precursor peptide, the core sequences can encode many different natural products, as long as certain conserved sequence elements are maintained outside of the cores; and 3) Inverse kinetic order. In the canonical cyanobactin pathway, the enzymes post-translationally modify the substrate in a defined order, and each step is slower than the previous one.
Based upon these principles and the extensive knowledge gained through biosynthetic and biochemical investigations described above, rules have been gleaned to select enzymes and to design suitable precursor peptides. Here, we will define the steps needed to synthesize designed products (Figure 16). Note that, although the enzymes are quite broad in substrate utilization, not every peptide-like compound will be a substrate. Current knowledge of substrate rules is defined in the text above.
Figure 16.
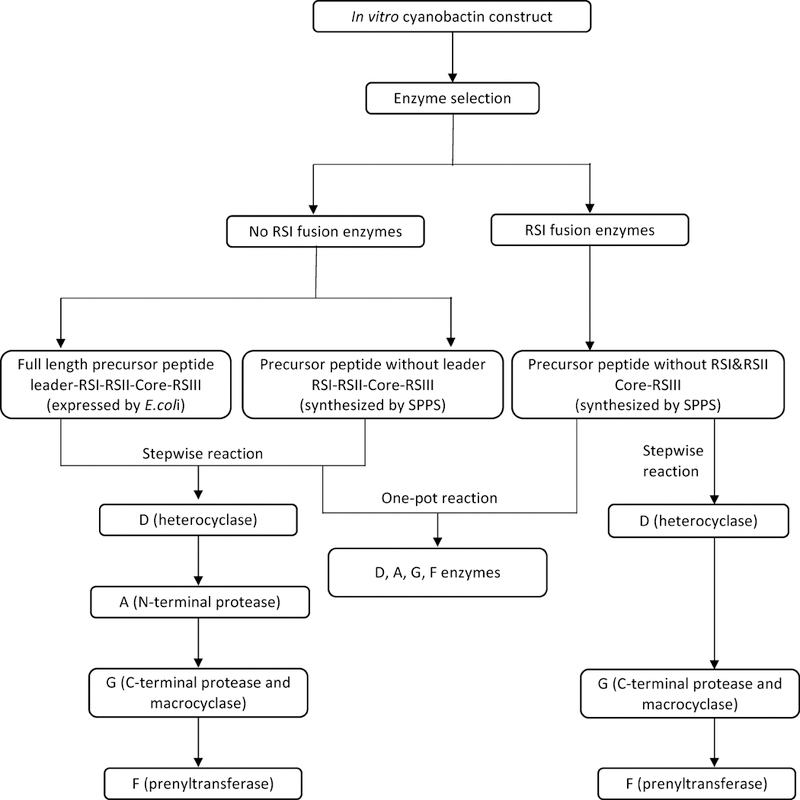
General steps for in vitro construction of artificial cyanobactins.
Step 1. Choose an enzyme
Step 1a. Enzyme selection.
Enzymes are chosen based upon the desired structural features of the products. A pool of well-characterized cyanobactin enzymes are available to program products with desired chemical features (Table 1). For example, PatD installs both thiazoline and oxazoline, whereas LynD, TruD, and ThcD install only thiazoline. The substrate selectivities of these enzymes, while quite broad and overlapping, are also subtly different.
Table 1.
Characterized cyanobactin enzymes, their GenBank or PDB accession number, post-translational modifications (PTM) and the paired recognition sequences (RS)
| Enzyme | GenBank Accession | PTMa | RSb |
|---|---|---|---|
| TruD | ACA04490.1 | Cys -> thioazoline | RSI: LAELSEEAL |
| LynD | 4V1T (PDB) | ||
| ThcD | WP_012626011.1 | ||
| PatD | AAY21153.1 | Cys -> thioazoline Thr/Ser -> oxazoline |
|
| TruA | ACA04487.1 | N-terminal proteolysis | RSII: GVDAS |
| PatA | AAY21150.1 | RSII: GLEAS / GVEPS |
|
| PagA | AED99426.1 | RSII: GLTPH | |
| ThcA | WP_041236525.1 | RSII: AVLAS | |
| PatG (protease domain) |
AAY21156.1 (amino acids 513–866) |
C-terminal proteolysis macrocyclization |
RSIII: AYD/AYDGE |
| TruG | ACA04494.1 | RSIII: SYD/SYDD | |
| AgeG (protease domain) |
CCH92964 (amino acids 609–960) |
C-terminal proteolysis | |
| TruF1 | ACA04492.1 | prenlyation (isoprene) | Thr/Ser |
| PagF | AED99429.1 | O-prenylation | Tyr |
| LynF | WP_083798475.1 | Claisen rearrangement -> C-prenlyation | |
| KgpF | KXS89935.1 | Trp prenylation | Trp |
| AgeMTPT | CCH92966 | N-terminal prenylation C-terminal methylation |
N and C terminus |
| ThcOxi | 5lq4 (PDB) | thioazoline -> thioazole | Thioazoline (cyclic substrate) |
PTM: Post-translational modifications processed by individual cyanobactin enzymes.
