

Abstract
Reductionist in vitro T cell assays have identified metabolic pathways critical for T cell function within the tumor microenvironment. We discuss the challenges of testing these concepts using in vivo tumor models.
Exciting new technologies and conceptual advancements have re-energized the study of the metabolic competition between tumors and the adaptive immune system (Chang et al., 2015; Ho et al., 2015; Sugiura and Rathmell, 2018). Despite these advancements, there remains little discussion in the literature on how investigators should model nutrient depletion in the tumor microenvironment when studying immune cell/tumor cell metabolic interactions and competition. How can we more accurately distinguish and dissociate nutrient deprivation’s effects on immune cells from other immunosuppressive components of the tumor microenvironment in vivo? Are T cells engineered with altered metabolic programming specifically overcoming a nutrient limitation or are they simply better tumor-specific T cells that function better in all nutrient environments? This essay will lay out some of the critical issues facing the field and hopefully spur the development of improved approaches for testing new immune therapies. We believe that greater mechanistic understanding of immunometabolism will fully harness the clinical potential of immune therapies.
Challenges T cells face in the solid tumor microenvironment
In vivo, tumors alter the abundance of dozens of metabolites in the interstitial fluid relative to healthy tissue (Kamphorst et al., 2015; Pavlova and Thompson, 2016). Most commonly solid tumor environments have been reported to be deprived of oxygen, glucose, glutamine, multiple amino acids (e.g. arginine, tryptophan) and is highly acidic (Fig 1A). Different tumors and even separate regions of the same tumor can host diverse nutrient environments and can experience intermittent blood flow and nutrient supply (DeBerardinis and Chandel, 2016; Vaupel et al., 1987). An abundance of pro-angiogenic factors (like vascular endothelial growth factor-A) promote disturbed vasculature characterized by large and leaky vessels, erratic branching, and irregular and slow blood flow which inhibits efficient nutrient delivery (Carmeliet, 2005; Lanitis et al., 2015). In addition metabolites can be altered by elevated expression of enzymes expressed by tumor cells or tumor-associated antigen-presenting cells. For example, high expression of the enzyme indoleamine 2,3-dioxygenase in the tumor microenvironment can lead to local depletion of tryptophan and regulate T cell proliferation and induce apoptosis (Fallarino et al., 2002; Platten et al., 2012; Schafer et al., 2016). Furthermore, the protein kinase, general control nonderepressible 2 (GCN2), also can sense amino acid deprivation in the tumor microenvironment and induce signaling that promotes T cell anergy (Munn et al., 2005), highlighting that signaling pathways induced by low levels of nutrients can be as damaging to the anti-tumor response as the lack of nutrients to support immune cell function and growth. T cells require glucose, glutamine, and mitochondrial pathways for activation, maximal proliferation and/or effector function (Cham and Gajewski, 2005; Macintyre et al., 2014; Procaccini et al., 2016; Ron-Harel et al., 2016; Sena et al., 2013). Aerobic glycolysis serves the increased biosynthetic demands of highly proliferating cells. Glycolytic intermediates can produce nucleotides, lipids, and amino acids necessary for cellular proliferation in both T cells and cancer cells (Vander Heiden et al., 2009). Similar reliance on aerobic glycolysis between proliferating cancer cells and activated T cells intensifies a competition for limited nutrients within the tumor microenvironment (Frauwirth et al., 2002). Competition for glucose also plays a clear role in limiting effective anti-tumor responses in vivo (Chang et al., 2015; Ho et al., 2015). Aerobic glycolysis is required for maximal IFN-γ production by effector T cells though at least two independent mechanisms. The glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) can inhibit translation of IFN-γ mRNA, while another study has demonstrated that the glycolytic enzyme, lactate dehydrogenase A (LDHA), promotes histone acetylation along the IFN-γ locus (Chang et al., 2013; Peng et al., 2016). Although some attempts to increase nutrient availability have been successful, we have little understanding how to make T cells more fit in a nutrient limiting environment (Chang et al., 2015; Lanitis et al., 2015).
Figure 1: Metabolic challenges T cells encounter in the solid tumor microenvironment.