RS: recognition sequences known to be processed by the enzyme.
In addition to wild-type enzymes, a series of artificial constructs have been designed. For example, PatG protease domain is fully functional without addition of other elements (Lee et al., 2009). PatG-subtiligase has been designed to increase the efficiency of transamination reactions and some types of circularizations, particularly where there is a flexible linker present. Another subtlety is that RSI can be fused to TruD or LynD, enabling N-terminal cleavage of RSI to be skipped during the process.
Step 1b. Enzyme expression and purification.
Methods have been previously described in detail (Sardar, Pierce, et al., 2015). Briefly, genes are codon optimized for the E. coli host. All protein sequences are available in GenBank (Table 1).
Step 2. Design the substrate peptide
Step 2a. Synthetic versus E. coli-expressed substrates.
Both synthetic and recombinant methodologies are suitable for generating substrate peptides. The substrate should at least contain the recognition sequences for the enzymes and a core sequence encoding the final product. The recognition sequences should be in certain positions to make the substrate fully functions: 1) RSI should be placed in the front; 2) RSII should be right in front of the core sequence; 3) RSIII should be right after the core sequence.
When synthesized in E. coli, full-length leader sequences should be used, with an N-terminal His6-tag. An example of such a construct is given as sequence 1 (core peptide in bold):
Sequence 1. MGSSHHHHHHSSGLVPRGSHMNKKNILPQLGQPVIRLTAGQLSSQ LAELSEEALGGVDASTSIAPFCAYDGE
In this sequence, the first residues through QLSSQ (underlined) are not essential for enzymatic reaction but are very useful in stable expression in E. coli. Methods to express the constructs have been previously described (Sardar, Pierce, et al., 2015).
By contrast, synthetic peptides can lack such a sequence. An example is given in sequence 2, wherein recognition sequence elements are present, but much of the precursor peptide has been excised:
Sequence 2. LAELSEEALGGVDASTSIAPFCAYDGE
The advantage of such a sequence is clearly in its brevity. Moreover, if heterocyclization is not desired (core sequence ending with Pro), or if RSI has been fused in cis with TruD/PatD, then an even shorter sequence lacking RSI and RSII is possible, as shown in sequence 3:
Sequence 3. TSIAPFCAYDGE
Finally, only a few residues of RSIII are truly required, so that a reasonable substrate is provided in sequence 4:
Sequence 4. TSIAPFCAYD
Moreover, synthetic substrates are more readily amenable to modifications with non-protein elements. A good example of such a synthetic substrate is given by sequence 5, with more details described in Section 4 on PatG substrates above:
Sequence 5. aminohexanoic acid-TPFCAYD
This substrate, although short and highly artificial, is fully suitable for reaction by heterocyclase, macrocyclase, and prenyltransferase.
Some enzymes can accept non-native recognition sequences from other enzymes in the same class. For example, sequence 6 contains the native TruG recognition sequence, but is still a good substrate for reaction with PatG:
Sequence 6. TSIAPFCSYD
There are several possible variations of these methods. Most importantly, multiple core peptides can be incorporated into a single precursor. In the case of in vitro synthesis, this will have the effect of increasing the yield per mole of precursor, or of generating multiple different compounds in a single reaction. In these cases, additional core peptides are each flanked by additional RSII and RSIII sequences. An example is given by sequence 7, encoding three different core peptides shown in bold:
Sequence 7. MGSSHHHHHHSSGLVPRGSHMNKKNILPQLGQPVIRLTAGQLSSQ LAELSEEALGGVDASTSIAPFCSYDGVDASTSLAPFCSYDGVDASSSIAPFCSYDD
Step 2b. Pairing recognition sequences with enzymes.
The recognition sequences required for each enzymatic reaction are given in Table 1. For example, if cleavage by protease PatA is required, the sequence GVDAS or closely related sequences (e.g. GLEAS) shown in the table should be appended prior to the core, as shown in Sequence 7 above.
Step 2c. Synthetic methods.
In vivo expression methods have been described in detail (Sardar, Pierce, et al., 2015; Tianero et al., 2016). Substrate synthesis is generally performed by standard solid-phase synthetic methods, although the approach is compatible with many other types of synthetic techniques.
Step 3. Perform reactions
The detailed requirements of each enzyme for reactivity has been previously described (Sardar, Tianero, & Schmidt, 2016). For in vitro cyanobactin construction, the enzyme reaction can undergo either step by step reactions or one-pot reaction.
Step 3a. Stepwise method.
The order of enzyme addition should follow the canonical order of enzymatic reactions: D -> A -> G -> F/MTPT -> Oxi. If one of these enzymatic reactions is not desired, the step can be skipped. For example, if prenylation is not required, the F enzyme step can be left out. Most of the reaction conditions have been described previously (Sardar, Pierce, et al., 2015).
Reactions using AgeMTPT require a slightly different order. When used as a prenyltransferase, dimethylallyl pyrophosphate (DMAPP, 5 mM) is the isoprene donor. This step should be performed before the C-terminal proteolysis step. DMAPP is chemically synthesized with the established method (Davisson et al., 1986). When used as a methyltransferase, S-adenosyl-L-methionine (SAM, Sigma-Aldrich, 5–10 mM) is used as the methyl donor. This step should follow the C-terminal proteolysis and before the oxidation of the thiazoline. Enzymatic reaction mixtures contain Tris pH 7.5 (50 mM), MgCl2 (5 mM), substrate (50 μM) and enzyme (2–5 μM). The mixture is incubated at 37 °C for up to 2 h for prenyl transfer and up to 18 h for methyl transfer.
The AgeG reaction condition is very similar to that of PatG: enzyme (5–10 μM), substrate (50 μM), Tris pH 7.5 (50 mM), and MgCl2 (5 mM) at 37 °C for 24 h (Sardar et al., 2017).
For Trp prenyltransferase KgpF reaction, enzyme (5 μM) is added to the reaction mixture containing substrate (100 μM), 1% dimethyl sulfoxide (DMSO), DMAPP or isopentenyl pyrophosphate (IPP, 1 mM), MgCl2 (12 mM), NaCl (150 mM), HEPES pH 7.5 (10 mM), and tris(2-carboxyethyl)phosphine (TCEP) (3 mM).(Parajuli et al., 2016) Since the prenyltransferase is a slower enzyme, the reactions should be incubated at 37 ˚C for 40 h. DMAPP is a better cofactor than IPP for the native substrate kawaguchipeptin B.
Oxidase Thcoxi has been used to oxidize thiazoline to thiazole in cyclic peptide substrates (Houssen et al., 2014). Cyclic peptide substrate dissolved in DMSO are diluted to 20 μM final concentration with 0.5% DMSO, in a reaction mixture containing FMN cofactor (50 μM), Thcoxi (10 μM), NaCl (500 mM) and Tris pH 8.0 (20 mM). The reaction mixture is incubated overnight at 37 °C with shaking. Oxidase ApOxi from a microcin pathway has been used to oxidize thiazoline into thiazole on either linear or cyclic cyanobactin precursors (Houssen et al., 2014). Enzyme (20 μM) is incubated with heterocyclic peptide (200 μM) and FMN cofactor (1 mM) overnight at 37 °C.
Alternatively, thiazoline rings can be oxidized by a chemical method using MnO2: dissolve product (500 μg) in anhydrous DMSO (0.25 mL) and add activated MnO2 (3 mg) (Houssen et al., 2014; Sardar et al., 2017). Stir the mixture at 150 rpm at 30 °C for 72–96 h. Remove MnO2 by centrifugation and wash the pellet three times with methanol (1 mL each). Combine DMSO and methanol fractions and dry under vacuum. Purify the mixture using HPLC. Other chemical oxidation methods, such as DDQ/DCM method and K2CO3/DMF sometimes result in unstable products or side products and are not recommended.
It is not necessary to purify the product after each step except in the case of N-terminal protease A, where yield is improved if the product of PatA cleavage is purified. In the heterocyclization step, reducing agent dithiothreitol (DTT) is required to prevent intramolecular disulfide bridge formation in the substrate peptide. However, DTT will inhibit the activity of the following modifying enzyme protease A. To maintain the activity of protease A, several different methods can be used to remove DTT:
HPLC method. The mobile phase generally consists of acetonitrile (1% to 99% in water over 20 min). The solid phase is C4 for longer peptides (i.e., over 60 amino acids) and C18 for shorter peptides. In the case of azoline-containing peptides, a diagnostic 255-nm shoulder in the UV spectrum aids in purification.
Dialysis method. Dialysis tubing such as SnakeSkin, 3.5K MWCO (Thermofisher) is soaked in a buffer (1 L) consisting of the same composition as the reaction buffer: Tris pH 7.5 (50 mM) and MgCl2 (5 mM). The dialysis is performed at 4 °C with stirring for 6 hours, followed by placement in fresh buffer for a total of 18 hours of dialysis. For smaller-scale reactions, a 96-well microdialysis plate (Thermofisher, Pierce) can be used with similar conditions.
Desalting method. The reaction mixture is passed through a size-exclusion desalting column such as Bio-Gel P-6 (Bio-rad) following the manufacturer’s protocol. After removing the storage buffer, the reaction mixture is passed through the column, and the flow-through is collected.