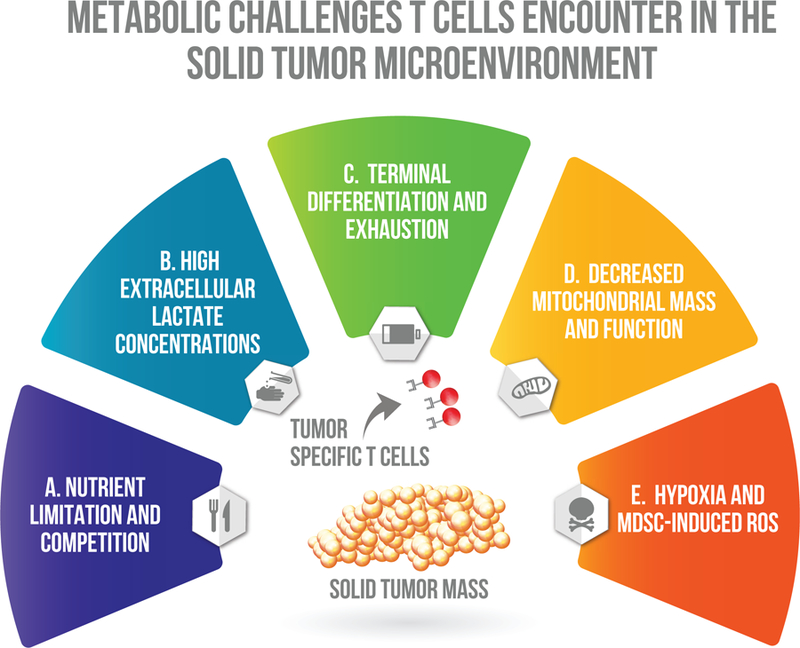
A. Nutrient limitation and competition. Disturbed vasculature of solid tumors are often unable to provide nutrients critical for T cell activation. Activated T cells and proliferating tumor cells rely on aerobic glycolysis and compete for extracellular glucose. B. High extracellular lactate concentrations. Reliance on aerobic glycolysis by tumor cells promotes a large buildup of the waste product lactate in the extracellular milieu of solid tumors. High extracellular concentrations of lactate hinder T cell activation by inhibiting efficient secretion of lactic acid from the cytoplasm and acidifying intracellular pH. C. Hypoxia and MDSC-induced ROS. T cells in the solid tumor microenvironment are constantly exposed to reactive oxygen species, which are highly reactive and damage cellular structures. Hypoxia rapidly induces ROS production by T cells and myeloid derived suppressor cells frequently suppress effector T cells by secreting ROS. D. Decreased mitochondrial mass and function. Tumor specific T cells have defects in mitochondria biogenesis, size, cristae structure, voltage potential and function. E. Terminal differentiation and exhaustion. Chronic antigen exposure causes loss of co-stimulatory receptors while promoting inhibitory co-receptors that inhibit glycolytic metabolism and activation signaling.
Due to dysfunctional vasculture and the high glycolytic rate of many tumor cells, the tumor microenvrionment is often acidic because of high lactate concentrations (Fig 1B). Both tumor and T cells rely on monocarboxylate transporters to secrete and uptake lactate generated from glycolysis and pyruvate. High lactate concentrations in the environment prevent T cells from efficiently transporting lactate out of the cell and increase intracellular acidity and inhibit functionality and NFAT activity (Brand et al., 2016; Fischer et al., 2007). Brand et al. demonstrated that melanoma tumor cells engineered to have low lactate dehydrogenase A (LDHA) activity, have greater immune infiltration and activity than tumor cells with normal LDHA expresion. LDHA is the glycolytic enzyme required for conversion of pyruvate to lactate. Furthermore the administration of the proton pump inhibitor, esomeprazole, can normalize tumor pH in vivo and when combined with immunotherapy can promote tumor clearance (Calcinotto et al., 2012). While glucose deprivation and high lactate concentrations inhibit anti-tumor responses, they also promote immunosuppressive T regulatory cell and macrophage polarization and function (Angelin et al., 2017; Colegio et al., 2014).