Although the HPLC method is useful and provides a pure compound, recovery suffers due to the intrinsic limitations of chromatography. Dialysis is useful for larger substrates (3.5 kDa) but is time consuming. Desalting columns using size-exclusion resins are often ideal, as they are faster and provide higher recovery, although not of pure compounds.
Step 3b. One-pot synthesis method.
One-pot reaction provides a good alternative approach to quickly produce the cyanobactins. Enzymes (D + A + G) are mixed with the substrate in the same concentrations as described in the stepwise methods above. When the substrate does not tend to form the disulfide bridge, DTT can be left out from the reaction mixture, simplifying the process, but it is essential if disulfide bridges might be formed in the substrate. With DTT, N-terminal protease A is partially inhibited and results in a side product. This side product is formed by modifications only with heterocyclase D and N-terminal protease / macrocyclase G. In a typical one-pot synthesis reaction, the following conditions are used: Tris pH 7.5 (50 mM), MgCl2 (5 mM), CaCl2 (10 mM), ATP (1 mM), heterocyclase D (2 μM), protease A (2 μM), and protease and macrocyclase G (10 μM). If prenyltransferase is used, then 10 μM of the enzyme and DMAPP (10 mM) would be added. The reaction mixture is commonly incubated at 37 °C for 18 hours (Sardar, Lin, et al., 2015). Up till now only monoprenlyated product can be observed.
8. Conclusion
Cyanobactin biosynthetic pathways are highly promiscuous and useful in biotechnology, where they have been used to synthesize many natural products, designed compounds, and libraries in vivo and in vitro. Ultimately, such tools will be useful in the directed genetic synthesis of desired compounds without constraints. In order for this goal to be realized, many different enzymes and biosynthetic pathways are needed, and the rules underlying their combination will need to be elucidated. These goals, and progress toward them with cyanobactin enzymes, have recently been reviewed (Gu & Schmidt, 2017; Sardar & Schmidt, 2016).
Since the discovery of pat in 2005, much has been learned about the genetics, chemistry, and biochemistry of this intriguing group of compounds. Here, we hope that we have provided an unbiased catalog of what is known about cyanobactin enzyme structure and function. In turn, we hope this facilitates the broad accessibility of cyanobactin enzymes to non-specialists for use in synthetic chemistry and synthetic biology applications.
References
- Agarwal V, Pierce E, McIntosh J, Schmidt EW, & Nair SK (2012). Structures of cyanobactin maturation enzymes define a family of transamidating proteases. Chem Biol, 19(11), 1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandru-Crivac CN, Umeobika C, Leikoski N, Jokela J, Rickaby KA, Grilo AM, … Houssen WE (2017). Cyclic peptide production using a macrocyclase with enhanced substrate promiscuity and relaxed recognition determinants. Chem Commun (Camb), 53(77), 10656–10659. [DOI] [PubMed] [Google Scholar]
- Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, … Chang G (2009). Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science, 323(5922), 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, … van der Donk WA (2013). Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep, 30(1), 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw PJ, Roy RS, Kelleher NL, & Walsh CT (1998). Kinetics and regioselectivity of peptide-to-heterocycle conversions by microcin B17 synthetase. Chem Biol, 5(7), 373–384. [DOI] [PubMed] [Google Scholar]
- Bent AF, Koehnke J, Houssen WE, Smith MC, Jaspars M, & Naismith JH (2013). Structure of PatF from Prochloron didemni. Acta Crystallogr Sect F Struct Biol Cryst Commun, 69(Pt 6), 618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bent AF, Mann G, Houssen WE, Mykhaylyk V, Duman R, Thomas L, Naismith JH (2016). Structure of the cyanobactin oxidase ThcOx from Cyanothece sp. PCC 7425, the first structure to be solved at Diamond Light Source beamline I23 by means of S-SAD. Acta Crystallogr D Struct Biol, 72(Pt 11), 1174–1180. doi: 10.1107/S2059798316015850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram A, & Pattenden G (2007). Marine metabolites: metal binding and metal complexes of azole-based cyclic peptides of marine origin. Nat Prod Rep, 24(1), 18–30. doi: 10.1039/b612600f [DOI] [PubMed] [Google Scholar]
- Booth J, Alexandru-Crivac CN, Rickaby KA, Nneoyiegbe AF, Umeobika U, McEwan AR, … Shalashilin DV (2017). A blind test of computational technique for predicting the likelihood of peptide sequences to cyclize. J Phys Chem Lett, 8(10), 2310–2315. doi: 10.1021/acs.jpclett.7b00848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braisted AC, Judice JK, & Wells JA (1997). Synthesis of proteins by subtiligase. Methods Enzymol, 289, 298–313. [DOI] [PubMed] [Google Scholar]
- Burkhart BJ, Hudson GA, Dunbar KL, & Mitchell DA (2015). A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat Chem Biol, 11(8), 564–570. doi: 10.1038/nchembio.1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart BJ, Kakkar N, Hudson GA, van der Donk WA, & Mitchell DA (2017). Chimeric leader peptides for the generation of non-natural hybrid RiPP products. ACS Cent Sci, 3(6), 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart BJ, Schwalen CJ, Mann G, Naismith JH, & Mitchell DA (2017). YcaO-dependent posttranslational amide activation: biosynthesis, structure, and function. Chem Rev, 117(8), 5389–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll A, Coll J, Bourne D, Macleod J, Zabriskie T, Ireland C, & Bowden B (1996). Patellins 1–6 and trunkamide A: novel cyclic hexa-, hepta- and octa-peptides from colonial ascidians, Lissoclinum sp. Australian Journal of Chemistry, 49(6), 659–667. [Google Scholar]
- Chang TK, Jackson DY, Burnier JP, & Wells JA (1994). Subtiligase: a tool for semisynthesis of proteins. Proc Natl Acad Sci U S A, 91(26), 12544–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comba P, Eisenschmidt A, Gahan LR, Herten DP, Nette G, Schenk G, & Seefeld M (2017). Is Cu(II) coordinated to patellamides inside Prochloron cells? Chemistry, 23(50), 12264–12274. [DOI] [PubMed] [Google Scholar]
- Cox CL, Doroghazi JR, & Mitchell DA (2015). The genomic landscape of ribosomal peptides containing thiazole and oxazole heterocycles. BMC Genomics, 16, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davagnino J, Herrero M, Furlong D, Moreno F, & Kolter R (1986). The DNA replication inhibitor microcin B17 is a forty-three-amino-acid protein containing sixty percent glycine. Proteins, 1(3), 230–238. [DOI] [PubMed] [Google Scholar]
- Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacher M, & Poulter CD (1986). Phosphorylation of isoprenoid alcohols. The Journal of Organic Chemistry, 51(25), 4768–4779. [Google Scholar]
- Degnan BM, Hawkins CJ, Lavin MF, McCaffrey EJ, Parry DL, van den Brenk AL, & Watters DJ (1989). New cyclic peptides with cytotoxic activity from the ascidian Lissoclinum patella. J Med Chem, 32(6), 1349–1354. [DOI] [PubMed] [Google Scholar]
- Donia MS, Fricke WF, Ravel J, & Schmidt EW (2011). Variation in tropical reef symbiont metagenomes defined by secondary metabolism. PLoS One, 6(3), e17897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, Hathaway BJ, Sudek S, Haygood MG, Rosovitz MJ, Ravel J, & Schmidt EW (2006). Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat Chem Biol, 2(12), 729–735. [DOI] [PubMed] [Google Scholar]
- Donia MS, Ravel J, & Schmidt EW (2008). A global assembly line for cyanobactins. Nat Chem Biol, 4(6), 341–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, Ruffner DE, Cao S, & Schmidt EW (2011). Accessing the hidden majority of marine natural products through metagenomics. Chembiochem, 12(8), 1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, & Schmidt EW (2010). Cyanobactins - ubiquitous cyanobacterial ribosomal peptide metabolites. In Comprehensive Natural Products II: Chemistry and Biology (Vol. 2, pp. 539–558). [Google Scholar]
- Donia MS, & Schmidt EW (2011). Linking chemistry and genetics in the growing cyanobactin natural products family. Chem Biol, 18(4), 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar KL, Chekan JR, Cox CL, Burkhart BJ, Nair SK, & Mitchell DA (2014). Discovery of a new ATP-binding motif involved in peptidic azoline biosynthesis. Nat Chem Biol, 10(10), 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar KL, Melby JO, & Mitchell DA (2012). YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations. Nat Chem Biol, 8(6), 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar KL, Tietz JI, Cox CL, Burkhart BJ, & Mitchell DA (2015). Identification of an auxiliary leader peptide-binding protein required for azoline formation in ribosomal natural products. J Am Chem Soc, 137(24), 7672–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher JD, & Vandeputte J (1955). Thiostrepton, a new antibiotic. II. Isolation and chemical characterization. Antibiot Annu, 3, 560–561. [PubMed] [Google Scholar]