As T cells become exposed to chronic antigen in the tumor microenvironment, they begin to terminally differentiate and become exhausted. As exhaustion becomes more severe, T cells increase expression of multiple inhibitory receptors, while simultaneously inhibiting costimulatory receptor expression (Fig 1C). Inhibitory receptors can inhibit co-stimulation and phosphorylation events downstream of T cell receptor (TCR) activation, and prevent activation signals (Akt and mTORC1) necessary for increasing surface expression of the major glucose transporter, Glut-1, in T cells (Jacobs et al., 2008; Parry et al., 2005; Siska et al., 2016). Without sufficient surface expression of Glut-1, the activated T cells are unable to properly upregulate glycolysis and have inhibited proliferation and effector function.
Upregulation of mitochondrial pathways (oxidative phosphorylation and one-carbon metabolism) are essential for proper T cell proliferation. (Chang et al., 2013; Procaccini et al., 2016; Ron-Harel et al., 2016; Ron-Harel et al., 2015). In addition mitochondria play roles beyond producing energy and biosynthesis, though epigenetic and signaling roles during T cell activation (Minocherhomji et al., 2012; Sena et al., 2013). Chronic antigen exposure affects metabolic pathways beyond glycolysis, by inhibiting mitochondrial functions (Fig 1D). Exhausted T cells in models of cancer or chronic virus infection often have defects in mitochondria number, size, and voltage potential and function (Bengsch et al., 2016; Scharping et al., 2016; Siska et al., 2017). One study has proposed that the tumor microenvironment downregulates mitochondrial biosynthesis by inhibiting expression of PPAR-gamma coactivator 1α (PGC1α), while another has suggested ROS induced damage may induce decreased mitochondrial mass (Scharping et al., 2016; Siska et al., 2017). Scharping et al. has successfully demonstrated that overexpression of PGC1α in tumor-specific T cells is able to prevent downregulation of mitochondrial mass and enhance T cell functionality in the tumor microenvironment.
Tumor infiltrating lymphocytes are exposed to large amounts of reactive oxygen species (ROS, Fig 1E). ROS are highly chemically reactive and can damage cellular structures, and inhibit T cell activation. Production of ROS drastically increases in T cells exposed to hypoxia and is one of the major mechanisms of immune suppression by myeloid derived suppressor cells (Kusmartsev et al., 2004; Tafani et al., 2016). Myeloid derived suppressor cells are a heterogeneous immunosuppressive subset of cells overexpressed in patients and mice with cancer. While ROS are necessary for IL-2 production and proliferation by T cells, it remains unclear the quantities in which ROS will become harmful to T cells (Sena et al., 2013). Exposure to oxidative stress by low doses of exogenous hydrogen peroxide are sufficient to inhibit T cell functionality and survival (Ligtenberg et al., 2016).
Challenges of applying immunometabolism findings to tumor models in vivo.
Given the metabolic challenges tumor-specific T cells face, there have been numerous attempts to mitigate these effects via pharmacological or genetic modifications to improve tumor-specific T cell therapy in vivo. However our interpretation of in vivo studies has been hindered by numerous technical and biological challenges.
It is incredibly difficult to distinguish whether newly designed immune interventions targeting the metabolic challenges of solid tumors have generated T cells that operate better in nutrient depleted environments or simply have made a better T cell (that operates better in all environments). The key to distinguishing these possibilities are 1) identifying how a modification improves the anti-tumor response and 2) understanding how individual metabolic interactions lead to distinct functional outcomes. For example, enhancing mitochondrial fusion, increasing L-Arginine media concentrations, or inhibiting glycolytic metabolism while T cells are being expanded for adoptive cell therapy all enhance the therapeutic activity of the infused T cells (Buck et al., 2016; Geiger et al., 2016; Sukumar et al., 2013). All of these interventions alter T cell metabolism, but do they enable T cells to function better in the tumor microenvironment? It is challenging to envision a mechanism by which temporarily expanding T cells in low glucose empowers them to function better in the tumor microenvironment. Rather, previous studies demonstrate that T cells expanded in low glucose retain a less differentiated phenotype which enables improved engraftment of the expanded T cells which correlates with improved tumor control (Gattinoni et al., 2011). Expansion in the presence of higher levels of L-arginine also results in T cells that are less differentiated and more reliant on oxidative phosphorylation instead of glycolysis (Geiger et al., 2016). These cells have higher intracellular concentrations of L-arginine, which may make them resistant to low arginine levels in the tumor microenvironment. Likewise, T cells with enhanced mitochondrial fusion are less differentiated in culture and exhibit greater reliance on oxidative phosphorylation (Buck et al., 2016). Enhanced fusion may enhance mitochondrial function and combat mitophagy frequently observed during nutrient deprivation (Rambold et al., 2011). How do we determine to what extent differentiation or improved fitness in the tumor microenvironment results in improved tumor clearance? Furthermore, any attempts to modulate metabolism in the tumor microenvironment is likely going to cascade and affect many of the surrounding cells and may have cell-specific effects. For example, enhancing tumor glycolysis is sufficient to decrease available glucose for T cells, but may also create a better environment for tumor associated macrophages and T regulatory cells (Angelin et al., 2017; Chang et al., 2015; Netea-Maier et al., 2018). In addition, simply increasing T cell activity is sufficient to modulate metabolites in the serum of mice, and the effects of these changes on other cell types is not understood (Miyajima et al., 2017). Future studies must work to better distinguish mechanisms of metabolic immune interventions to provide mechanistic understanding of immunometabolism (Fig 2A).
Figure 2: Challenges of applying immunometabolism findings to tumor models in vivo.
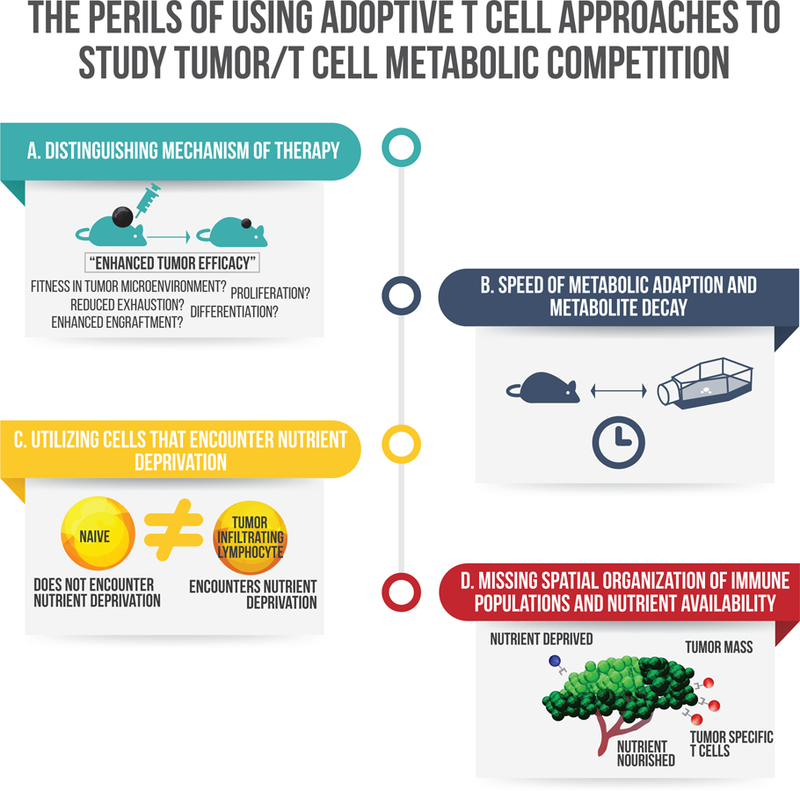
A. Many researchers have found ways to improve anti-tumor efficacy of adoptively transferred T cells through metabolic manipulation or genetic engineering. Too often though we lack the resolution to distinguish the mechanism by which enhanced tumor efficacy is reached. B. The speed by which cells metabolically adapt to changes in the extracellular environment during extraction procedures, and how quickly some metabolites can decay or be lost in commonly used isolation techniques hinder our understanding of in vivo biology. C. The most accessible cells in the blood or spleen (that are commonly used in metabolic studies of nutrient limitation) do not reflect the metabolic traits or adaptive properties of terminally differentiated cells found in most tumors. D. The field has not explored the spatial organization of nutrient depletion and immune cell infiltration in solid tumors.