- Genilloud O, Moreno F, & Kolter R (1989). DNA sequence, products, and transcriptional pattern of the genes involved in production of the DNA replication inhibitor microcin B17. J Bacteriol, 171(2), 1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Pastor JE, San Millan J. L., Castilla MA, & Moreno F (1995). Structure and organization of plasmid genes required to produce the translation inhibitor microcin C7. J Bacteriol, 177(24), 7131–7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Ito Y, Kato Y, Tsunoda S, & Suga H (2014). One-pot synthesis of azoline-containing peptides in a cell-free translation system integrated with a posttranslational cyclodehydratase. Chem Biol, 21(6), 766–774. [DOI] [PubMed] [Google Scholar]
- Goto Y, & Suga H (2016). A post-translational cyclodehydratase, PatD, tolerates sequence variation in the C-terminal region of substrate peptides. Chemistry Letters, 45(11), 1247–1249. [Google Scholar]
- Gu W, & Schmidt EW (2017). Three principles of diversity-generating biosynthesis. Acc Chem Res, 50(10), 2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Pierce E, Roe D, Morita M, McIntosh JA, Agarwal V, … Nair SK (2016). Molecular basis for the broad substrate selectivity of a peptide prenyltransferase. Proc Natl Acad Sci U S A, 113(49), 14037–14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houssen WE, Bent AF, McEwan AR, Pieiller N, Tabudravu J, Koehnke J, … Jaspars M (2014). An efficient method for the in vitro production of azol(in)e-based cyclic peptides. Angew Chem Int Ed Engl, 53(51), 14171–14174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houssen WE, Koehnke J, Zollman D, Vendome J, Raab A, Smith MC, … Jaspars M (2012). The discovery of new cyanobactins from Cyanothece PCC 7425 defines a new signature for processing of patellamides. Chembiochem, 13(18), 2683–2689. [DOI] [PubMed] [Google Scholar]
- Ishida K, Matsuda H, Murakami M, & Yamaguchi K (1997). Kawaguchipeptin B, an antibacterial cyclic undecapeptide from the cyanobacterium Microcystis aeruginosa. J Nat Prod, 60(7), 724–726. [DOI] [PubMed] [Google Scholar]
- Kelleher NL, Hendrickson CL, & Walsh CT (1999). Posttranslational heterocyclization of cysteine and serine residues in the antibiotic microcin B17: distributivity and directionality. Biochemistry, 38(47), 15623–15630. [DOI] [PubMed] [Google Scholar]
- Koehnke J, Bent A, Houssen WE, Zollman D, Morawitz F, Shirran S, Naismith JH (2012). The mechanism of patellamide macrocyclization revealed by the characterization of the PatG macrocyclase domain. Nat Struct Mol Biol, 19(8), 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J, Bent AF, Zollman D, Smith K, Houssen WE, Zhu X, Naismith JH (2013). The cyanobactin heterocyclase enzyme: a processive adenylase that operates with a defined order of reaction. Angew Chem Int Ed Engl, 52(52), 13991–13996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J, Mann G, Bent AF, Ludewig H, Shirran S, Botting C, … Naismith JH (2015). Structural analysis of leader peptide binding enables leader-free cyanobactin processing. Nat Chem Biol, 11(8), 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J, Morawitz F, Bent AF, Houssen WE, Shirran SL, Fuszard MA, … Naismith JH (2013). An enzymatic route to selenazolines. Chembiochem, 14(5), 564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuyama T, Noel JP, & Richard SB (2005). Structural basis for the promiscuous biosynthetic prenylation of aromatic natural products. Nature, 435(7044), 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, McIntosh J, Hathaway BJ, & Schmidt EW (2009). Using marine natural products to discover a protease that catalyzes peptide macrocyclization of diverse substrates. J Am Chem Soc, 131(6), 2122–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Mitchell DA, Markley AL, Hensler ME, Gonzalez D, Wohlrab A, Dixon JE (2008). Discovery of a widely distributed toxin biosynthetic gene cluster. Proc Natl Acad Sci U S A, 105(15), 5879–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikoski N, Fewer DP, Jokela J, Alakoski P, Wahlsten M, & Sivonen K (2012). Analysis of an inactive cyanobactin biosynthetic gene cluster leads to discovery of new natural products from strains of the genus Microcystis. PLoS One, 7(8), e43002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikoski N, Fewer DP, Jokela J, Wahlsten M, Rouhiainen L, & Sivonen K (2010). Highly diverse cyanobactins in strains of the genus Anabaena. Appl Environ Microbiol, 76(3), 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikoski N, Fewer DP, & Sivonen K (2009). Widespread occurrence and lateral transfer of the cyanobactin biosynthesis gene cluster in cyanobacteria. Appl Environ Microbiol, 75(3), 853–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikoski N, Liu L, Jokela J, Wahlsten M, Gugger M, Calteau A, … Fewer DP (2013). Genome mining expands the chemical diversity of the cyanobactin family to include highly modified linear peptides. Chem Biol, 20(8), 1033–1043. [DOI] [PubMed] [Google Scholar]