Unlike standard epigenetic or transcriptional changes that often occur over several hours or days, many metabolic changes occur incredibly quickly in response to different environments. This may even be more pronounced in immune cells, because these cells readily circulate and traffic to diverse areas of the body, and thus must be able to adapt rapidly in diverse metabolic environments. While researchers can observe cell intrinsic differences of different T cell subsets in vitro, how much information is lost because of isolation and prolonged in vitro expansion (Dimeloe et al., 2016; Pan et al., 2017; Procaccini et al., 2016; van der Windt et al., 2012)? Quantification is further complicated during conventional in vitro extraction techniques because of metabolite loss, leakage, or decay (Chen et al., 2016; Van Gulik et al., 2012; Vuckovic et al., 2011; Yang et al., 2017). Thus, during commonly used isolation procedures and before any assay can be performed in vitro, metabolic changes are occurring (Fig 2B). Alternative approaches such as utilizing isotopic tracers are highly compatible with in vivo models, can be easily administered, and may better recapitulate in vivo biology (Hensley et al., 2016; Sun et al., 2017).
Immune cells isolated for nutrient depletion studies should represent the cells that would traffic to sites of inflammation and nutrient depletion in the tumor microenvironment (Fig 2C). Many in vitro T cell studies have examined nutrient depletion on total T cells isolated from the peripheral blood or spleens of humans or mice. However, naïve cells that often represent the majority of T cells in circulation or in the spleen do not travel to sites of inflammation where vasculature is often disturbed or injured (Klebanoff et al., 2006; Thome et al., 2014). Differentiated effector memory T cells or tissue resident memory T cells are the cells that reside in the environments that actually encounter nutrient depletion and have distinct responses to metabolic stress (Dimeloe et al., 2016; Ecker et al., 2018). These studies have suggested that effector memory T cells may prioritize effector functions over proliferation during nutrient limitation. More work is needed to titer nutrient availabilities to better delineate how they affect specific aspects of T cell function and proliferation in different T cell populations. Thus, many previous studies have lacked sufficient resolution to determine whether T cells in the blood mimic the same metabolic and adaptive features of T cells that reside in the tissue. Furthermore, there is on-going discussion on which cell populations represent the best control to tumor infiltrating lymphocytes (TILs). Are cells in the blood or spleen, effector memory T cells, or tissue-resident cells from the tissue of choice the best cells to compare to TILs? While tissue-resident cells are often thought of as the best control because they reside in the same niche and similar effector phenotype, their limited numbers and technically difficult isolation have hindered their wide-spread use.
There remain large gaps in understanding of immune cell localization within hypoxic or glucose depleted regions of tumors. Do we truly observe anti-tumor function and immune cell proliferation only in sites of nutrient availability? Do immune cells have higher rates of death in sites of nutrient depletion or do they simply traffic away from those sites? Which nutrients are the most essential for function in vivo? Regions of nutrient depletion can often be examined through florescent analogues like 2-NBDG for glucose distribution, staining with pimonidazole for regions experiencing hypoxia, or through distinct protein expression of hypoxic or nutrient depleted cells (Airley et al., 2001). These are well validated, and compatible with commonly used methods to identify immune cell localization and activity (Bennewith and Durand, 2004; He et al., 2008; Sukumar et al., 2013). Improvements in the resolution of mass spectrometry imaging techniques have also identified tissue-resident immune populations and metabolite gradients in tumors (Dilillo et al., 2017; Holzlechner et al., 2017). Additionally, sensitive reporter constructs could be useful to map the duration and regional location of nutrient limiting milieus. Elucidating the spatial organization of nutrient availability and immune populations will be crucial for greater insight of immune surveillance in the tumor microenvironment (Fig 2D).
The last decade of research has led to incredible findings and a growing interest in immunometabolism. Researchers have only just begun to understand how to translate the basic findings into in vivo tumor models and face many challenges. Future studies designed to overcome these challenges will be essential for providing mechanistic understanding of in vivo immunometabolism and for translating these approaches into the clinical arena.
Footnotes
Declaration of Interests: JLR is a founder of Tmunity Therapeutics and owns equity in the company. CE declares no competing interests
References
- Airley R, Loncaster J, Davidson S, Bromley M, Roberts S, Patterson A, Hunter R, Stratford I, and West C (2001). Glucose transporter glut-1 expression correlates with tumor hypoxia and predicts metastasis-free survival in advanced carcinoma of the cervix. Clin Cancer Res 7, 928–934. [PubMed] [Google Scholar]
- Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, Kopinski PK, Wang L, et al. (2017). Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 25, 1282–45098112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, et al. (2016). Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 45, 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennewith KL, and Durand RE (2004). Quantifying transient hypoxia in human tumor xenografts by flow cytometry. Cancer Res 64, 6183–6189. [DOI] [PubMed] [Google Scholar]
- Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. (2016). LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 24, 657–671. [DOI] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang C-HH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, et al. (2016). Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 166, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, Cova A, Canese R, Jachetti E, Rossetti M, et al. (2012). Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res 72, 2746–2756. [DOI] [PubMed] [Google Scholar]
- Carmeliet P (2005). VEGF as a key mediator of angiogenesis in cancer. Oncology 69 Suppl 3, 4–10. [DOI] [PubMed] [Google Scholar]
- Cham CM, and Gajewski TF (2005). Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol 174, 4670–4677. [DOI] [PubMed] [Google Scholar]
- Chang C-HH, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-HH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Freinkman E, Wang T, Birsoy K, and Sabatini DM (2016). Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 166, 1324–1963042816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio OR, Chu N-QQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilillo M, Ait-Belkacem R, Esteve C, Pellegrini D, Nicolardi S, Costa M, Vannini E, Graaf EL, Caleo M, and McDonnell LA (2017). Ultra-High Mass Resolution MALDI Imaging Mass Spectrometry of Proteins and Metabolites in a Mouse Model of Glioblastoma. Sci Rep 7, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimeloe S, Mehling M, Frick C, Loeliger J, Bantug GR, Sauder U, Fischer M, Belle R, Develioglu L, Tay S, et al. (2016). The Immune-Metabolic Basis of Effector Memory CD4+ T Cell Function under Hypoxic Conditions. J Immunol 196, 106–114. [DOI] [PubMed] [Google Scholar]
- Ecker C, Guo L, Voicu S, Gil-de-Gomez L, Medvec A, Cortina L, Pajda J, Andolina M, Torres-Castillo M, Donato JL, et al. (2018). Differential Reliance on Lipid Metabolism as a Salvage Pathway Underlies Functional Differences of T Cell Subsets in Poor Nutrient Environments. Cell Rep 23, 741–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, Fioretti MC, and Puccetti P (2002). T cell apoptosis by tryptophan catabolism. Cell Death Differ 9, 1069–1077. [DOI] [PubMed] [Google Scholar]
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. (2007). Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819. [DOI] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, and Thompson CB (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MFF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. (2011). A human memory T cell subset with stem cell-like properties. Nat Med 17, 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et al. (2016). L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167, 829–84033536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Deng X, Wen B, Liu Y, Sun X, Xing L, Minami A, Huang Y, Chen Q, Zanzonico PB, et al. (2008). Noninvasive molecular imaging of hypoxia in human xenografts: comparing hypoxia-induced gene expression with endogenous and exogenous hypoxia markers. Cancer Res 68, 8597–8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho P-CC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui Y-CC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzlechner M, Strasser K, Zareva E, Steinhäuser L, Birnleitner H, Beer A, Bergmann M, Oehler R, and Marchetti-Deschmann M (2017). In Situ Characterization of Tissue-Resident Immune Cells by MALDI Mass Spectrometry Imaging. J Proteome Res 16, 65–76. [DOI] [PubMed] [Google Scholar]
- Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, and Rathmell JC (2008). Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol 180, 4476–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, Gattinoni L, and Restifo NP (2006). CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev 211, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusmartsev S, Nefedova Y, Yoder D, and Gabrilovich DI (2004). Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol 172, 989–999. [DOI] [PubMed] [Google Scholar]