- Li YM, Milne JC, Madison LL, Kolter R, & Walsh CT (1996). From peptide precursors to oxazole and thiazole-containing peptide antibiotics: microcin B17 synthase. Science, 274(5290), 1188–1193. [DOI] [PubMed] [Google Scholar]
- Mann G, Koehnke J, Bent AF, Graham R, Houssen W, Jaspars M, … Naismith JH (2014). The structure of the cyanobactin domain of unknown function from PatG in the patellamide gene cluster. Acta Crystallogr F Struct Biol Commun, 70(Pt 12), 1597–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, Donia MS, Nair SK, & Schmidt EW (2011). Enzymatic basis of ribosomal peptide prenylation in cyanobacteria. J Am Chem Soc, 133(34), 13698–13705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, Donia MS, & Schmidt EW (2010). Insights into heterocyclization from two highly similar enzymes. J Am Chem Soc, 132(12), 4089–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, Lin Z, Tianero MD, & Schmidt EW (2013). Aestuaramides, a natural library of cyanobactin cyclic peptides resulting from isoprene-derived Claisen rearrangements. ACS Chem Biol, 8(5), 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, Robertson CR, Agarwal V, Nair SK, Bulaj GW, & Schmidt EW (2010). Circular logic: nonribosomal peptide-like macrocyclization with a ribosomal peptide catalyst. J Am Chem Soc, 132(44), 15499–15501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, & Schmidt EW (2010). Marine molecular machines: heterocyclization in cyanobactin biosynthesis. Chembiochem, 11(10), 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeever B, & Pattenden G (2003). Total synthesis of trunkamide A, a novel thiazoline-based prenylated cyclopeptide metabolite from Lissoclinum sp. Tetrahedron, 59(15), 2713–2727. [Google Scholar]
- Metzger U, Schall C, Zocher G, Unsold I, Stec E, Li SM, … Stehle T (2009). The structure of dimethylallyl tryptophan synthase reveals a common architecture of aromatic prenyltransferases in fungi and bacteria. Proc Natl Acad Sci U S A, 106(34), 14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocek U, Chen LC, Keller PJ, Houck DR, Beale JM, & Floss HG (1989). 1H and 13C NMR assignments of the thiopeptide antibiotic nosiheptide. J Antibiot (Tokyo), 42(11), 1643–1648. [DOI] [PubMed] [Google Scholar]
- Morris RP, Leeds JA, Naegeli HU, Oberer L, Memmert K, Weber E, … Krastel P (2009). Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J Am Chem Soc, 131(16), 5946–5955. [DOI] [PubMed] [Google Scholar]
- Okada M, Sugita T, Akita K, Nakashima Y, Tian T, Li C, … Abe I (2016). Stereospecific prenylation of tryptophan by a cyanobacterial post-translational modification enzyme. Org Biomol Chem, 14(40), 9639–9644. [DOI] [PubMed] [Google Scholar]
- Oman TJ, Knerr PJ, Bindman NA, Velasquez JE, & van der Donk WA (2012). An engineered lantibiotic synthetase that does not require a leader peptide on its substrate. J Am Chem Soc, 134(16), 6952–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onaka H, Nakaho M, Hayashi K, Igarashi Y, & Furumai T (2005). Cloning and characterization of the goadsporin biosynthetic gene cluster from Streptomyces sp. TP-A0584. Microbiology, 151(Pt 12), 3923–3933. doi: 10.1099/mic.0.28420-0 [DOI] [PubMed] [Google Scholar]
- Ortega MA, Hao Y, Zhang Q, Walker MC, van der Donk WA, & Nair SK (2015). Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB. Nature, 517(7535), 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueis E, Adamson C, Mann G, Ludewig H, Redpath P, Migaud M, … Naismith JH (2015). Derivatisable Cyanobactin Analogues: A Semisynthetic Approach. Chembiochem, 16(18), 2646–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueis E, Jaspars M, Westwood NJ, & Naismith JH (2016). Enzymatic Macrocyclization of 1,2,3-triazole peptide mimetics. Angew Chem Int Ed Engl, 55(19), 5842–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueis E, Nardone B, Jaspars M, Westwood NJ, & Naismith JH (2017). Synthesis of hybrid cyclopeptides through enzymatic macrocyclization. ChemistryOpen, 6(1), 11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueis E, Stevenson H, Jaspars M, Westwood NJ, & Naismith JH (2017). Bypassing the proline/thiazoline requirement of the macrocyclase PatG. Chem Commun (Camb), 53(91), 12274–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T, Yamashita K, Goto Y, Shimomura M, Hayashi S, Asamizu S, … Onaka H (2017). Dissection of goadsporin biosynthesis by in vitro reconstitution leading to designer analogues expressed in vivo. Nat Commun, 8, 14207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli A, Kwak DH, Dalponte L, Leikoski N, Galica T, Umeobika U, … Fewer DP (2016). A unique tryptophan C-prenyltransferase from the Kawaguchipeptin biosynthetic pathway. Angew Chem Int Ed Engl, 55(11), 3596–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascard C, Ducruix A, Lunel J, & Prange T (1977). Highly modified cysteine-containing antibiotics. Chemical structure and configuration of nosiheptide. J Am Chem Soc, 99(19), 6418–6423. [DOI] [PubMed] [Google Scholar]