- Lanitis E, Irving M, and Coukos G (2015). Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol 33, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, Riet T, Abken H, and Kiessling R (2016). Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J Immunol 196, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, et al. (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Tollefsbol TO, and Singh KK (2012). Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 7, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima M, Zhang B, Sugiura Y, Sonomura K, Guerrini MM, Tsutsui Y, Maruya M, Vogelzang A, Chamoto K, Honda K, et al. (2017). Metabolic shift induced by systemic activation of T cells in PD-1-deficient mice perturbs brain monoamines and emotional behavior. Nature Immunol 18, 1342–1352. [DOI] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, and Mellor AL (2005). GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22, 633–642. [DOI] [PubMed] [Google Scholar]
- Netea-Maier RT, Smit JWAWA, and Netea MG (2018). Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship. Cancer Lett 413, 102–109. [DOI] [PubMed] [Google Scholar]
- Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, et al. (2017). Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, and Riley JL (2005). CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25, 9543–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, and Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platten M, Wick W, and Van den Eynde BJJ (2012). Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res 72, 5435–5440. [DOI] [PubMed] [Google Scholar]
- Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, Faicchia D, Marone G, Tramontano D, Corona M, et al. (2016). The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity 44, 406–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, and Lippincott-Schwartz J (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci 108, 10190–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, Dephoure N, Satterstrom FK, Sheffer M, Spinelli JB, et al. (2016). Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab 24, 104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron-Harel N, Sharpe AH, and Haigis MC (2015). Mitochondrial metabolism in T cell activation and senescence: a mini-review. Gerontology 61, 131–138. [DOI] [PubMed] [Google Scholar]
- Schafer CC, Wang Y, Hough KP, Sawant A, Grant SC, Thannickal VJ, Zmijewski J, Ponnazhagan S, and Deshane JS (2016). Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget 7, 75407–75424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang C-RR, Schumacker PT, Licht JD, Perlman H, et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, Chiang YJ, Corona AL, Gemta LF, Vincent BG, et al. (2017). Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siska PJ, van der Windt GJ, Kishton RJ, Cohen S, Eisner W, MacIver NJ, Kater AP, Weinberg JB, and Rathmell JC (2016). Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J Immunol 197, 2532–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, and Rathmell JC (2018). Metabolic Barriers to T Cell Function in Tumors. J Immunol 200, 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. (2013). Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun RC, Fan TW, Deng P, Higashi RM, Lane AN, Le A-TT, Scott TL, Sun Q, Warmoes MO, and Yang Y (2017). Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat Commun 8, 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafani M, Sansone L, Limana F, Arcangeli T, De Santis E, Polese M, Fini M, and Russo MA (2016). The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxid Med Cell Longev 2016, 3907147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, Kato T, Lerner H, Shen Y, and Farber DL (2014). Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 159, 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang C-HH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, and Pearce EL (2012). Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gulik WM, Canelas AB, Taymaz-Nikerel H, Douma RD, de Jonge LP, and Heijnen JJ (2012). Fast sampling of the cellular metabolome. Methods Mol Biol 881, 279–306. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P, Fortmeyer HP, Runkel S, and Kallinowski F (1987). Blood flow, oxygen consumption, and tissue oxygenation of human breast cancer xenografts in nude rats. Cancer Res 47, 3496–3503. [PubMed] [Google Scholar]
- Vuckovic D, de Lannoy I, Gien B, Shirey RE, Sidisky LM, Dutta S, and Pawliszyn J (2011). In vivo solid-phase microextraction: capturing the elusive portion of metabolome. Angew Chem Int Ed 50, 5344–5348. [DOI] [PubMed] [Google Scholar]
- Yang X, Ma Y, Li N, Cai H, and Bartlett MG (2017). Development of a Method for the Determination of Acyl-CoA Compounds by Liquid Chromatography Mass Spectrometry to Probe the Metabolism of Fatty Acids. Anal Chem 89, 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]