- Regni CA, Roush RF, Miller DJ, Nourse A, Walsh CT, & Schulman BA (2009). How the MccB bacterial ancestor of ubiquitin E1 initiates biosynthesis of the microcin C7 antibiotic. EMBO J, 28(13), 1953–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffner DE, Schmidt EW, & Heemstra JR (2015). Assessing the combinatorial potential of the RiPP cyanobactin tru pathway. ACS Synth Biol, 4(4), 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Millan JL, Kolter R, & Moreno F (1985). Plasmid genes required for microcin B17 production. J Bacteriol, 163(3), 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar D, Hao Y, Lin Z, Morita M, Nair SK, & Schmidt EW (2017). Enzymatic N- and C-protection in cyanobactin RiPP natural products. J Am Chem Soc, 139(8), 2884–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar D, Lin Z, & Schmidt EW (2015). Modularity of RiPP enzymes enables designed synthesis of decorated peptides. Chem Biol, 22(7), 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar D, Pierce E, McIntosh JA, & Schmidt EW (2015). Recognition sequences and substrate evolution in cyanobactin biosynthesis. ACS Synth Biol, 4(2), 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar D, & Schmidt EW (2016). Combinatorial biosynthesis of RiPPs: docking with marine life. Curr Opin Chem Biol, 31, 15–21. doi: 10.1016/j.cbpa.2015.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardar D, Tianero MD, & Schmidt EW (2016). Directing biosynthesis: practical supply of natural and unnatural cyanobactins. Methods Enzymol, 575, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter I, & Berger A (1967). On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun, 27(2), 157–162. [DOI] [PubMed] [Google Scholar]
- Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, & Ravel J (2005). Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc Natl Acad Sci U S A, 102(20), 7315–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudek S, Haygood MG, Youssef DT, & Schmidt EW (2006). Structure of trichamide, a cyclic peptide from the bloom-forming cyanobacterium Trichodesmium erythraeum, predicted from the genome sequence. Appl Environ Microbiol, 72(6), 4382–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeaux CJ, Ha T, & van der Donk WA (2014). A price to pay for relaxed substrate specificity: a comparative kinetic analysis of the class II lanthipeptide synthetases ProcM and HalM2. J Am Chem Soc, 136(50), 17513–17529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tianero MD, Donia MS, Young TS, Schultz PG, & Schmidt EW (2012). Ribosomal route to small-molecule diversity. J Am Chem Soc, 134(1), 418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tianero MD, Pierce E, Raghuraman S, Sardar D, McIntosh JA, Heemstra JR, …Schmidt EW (2016). Metabolic model for diversity-generating biosynthesis. Proc Natl Acad Sci U S A, 113(7), 1772–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland Brown L. C., Acker MG, Clardy J, Walsh CT, & Fischbach MA (2009). Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc Natl Acad Sci U S A, 106(8), 2549–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AB, & Jacobs RS (1993). A marine natural product, patellamide D, reverses multidrug resistance in a human leukemic cell line. Cancer Lett, 71(1–3), 97–102. [DOI] [PubMed] [Google Scholar]
- Wipf P, & Uto Y (2000). Total synthesis and revision of stereochemistry of the marine metabolite trunkamide A. J Org Chem, 65(4), 1037–1049. [DOI] [PubMed] [Google Scholar]
- Yu Y, Duan L, Zhang Q, Liao R, Ding Y, Pan H, … Liu W (2009). Nosiheptide biosynthesis featuring a unique indole side ring formation on the characteristic thiopeptide framework. ACS Chem Biol, 4(10), 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabriskie TM, Foster MP, Stout TJ, Clardy J, & Ireland CM (1990). Studies on the solution- and solid-state structure of patellin 2. Journal of the American Chemical Society, 112(22), 8080–8084. [Google Scholar]
- Zhang F, Li C, & Kelly WL (2016). Thiostrepton Variants Containing a Contracted Quinaldic Acid Macrocycle Result from Mutagenesis of the Second Residue. ACS Chem Biol, 11(2), 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemert N, Ishida K, Quillardet P, Bouchier C, Hertweck C, de Marsac NT, & Dittmann E (2008). Microcyclamide biosynthesis in two strains of Microcystis aeruginosa: from structure to genes and vice versa. Appl Environ Microbiol, 74(6), 1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]